
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-centered monocyclic carbon wheel clusters with record coordination numbers in planar species†
Xiao-Qin Lu,
Hai-Gang Lu * and
Si-Dian Li*
* and
Si-Dian Li*
Nanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: luhg@sxu.edu.cn; lisidian@sxu.edu.cn
First published on 9th August 2021
Abstract
The highest coordination number identified to date in planar species is CN = 10 in metal-centered monocyclic boron wheel clusters D10h M©B10− (M = Ta and Nb) (Galeev et. al., Angew. Chem. Int. Ed., 2012, 51, 2101). Extensive global minimum searches and first-principles theory calculations performed herein indicate that the experimentally observed LaC13+ and LaC14+ possess the well-defined global minima of perfect metal-centered monocyclic carbon wheel D13h La©C13+ (1)  and slightly off-centered C2v La©C14+ (4) (1A1) with record coordination numbers of CN = 13 and 11 in planar structures, respectively, further pushing the boundary of our understanding of chemical structures and bonding. Detailed molecular orbital, nucleus-independent chemical shift, and ring current analyses indicate that D13h La©C13+ (1) is σ + π dually aromatic in nature, with 14 totally delocalized in-plane σ electrons and 14 totally delocalized out-of-plane π electrons each matching the 4N + 2 aromatic rule (Nσ = Nπ = 3). Similar σ + π dually aromatic metal-centered monocyclic wheel clusters D13h Ca©C13 (2), C13v Ac©C13+ (3), C2v Y©B6C6+ (5), and C2v Sc©B5C6 (6) have also been obtained with CN = 13, 13, 12, and 11, respectively. The results obtained in this work effectively enrich the chemical structures and bonding patterns of planar hypercoordinated complexes.
and slightly off-centered C2v La©C14+ (4) (1A1) with record coordination numbers of CN = 13 and 11 in planar structures, respectively, further pushing the boundary of our understanding of chemical structures and bonding. Detailed molecular orbital, nucleus-independent chemical shift, and ring current analyses indicate that D13h La©C13+ (1) is σ + π dually aromatic in nature, with 14 totally delocalized in-plane σ electrons and 14 totally delocalized out-of-plane π electrons each matching the 4N + 2 aromatic rule (Nσ = Nπ = 3). Similar σ + π dually aromatic metal-centered monocyclic wheel clusters D13h Ca©C13 (2), C13v Ac©C13+ (3), C2v Y©B6C6+ (5), and C2v Sc©B5C6 (6) have also been obtained with CN = 13, 13, 12, and 11, respectively. The results obtained in this work effectively enrich the chemical structures and bonding patterns of planar hypercoordinated complexes.
1. Introduction
Searching for the maximum coordination number in planar species has fascinated chemists for many years and continuously pushed the boundary of our understanding of chemical structures and bonding.1,2 The central atom and periphery atoms around it in the ligand in stable planar hypercoordinated structures must match both geometrically and electronically, i.e., they must have the right atomic sizes and electronic configurations. The experimentally observed boron-centered monocyclic boron wheel clusters D7h B©B72− and D8h B©B8− are good examples in which the central B atoms have the coordination numbers of CN = 7 and 8, respectively.3 These boron cluster monoanions prove to be σ + π dually aromatic in nature with six delocalized σ and six delocalized π electrons (6 σ + 6 π) each conforming to the 4N + 2 aromatic rule (Nσ = Nπ = 1). Based on the double aromaticity requirement in bare boron wheel clusters, a general electronic design principle x + n + k = 12 or 16 was developed for metal-centered monocyclic boron wheel clusters M©Bnk− by the groups of Wang and Boldyrev,1,4–6 where x stands for the formal valence of the metal center M. This design principle has been successfully applied to the experimentally characterized octacoordinated D8h Co©B8− with CN = 8,5 nonacoordianted D9h Ru©B9−, D9h Rh©B9−, and D9h Ir©B9− with CN = 9,7,8 and, finally, to the decacoordinated D10h Ta©B10− and D10h Nb©B10− with CN = 10 which has proven to be the highest coordination number in planar species observed to date.1,2However, planar hypercoordination chemistry may go well beyond metal-centered monocyclic boron wheel clusters. The recent experimental characterization of perfect planar D9h C18 by atom manipulations and high-resolution atomic force microscopy9 inspires us to coordinate transition metal centers with bare cyclo[n]carbon ring-like clusters (Cn, n ≥ 11) to form metal-centered monocyclic carbon wheel complexes M©Cnk+. Very recently, the electronic properties of metal-carbon ring complexes such as Li@C18 and MC16 have been reported.10,11 Carbon ([He]2s22p2) is known to be effective ligand to various transition metals with a smaller covalent radius than boron ([He]2s22p1), while group IIIB metals La, Y, and Sc with the valence electronic configurations of (n-1)d1ns2 possess the largest covalent radii in the periodic table.12 Metal-centered monocyclic carbon wheel clusters M©Cnk+ (M = La, Y, Sc) are thus possible to have even higher coordination numbers than their boron counterparts M©Bnk−. In fact, two families of LaCn+ monocations (n = 12–40) were observed in mobility measurements as early as in 1994, with a La atom inserted into the carbon ring for even-numbered clusters or attached to the inside or outside of the carbon ring for odd-numbered clusters.13 More detailed mass spectra were reported late on LaCn+ in which LaC13+ appeared to be a prominent species with the La center most likely attached inside a C13 ring (ring Ib), while LaC14+ was the only LaC2n+ cluster with even number of C atoms for which ring Ib structure had a higher mass intensity than its competing isomer with a La atom inserted into a C14 ring (ring Ia).14 Similar prominent mass peaks were also observed for YC13+, CeC13+, and ScC13+.15 Early density functional theory (DFT) calculations indicated that the most stable isomer of LaC13+ possessed a nearly-cumulenic structure with the La atom located at the center of the carbon ring.16,17 However, the unique role these experimentally observed LaCn+ species (n = 13, 14) play in planar hypercoordination chemistry has largely omitted in previous investigations and their accurate geometrical and electronic structures and detailed La–C coordination bonding patterns remain to be fully evaluated using the state-of-the-art theoretical approaches to interpret their behaviors observed in experiments.
Detailed first-principles theory calculations performed in this work indicate that the experimentally observed La©C13+ (1) with a perfect D13h symmetry and slightly off-centered La©C14+ (4) with a C2v geometry achieve the record coordination numbers of CN = 13 and 11 in planar species reported to date. The enhanced stability of La©C13+ (1) originates from its σ + π dual aromaticity with 14 delocalized σ electrons and 14 delocalized π electrons (14 σ + 14 π) each matching the 4N + 2 aromatic rule (Nσ = Nπ = 3). σ + π dually aromatic D13h Ca©C13 (2), C13v Ac©C13+ (3), C2v Y©B6C6+ (5), and C2v Sc©B5C6 (6) have also been obtained at first-principles theory level with CN = 13, 13, 12, and 11, respectively. The highly stable La©C13+ (1) is found to behave like a super-hydrogen monocation (H+) in its substituted complex compounds.
2. Theoretical procedure
Extensive global-minimum (GM) searches were performed on LaC13+, LaC14+, CaC13, AcC13+, YB6C6+, ScB5C6, and CeC13+/2+ using the TGmin2 code18 at DFT level based on the constraint basin-hopping algorithm.19 In total, more than 1000 stationary points with different spin multiplicities were probed for each species at PBE/DZVP level using the CP2K program.20,21 Low-lying isomers were then fully optimized at the M06-2X and PBE0 level,22,23 with the 6-311+G(d,p) basis sets for C, B, N, Ca, and Sc24 and Stuttgart relativistic small-core pseudopotentials for La, Y, Ce, and Ac,25,26 using the Gaussian-16 program suite.27 Frequency analyses were performed to make sure all the optimized structures are true minima of the systems. Relative energies for the five lowest-lying isomers were further refined using the more accurate coupled cluster method with triple excitations CCSD(T)28–30 implemented in Molpro31 with the basis set of cc-pVTZ for C, B, N, Ca, and Sc and the Stuttgart small-core pseudopotential for La, Y, and Ac. The optimized GM structures are summarized in Fig. 1 and more alternative isomers tabulated in Fig. S1–S6.† Natural bonding orbital (NBO) analyses were performed using the NBO 6.0 program.32 Born–Oppenheimer molecular dynamics (BOMD) simulations were performed on LaC13+ (1) for 30 ps using the CP2K software suite at 300, 800, and 1000 K.21 The anisotropy of the current-induced density (ACID)33 analyses were performed using the ACID code, with the ring-current maps generated using POV-Ray 3.7.34 The iso-chemical shielding surfaces (ICSSs)35,36 were generated with the Multiwfn 3.8 code.37 The UV-vis spectra were simulated using the time-dependent TD-DFT-M06-2X approach.38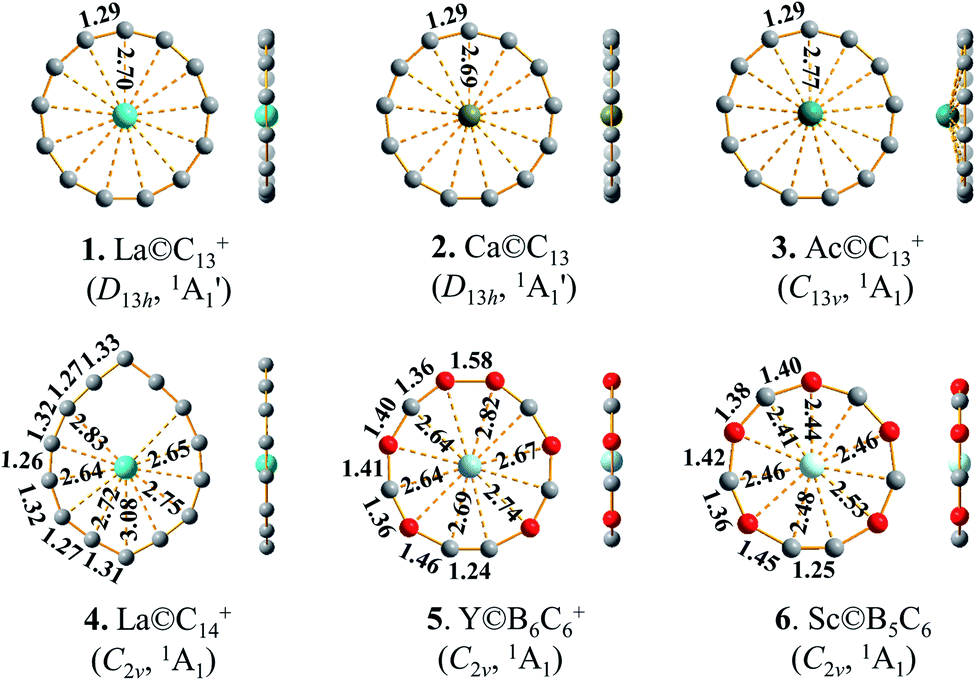 | ||
| Fig. 1 Top and side views of the optimized La©C13+ (1), Ca©C13 (2), Ac©C13+ (3), La©C14+ (4), Y©B6C6+ (5), and Sc©B5C6 (6) at M06-2X level, with bond lengths indicated in Å. | ||
3. Results and discussion
We start from LaC13+, the most concerned species observed in LaCn+ series (n = 12–40).13 Encouragingly and interestingly, as shown in Fig. 1 and S1,† extensive GM searches indicate that LaC13+ possesses the well-defined perfect planar GM of D13h La©C13+ (1) which contains a La atom located exactly at the center of the C13 wheel ligand, with the optimized La–C coordination bond lengths of rLa–C = 2.70 Å which are slightly longer than the sum of the self-consistent single-bond covalent radii of La and C (2.55 Å)12 and C–C bond lengths of rC–C = 1.29 Å which lie between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double-bond (1.34 Å) and C
C double-bond (1.34 Å) and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond (1.20 Å), setting up the highest coordination number of CN = 13 in planar species reported to date. La©C13+ (1) possesses the huge HOMO–LUMO gap of ΔEgap = 5.33 eV at M06-2X level, well underlying its high chemical stability. The slightly distorted triplet Cs LaC13+ (3A′) appears to be the second lowest-lying isomer lying 2.41 eV above the GM at CCSD(T). The seventh C2v isomer with a La inserted into the C13 ring and the fifth C2v isomer with a La attached to the outside of the C13 ring are found to lie 3.36 eV and 3.09 eV higher in energy than the GM at M06-2X, respectively. Extensive BOMD simulations indicate that La©C13+ (1) is highly dynamically stable at both 800 K and 1000 K, with the small average root-mean-square-deviations of RMSD = 0.11 and 0.13 Å and maximum bond length deviations of MAXD = 0.31 and 0.36 Å, respectively (Fig. S7†). No high-lying isomers were observed during the dynamical simulations.
C triple bond (1.20 Å), setting up the highest coordination number of CN = 13 in planar species reported to date. La©C13+ (1) possesses the huge HOMO–LUMO gap of ΔEgap = 5.33 eV at M06-2X level, well underlying its high chemical stability. The slightly distorted triplet Cs LaC13+ (3A′) appears to be the second lowest-lying isomer lying 2.41 eV above the GM at CCSD(T). The seventh C2v isomer with a La inserted into the C13 ring and the fifth C2v isomer with a La attached to the outside of the C13 ring are found to lie 3.36 eV and 3.09 eV higher in energy than the GM at M06-2X, respectively. Extensive BOMD simulations indicate that La©C13+ (1) is highly dynamically stable at both 800 K and 1000 K, with the small average root-mean-square-deviations of RMSD = 0.11 and 0.13 Å and maximum bond length deviations of MAXD = 0.31 and 0.36 Å, respectively (Fig. S7†). No high-lying isomers were observed during the dynamical simulations.
Replacing the La center in La©C13+ (1) with a Ca atom generates the charge-transfer neutral complex D13h Ca©C13 (2). The Ca center in Ca©C13 (2) matches the C13 wheel ligand perfectly both electronically and geometrically though it has a much lower Wiberg bond index of WBICa = 0.26 than La in La©C13+ (1) where WBILa = 1.76 (Table 1). The Ca–C coordination interaction with the low bond order of WBICa–C = 0.02 in Ca2+©C132− (2) is thus almost purely ionic. Using an Ac atom which has a larger atomic radius than La to replace La in La©C13+ (1), the slightly buckled C13v Ac©C13+ (3) is generated in which the Ac atom lies 0.59 Å above the C13 ring, with the periphery C–C distances of rC–C = 1.29 Å remaining basically unchanged. The experimentally observed prominent ScC13+ and YC13+ monocations15 have severely off-centered C2v Sc©C13+ (1A1) (with CN = 8) and C2v Y©C13+ (1A1) (with CN = 9) GM structures due to strong ring strains, respectively, while the open-shell CeC13+ possesses a slightly distorted GM C2v Ce©C13+ (2B2) which has practically a D13h symmetry (Fig. S8†). With one more valence electron detached, the Ce©C132+ dication isovalent with La©C13+ (1) has indeed a perfect D13h GM (Fig. S8†).
| M(x)©BmCnk± | CN | ΔEgap/eV | qM | Electronic configurations of central metal M | WBIM | WBIM–C | WBIM–B | NICS (1) |
|---|---|---|---|---|---|---|---|---|
| D13h La©C13+ (1) | 13 | 5.33 | 2.08 | [Xe] 4f0.435d0.416s0.02 | 1.76 | 0.14 | — | −58.54 |
| D13h Ca©C13 (2) | 13 | 5.63 | 1.87 | [Ar] 3d0.094s0.034p0.01 | 0.26 | 0.02 | — | −44.68 |
| C13v Ac©C13+ (3) | 13 | 5.26 | 2.38 | [Rn] 5f0.196d0.357s0.03 | 1.22 | 0.10 | — | −55.16 |
| C2v La©C14+ (4) | 11 | 3.72 | 2.15 | [Xe] 4f0.285d0.506s0.03 | 1.64 | 0.07–0.16 | — | −29.21 |
| C2v Y©B6C6+ (5) | 12 | 5.70 | 2.12 | [Kr] 5s0.064d0.705p0.01 | 1.67 | 0.10–0.22 | 0.09–0.12 | −24.08 |
| C2v Sc©B5C6 (6) | 11 | 5.32 | 1.94 | [Ar] 3d0.874s0.074p0.02 | 1.94 | 0.13–0.29 | 0.13–0.14 | −29.62 |
It is natural to ask at current stage whether it is possible to form metal-centered monocyclic carbon wheel clusters with coordination numbers greater than thirteen (i.e., CN > 13). We carefully checked the hypercoordination chemistry of the experimentally observed La©C14+ in this work. Although a bare C14 has a perfect C7h acetylenic structure,39 with a La atom added in, the La-doped LaC14+ possesses the off-centered planar GM of C2v La©C14+ (4) which has the actual coordination number of CN = 11 (Fig. 1 and S4†). The three C atoms on the top part of La©C14+ (4) with La–C distances great than 3.1 Å have the practically negligible La–C coordination bond orders (with WBILa–C ≈ 0.00). Using Ac, the largest actinide metal in the periodical table, to replace La,12 an elongated planar D2h Ac©C14+ (Fig. S8†) with CN = 12 is generated. A C14 ring is obviously too big in size to host a transition-metal atom at its geometrical center comfortably to form a perfect wheel complex Dnh M©Cn with the same M–C coordination bonding distances. We conclude that CN = 13 is the highest coordination number in metal-centered monocyclic carbon wheel clusters M©Cn+ with effective M–C coordination interactions.
Introducing certain numbers of B atoms into the carbon rings generates more structural diversities in B–C binary wheel ligands BmCn with CN ≤ 13. As examples, the metal-centered monocyclic C–B binary wheel complexes Y©B6C6+ (5) and Sc©B5C6 (6) in Fig. 1 and C2v La©BC12 in Fig. S8† as the GMs of the systems possess the coordination numbers of CN = 12, 11, and 13, respectively. These planar C–B binary wheel complexes have larger C–B and B–B periphery distances than the corresponding C–C distances in La©C13+ (1) because B has a larger covalent radius than C.
To interpret the high stabilities of these hypercoordinated planar species, we performed detailed NBO and molecular orbital analyses on La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6). As tabulated in Table 1, the La center in La©C13+ (1) possesses the natural atomic charge of qLa = + 2.08 |e|, electronic configuration of La[Xe]4f0.435d0.416s0.02, and total Wiberg bond order of WBILa = 1.76. The C atoms on the C13 wheel ligand have the total Wiberg bond orders of WBIC = 3.99 and C–C bond orders of WBIC–C = 1.73, revealing the cumulenic nature of the complex. Obviously, the La center donates its 6s electron almost completely to the C13 ligand. The La–C13 coordination interactions mainly originate from contributions involving the 5d and 4f atomic orbitals of the La center, as indicated in the degenerated π-HOMO/HOMO′, π-HOMO-2/HOMO-2′, σ-HOMO-1/HOMO-1′, and σ-HOMO-3/HOMO-3 in Fig. 2(a). Although each La–C coordination interaction in La©C13+ (1) has a relatively low bond order (WBILa–C = 0.14), the thirteen equivalent La–C coordination bonds function together to effectively stabilize the hypercoordinated cluster, making it the well-defined GM of the system observed solely in gas-phase experiments.13,14 Similar situations happen in Ac©C13+ (3), Y©B6C6+ (5), and Sc©B5C6 (6).
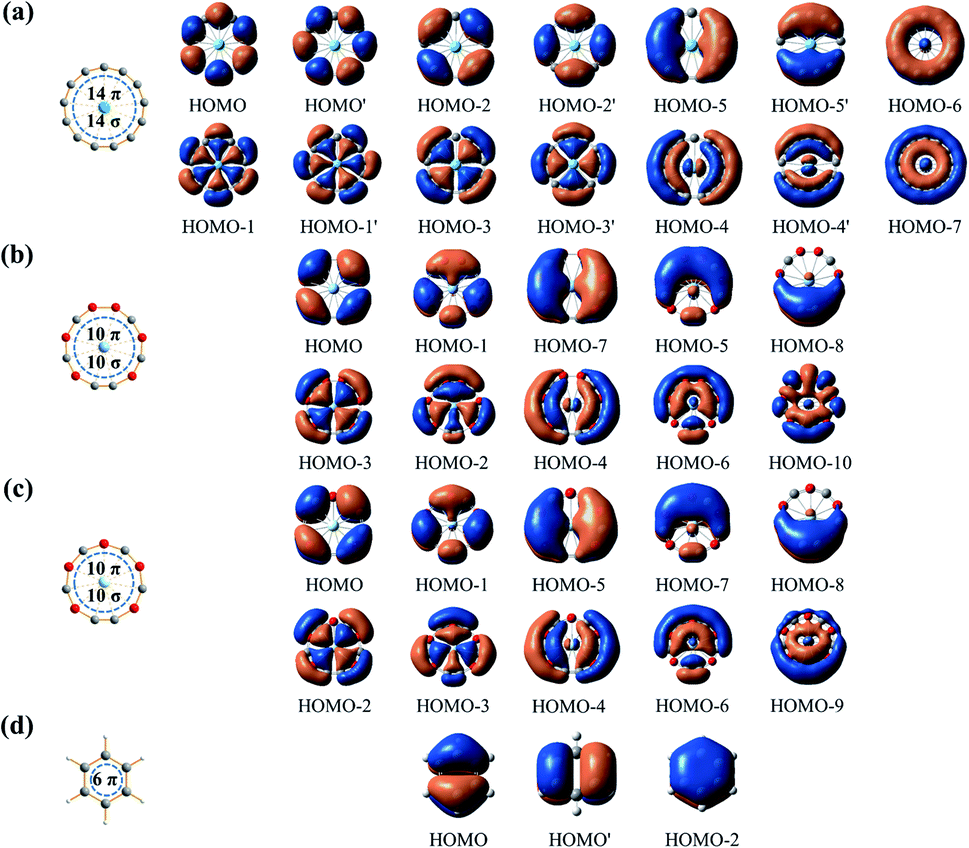 | ||
| Fig. 2 Delocalized π- and σ-CMOs of (a) La©C13+ (1), (b) Y©B6C6+ (5), and (c) Sc©B5C6 (6), in comparison with the delocalized π-CMOs of (d) D6h C6H6. | ||
The 14 totally delocalized canonical molecular orbitals (CMOs) of La©C13+ (1) are collectively shown in Fig. 2(a), including 7 delocalized π-CMOs and 7 delocalized σ-CMOs. Its remaining 13 σ-CMOs correspond to 13 two-center-two-electron (2c-2e) C–C σ bonds along the periphery of the C13 ligand (Fig. S9†). The degenerate f-type HOMO/HOMO′, d-type HOMO-2/HOMO-2′ and p-type HOMO-5/HOMO-5′ and non-degenerate s-type HOMO-6 possess three, two, one, and zero nodal surfaces, respectively, forming an out-of-plane 14 π electron system matching the 4Nπ + 2 aromatic rule with Nπ = 3. Similarly, the degenerate HOMO-1/HOMO-1′, HOMO-3/HOMO-3′, and HOMO-4/HOMO-4′ and non-degenerate HOMO-7 form an in-plane 14 σ electron system matching the 4Nσ + 2 aromatic rule with Nσ = 3. Such a unique 14 σ + 14 π electronic configuration renders σ + π dual aromaticity to La©C13+ (1), effectively stabilizing the monocation observed in experiments, similar to the situation in the σ + π dually aromatic D10h Ta©B10− which possesses a 10 σ + 6 π electronic configuration.1 It is noticed that metal-centered wheel D13h La©C13+ (1) has the same numbers of delocalized σ and π electrons (14 σ + 14 π) as the highly stable ring-like acetylenic C7h C14.39
As shown in Fig. 2(b) and (c), both the metal-centered monocyclic C–B binary wheel clusters Y©B6C6+ (5) and Sc©B5C6 (6) have 5 delocalized π-CMOs and 5 delocalized σ-CMOs, forming an out-of-plane 10 π electron system conforming to the 4Nπ + 2 π-aromatic rule (Nπ = 2) and an in-plane 10 σ electron system conforming to 4Nσ + 2 σ aromatic rule (Nσ = 2). Such 10 σ + 10 π electronic configurations make both Y©B6C6+ (5) and Sc©B5C6 (6) σ + π dually aromatic in nature, similar to but with more delocalized electrons than the previously reported Co©B8− and Ru©B9− with 6 σ + 6 π delocalized electrons and Ta©B10− and Nb©B10− with 10 σ + 6 π delocalized electrons.1,5–8
The La–C13 coordination interactions in La©C13+ (1) mainly originate from d–p and f–p σ-coordination interactions involving the in-plane La 5dx2−y2/5dxy and 4fx(x2−3y2)/4fy(y2−3x2) atomic orbitals and C 2px/2py hybridized atomic orbitals, as demonstrated in the degenerated HOMO-3/HOMO-3′ (5d–2p σ-coordination) and HOMO-1/HOMO-1′ (4f–2p σ-coordination) in Fig. 2(a), respectively. Quantitatively, the La center contributes more to the delocalized in-plane σ-CMOs (2.81–7.63%) than to the delocalized out-of-plane π-CMOs (0.08–3.66%). The delocalized in-plane σ-CMOs thus dominate the La–C coordination bonding interactions. Similarly, the in-plane 4d–2p σ-coordination interactions (HOMO-2 and HOMO-3) in Y©B6C6+ (5) and 3d–2p σ-coordination interactions (HOMO-2 and HOMO-3) in Sc©B5C6 (6) dominate the metal–ligand interactions in the two 10 σ + 10 π systems.
The aromatic nature of La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6) is further evidenced by their calculated nucleus-independent chemical shift (NICS) values. Based on the calculated NICS-ZZ components, Fig. 3(a) depicts the ICSS surfaces of 1, 5, and 6 with the Z-axis perpendicular to the molecular planes to illuminate the chemical shielding around the metal centers, in comparison with that of the prototypical aromatic benzene (C6H6). Obviously, the space inside the C13, B6C6, or B5C6 rings in horizontal direction or within about 1.0 Å above the metal centers in vertical direction belong to chemical shielding regions with negative NICS-ZZ values (highlighted in yellow), while the chemical de-shielding areas with positive NICS values (highlighted in green) are located outside the wheel ligands in horizontal direction. Fig. 3(a) clearly shows the ICSS surfaces of these metal-centered planar complexes are exactly similar to that of the aromatic benzene.
 | ||
| Fig. 3 (a) Calculated iso-chemical shielding surfaces (ICSSs) of La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6), compared with that of aromatic benzene C6H6. Yellow and green regions stand for chemical shielding and de-shielding areas, respectively. (b) Calculated π- and σ-ring current maps of La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6), respectively, in comparison with the π-ring current map of D6h C6H6. The external magnetic field is perpendicular to the wheel plane. The red arrows represent directions and magnitudes of the ring currents at various positions on the ACID iso-surfaces. | ||
Calculating the anisotropy of current-induced density (ACID)34 is an effective approach to graphically display the ring currents induced by an external magnetic field in vertical directions perpendicular to the molecular planes. ACID can be decomposed into σ and π components separately. Fig. 3(b) clearly indicates the π-ring current maps of La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6) are extremely similar to the corresponding π-ring current map of π-aromatic benzene C6H6. Besides, in contrast to benzene which possesses no delocalized σ-electrons, La©C13+ (1), Y©B6C6+ (5), and Sc©B5C6 (6) also exhibit strong σ-ring currents, rendering additional σ-aromaticity to stabilize the systems, as shown in Fig. 3(b) (see Fig. S10† for high-resolution π- and σ-ring current maps of 1, 5, and 6). The observation of both σ- and π-diatropic ring currents in 1, 5, and 6 well supports the σ + π dually aromatic nature of these planar hypercoordinated complexes.
Based on the planar hypercoordinated species discussed above, we develop a universal electronic design principle for σ + π dually aromatic metal-centered monocyclic boron, carbon, or boron-carbon binary wheel clusters M(x)©BmCnk±. There exist m + n 2c-2e σ bonds along the periphery of the monocyclic BmCn wheel ligand. With each C atom providing two delocalized electrons and each B atom contributing one delocalized electron, the total number of the delocalized electrons in M(x)©BmCnk± matches the requirement of σ + π dual aromaticity of the system:
| m + 2n + x ± k = L (L = 12, 16, 20, or 28) | (1) |
The highly stable La©C13+ (1) with a wide HOMO–LUMO gap and fully occupied bonding inner-shell CMOs can be used as a super-hydrogen monocation (H+) to form various substituted multi-nuclei complexes. Typical examples include C13v [La©C13]X (X = F, Cl, Br) (7), D13h [La©C13]+L2 (L = Ar, Kr) (8), C2v [La©C13]2O (9), and C3v N[La©C13]3 (10) (Fig. 4) which can be derived from the parent species C∞v HX, D∞h H+L2, C2v H2O, and C3v NH3 by substituting H+ monocation(s) with La©C13+ (1) unit(s), respectively, presenting the viable possibility to form complex compounds with multiple hypercoordinated metal centers.
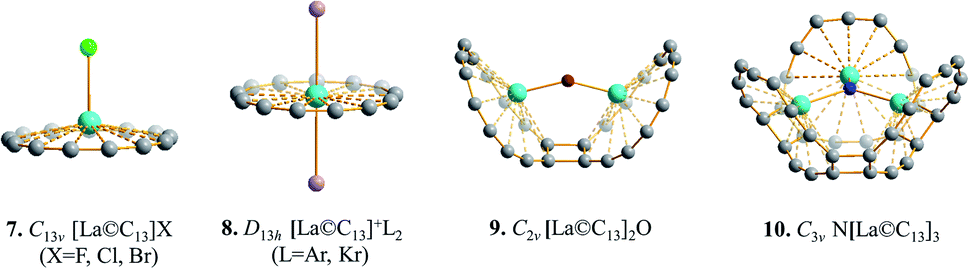 | ||
| Fig. 4 Optimized structures of [La©C13]X (7) (X = F, Cl, Br), [La©C13]+L2 (8) (L = Ar, Kr), [La©C13]2O (9), and N[La©C13]3 (10) at M06-2X level. | ||
Finally, as examples, the simulated IR, Raman, and UV-vis spectra of D13h La©C13+ (1) and C2v Y©B6C6+ (5) are presented in Fig. S11† to facilitate their spectroscopic characterizations. La©C13+ (1) possesses highly simplified spectra due to its perfect D13h symmetry. It has two main IR peaks at 39 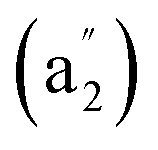 and 866
and 866 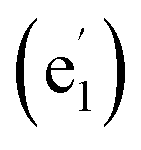 cm−1, three Raman peaks at 272
cm−1, three Raman peaks at 272 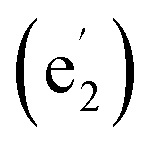 , 664
, 664 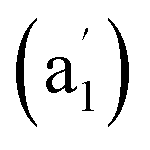 , and 1280
, and 1280 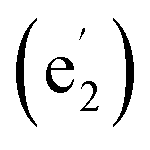 cm−1, and three UV absorption peaks at 156
cm−1, and three UV absorption peaks at 156 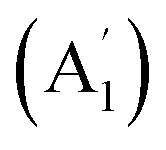 , 174
, 174 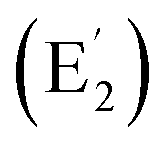 , and 200
, and 200 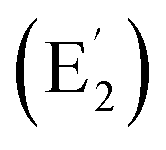 nm, respectively. C2v Y©B6C6+ (5) has more complicated spectra, with two main IR absorption peaks at 499 (b1) and 588 (b2) cm−1, two major Raman scattering bands at 211 (b2) and 623 (a1) cm−1, and five UV absorption bands around 134 (1B2), 161 (1A1), 191 (1B2), 207 (1A1), and 219 (1B2) nm, respectively.
nm, respectively. C2v Y©B6C6+ (5) has more complicated spectra, with two main IR absorption peaks at 499 (b1) and 588 (b2) cm−1, two major Raman scattering bands at 211 (b2) and 623 (a1) cm−1, and five UV absorption bands around 134 (1B2), 161 (1A1), 191 (1B2), 207 (1A1), and 219 (1B2) nm, respectively.
4. Conclusions
Extensive first-principles theory calculations performed in this work unveil the highest coordination numbers of CN = 13 in La©C13+ (1), Ca©C13 (2), and Ac©C13+ (3), CN = 12 in Y©B6C6+ (5), and CN = 11 in La©C14+ (4) and Sc©B5C6 (6) reported to date in planar species, effectively enriching the structural and bonding patterns in planar hypercoordination chemistry. σ + π dually aromatic metal-centered monocyclic B–C binary wheel complexes M(x)©BmCnk± follow the electronic design principle of m + 2n + x ± k = L (L = 12, 16, 20, or 28) which can be readily extended to more complicated systems containing other periphery atoms rather than B and C. The recent experimental identification of the sp-hybridized molecular carbon allotrope, π-aromatic C18, induced by atom manipulations,9 presents the possibility to synthesize and characterize the σ + π dually aromatic La©C13+ (1) and its complex compounds to open a new area in planar hypercoordination chemistry, catalysis, and materials science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21720102006 and 21973057 to S.-D. Li).Notes and references
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS.
- T. Heine and G. Merino, Angew. Chem., Int. Ed., 2012, 51, 4275–4276 CrossRef CAS.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 123, 9506–9509 CrossRef.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2013, 138, 134315 CrossRef PubMed.
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed.
- K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, Science, 2019, 365, 1299–1301 CrossRef CAS.
- Z. Liu, X. Wang, T. Lu, A. Yuan and X. Yan, ChemRxiv, 2021 DOI:10.26434/chemrxiv.14601168.v1.
- Y. Jiang, Y. Wu, J. Deng and Z. Wang, Phys. Chem. Chem. Phys., 2021, 23, 8817–8824 RSC.
- P. Pyykkç and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef.
- D. E. Clemmer, K. B. Shelimov and M. F. Jarrold, J. Am. Chem. Soc., 1994, 116, 5971–5972 CrossRef CAS.
- K. B. Shelimov, D. E. Clemmer and M. F. Jarrold, J. Phys. Chem., 1995, 99, 11376–11386 CrossRef CAS.
- R. Klingeler, P. S. Bechthold, M. Neeb and W. Eberhardt, J. Chem. Phys., 2000, 113, 4 CrossRef.
- S. Roszak and K. Balasubramanian, Chem. Phys. Lett., 1997, 264, 80–84 CrossRef CAS.
- D. L. Strout and M. B. Hall, J. Phys. Chem. A, 1998, 102, 641–645 CrossRef CAS.
- X. Chen, Y. F. Zhao, Y. Y. Zhang and J. Li, J. Comput. Chem., 2019, 40, 1105–1112 CAS.
- D. J. Wales and H. A. Scheraga, Science, 1999, 285, 1368–1372 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- J. Čížek, Adv. Chem. Phys., 1969, 14, 35 Search PubMed.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- H. J. Werner, et al., Molpro, version.1., 2012, (www.%20molpro.net) Search PubMed.
- P. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, 2013 Search PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS.
- Povray, Persistence of vision raytracer, POV-Ray 3.7, http://www.povray.org Search PubMed.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 1893–1898 Search PubMed.
- E. Kleinpeter, S. Klod and A. Koch, J. Mol. Struct., 2007, 811, 45–60 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- S. Arulmozhiraja and T. Ohno, J. Chem. Phys., 2008, 128, 114301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05367a |
| This journal is © The Royal Society of Chemistry 2021 |