
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-free Pd/Ag-mediated dehydrogenative alkynylation of imidazole derivatives†
Fabio Bellina *a,
Matteo Biagettib,
Sara Guarientob,
Marco Lessia,
Mattia Faustia,
Paolo Ronchib and
Elisabetta Rosadonia
*a,
Matteo Biagettib,
Sara Guarientob,
Marco Lessia,
Mattia Faustia,
Paolo Ronchib and
Elisabetta Rosadonia
aDipartimento di Chimica e Chimica Industriale, Università di Pisa, Via Moruzzi 13, 56124 Pisa, Italy. E-mail: fabio.bellina@unipi.it
bChemistry Research and Drug Design, Chiesi Farmaceutici S.p.A., Centro Ricerche, Largo Belloli 11/A, 43122 Parma, Italy
First published on 22nd July 2021
Abstract
A variety of 2-alkynyl(benzo)imidazoles have been synthesized by dehydrogenative alkynylation of (benzo)imidazoles with terminal alkyne in NMP under air in the presence of Ag2CO3 as the oxidant and Pd(OAc)2 as the catalyst precursor. The data obtained in this study support a reaction mechanism involving a non-concerted metalation deprotonation (n-CMD) pathway.
The development of synthetic protocols that enable the direct and selective functionalization of Csp2–H bonds are of primary importance in the decoration of the imidazole scaffold.1–10 The presence of two nitrogen atoms in the pentatomic structure of this important azole in fact introduces a differentiation in the chemical behaviour of the three C–H bonds, allowing their selective involvement in carbon–carbon bond-forming reactions as long as appropriate experimental conditions are identified.
During our studies dedicated to the synthesis and investigation of azole-based fluorophores featuring a π-conjugated backbone end-capped with electron-donating (EDG) and electron-acceptor (EWG) groups (the so-called “push–pull” systems),11–15 taking into account the structural characteristics and synthetic versatility of a triple carbon–carbon bond16–23 that make it an excellent π-spacer, we recently decided to evaluate the possibility of obtaining alkynyl-substituted imidazoles by transition metal-catalyzed Csp2–Csp bond-forming reactions. Among the synthetic procedures that allow the selective alkynylation of imidazoles and other five-membered heteroarenes,5,24 there is no doubt that the possibility of forming the new σ carbon–carbon bond through the double activation of two distinct carbon–hydrogen bonds through a cross-dehydrogenative alkynylation (CDA) represents the best synthetic approach in terms of atom economy and functional group tolerance (eqn (1), Scheme 1).25–34 In fact, when compared with the transition metal-catalyzed direct Csp2–H alkynylation protocols involving 1-haloalkynes (the so-called “inverse Sonogashira coupling”) (eqn (2), Scheme 1),35–45 hypervalent iodine reagents (eqn (3), Scheme 1),46–48 or α,β-ynoic acids (decarboxylative direct arylation) (eqn (4), Scheme 1),26,49,50 the CDA methodology makes it possible to use terminal alkynes without the need for preliminary activation.5,24
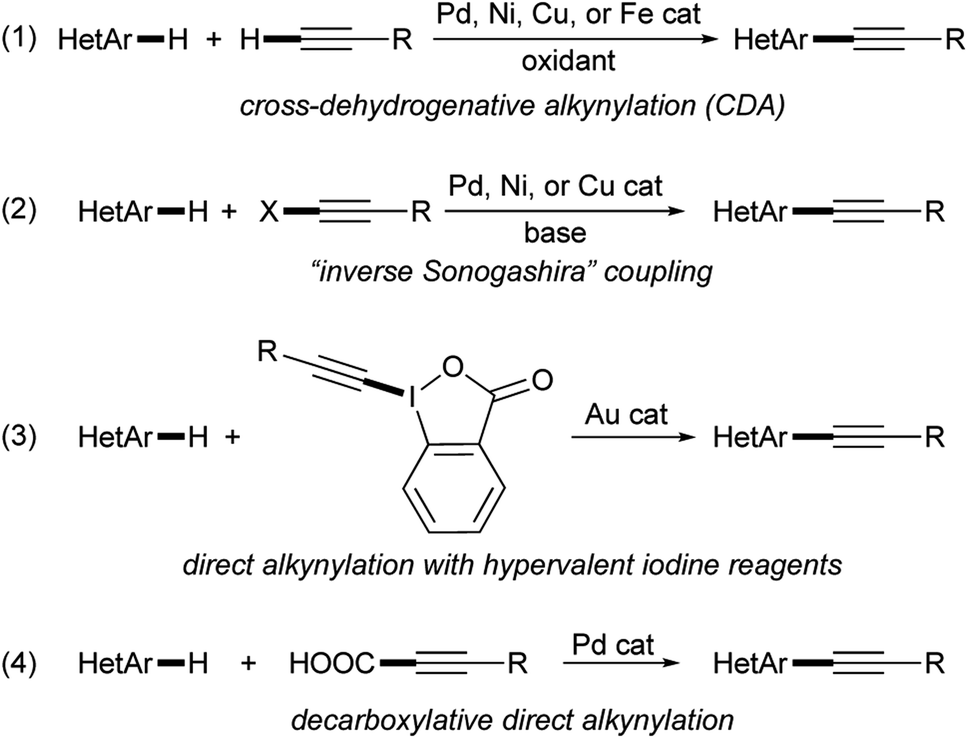 | ||
| Scheme 1 Synthetic protocols for the transition metal-catalyzed Csp2–H alkynylation of heteroarenes. | ||
Despite several papers having been dedicated to the dehydrogenative alkynylation of pyrroles,27 indoles,27,31 oxazoles,27,30,32–34 benzoxazoles,26,29,30,32,34 thiazoles,27,30 benzothiazoles,26,29,31,32,34 pyrazoles,25,27 1,3,4-oxadiazoles,33 imidazopyridines,27,34 and N-substituted sydnones,28 to the best of our knowledge a study specifically devoted to the dehydrogenative alkynylation of imidazoles has never been reported. However, careful reading the literature where alkynylation reactions involving azoles other than imidazole were described, it emerged that two papers reported the C-2 alkynylation of N-benzylimidazole and N-benzylbenzimidazole with phenylacetylene. In a study mainly devoted to the dehydrogenative alkynylation of imidazo[1,5-a]pyridine, Shihabara, Dohke and Murai employed the commercially unavailable Pd(II) complex bi(4-nitropyridinyl)Pd(OAc)2, Ag2CO3 as the oxidant and AcOH as additive, in a 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixture of DMF and DMSO for 2 h at 120 °C under argon.34 Due to the propensity of alkynes to give Glaser-type homocoupling side-products in the presence of silver(I) salts,51 phenylacetylene was slowly added over 90 min to the reaction mixture. In 2015, Likhar, Kantam and co-workers used a synthetic Pd(II) carbene complex. In this case Ag2O was used as the oxidant, Cs2CO3 base, performing the coupling in DMF at 85 °C under air.26
:5 mixture of DMF and DMSO for 2 h at 120 °C under argon.34 Due to the propensity of alkynes to give Glaser-type homocoupling side-products in the presence of silver(I) salts,51 phenylacetylene was slowly added over 90 min to the reaction mixture. In 2015, Likhar, Kantam and co-workers used a synthetic Pd(II) carbene complex. In this case Ag2O was used as the oxidant, Cs2CO3 base, performing the coupling in DMF at 85 °C under air.26
Encouraged by these results but with the intention of developing a simplified procedure that would allow the use of a commercial Pd(II) pre-catalyst in the absence of any palladium ligands, we decided to review the conditions of reaction and, in this work, we are pleased to summarize the results obtained in the synthesis of 2-alkynylimidazole derivatives by dehydrogenative alkynylation with terminal alkynes (Scheme 2). In particular, a careful screening allowed us to show how the reactivity of Csp2–H bond of imidazole derivatives and other azoles can be enhanced simply by performing the alkynylation using NMP as the reaction solvent, under air in non-anhydrous conditions, and without the need for palladium ligands.
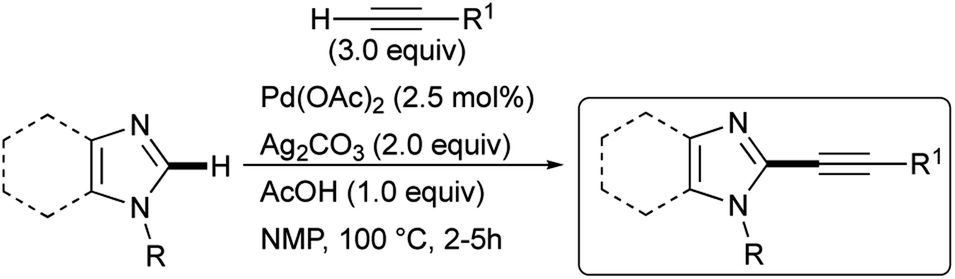 | ||
| Scheme 2 Pd/Ag-Promoted dehydrogenative alkynylation of imidazole derivatives. | ||
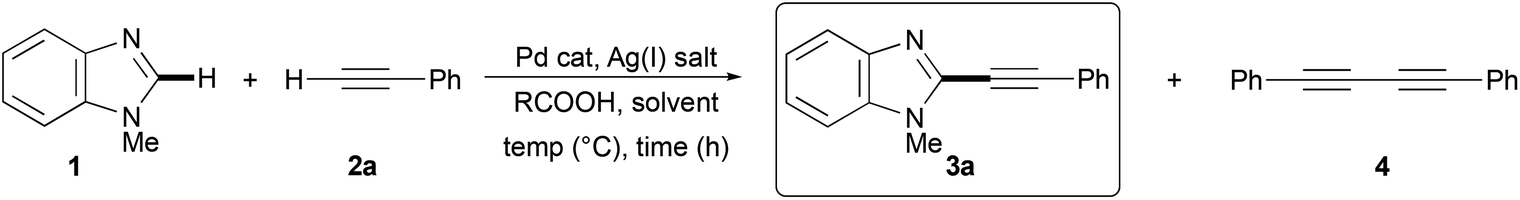
We decided to start our synthetic investigations by trying a cross-dehydrogenative coupling between N-methylbenzimidazole (1) and phenylacetylene (2a), chosen as typical coupling partners, under conditions very similar to those proposed by Murai and co-workers for the regioselective C-3 dehydrogenative alkynylation of 1-alkynyl-3-arylimidazo[1,5-a]-pyridines.34
Hence, to a mixture of 1.0 mmol 1, 5 mol% Pd(OAc)2, 1.5 equiv. Ag2CO3, and 1.0 equiv. AcOH in a 95:5 (v/v) mixture of DMF and DMSO under argon 3.0 equiv. of 2a were added dropwise over 90 min. However, after 2 h at 120 °C a 56% GLC conversion of 1 was recorded, and the required 2-phenylethynyl-N-methylbenzimidazole (3a) was obtained in only 34% GLC yield (entry 1, Table 1).
a
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Pd cat. (mol%) | Ag(I) salt (equiv.) | RCOOH | Solvent | Temp./react. timeb (°C/h) | 1 GLC conversionc (%) | 3a GLC yieldd (%) | 3a:4 AP% |
| a Reaction conditions: 2a (3.0 mmol) in the selected solvent (2.0 mL) was added dropwise over 45 min to a mixture of 1 (1.0 mmol), Pd cat., Ag(I) salt, RCOOH (1.0 mmol) in the selected solvent (2.0 mL) under air, unless otherwise reported.b After the reported reaction time the GLC conversion of 2a was quantitative.c GLC conversion of 1 vs. biphenyl.d GLC yield vs. biphenyl. In parentheses isolated yield.e This reaction was carried out under an argon atmosphere.f This reaction was carried out on a 10 mmol scale.g This reaction was carried out in the presence of TEMPO (1.0 equiv.) as radical scavenger. | ||||||||
| 1e | Pd(OAc)2 (5) | Ag2CO3 (1.5) | AcOH | DMF/DMSO (95:5) |
120/2 | 56 | 34 | 57:43 |
| 2 | Pd(OAc)2 (5) | Ag2CO3 (1.5) | AcOH | DMF/DMSO (95:5) |
100/5.5 | 67 | 66 | 32:68 |
| 3 | Pd(OAc)2 (5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
100/2 | 73 | 70 | 34:66 |
| 4 | Pd(OAc)2 (5) | — | AcOH | DMF/DMSO (95:5) |
100/3.5 | NR | — | — |
| 5 | Pd2(dba)3 (2.5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 73 | 69 | 30:70 |
| 6 | Pd(acac)2 (5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 67 | 63 | 27:73 |
| 7 | PdCl2 (5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 72 | 68 | 35:65 |
| 8 | Pd(OAc)2 (5) | Ag2CO3 (2.0) | — | DMF/DMSO (95:5) |
100/3.5 | 53 | 23 | 72:28 |
| 9 | Pd(OAc)2 (5) | Ag2CO3 (2.0) | — | AcOH | 100/3.5 | 0 | — | 0:100 |
| 10 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 76 | 69 | 30:70 |
| 11 | Pd(OAc)2 (2.5) | AgOAc (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 55 | 48 | 25:75 |
| 12 | Pd(OAc)2 (2.5) | Ag2O (2.0) | AcOH | DMF/DMSO (95:5) |
100/3.5 | 66 | 62 | 28:72 |
| 13 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | EtCOOH | DMF/DMSO (95:5) |
100/3.5 | 80 | 71 | 35:65 |
| 14 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | PivOH | DMF/DMSO (95:5) |
100/3.5 | 45 | 45 | 22:78 |
| 15 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | DMF/DMSO (95:5) |
80/3.5 | 43 | 40 | 17:83 |
| 16 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | DMF | 100/3.5 | 75 | 70 | 34:66 |
| 17 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | DMA | 100/3.5 | 81 | 75(68) | 33:67 |
| 18 | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | NMP | 100/3.5 | 85 | 81(69) | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :64 :64 |
| 19f | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | NMP | 100/3.5 | 82 | 66(51) | 40:60 |
| 20g | Pd(OAc)2 (2.5) | Ag2CO3 (2.0) | AcOH | NMP | 100/3.5 | 83 | 80 | 36:64 |
Taking note of this unexpected negative result, we decided to re-examine the reaction conditions, starting from the consideration that even if Ag2CO3 can theoretically serve as the oxidant, run the reaction in air (open flask) may be critical.28 Gratifyingly, when the coupling was performed under air there was a marked increase in GLC yield, which went from 34% to 66% (entry 2, Table 1). The use of a slightly more excess of Ag2CO3 resulted in a 70% GLC yield (entry 3, Table 1), while air alone resulted ineffective in promoting the alkynylation (entry 4, Table 1). The use of palladium pre-catalysts different from Pd(OAc)2 did not give any significant improvement in terms of conversion and yield (entries 5–7, Table 1). The presence of 1.0 equiv. AcOH revealed to be essential for the coupling (entry 8, Table 1), but when the coupling was carried out using AcOH as the reaction solvent 2a was entirely converted into the side-product 4 and no conversion of 1 into 3a was observed (entry 9, Table 1).
Continuing with the screening, we observed that halving the palladium load did not lead to a loss of reaction efficiency (entry 10, Table 1), and that the use of silver(I) salts different from Ag2CO3 gave slightly worse results (entries 11 and 12, Table 1).
Propionic acid as additive gave results similar to AcOH (entry 13, Table 1), while pivalic acid resulted less effective, giving rise to 3a in only 45% GLC yield (entry 14, Table 1). As already observed,31 the use of the right buffer system may be crucial for the deprotonation of the terminal alkyne, and also for the reoxidation of Pd(0) to Pd(II).
With our delight the chemical yield went up to 69% when the DMF/DMSO mixture was replaced by NMP (entry 18, Table 1), and the efficacy of this solvent was confirmed carrying out the coupling on a ten times higher scale (entry 19, Table 1). A quite similar isolated yield was obtained performing the coupling in DMA as the solvent (entry 17, Table 1), while DMF alone gave a poorer result (entry 16, Table 1).
In contrast to what observed by Murai and co-workers,34 it should be highlighted that the slow addition of phenylacetylene never avoided in our hands the formation of Glaser side-product 4.
Finally, when the reaction was performed in the presence of 1.0 equiv. of TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl as radical scavenger, it still proceeded smoothly to afford 2-alkynylazole 3a in 80% GLC yield (entry 20, Table 1). This experiment suggests that free radicals should be not involved in the plausible catalytic cycle (see later).
The satisfactory result obtained in the preparation of 3a from 1 and 2a under the experimental conditions summarized in entry 18 of Table 1 prompted us to extend this simple methodology to the selective synthesis of 2-arylethynyl-1-methyl-1H-imidazoles 6a–h and 2-arylethynyl-1-methyl-1H-benzimidazoles 3a–e starting from 5 or 1 and terminal alkynes 2a–h. In details, the slow addition over 45 min of 2 to a mixture of 5 or 1 in the presence of 2.5 mol% Pd(OAc)2, 2.0 equiv. Ag2CO3, 1.0 equiv. AcOH in NMP as the solvent allowed us to recover the required 2-alkynyl substituted derivatives 6a–h or 3a–e in 62–74% isolated yield after 3.5 h at 100 °C under air (Scheme 3).
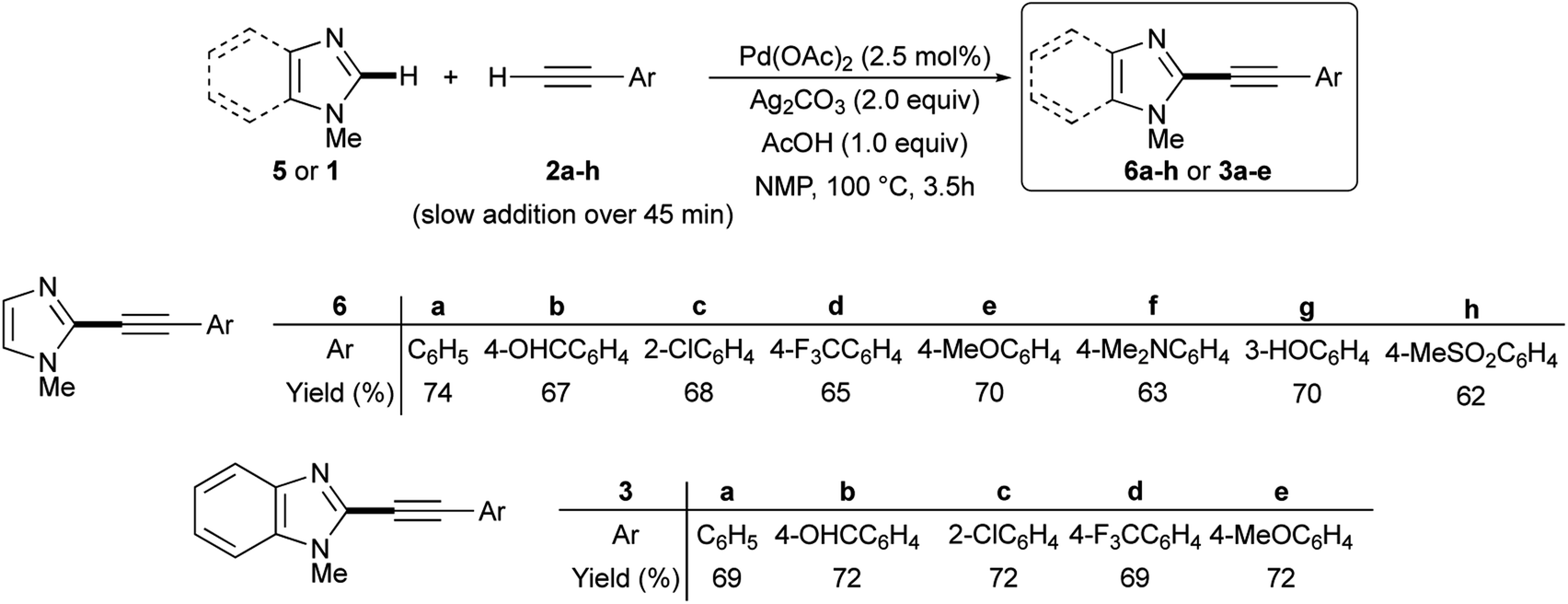 | ||
| Scheme 3 Ligandless Pd/Ag-promoted dehydrogenative alkynylation of 1-methylimidazole 5 or 1-methylbenzimidazole 1 with terminal alkynes 2a–h. | ||
Noteworthy, the reaction outcome was not influenced by the electronic nature of the aromatic moiety on the terminal alkyne. More importantly, several functional groups on the phenyl ring resulted well tolerated, including formyl and hydroxy groups that are potentially sensitive to the oxidative conditions required by dehydrogenative couplings.
While good results were obtained when arylacetylenes were used as coupling partners, worse results were observed when TIPS-acetylene 2i or 1-octyne (2j) were reacted with 5. In fact, the expected 2-alkynyl substituted imidazoles 6i and 6j were isolated in 30% and 11% yields, respectively (Scheme 4).25,31 The low reactivity of these two terminal alkynes in respect to their aryl analogues could be attributed, as already noted,31 to the decreased electrophilic nature of the corresponding alkynyl-palladium(II) intermediates being generated during the reaction. As a further proof of their lower reactivity, the slow addition of 2i and 2j was found to be unnecessary.
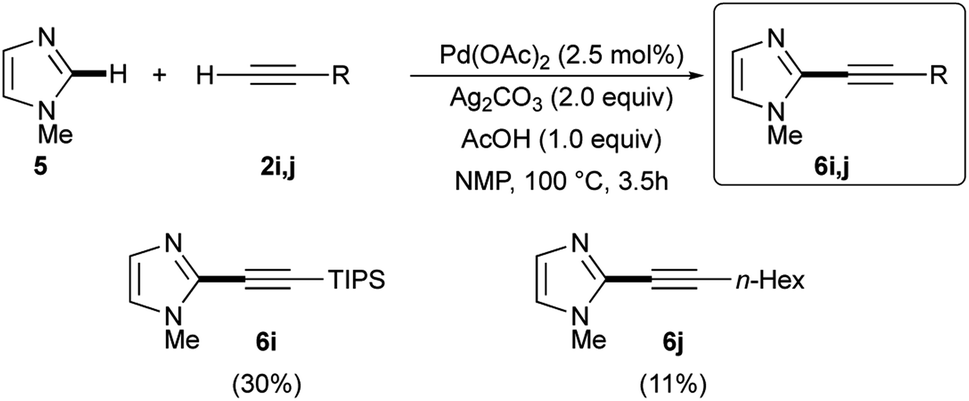 | ||
| Scheme 4 Ligandless Pd/Ag-promoted dehydrogenative alkynylation of 1-methylimidazole 5 with terminal alkynes 2i and 2j. | ||
Gratifyingly, the optimized Pd(OAc)2/Ag2CO3-promoted coupling conditions proved to be useful also for the C-2 dehydrogenative alkynylation of 4,5-diphenyl-1-methylimidazole 7. In fact, this disubstituted azole gave the expected C-2 alkynylation product 8 in 70% isolated yield when reacted with phenylacetylene (2a) (Scheme 5).
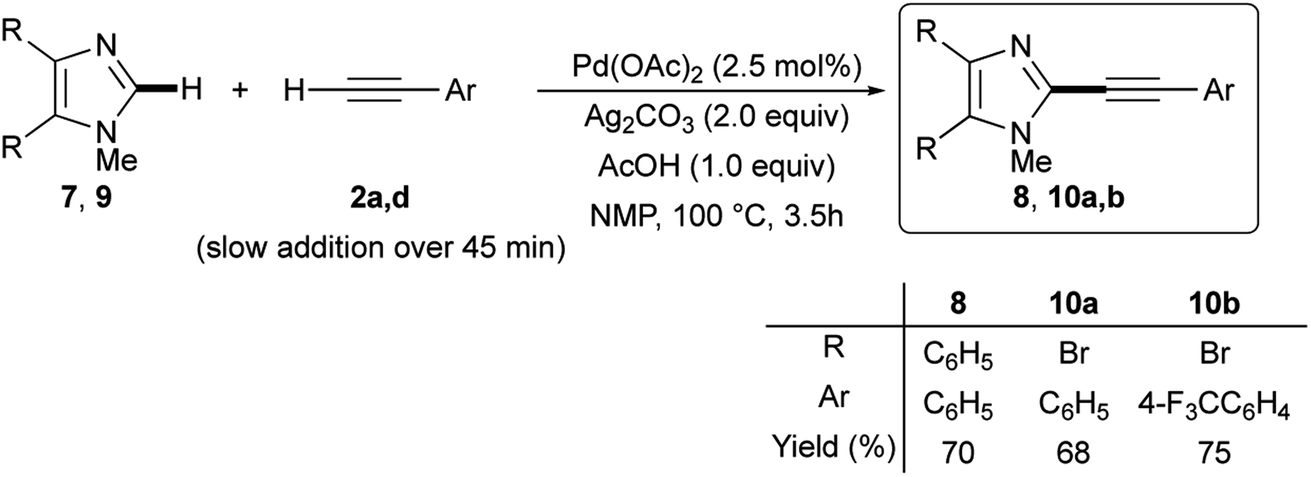 | ||
| Scheme 5 Ligandless Pd/Ag-promoted dehydrogenative alkynylation of 4,5-disubstituted 1-methylimidazole 7 or 9 with terminal alkynes 2a or 2d. | ||
One of the main advantages of dehydrogenative cross-coupling reactions is the tolerance, among others, of carbon–halogen bonds that, hence, are available for subsequent transformations. We were please to confirm this tolerance; in fact, 4,5-dibromo-1-methyl-1H-imidazole 9 was efficiently reacted with alkynes 2a and 2d, giving rise to the required 2-alkynyl-4,5-dibromoimidazoles 10a,b in 68 and 75% isolated yields, respectively (Scheme 5). Notably, no conventional Sonogashira-type coupling side-products were observed.
Similar satisfactory yields were obtained when 1-benzyl- and 1-phenyl-1H-imidazoles 11 and 12 when reacted with 2a. The corresponding 2-phenylethynylimidazoles 13 and 14 were recovered in 68% and 52% isolated yields, respectively (Scheme 6). In contrast, the C-2 alkynylation of thiazole (15) and oxazole (16) with 2a gave the required products 17 and 18 in somewhat lower yields (48 and 42%, respectively). It is noteworthy, however, that the observed reactivity of 1-substituted imidazole 6, 11 and 12, i.e. 1-methyl > 1-benzyl > 1-phenyl, and that found for the three 1,3-azoles 6, 13 and 14, i.e. 1-methylimidazole > thiazole > oxazole, parallels that reported in the literature for classical SEAr.52,53 As regards the regioselectivity, when the reaction involved 1-substituted imidazoles 5, 11, and 12, thiazole 15 and oxazole 16 a clean C-2 alkynylation was observed, while the corresponding C-5 or even the less probable C-4 alkynyl-substituted regioisomers were never detected in the crude reaction mixtures.54 However, it is worth mentioning that when the alkynylation was tempted on 1,2-dimethyl-1H-imidazole 19, the C-5 alkynylated product 20 was obtained in 42% isolated yield (eqn (1)).
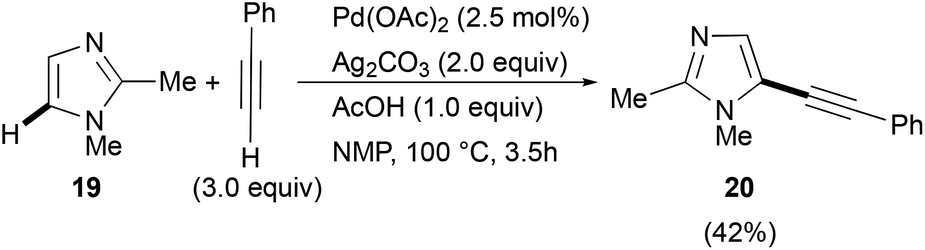 | (1) |
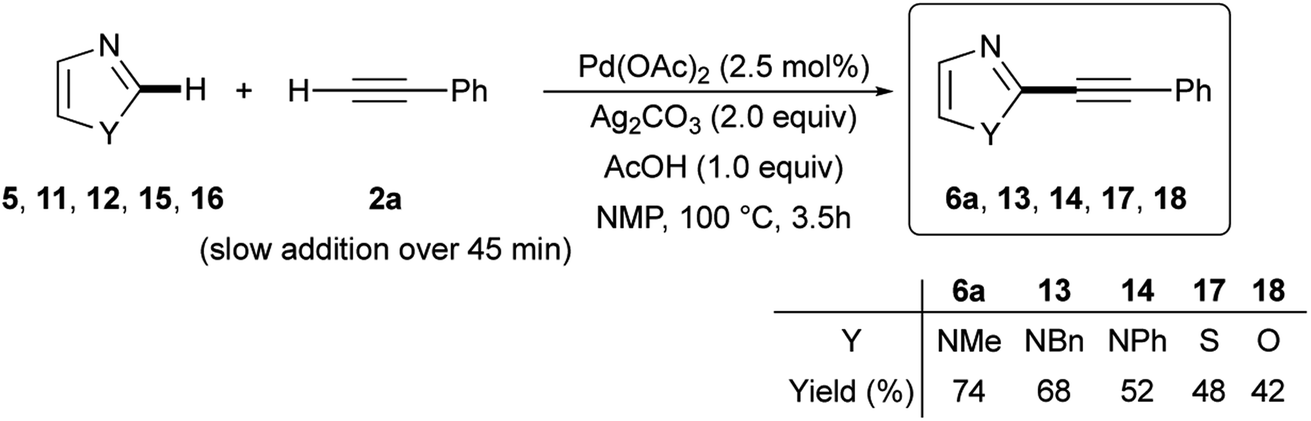 | ||
| Scheme 6 Ligandless Pd/Ag-promoted dehydrogenative alkynylation of 1-substituted imidazoles 5, 11, 12, thiazole (15), and oxazole (16) with phenylacetylene (2a). | ||
Based on earlier reports and our observations, a possible reaction mechanism for this Pd/Ag-promoted dehydrogenative alkynylation of imidazoles is summarized in Fig. 1, using 1-methylimidazole 5 as an example. Initial transmetallation involving a Pd(II) complex and a (presumed) silver(I) acetylide generates the alkynyl Pd(II) species A, which then affords the imidazole–Pd–alkyne complex E through a sequence of base-assisted carbanion generation from an azole–Pd complex (B → C), followed by a C-palladation step via carbene D. This particular C–H palladation mechanism, known as non-concerted metalation-deprotonation (n-CMD) pathway, was initially proposed by Hoarau and co-workers to justify the observed C-2 regioselectivity in the copper-free Pd-catalyzed direct arylation of oxa(thia)zoles-4-carboxylate with aryl bromides.55–57
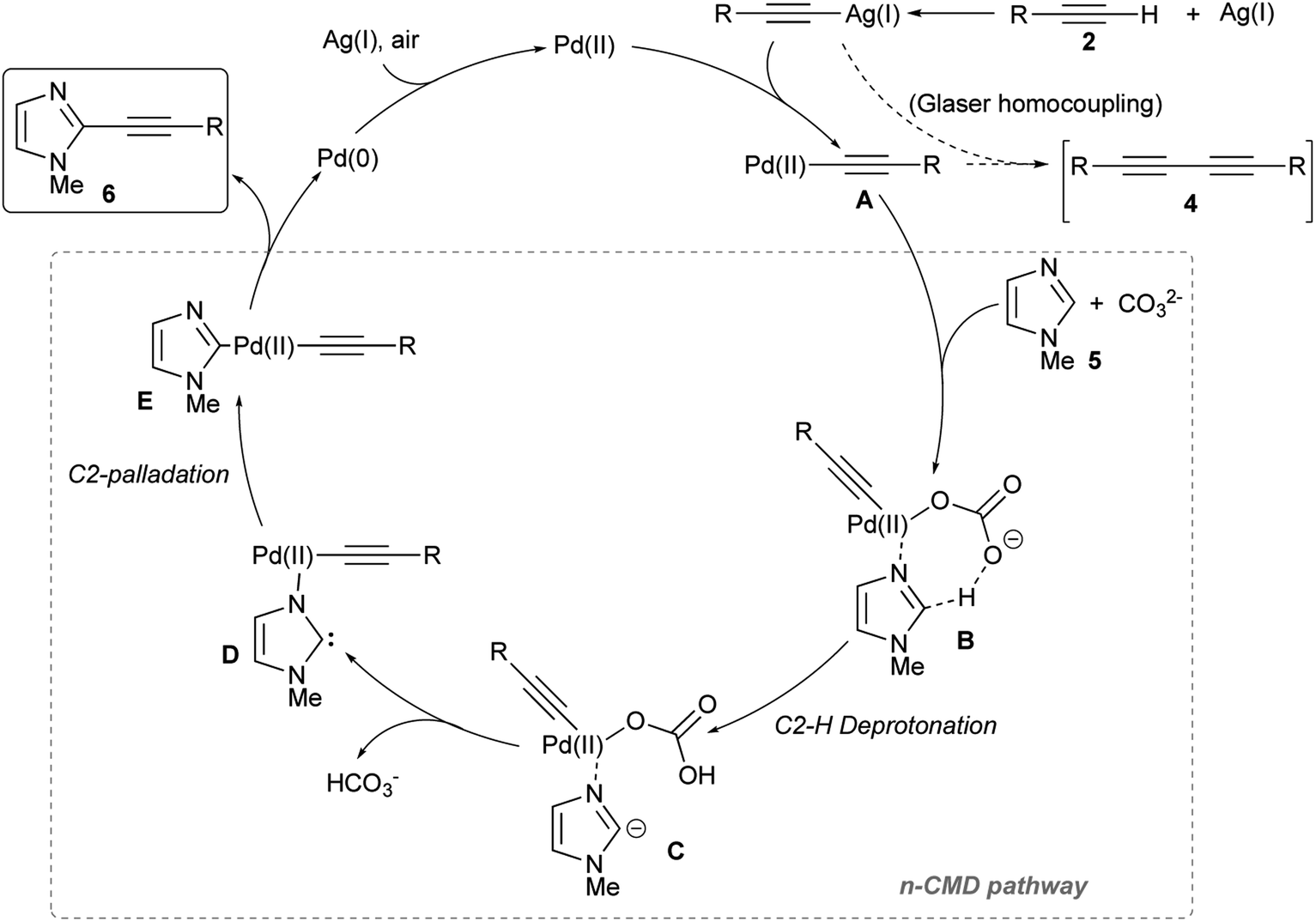 | ||
| Fig. 1 Proposed n-CMD mechanistic pathway for the Pd(OAc)2/Ag2CO3-promoted dehydrogenative alkynylation of 1-methylimidazole 5. | ||
Although the comparison of the reactivity of 1-methylimidazole 5 with both the other two 1-substituted imidazoles 11 and 12, and with thiazole (15) and oxazole (16) suggested a SEAr mechanism (hence via a classic Wheland intermediate),58–60 it should be noted that the most reactive position should have been C-5, and not the position C-2 that instead has the most acidic C–H bond. Similarly, a pure concerted metalation–deprotonation (CMD) pathway can reasonably be excluded considering that DFT calculations have shown that in the case of 1,3-azoles position C-5 is, again, the most reactive.61 The n-CMD mechanism hypothesizes that the deprotonation occurs through the formation of an azole–Pd(II) complex and, therefore, justifies the observed reactivity by the most acidic C–H bond, i.e. the one in position 2 on the azole nucleus. On the other hand, one key step of this mechanism is the formation of N3–Pd complex C, which is the more effective the more the terminal alkyne is electron-poor (hence having the Csp–H bond more acid). That the steps of the mechanism from A to E are certainly critical for the success of the reaction is also proved by the beneficial effect resulting from the slow addition of the terminal alkyne 2 to the reaction mixture. Finally, reductive elimination involving complex E gave the coupling product 6, and reoxidation of Pd(0) to Pd(II) by Ag(I) or air closed the catalytic cycle.
Conclusions
In this study we developed a simple and efficient ligandless Pd(II) catalyzed dehydrogenative alkynylation of imidazoles with terminal alkynes under air in non-anhydrous conditions, starting from a preliminary screening of the role of silver(I) oxidant, catalyst precursors, solvents, and reaction temperature on the efficiency and selectivity of the alkynylation of 1-methylbenzimidazole (1). The optimized reaction conditions allowed us to easily prepare several 2-alkynyl-substituted 1,3-azoles in good isolated yields, working in an open vessel without any added ligand. The experimental results point towards a non-concerted metalation–deprotonation mechanistic pathway, that allows us to justify both the C-2 regioselectivity and the “SEAr-like” reactivity of the azoles, as well as the higher reactivity of arylacetylenes when compared with that of alkyl- or silyl-substituted analogues. From the proposed mechanistic machinery it also emerged that Ag2CO3 plays not only as Pd(0) oxidant, but also has an active role in generating Ag(I) acetylide, and acts as an inner-sphere base in the C2–H deprotonation step. The application of this procedure to the preparation of new push–pull heteroaromatic fluorophores as functional materials is undergoing and will be published in the next future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Chiesi Farmaceutici (Parma, Italy) and Università di Pisa (PRA_2020_219).References
- J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS.
- F. Bellina, R. Rossi, M. Lessi, C. Manzini and L. Perego, Synthesis, 2014, 46, 2833–2883 CrossRef.
- F. Bellina, in C-H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer, Cham, 2016, vol. 55, pp. 77–102 Search PubMed.
- R. Rossi, M. Lessi, C. Manzini, G. Marianetti and F. Bellina, Adv. Synth. Catal., 2015, 357, 3777–3814 CrossRef CAS.
- B. Panda, Asian J. Org. Chem., 2020, 9, 492–507 CrossRef CAS.
- F. Bellina, Synthesis, 2021, 53(15), 2517–2544 CrossRef CAS.
- W. C. Lee, T. H. Wang and T. G. Ong, Chem. Commun., 2014, 50, 3671–3673 RSC.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- H. Kim, Y. J. Hwang, I. Han and J. M. Joo, Chem. Commun., 2018, 54, 6879–6882 RSC.
- J. Le Bras and J. Muzart, Catalysts, 2020, 10, 571–618 CrossRef CAS.
- M. Lessi, C. Manzini, P. Minei, L. A. Perego, J. Bloino, F. Egidi, V. Barone, A. Pucci and F. Bellina, Chempluschem, 2014, 79, 366–370 CrossRef CAS PubMed.
- V. Barone, F. Bellina, M. Biczysko, J. Bloino, T. Fornaro, C. Latouche, M. Lessi, G. Marianetti, P. Minei, A. Panattoni and A. Pucci, Phys. Chem. Chem. Phys., 2015, 17, 26710–26723 RSC.
- F. Bellina, C. Manzini, G. Marianetti, C. Pezzetta, E. Fanizza, M. Lessi, P. Minei, V. Barone and A. Pucci, Dyes Pigm., 2016, 134, 118–128 CrossRef CAS.
- M. Lessi, G. Panzetta, G. Marianetti and F. Bellina, Synthesis, 2017, 49, 4676–4686 CrossRef CAS.
- G. Marianetti, M. Lessi, V. Barone, F. Bellina, A. Pucci and P. Minei, Dyes Pigm., 2018, 157, 334–341 CrossRef CAS.
- Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, 1995 Search PubMed.
- E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3159 CrossRef CAS PubMed.
- S. Toyota, Chem. Rev., 2010, 110, 5398–5424 CrossRef CAS PubMed.
- B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC.
- R. Chinchilla and C. Najera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- F. Bellina, M. La Manna and E. Rosadoni, Curr. Org. Chem., 2021, 25, 1–26 CrossRef.
- H. Ha, C. Shin, S. Bae and J. M. Joo, Eur. J. Org. Chem., 2018, 2018, 2645–2650 CrossRef CAS.
- T. Parsharamulu, P. Vishnuvardhan Reddy, P. R. Likhar and M. Lakshmi Kantam, Tetrahedron, 2015, 71, 1975–1981 CrossRef CAS.
- X. Jie, Y. Shang, P. Hu and W. Su, Angew. Chem., Int. Ed., 2013, 52, 3630–3633 CrossRef CAS PubMed.
- C. Wu, P. Li, Y. Fang, J. Zhao, W. Xue, Y. Li, R. C. Larock and F. Shi, Tetrahedron Lett., 2011, 52, 3797–3801 CrossRef CAS.
- S. S. Patil, R. P. Jadhav, S. V. Patil and V. D. Bobade, Tetrahedron Lett., 2011, 52, 5617–5619 CrossRef CAS.
- S. H. Kim, J. Yoon and S. Chang, Org. Lett., 2011, 13, 1474–1477 CrossRef CAS PubMed.
- L. Yang, L. Zhao and C. J. Li, Chem. Commun., 2010, 46, 4184–4186 RSC.
- N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358–2361 CrossRef CAS PubMed.
- M. Kitahara, K. Hirano, H. Tsurugi, T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 1772–1775 CrossRef CAS PubMed.
- F. Shibahara, Y. Dohke and T. Murai, J. Org. Chem., 2012, 77, 5381–5388 CrossRef CAS PubMed.
- I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742–7743 CrossRef CAS PubMed.
- Y. Gu and X.-m. Wang, Tetrahedron Lett., 2009, 50, 763–766 CrossRef CAS.
- E. Brachet and P. Belmont, J. Org. Chem., 2015, 80, 7519–7529 CrossRef CAS PubMed.
- S. H. Kim and S. Chang, Org. Lett., 2010, 12, 1868–1871 CrossRef CAS PubMed.
- A. Mondal, H. Chen, L. Flamig, P. Wedi and M. van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662–18667 CrossRef CAS PubMed.
- A. Mondal and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 742–746 CrossRef CAS PubMed.
- U. N. Patel and B. Punji, Asian J. Org. Chem., 2018, 7, 1390–1395 CrossRef CAS.
- N. Matsuyama, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 4156–4159 CrossRef CAS PubMed.
- J. Aziz, T. Baladi and S. Piguel, J. Org. Chem., 2016, 81, 4122–4133 CrossRef CAS PubMed.
- T. Kawano, N. Matsuyama, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2010, 75, 1764–1766 CrossRef CAS PubMed.
- F. Besselievre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553–9556 CrossRef CAS PubMed.
- J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346–9349 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304–7307 CrossRef CAS PubMed.
- Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743–6747 CrossRef CAS PubMed.
- B. Zhao, Org. Biomol. Chem., 2012, 10, 7108–7119 RSC.
- J. Kim, D. Kang, E. J. Yoo and P. H. Lee, Eur. J. Org. Chem., 2013, 2013, 7902–7906 CrossRef CAS.
- X. Feng, Z. Zhao, F. Yang, T. Jin, Y. Ma and M. Bao, J. Organomet. Chem., 2011, 696, 1479–1482 CrossRef CAS.
- L. I. Belen'kii and N. D. Chuvylkin, Chem. Heterocycl. Compd., 1997, 32, 1319–1343 CrossRef.
- J. A. Joule and K. Mills, in Heterocyclic Chemistry at a Glance, John Wiley & Sons, 2nd edn, 2013, ch. 12, pp. 107–121, DOI: DOI:10.1002/9781118380208.ch12.
- M. R. Grimmett, in Advances in Heterocyclic Chemistry, 1970, vol. 12, pp. 103–183, DOI: DOI:10.1016/s0065-2725(08)60973-3.
- L. Theveau, C. Verrier, P. Lassalas, T. Martin, G. Dupas, O. Querolle, L. Van Hijfte, F. Marsais and C. Hoarau, Chem.–Eur. J., 2011, 17, 14450–14463 CrossRef CAS PubMed.
- L. Théveau, O. Querolle, G. Dupas and C. Hoarau, Tetrahedron, 2013, 69, 4375–4380 CrossRef.
- V. Gandon and C. Hoarau, J. Org. Chem., 2021, 86, 1769–1778 CrossRef CAS PubMed.
- S. Pivsa-Art, T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 467–473 CrossRef CAS.
- F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, Eur. J. Org. Chem., 2006, 2006, 693–703 CrossRef.
- C. H. Park, V. Ryabova, I. V. Seregin, A. W. Sromek and V. Gevorgyan, Org. Lett., 2004, 6, 1159–1162 CrossRef CAS PubMed.
- S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures as well as 1H and 13C NMR spectra of the substrates and the products. See DOI: 10.1039/d1ra05303e |
| This journal is © The Royal Society of Chemistry 2021 |