
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuran oxidation by Mn(III)/Co(II) catalysts – application to benzofuran synthesis†
Tingshu Wang‡
ab,
Miao Zhang‡ab,
Yifan Zhengac,
Junmo Seongd,
Myoung Soo Lah d and
Sangho Koo*abc
d and
Sangho Koo*abc
aSchool of Pharmacy, East China University of Science and Technology, Meilong Road 130, Shanghai 200237, P. R. of China
bDepartment of Energy Science and Technology, Myongji University, Myongji-Ro 116, Cheoin-Gu, Yongin, Gyeonggi-Do 17058, Korea. E-mail: sangkoo@mju.ac.kr
cDepartment of Chemistry, Myongji University, Myongji-Ro 116, Cheoin-Gu, Yongin, Gyeonggi-Do 17058, Korea
dDepartment of Chemistry, Ulsan National Institute of Science and Technology, UNIST-gil 50, Ulsan, 44919, Korea
First published on 22nd September 2021
Abstract
Furans containing a β-ketoester group at 2-position undergo oxidative ring-opening by Mn(III)/Co(II) catalysts under an O2 atmosphere to produce 1,4-dicarbonyl moieties through an endoperoxide intermediate, which consecutively cyclized with the β-ketoester unit to afford 4-hydroxy-2-cyclohexen-1-ones. This oxidation/cyclization products were efficiently transformed into versatile benzofuran derivatives after consecutive aromatization and Paal–Knorr reaction.
Furan is a hetero-aromatic compound, ready to react with various oxidants or reactive electrophiles to serve as versatile C4 building blocks in organic synthesis.1 Furan can produce a conjugated 1,4-dicarbonyl unit by singlet oxygen through an endoperoxide intermediate,2 which may be trapped by a nearby internal hydroxy group to generate a pyranose structure as was reported by Achmatowicz.3 This oxidative conversion may also be attained through the epoxide intermediate by other oxidants including dioxiranes, metal oxides, N-bromosuccinimide, H2O2, peroxy acids etc.4 Lallemand et al. demonstrated that the furan oxidation product by MCPBA would be trapped by a nearby enolic carbon of β-ketoester leading to a polyoxygenated decalin system (Scheme 1).5
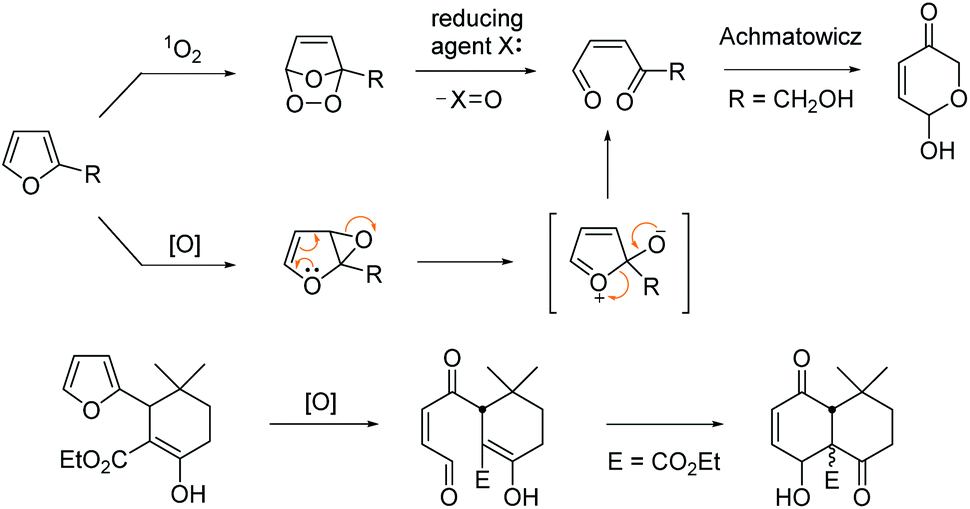 | ||
| Scheme 1 Furan as versatile C4 building blocks for pyranose and cyclohexenone by oxidation. | ||
We recently reported that the MnIII-mediated peroxy radical in β-ketoester A induced oxidation of the acetyl group to α-ketoester C through the dioxetane intermediate B, which was eventually transformed into bichalcophen D by hetero-aromatization (Paal–Knorr reaction).6a In the present work, we want to report that β-ketoester 2a containing a proximal furan moiety undergoes the furan oxidation to the conjugated 1,4-dicarbonyl intermediate E under the same reaction conditions, and is trapped by the active α-methinyl carbon radical of β-ketoester to produce 4-hydroxy-2-cyclohexen-1-ones 3a-1 and 3a-2 (Scheme 2). Whereas the distal electron-deficient furan ring in A was not affected, the proximal furan ring in 2a was smoothly oxidized by MnIII/CoII catalysts under aerobic condition. Cyclohexenones 3a-1 and 3a-2, stereoisomers at the α-carbon of β-ketoester with both hydroxy and benzoyl substituents in equatorial positions (vide infra), were produced in equal amounts, which would be perfect substates for the conversion into versatile benzofuran derivative 4a after decarbonylative aromatization and Paal–Knorr reaction.
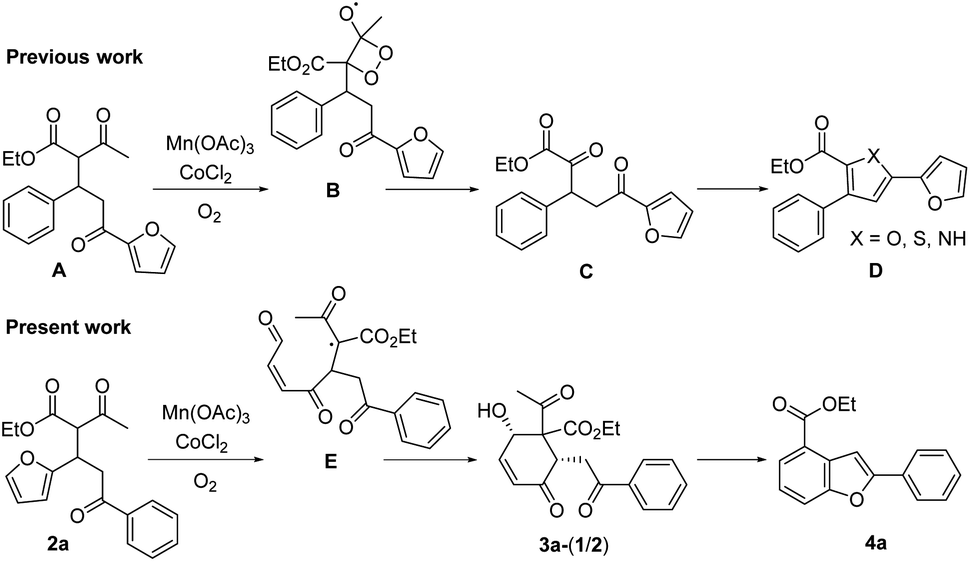 | ||
| Scheme 2 Disparate reactivity of β-ketoesters A and 2a under the MnIII/CoII catalyzed oxidation. | ||
We herein delineated the optimal condition for the furan oxidation of β-ketoester 2a to produce 4-hydroxy-2-cyclohexen-1-ones 3a-(1/2). Generality of the furan oxidation was demonstrated for β-ketoesters 2 with various aroyl substituents. Finally, the transformation of the above oxidative cyclization products 3 into versatile benzofuran derivatives 4 was described.
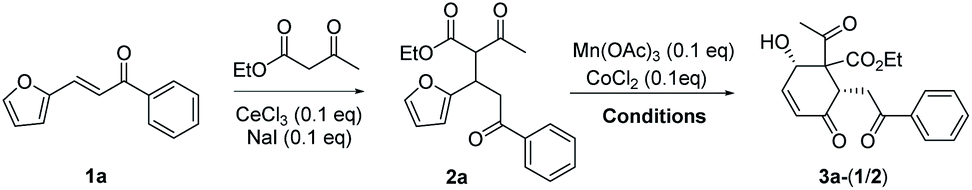
The substrate 2a for furan oxidation was prepared by conjugate addition of ethyl acetoacetate to chalcone derivative 1a containing a furan-2-yl group at β-position. Ethyl acetoacetate was utilized as reagent and solvent at 60 °C under CeCl3 and NaI catalysts (10 mol% each).7 The Michael adduct 2a was obtained in 58% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture, which was utilized for the oxidation reaction without separation. The typical condition for the previous oxidative deacetylation of chalcone derivative A under Mn(OAc)3 and CoCl2 catalysts (10 mol% each) in AcOH was utilized as a standard condition for the optimization study of the furan oxidation of 2a (Table 1).6a
:1 diastereomeric mixture, which was utilized for the oxidation reaction without separation. The typical condition for the previous oxidative deacetylation of chalcone derivative A under Mn(OAc)3 and CoCl2 catalysts (10 mol% each) in AcOH was utilized as a standard condition for the optimization study of the furan oxidation of 2a (Table 1).6a
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Atmosphere | Light | Yield (%) 3a-(1/2) |
| a The catalytic oxidation reactions were carried out in 0.5–1.0 g scale (1.5–3.0 mmol) of the Michael adduct 2a.b UV at 365 nm was irradiated to the reaction flask in a darkroom lamp.c The reaction was carried out only with CoII (without MnIII).d The reaction was carried out in the presence of TEMPO (1 equiv.).e CF3CO2H (15% of total volume) was added to lower the melting point of the solution. | ||||||
| 1 | AcOH | 25 | 48 | Air | Normal | 44 |
| 2 | AcOH | 25 | 13 | Air | UVb | 57 |
| 3 | AcOH | 25 | 18 | O2 | Normal | 55 |
| 4 | AcOH | 25 | 5 | O2 | UVb | 63 |
| 5c | AcOH | 25 | 24 | O2 | Normal | 0 |
| 6c | AcOH | 25 | 24 | O2 | UVb | 44 |
| 7 | AcOH | 25 | 24 | Argon | Normal | 0 |
| 8d | AcOH | 25 | 48 | O2 | UVb | 0 |
| 9e | AcOH | 0 | 24 | Air | Normal | 20 |
| 10 | AcOH | 40 | 24 | Air | Normal | 16 |
| 11 | Ac2O | 25 | 43 | Air | Normal | 30 |
| 12 | EtOH | 25 | 120 | Air | Normal | Trace |
The furan oxidation/cyclization product, 4-hydroxy-2-cyclohexen-1-ones 3a-(1/2), was obtained in 44% yield at 25 °C under the above standard condition in 48 h (entry 1). UV irradiation at 365 nm wavelength not only speed up the rate (13 h), but also increase the yield of the reaction (57%, entry 2). Similar improvement on the yield (55%) and the reaction time (18 h) was observed under O2 atmosphere, supplied by a balloon filled with O2 (entry 3). These two effects can be combined to further speed up the oxidation reaction (5 h), but the yield remained almost the same (63%) within experimental errors (entry 4). The positive O2 effect was obvious in the MnIII catalyst regeneration as well as in the peroxy radical formation.8 UV irradiation would generate singlet oxygen for the furan oxidation in the presence of metal catalysts just as in organic dye sensitizer.9 In the absence of MnIII catalyst (only in the presence of CoII catalyst), normal illumination was not enough to generate singlet oxygen (entry 5), and UV irradiation was necessary to produce the oxidation products 3a-(1/2) in 44% yield (entry 6). The reactions under argon8b atmosphere as well as in the presence of TEMPO10 (1 equiv.) were unable to generate the peroxy radical or singlet oxygen as was expected (entries 7 and 8).
Temperature and solvent effects on the furan oxidation by peroxy radical were then studied (entries 9–12). Low temperature would be beneficial to the reaction utilizing a gaseous reagent because of the increased solubility of gas in solution. When the reaction was proceeded at 0 °C (15% volume of CF3CO2H was added to lower the m.p. of AcOH solution),11 however, only 20% yield of products 3a-(1/2) was obtained presumably due to insufficient activation energy for the reaction. Higher temperature reaction (at 40 °C) produced even lower 16% product yield, presumably because of lower solubility of O2 in solution. AcOH was superior to Ac2O as solvent, but only a trace amount of the product was obtained in EtOH even for a prolonged reaction time. Therefore, we determined the optimal condition for the furan oxidation utilizing Mn(OAc)3 and CoCl2 (10 mol% each) as that of entry 4 in Table 1 (in AcOH at 25 °C under O2 with 365 nm UV irradiation).
Singlet oxygen mechanism would partly explain the oxidation of 2a to 3a-(1/2).2 We were also inclined to suggest the peroxy radical mechanism based on the optimization study as well as our previous work (Scheme 3).8a The initially formed α-carbon radical of β-ketoester unit in 2a by Mn(OAc)3 captured O2 to afford the peroxy radical F, which would oxidize the proximal furan moiety by peroxy radical transfer. The endoperoxide G would be a common intermediate in both mechanisms, which would be reductively ring-opened by the assistance of Mn(OAc)2. Mn(OAc)3 can be regenerated in this way together with the general oxidation by CoCl2 catalyst and O2. Cyclization between methinyl carbon radical and the formyl group in E produced 4-hydroxy-2-cyclohexen-1-ones 3a-(1/2).
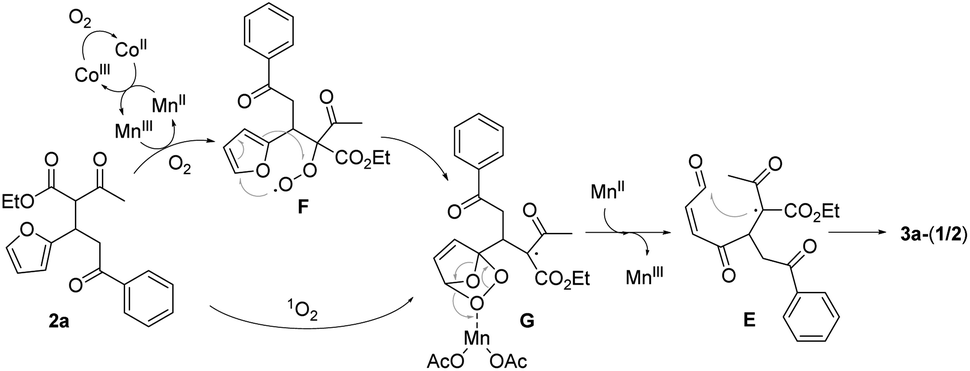 | ||
| Scheme 3 Mechanism of the oxidation of 2a to 3a-(1/2) by MnIII/CoII catalysts. | ||
Generality of the furan oxidation under MnIII/CoII catalysts was demonstrated for various β-ketoesters 2, prepared in 58–94% yields by conjugate addition of ethyl acetoacetate to β-furyl-α,β-unsaturated aryl ketones 1 of diverse aromatic moieties (Table 2). Two diastereomeric 4-hydroxy-2-cyclohexen-1-ones 3-1 and 3-2 were obtained in most cases at the quaternary carbon center, while the substituents at the other two chiral centers were fixed as syn in equatorial positions. The structures of both racemates were identified by X-ray diffraction experiments on 3j-1 (Ar = 2-naphthalene) and 3a-2 (Ar = Ph).12 The ORTEP diagrams of both structures were included in the ESI.† The 1H-NMR spectra of diastereomers were very similar in each group, and the stereochemistry of the other products was decided unambiguously by comparison of the 1H-NMR with that of the above authentic.
|
|||||
|---|---|---|---|---|---|
| Entry | Compd. | Ar– | Yield (%) 3-1b | Yield (%) 3-2c | Yield (%) 4 |
| a The catalytic oxidation reactions were carried out in 0.5–1.0 g scale (1.5–3.0 mmol) of the Michael adducts 2a–m.b The stereochemistry of 3-1 was assigned by X-ray diffraction analysis of compound j.c The stereochemistry of 3-2 was assigned by X-ray diffraction analysis of compound a.d Compounds 5-1 and 5-2 (1:1) were also obtained in 20% yield. See Scheme 4 for the structure and the mechanism of formation. |
|||||
| 1 | a | C6H5– | 30 | 33 | 41 |
| 2 | b | p-F–C6H4– | 25 | 23 | 38 |
| 3 | c | p-Cl–C6H4– | 22 | 24 | 56 |
| 4 | d | p-Br–C6H4– | 19 | 18 | 46 |
| 5d | e | p-NO2–C6H4– | 31 | 31 | 11 |
| 6 | f | p-MeO–C6H4– | 27 | 36 | 52 |
| 7 | g | m-MeO–C6H4- | 35 | 9 | 43 |
| 8 | h | o-MeO–C6H4- | 21 | 21 | 42 |
| 9 | i | p-Me–C6H4– | 31 | 23 | 45 |
| 10 | j | 2-Naphthyl– | 24 | 21 | 56 |
| 11 | k | 2-Furyl– | 20 | 16 | 33 |
| 12 | l | 2-Thiophenyl– | 19 | 20 | 47 |
| 13 | m | 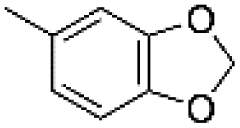 |
44 | — | 44 |
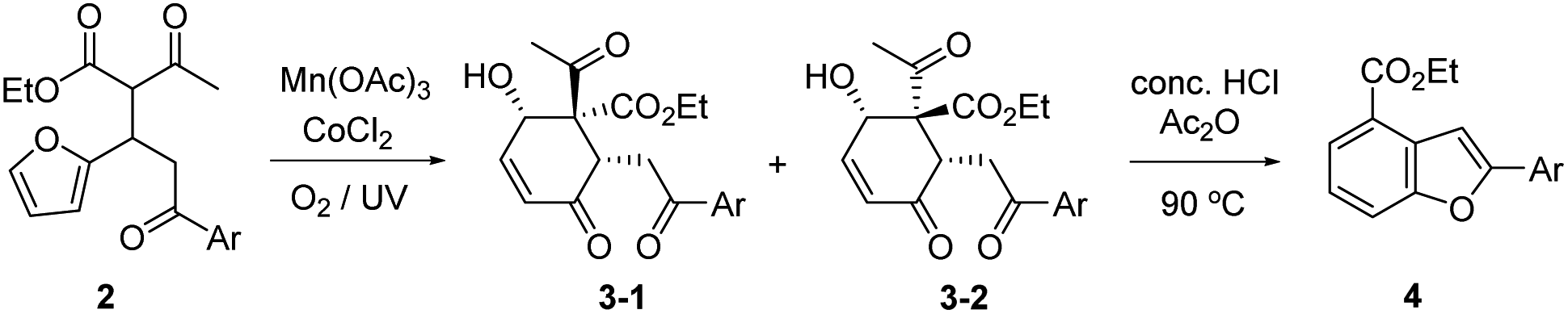
Furan oxidation and subsequent cyclization under the above optimized condition were progressed smoothly for β-ketoesters 2 with various aroyl substituents to produce 4-hydroxy-2-cyclohexen-1-ones 3-1 and 3-2 in total 36–63% yields. The furan oxidation/cyclization seems to be insensitive to the identity of distal aromatic moiety. Highest yields (total 62–63%) of 3 were obtained for 4-nitrophenyl (2e) and 4-methoxyphenyl (2f) as well as the parent phenyl (2a) cases. It is interesting to note that further cyclization to 1,3-dioxolanes 5-1 and 5-2 (20% yield, 1:1 diastereomers, see Scheme 4) was observed for the electron-withdrawing 4-nitrophenyl case (vide infra). The structure of 5-2 was also identified by X-ray analysis (see ORTEP diagram in ESI†).12 Halogen substituents survived during this radical reaction even though somewhat lower yields of 3 (total 37–48%) were obtained (entries 2–4). Furan oxidation also progressed smoothly for the phenyl group with ortho- and meta-alkoxy substitutions with a little diminished yield, in which diastereomer 3-1 was obtained as the major isomer in meta cases (entries 7 and 13). Diversity of the aromatic substituents included 2-naphthalene, 2-furan, 2- thiophene, and 2-benzodioxolane (entries 10–13). It was noteworthy that the proximal furan in 2k was readily oxidized, while the remote electron-deficient (less reactive) furan survived under this condition (entry 11).
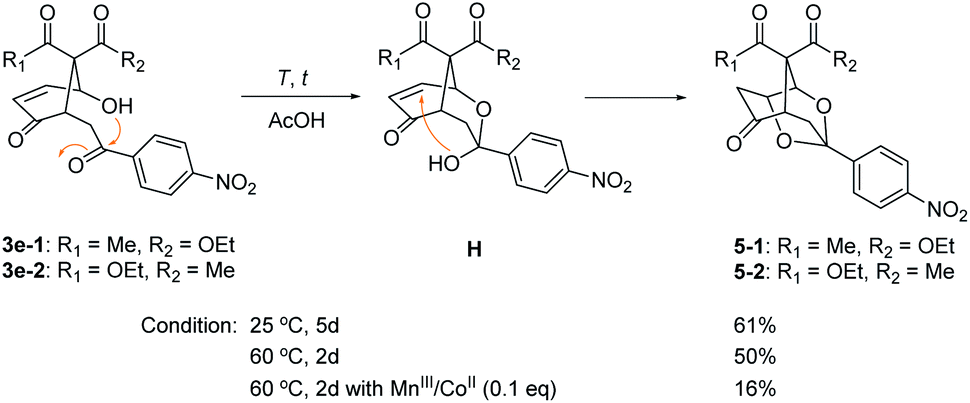 | ||
| Scheme 4 1,3-Dioxolanes 5 by further cyclization of furan oxidation product 3e. | ||
The benzofuran structure is widely distributed in biologically active nature products and regarded as a core skeleton for the drug discovery.13 Fluorescent 2-arylbenzofurans might be important as labelling and optoelectrical materials (see ESI† for UV/FL spectra of 4a), for which efficient synthetic methods have been studies.14 The above furan oxidation/cyclization product, 4-hydroxy-2-cyclohexen-1-ones 3 with 6-aroyl substituent, provided versatile 2-arylbenzofuran derivatives 4 after decarbonylative aromatization and Paal–Knorr reaction (Table 2). Various conditions for Paal–Knorr reaction of 4-hydroxy-2-cyclohexen-1-ones 3-1 and 3-2 were tried, among which the one using concentrated HCl in acetic anhydride at 90 °C produced highest yields of the desired 2-arylbenzofuran derivatives 4. The conversion requires a strong acid at high temperature for decarbonylative aromatization as well as Paal–Knorr reaction. An appreciable amount of tar formation seemed to be indispensable under the condition using benzene solvent, which was significantly reduced in acetic anhydride solvent. The corresponding benzofuran derivatives 4 were obtained in 33–56% yield ranges except the electron-withdrawing 4-nitrophenyl case (entry 5), where the Paal–Knorr reaction might be slowed down to produce 4e in only 11% yield presumably due to the competition to form 1,3-dioxolane 5.
The caged 1,3-dioxolanes 5-1 and 5-2 (1:1 stereoisomers) were obtained in 20% yield as by-product from β-ketoester 2e with p-nitrophenyl substituent (vide supra). It was presumed to be formed by intramolecular Michael addition of the hemiacetal H, which was derived from the nucleophilic addition of syn 4-hydroxyl group onto the electron-deficient carbonyl group in 3e (Scheme 4). Further cyclization to 1,3-dioxolane 5 was observed only for 3e with p-nitrophenyl group. This structure is similar to anticonvulsant paeonimetabolin I, a metabolite from paeoniflorin, which is an important ingredient of traditional Chinese medicine curing for abdominal pain.15 An optimal condition for intramolecular cyclic acetalization of 3e (a 1:1 mixture of stereoisomers) has been screened. 1,3-Dioxolane 5 (a 1:1 mixture of stereoisomers) was produced in 61% yield at 25 °C for 5 days and in 50% yield at 60 °C for 2 days in AcOH. The presence of MnIII/CoII catalysts at 60 °C deteriorated the acetalization reaction to give 5 in only 16% yield.
In conclusion, β-ketoesters 2, easily prepared from chalcones 1 containing a β-furan-2-yl substituent by conjugate addition of ethyl acetoacetate, undergo facile furan oxidation by MnIII/CoII catalyst under O2 atmosphere. The resulting 1,4-dicarbonyl moiety participates in consecutive intramolecular cyclization with the β-ketoester unit to produce 4-hydroxy-2-cyclohexen-1-ones 3. The furan oxidation may proceed through the endoperoxide intermediate formed by the α-peroxy radical of the β-ketoesters and partly by singlet oxygen. Generality of the furan oxidation by MnIII/CoII catalyst was demonstrated for β-ketoesters 2 with various aroyl substituents. 4-Hydroxy-2-cyclohexen-1-one 3e with 4-nitrophenyl substituent underwent further intramolecular acetalization to produce caged 1,5-dioxolane 5. 4-Hydroxy-2-cyclohexen-1-ones 3 were smoothly converted into versatile 4′-carboethoxy-5-arylbenzofurans 4 after simultaneous decarbonylative aromatization and Paal–Knorr reaction under conc. HCl in acetic anhydride at 90 °C. The new inventions described in this paper undoubtedly contribute to the valuable addition to the repertoires of furan oxidation in organic synthesis.
Author contributions
T. Wang: synthesis (lead), analysis (supporting); M. Zhang: synthesis (equal), analysis (supporting); Y. Zheng: synthesis (supporting); J. Seong: data curation (supporting), M.-S. Lah: data curation (lead), S. Koo: project administration (lead), funding acquisition (lead), investigation (lead), formal analysis (lead), writing manuscript (lead).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) through the grant funded by the Korean government, Ministry of Science and ICT (NRF-2020R1A2C1010724) and partly by Basic Science Research Program funded by the Ministry of Education (NRF-2020R1A6A1A03038817). This paper is dedicated to my dear Professor Lanny S. Liebeskind.Notes and references
- (a) P. Merino, Org. React., 2015, 87, 1–256 CAS; (b) P. Merino, T. Tejero, J. I. Delso and R. Matute, Curr. Org. Chem., 2007, 11, 1076–1091 CrossRef CAS; (c) G. Piancatelli, M. D'Auria and F. D'Onofrio, Synthesis, 1994, 867–889 CrossRef CAS; (d) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef CAS.
- (a) W. Adam and K. Takayama, J. Org. Chem., 1979, 44, 1727–1728 CrossRef CAS; (b) B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469–488 CrossRef CAS.
- O. Achmatowicz, Jr, P. Bukowski, B. Szechner, Z. Zwierzchowska and A. Zamojski, Tetrahedron, 1971, 27, 1973–1996 CrossRef.
- (a) L. Zhang, Y. Zhang, W. Li and X. Qi, Angew. Chem., 2019, 131, 5042–5045 CrossRef; (b) Y. Kobayashi, G. B. Kumar, T. Kurachi, H. P. Acharya, T. Yamazaki and T. Kitazume, J. Org. Chem., 2001, 66, 2011–2018 CrossRef CAS PubMed; (c) A. Massa, M. R. Acocella, M. De Rosa, A. Soriente, R. Villano and A. Scettri, Tetrahedron Lett., 2003, 44, 835–837 CrossRef CAS; (d) J. Finlay, M. A. Mckervey and H. Q. N. Gunaratne, Tetrahedron Lett., 1998, 39, 5651–5654 CrossRef CAS; (e) L. A. Badovskaya, V. V. Poskonin and L. V. Povarova, Russ. Chem. Bull., 2017, 66, 593–599 CrossRef CAS.
- (a) P.-Y. Renard and J.-Y. Lallemand, Synlett, 1993, 163–164 CrossRef CAS; (b) C. Descoins, Jr, G. V. Thanh, F.-D. Boyer, P.-H. Ducrot, C. Descoins and J.-Y. Lallemand, Synlett, 1999, 240–242 CrossRef.
- (a) X. Jiang, H. Jin, T. Wang, H. Yoo and S. Koo, Synthesis, 2019, 51, 3259–3268 CrossRef CAS; (b) H. Jin, X. Jiang, H. Yoo, T. Wang, C. G. Sung, U. Choi, C.-R. Lee, H. Yu and S. Koo, ChemistrySelect, 2020, 5, 12421–12424 CrossRef CAS.
- G. Bartoli, M. Bosco, M. C. Bellucci, E. Marcantoni, L. Sambri and E. Torregiani, Eur. J. Org. Chem., 1999, 617–620 CrossRef CAS.
- (a) Y. Ju, D. Miao, J. G. Seo and S. Koo, Adv. Synth. Catal., 2014, 356, 3059–3066 CrossRef CAS; (b) Z. Li, H. Jung, M. Park, M. S. Lah and S. Koo, Adv. Synth. Catal., 2011, 353, 1913–1917 CrossRef CAS.
- (a) K. Gollnick and A. Griesbeck, Angew. Chem., Int. Ed., 1983, 22, 726–727 CrossRef; (b) M. R. Iesce, F. Cermola, A. Guitto, R. Scarpati and M. L. Graziano, J. Org. Chem., 1995, 60, 5324–5327 CrossRef CAS; (c) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS PubMed.
- M. V. Baldovi, M. Mohtat and J. C. Scaiano, Macromolecules, 1996, 29, 5497–5499 CrossRef CAS.
- Y. Ju, D. Miao, R. Yu and S. Koo, Org. Biomol. Chem., 2015, 13, 2588–2599 RSC.
- CCDC deposition numbers: 2076252 for 3j-1, 2076253 for 3a-2, and 2076254 for 5-2.†.
- (a) Y.-h. Miao, Y.-h. Hu, J. Yang, T. Liu, J. Sun and X.-j. Wang, RSC Adv., 2019, 9, 27510–27540 RSC; (b) H. Khanam and Shamsuzzam, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed; (c) R. Naik, D. S. Harmalkar, X. Xu, K. Jang and K. Lee, Eur. J. Med. Chem., 2015, 90, 379–393 CrossRef CAS PubMed.
- L.-Y. Liao, G. Shen, X. Zhang and X.-F. Duan, Green Chem., 2012, 14, 695–701 RSC.
- (a) S. Hatakeyama, M. Kawamura, E. Shimanuki and S. Takano, Tetrahedron Lett., 1992, 33, 333–336 CrossRef CAS; (b) O. A. Heikal, T. Akao, S. Takeda and M. Hattori, Biol. Pharm. Bull., 1997, 20, 517–521 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, 1H/13C-NMR spectra, ORTEP diagrams for 3j-1, 3a-2, and 5-2, UV/FL spectra for 4a. CCDC 2076252–2076254. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05305a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |