
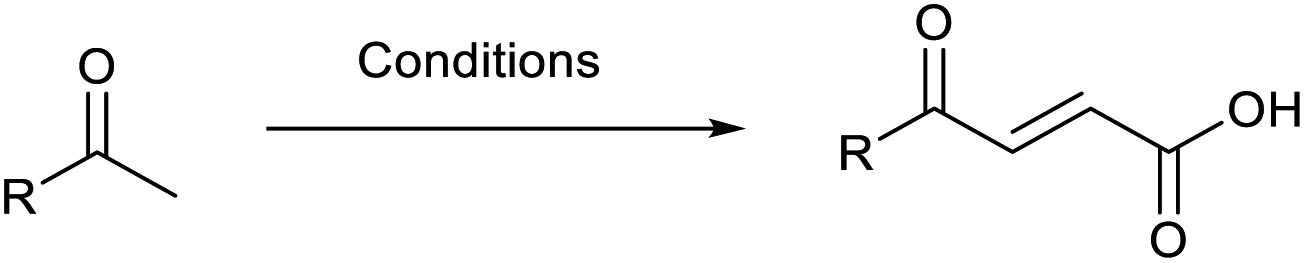
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of 4-oxo-2-butenoic acids by aldol-condensation of glyoxylic acid†
Mélanie Uguen‡
 *a,
Conghao Gai‡b,
Lukas J. Sprengera,
Hang Liub,
Andrew G. Leachc and
Michael J. Waring*a
*a,
Conghao Gai‡b,
Lukas J. Sprengera,
Hang Liub,
Andrew G. Leachc and
Michael J. Waring*a
aCancer Research UK Drug Discovery Unit, Newcastle University Centre for Cancer, Chemistry, School of Natural and Environmental Sciences, Newcastle University, Bedson Building, Newcastle upon Tyne NE1 7RU, UK. E-mail: mike.waring@ncl.ac.uk
bOrganic Chemistry Group, College of Pharmacy, Naval Medical University, Shanghai, 200433, P. R. China
cDivision of Pharmacy and Optometry, School of Health Sciences, University of Manchester, Manchester M13 9PT, UK
First published on 5th October 2021
Abstract
4-Oxobutenoic acids are useful as biologically active species and as versatile intermediates for further derivatisation. Currently, routes to their synthesis can be problematic and lack generality. Reaction conditions for the synthesis of 4-oxo-2-butenoic acid by microwave-assisted aldol-condensation between methyl ketone derivatives and glyoxylic acid have been developed. They provide the desired products in moderate to excellent yields for a wide range of substrates, by applying a simple procedure to accessible starting materials. The investigation revealed different conditions are required depending on the nature of the methylketone substituent, with aryl derivatives proceeding best using tosic acid and aliphatic substrates reacting best with pyrrolidine and acetic acid. This substituent effect is rationalised by frontier orbital calculations. Overall, this work provides methods for synthesis of 4-oxo-butenoic acids across a broad range of substrates.
Introduction
4-Oxo-2-butenoic acids are interesting building blocks for drug discovery. Several derivatives have shown biological activity in their own right, for example to treat cancer,1,2 neurodegenerative,3 metabolic,4 and gastric5 conditions as well as antimicrobial6,7 or antifungal6 properties (Fig. 1). In addition, their high reactivity makes them versatile intermediates for further derivatisation. | ||
| Fig. 1 Examples of biologically active 4-oxo-2-butenoic acids and analogues. | ||
Preparation of 4-oxo-2-butenoic acids has often proven to be scope-limited, with Friedel–Crafts acylations1 used for aromatic substrates (Fig. 2) and oxidative furan-opening8 for aliphatic ones (Fig. 2). Although we managed to obtain the desired 4-oxo-2-butenoic acid product when applying the oxidative furan oxidative conditions to an electron-rich aromatic example, we were unable to identify conditions compatible with electron-deficient aromatic substrates.
 | ||
| Fig. 2 Synthetic routes for the preparation of 4-oxo-2-butenoic acid derivatives. | ||
Aldol-condensation with glyoxylic acid is compatible with a larger range of starting materials with a few examples available in the literature, mainly using acetophenone derivatives as substrates (Fig. 2). Literature reported conditions are typically acid-promoted, most commonly by acetic acid,7,9–12 sulphuric acid,13–17 phosphoric acid,13,18 toluene-4-sulfonic acid19 and formic acid,20 with two acids frequently used together.12,19,21–28 The acid promotors are usually used neat or in large excess under reflux for relatively long periods of time. A few base-promoted procedures are also described in the literature, using potassium carbonate29,30 or sodium hydroxide at reflux or under reduced pressure.31 All these conditions are quite harsh on the reactants and resulting products, thus limiting the scope of the transformation.
Therefore, we decided to investigate the aldol-condensation with glyoxylic acid to identify efficient conditions for the preparation of 4-oxo-2-butenoic acid derivatives. We aimed to develop operationally simple conditions compatible with a large range of substrates by using microwave heating, which had the potential to reduce reaction times and increase yields.
Results and discussion
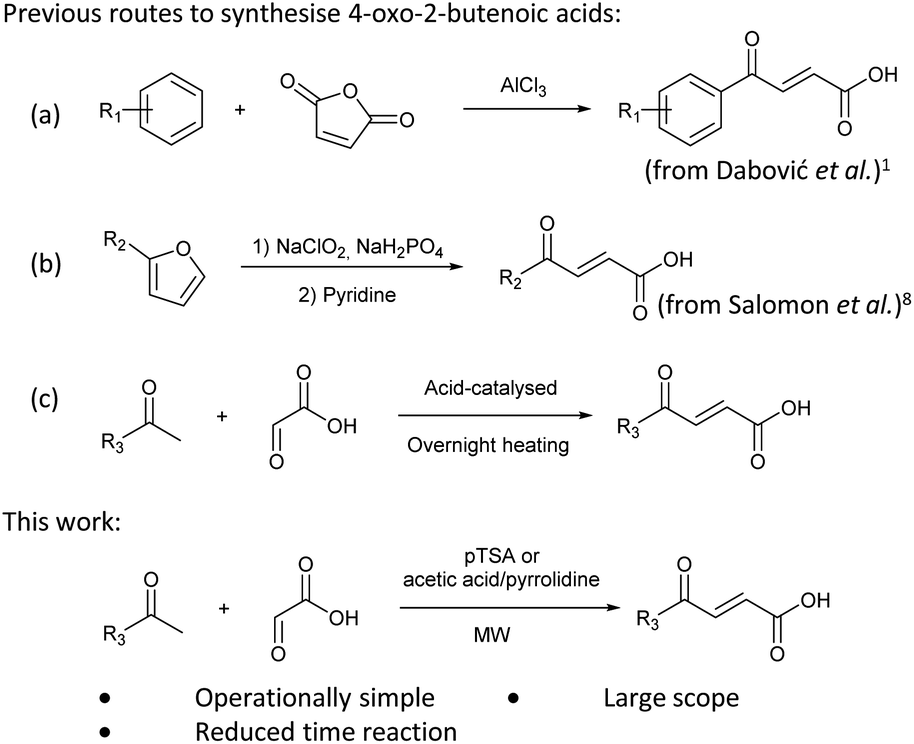
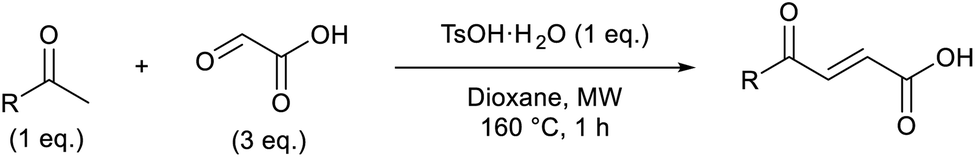
Initially, 4-methoxyacetophenone was used as a test substrate. Reactions with glyoxylic acid under basic conditions were first attempted but showed moderate conversion to the desired product (1) (Table S1†). Reaction via the pyrrolidine-derived enamine also showed moderate conversion, with the initial aldol adduct obtained in larger amount than the desired aldol-condensation product. Attempts to force the water elimination by addition of tosyl chloride led to an improved conversion of the starting material. It was proposed that the tosyl chloride was hydrolysed in situ and the reaction was catalysed by tosic acid. Accordingly, carrying out the direct tosic acid-promoted aldol-condensation provided the desired product in good yield (70%), confirming the previous hypothesis (Table 1, entry 1). These conditions were applied to the more electron deficient 4-cyanoacetophenone, in which case, even after prolonged heating, some starting ketone remained (Table 1, entry 2). To improve the conversion towards the formation of the product 2 as well as reducing the heating time, microwave-assisted heating was performed using similar conditions, leading to a moderate isolated yield of 2 (32%, Table 1, entry 3). Optimisation of the irradiation time and temperature allowed an increase of the yield to 45% with a reduced reaction time of 1 h (Table 1, entry 4). Shorter reaction time led to incomplete conversion of the starting material whereas increase of the temperature from 160 °C to 180 °C led to partial or total degradation of the desired product (Table 1, entries 5–7), suggesting that heating at 160 °C for 1 hour were giving the best results. These findings suggest that increased temperature and pressure allowed by the microwave reactor are key to drive the reaction while allowing a simple reaction set-up. Therefore, these optimised conditions were also applied to 4-methoxyacetophenone and showed an improved yield of 94% (Table 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Cond.a | Heat. | Time | Temp. (°C) | Isol. yield |
| a Conditions A: 3.0 eq. glyoxylic acid monohydrate, 1.0 eq. TsOH monohydrate, dioxane; conditions B: 3.0 eq. glyoxylic acid monohydrate, 1.0 eq. pyrrolidine, 1.0 eq. acetic acid, MeOH.b Products not isolated. | ||||||
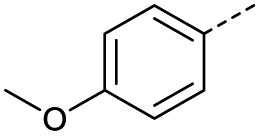
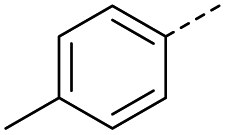
| 1 | 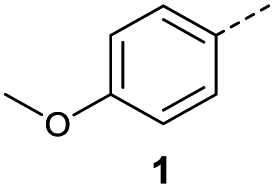 |
A | Conv. | 48 h | 80 | 70% |
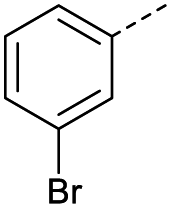
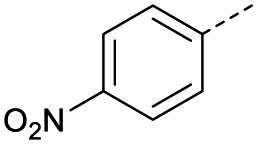
| 2 | 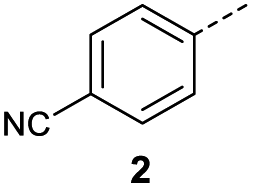 |
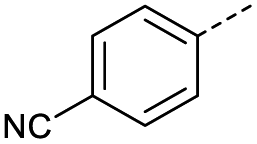
A | Conv. | 72 h | 80 | n.c.b |
| 3 | A | MW | 16 h | 100 | 32% | |
| 4 | A | MW | 1 h | 160 | 45% | |
| 5 | A | MW | 10 min | 180 | <20%b | |
| 6 | A | MW | 15 min | 180 | 40% | |
| 7 | A | MW | 5 min | 160 | 0% | |
| 8 |  |
A | MW | 16 h | 100 | 0% |
| 9 | B | MW | 8 h | 80 | 25% | |
| 10 | B | MW | 10 min | 100 | 14% | |
| 11 | B | MW | 8 h | 60 | 52% | |

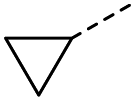


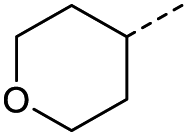


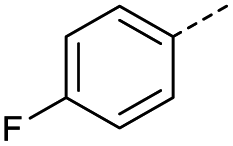
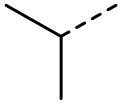
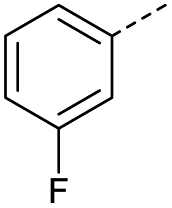
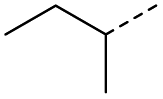

When applying these conditions to cyclohexylmethyl ketone starting material, no desired product 3 was formed (Table 1, entry 8). The major product was the aldol adduct intermediate. However, treatment of cyclohexylmethyl ketone with glyoxylic acid, in the presence of pyrrolidine and acetic acid (Table 1, entry 9), using microwave-assisted heating, enabled the isolation of the desired product 3 in 25% yield. Increasing the temperature from 80 °C to 100 °C led to product degradation, even with decreased reaction time (Table 1, entry 10), but decreasing the temperature to 60 °C improved the yield to 52% (Table 1, entry 11).
Hence, two sets of microwave-assisted conditions for the formation of 4-oxo-2-butenoic acid by aldol-condensation of glyoxylic acid with acetyl derivative have been developed. A scope evaluation of these conditions was performed on electron-rich aromatic (1), electron-poor aromatic (2, 6–9), halogen-containing electron-neutral aromatic (10 and 11), aliphatic ring (3, 12–13), heteroatom-containing (14) and aliphatic chain (15–18) 4-oxo-2-butenoic acid derivatives (Tables 2 and 3).
TsOH-promoted aldol-condensation provided the desired products in good to excellent yields for aromatic substrates generating products 1, 2 and 4–11 in 45–94% yield (Table 2). Electron donating and withdrawing substituents and ortho, meta and para substitution patterns were well tolerated. No desired product formation was observed when these conditions were applied to aliphatic substrates 3, 12–14 and 16–18. Pentan-2-one, however, yielded the desired product 15 under the TsOH-promoted conditions (by NMR) as a minor component relative to the expected internal aldol-condensation product 19 (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, Fig. S1†). After heating at 100 °C for 16 hours, 19 was the only product formed (99% yield). Hence, this suggests, as expected, that the tosic acid mediated reaction may not be compatible with methyl ketones bearing an additional enolisable centre.
:1.5, Fig. S1†). After heating at 100 °C for 16 hours, 19 was the only product formed (99% yield). Hence, this suggests, as expected, that the tosic acid mediated reaction may not be compatible with methyl ketones bearing an additional enolisable centre.
The pyrrolidine-acetic acid conditions were also applied to the selected substrates (Table 3). With aliphatic substrates, these conditions yielded the desired products 3 and 12–17 with yields between 43% and 92%, in which no product was obtained with the TsOH-promoted conditions. In this case, substrates with additional enolisable centres were tolerated (3, 12–17) and the internal aldol adduct was not observed. No desired product was obtained for the t-butyl ketone 18, presumably due to increased steric hindrance. Aromatic products 1, 4, 5 and 7–11 were obtained in poor 4 to 12% yields, much lower than with the TsOH-promoted conditions. However, no product was observed for the electron-poor examples 2 and 6 nor for the aliphatic chain 15 (Table 3).
|
|||||
|---|---|---|---|---|---|
| R | Product | Yield | R | Product | Yield |
| a Typical scale: 2.4 mmol, 6 mL of solvent. | |||||
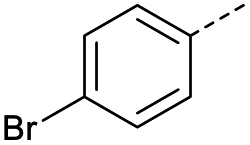
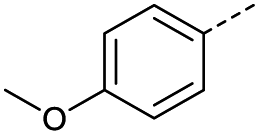 |
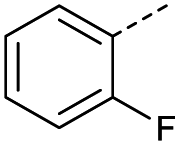
1 | 12% | 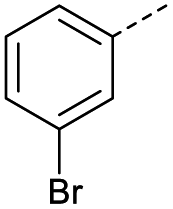 |
11 | <4% |
 |
2 | 0% |  |
3 | 52% |
 |
4 | 5% |  |
12 | 63% |
 |
5 | 5% | 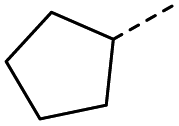 |
13 | 52% |
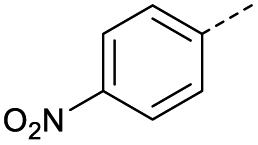 |
6 | 0% | 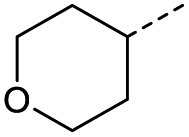 |
14 | 43% |
 |
7 | 5% |  |
15 | 0% |
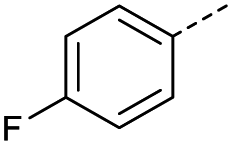 |
8 | 5% | 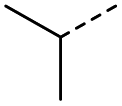 |
16 | 71% |
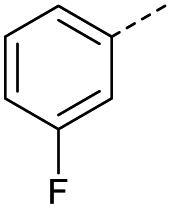 |
9 | 4% | 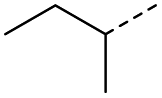 |
17 | 92% |
 |
10 | 4% | 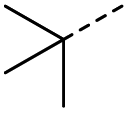 |
18 | 0% |
For all the examples above, the obtained 4-oxo-2-butenoic acids all had the E conformation for the alkene bond confirmed by NMR. No trace of the Z alkene was observed, demonstrating the stereoselectivity of this transformation.
Finally, scale-up of the synthesis of 2 from 0.69 mmol to 6.9 mmol demonstrated the scalability of the TsOH-promoted reaction with no change in yield. Scale up of the synthesis of 3 using the pyrrolidine-acetic acid conditions from 4.0 mmol to 7.2 mmol gave increased yield (from 38% to 52%).
Based on the assumption that the reaction proceeds by attack of the protonated glyoxylic acid by the enol form of the methylketone under the tosic acid promoted conditions, and by either the enol or the enamine in the presence of pyrrolidine/acetic acid (Scheme 1) as the rate determining step, the observed differences in reactivity were rationalised from the calculated energy gaps between the protonated glyoxylic acid LUMO and the enol or enamine HOMOs (Fig. 3). Calculations employed the RHF/6-31+G** level of theory in the Gaussian09 suite of programs.32 Geometries were optimised and frequencies computed to verify that they are minima.
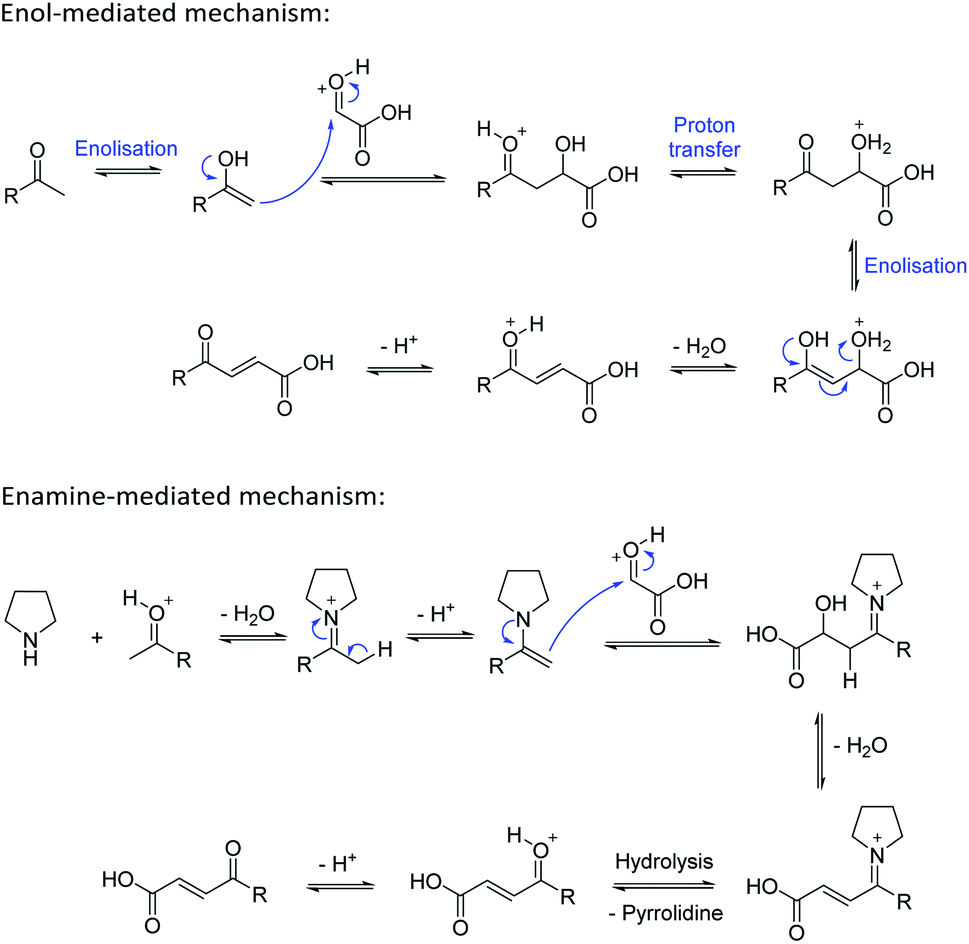 | ||
| Scheme 1 Mechanisms for the aldol condensation reaction. | ||
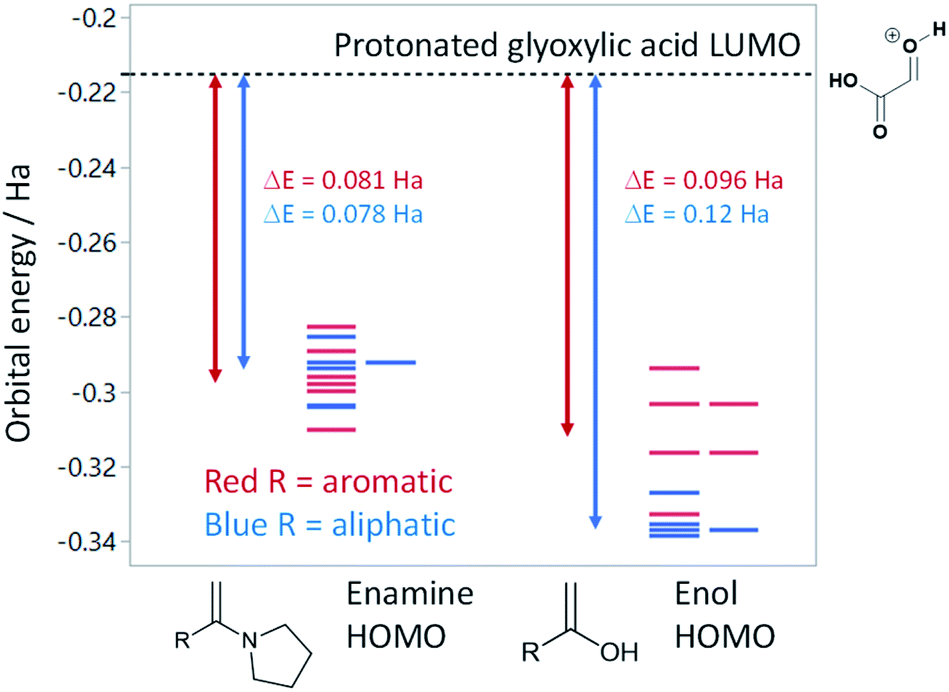 | ||
| Fig. 3 Calculated HOMO energies at the RHF/6-31+G** level for selected enamines and enols relative to the protonated glyoxylic acid LUMO. Double-headed arrows show the average HOMO–LUMO gap for aromatic (red) and aliphatic (blue) substituents. | ||
HOMOs of the enol form of the aromatic ketones are consistently higher in energy than those of the aliphatic examples. This explains why the TsOH conditions work better on aromatic compounds as the HOMO–LUMO energy gap is significantly reduced (ΔE (aromatic) = 0.096 Ha, ΔE (aliphatic) = 0.12 Ha, ΔΔE = 0.024 Ha). For the aliphatic examples, HOMOs of the enamines are higher in energy compared to their corresponding enols (average HOMO–LUMO ΔE = 0.078 Ha). This provides an explanation as to why the pyrrolidine-acetic acid conditions work better for aliphatic substrates, and that these reactions likely proceed via the enamine as the predominant pathway.
Conclusions
We successfully identified two sets of conditions that provide E-4-oxo-2-butenoic acids using microwave-assisted aldol-condensation with glyoxylic acid. These provide the desired products in good to excellent yields for substrate with various electronic and steric properties (electron-rich, -neutral and -poor aromatic, ortho-, meta-, and para-substituted aromatic, linear and cyclic aliphatic). The reaction can be scaled-up to mmol scale with no effect on the yield. It was observed that tosic acid promoted conditions work better for aromatic substrates whereas acid acetic and pyrrolidine are preferred for aliphatic substrates. These findings can be rationalised by the relative differences in HOMO–LUMO energy gaps for the enol and enamine intermediates. Overall, we describe a convenient and efficient synthesis for 4-oxo-2-butenoic acids which should allow an easier access to these biologically-relevant molecules and their derivatives.Experimental
General procedure A for the synthesis of 1, 2 and 4–11
In a Biotage microwave vial, acetyl derivative (1 eq.), glyoxylic acid monohydrate (3 eq.) and TsOH monohydrate (1 eq.) were dissolved in dioxane (2.5 mL mmol−1). The vial was closed with a 20 mm aluminium crimp cap with a PTFE/silicone septum and heated in the microwave for 1 h at 160 °C using the low absorption mode while stirred at 600 rpm with a PTFE stirring bar. 2 M HCl aqueous solution was added to the mixture. This was extracted 3 times with CH2Cl2. Combined organic phases were dried over MgSO4. The solvent was removed under vacuum. A typical scale was 2.4 mmol but the reaction was successfully scaled up and down.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acid), 1661s (CO ketone), 1592s (CC alkene), 1511s (CC aromatic), 1420s (O–H acid), 1167 (s, C–O methoxy); 1H NMR (500 MHz; CDCl3; Me4Si) δH 3.90 (3H, s, CH3), 6.88 (1H, d, J = 15.5 Hz, CHCH), 6.97–7.02 (2H, m, CH-Ar), 7.97–8.05 (3H, m, CHCH and CH-Ar); 13C NMR (126 MHz; CDCl3; Me4Si) δ 55.77 (CH3), 114.39 (C-Ar), 129.68 (C-Ar), 130.61 (CC), 131.55 (C-Ar), 138.77 (CC), 164.57 (C-Ar), 169.84 (COOH), 187.48 (CO); MS(ES+) m/z 207.2; HRMS calcd for C11H10O4 [M + H]+ 207.0579, found 207.0555.
O acid), 1661s (CO ketone), 1592s (CC alkene), 1511s (CC aromatic), 1420s (O–H acid), 1167 (s, C–O methoxy); 1H NMR (500 MHz; CDCl3; Me4Si) δH 3.90 (3H, s, CH3), 6.88 (1H, d, J = 15.5 Hz, CHCH), 6.97–7.02 (2H, m, CH-Ar), 7.97–8.05 (3H, m, CHCH and CH-Ar); 13C NMR (126 MHz; CDCl3; Me4Si) δ 55.77 (CH3), 114.39 (C-Ar), 129.68 (C-Ar), 130.61 (CC), 131.55 (C-Ar), 138.77 (CC), 164.57 (C-Ar), 169.84 (COOH), 187.48 (CO); MS(ES+) m/z 207.2; HRMS calcd for C11H10O4 [M + H]+ 207.0579, found 207.0555. CH), 7.91 (1H, d, J = 15.6 Hz, CHCH), 7.93 (2H, d, J = 8.4 Hz, CH-Ar), 8.17 (2H, d, J = 8.4 Hz, CH-Ar); 13C NMR (methanol-d4, 126 MHz) δC 117.86 (CN), 118.87 (C-Ar), 130.43 (C-Ar), 133.93 (C-Ar), 135.05 (CC), 136.84 (CC), 141.19 (C-Ar), 168.23 (COOH), 190.30 (CO); MS (ES+) m/z = 201.1 [M–H]−; HRMS calcd for C11H7NO3 200.0348 [M + H]+ found 200.0363.
CH), 7.91 (1H, d, J = 15.6 Hz, CHCH), 7.93 (2H, d, J = 8.4 Hz, CH-Ar), 8.17 (2H, d, J = 8.4 Hz, CH-Ar); 13C NMR (methanol-d4, 126 MHz) δC 117.86 (CN), 118.87 (C-Ar), 130.43 (C-Ar), 133.93 (C-Ar), 135.05 (CC), 136.84 (CC), 141.19 (C-Ar), 168.23 (COOH), 190.30 (CO); MS (ES+) m/z = 201.1 [M–H]−; HRMS calcd for C11H7NO3 200.0348 [M + H]+ found 200.0363. O acid), 1410s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 2.40 (3H, s, CH3), 6.67 (1H, d, J = 15.6 Hz, CHCH), 7.38 (2H, d, J = 7.8 Hz, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 7.94 (2H, d, J = 7.8 Hz, CH-Ar), 13.14 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 21.67 (CH3), 129.34 (C-Ar), 130.04 (C-Ar), 133.09 (CC), 134.18 (C-Ar), 136.65 (CC), 145.10 (C-Ar), 166.78 (COOH), 189.30 (CO); MS(ES+) m/z 191.1 [M + H]+; HRMS calcd for C11H11O3 [M + H]+ 191.0629, found 191.0697.
O acid), 1410s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 2.40 (3H, s, CH3), 6.67 (1H, d, J = 15.6 Hz, CHCH), 7.38 (2H, d, J = 7.8 Hz, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 7.94 (2H, d, J = 7.8 Hz, CH-Ar), 13.14 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 21.67 (CH3), 129.34 (C-Ar), 130.04 (C-Ar), 133.09 (CC), 134.18 (C-Ar), 136.65 (CC), 145.10 (C-Ar), 166.78 (COOH), 189.30 (CO); MS(ES+) m/z 191.1 [M + H]+; HRMS calcd for C11H11O3 [M + H]+ 191.0629, found 191.0697.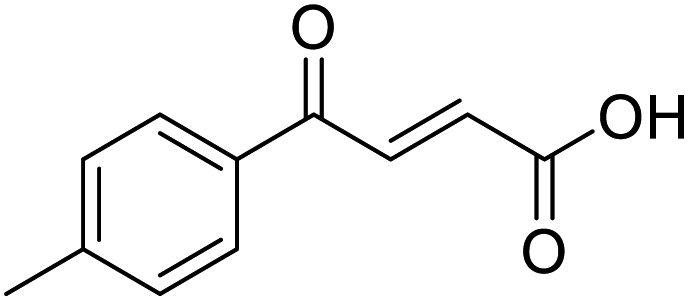 O acid), 1415s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 1.20 (3H, t, J = 7.2 Hz, CH3), 2.70 (2H, q, J = 7.2 Hz, CH2), 6.68 (1H, d, J = 15.6 Hz, CHCH), 7.41 (2H, d, J = 7.8 Hz, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 7.96 (2H, d, J = 7.8 Hz, CH-Ar), 13.15 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 15.48 (CH3), 28.68 (CH2), 128.87 (C-Ar), 129.45 (C-Ar), 133.18 (CC), 134.43 (C-Ar), 136.62 (CC), 151.07 (C-Ar), 166.80 (COOH), 189.34 (CO); MS(ES+) m/z 205.1 [M + H]+; HRMS calcd for C12H13O3 [M + H]+ 205.0791, found 205.0865.
O acid), 1415s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 1.20 (3H, t, J = 7.2 Hz, CH3), 2.70 (2H, q, J = 7.2 Hz, CH2), 6.68 (1H, d, J = 15.6 Hz, CHCH), 7.41 (2H, d, J = 7.8 Hz, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 7.96 (2H, d, J = 7.8 Hz, CH-Ar), 13.15 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 15.48 (CH3), 28.68 (CH2), 128.87 (C-Ar), 129.45 (C-Ar), 133.18 (CC), 134.43 (C-Ar), 136.62 (CC), 151.07 (C-Ar), 166.80 (COOH), 189.34 (CO); MS(ES+) m/z 205.1 [M + H]+; HRMS calcd for C12H13O3 [M + H]+ 205.0791, found 205.0865.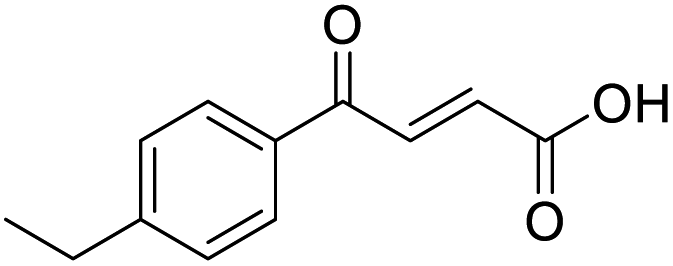 O acid), 1530s (NO2), 1419s (O–H acid), 1350s (NO2); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.71 (1H, d, J = 15.6 Hz, CHCH), 7.87 (1H, d, J = 15.6 Hz, CHCH), 8.25 (2H, d, J = 9.0 Hz, CH-Ar), 8.36 (2H, d, J = 9.0 Hz, CH-Ar), 13.25 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 124.42 (C-Ar), 130.65 (C-Ar), 134.36 (CC), 136.23 (CC), 141.23 (C-Ar), 150.63 (C-Ar), 166.56 (COOH), 189.40 (CO); MS(ES+) m/z 222.1 [M + H]+; HRMS calcd for C10H6NO5 [M–H]− 220.0329, found 220.0257.
O acid), 1530s (NO2), 1419s (O–H acid), 1350s (NO2); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.71 (1H, d, J = 15.6 Hz, CHCH), 7.87 (1H, d, J = 15.6 Hz, CHCH), 8.25 (2H, d, J = 9.0 Hz, CH-Ar), 8.36 (2H, d, J = 9.0 Hz, CH-Ar), 13.25 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 124.42 (C-Ar), 130.65 (C-Ar), 134.36 (CC), 136.23 (CC), 141.23 (C-Ar), 150.63 (C-Ar), 166.56 (COOH), 189.40 (CO); MS(ES+) m/z 222.1 [M + H]+; HRMS calcd for C10H6NO5 [M–H]− 220.0329, found 220.0257.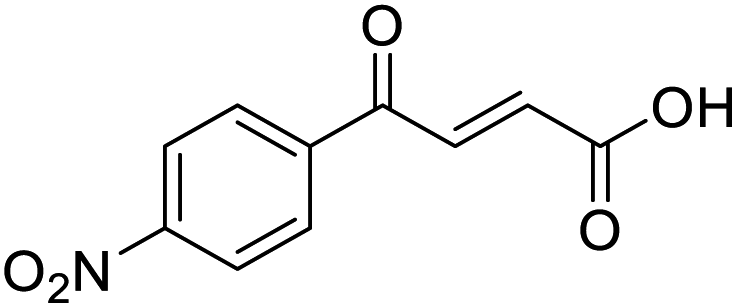 O acid), 1421s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.62 (1H, dd, J = 15.6 Hz, JHF = 0.6 Hz, CHCH), 7.37–7.41 (2H, m, CH-Ar), 7.57 (1H, dd, J = 15.6 Hz, JHF = 3.0 Hz, CHCH), 7.70–7.74 (1H, m, CH-Ar), 7.80–7.83 (1H, m, CH-Ar), 13.21 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 117.30 (d, JCF = 22.2 Hz, C-Ar), 125.50 (d, JCF = 2.8 Hz, C-Ar), 125.54 (d, JCF = 16.6 Hz, C-Ar), 131.22 (CC), 133.33 (CC), 136.06 (d, JCF = 9.0 Hz, C-Ar), 139.14 (d, JCF = 5.1 Hz, C-Ar), 161.40 (d, JCF = 252.0 Hz, CF-Ar), 166.63 (COOH), 188.54 (CO); 19F NMR (282 MHz, DMSO-d6) δF −111.29; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H8FO3 [M + H]+ 195.0379, found 195.0452.
O acid), 1421s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.62 (1H, dd, J = 15.6 Hz, JHF = 0.6 Hz, CHCH), 7.37–7.41 (2H, m, CH-Ar), 7.57 (1H, dd, J = 15.6 Hz, JHF = 3.0 Hz, CHCH), 7.70–7.74 (1H, m, CH-Ar), 7.80–7.83 (1H, m, CH-Ar), 13.21 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 117.30 (d, JCF = 22.2 Hz, C-Ar), 125.50 (d, JCF = 2.8 Hz, C-Ar), 125.54 (d, JCF = 16.6 Hz, C-Ar), 131.22 (CC), 133.33 (CC), 136.06 (d, JCF = 9.0 Hz, C-Ar), 139.14 (d, JCF = 5.1 Hz, C-Ar), 161.40 (d, JCF = 252.0 Hz, CF-Ar), 166.63 (COOH), 188.54 (CO); 19F NMR (282 MHz, DMSO-d6) δF −111.29; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H8FO3 [M + H]+ 195.0379, found 195.0452.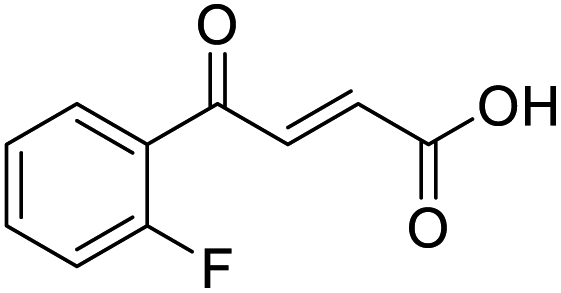 O acid), 1411s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.68 (1H, d, J = 15.6 Hz, CHCH), 7.36–7.40 (2H, m, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 8.10–8.14 (2H, m, CH-Ar), 13.07 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 116.51 (d, JCF = 21.9 Hz, C-Ar), 132.33 (d, JCF = 9.6 Hz, C-Ar), 133.37 (d, JCF = 2.0 Hz, C-Ar), 133.49 (CC), 136.40 (CC), 165.90 (d, JCF = 251.6 Hz, CF-Ar), 166.70 (COOH), 188.54 (CO); 19F NMR (282 MHz, DMSO-d6) δF −104.52; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H8FO3 [M + H]+ 195.0379, found 195.0468.
O acid), 1411s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.68 (1H, d, J = 15.6 Hz, CHCH), 7.36–7.40 (2H, m, CH-Ar), 7.87 (1H, d, J = 15.6 Hz, CHCH), 8.10–8.14 (2H, m, CH-Ar), 13.07 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 116.51 (d, JCF = 21.9 Hz, C-Ar), 132.33 (d, JCF = 9.6 Hz, C-Ar), 133.37 (d, JCF = 2.0 Hz, C-Ar), 133.49 (CC), 136.40 (CC), 165.90 (d, JCF = 251.6 Hz, CF-Ar), 166.70 (COOH), 188.54 (CO); 19F NMR (282 MHz, DMSO-d6) δF −104.52; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H8FO3 [M + H]+ 195.0379, found 195.0468.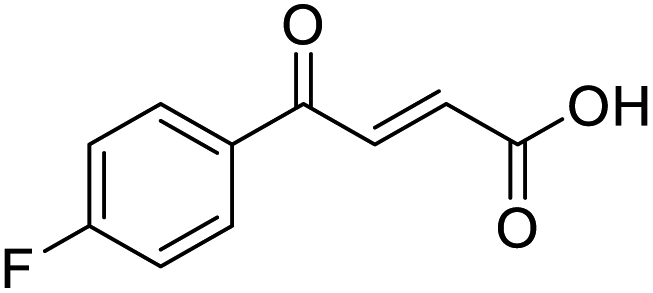 O acid), 1416s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.70 (1H, d, J = 15.6 Hz, CHCH), 7.54–7.58 (1H, m, CH-Ar), 7.62–7.65 (1H, m, CH-Ar), 7.79–7.81 (1H, m, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.90 (1H, dd, J = 7.8 Hz, JHF = 0.6 Hz, CH-Ar), 13.18 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 115.56 (d, JCF = 22.4 Hz, C-Ar), 121.35 (d, JCF = 21.3 Hz, C-Ar), 125.55 (d, JCF = 1.4 Hz, C-Ar), 131.68 (d, JCF = 7.8 Hz, C-Ar), 133.94 (CC), 136.23 (CC), 138.80 (d, JCF = 6.2 Hz, C-Ar), 162.72 (d, JCF = 244.5 Hz, CF-Ar), 166.64 (COOH), 189.02 (CO); 19F-NMR (282 MHz, DMSO-d6) δF −111.78; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H6FO3 [M–H]− 193.0379, found 193.0306.
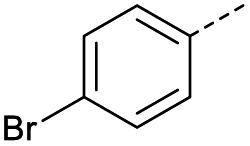
O acid), 1416s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.70 (1H, d, J = 15.6 Hz, CHCH), 7.54–7.58 (1H, m, CH-Ar), 7.62–7.65 (1H, m, CH-Ar), 7.79–7.81 (1H, m, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.90 (1H, dd, J = 7.8 Hz, JHF = 0.6 Hz, CH-Ar), 13.18 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 115.56 (d, JCF = 22.4 Hz, C-Ar), 121.35 (d, JCF = 21.3 Hz, C-Ar), 125.55 (d, JCF = 1.4 Hz, C-Ar), 131.68 (d, JCF = 7.8 Hz, C-Ar), 133.94 (CC), 136.23 (CC), 138.80 (d, JCF = 6.2 Hz, C-Ar), 162.72 (d, JCF = 244.5 Hz, CF-Ar), 166.64 (COOH), 189.02 (CO); 19F-NMR (282 MHz, DMSO-d6) δF −111.78; MS(ES+) m/z 195.1 [M + H]+; HRMS calcd for C10H6FO3 [M–H]− 193.0379, found 193.0306.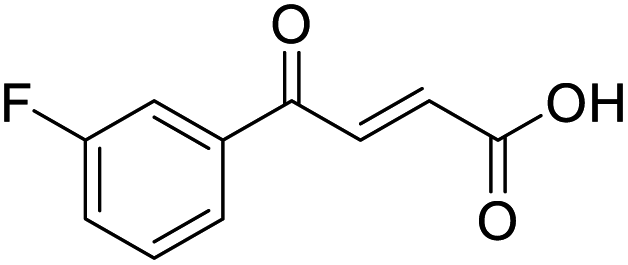 O acid), 1418s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.69 (1H, d, J = 15.6 Hz, CHCH), 7.78–7.81 (2H, m, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.96–7.98 (2H, m, CH-Ar), 13.09 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 128.65 (CBr-Ar), 131.21 (C-Ar), 132.55 (C-Ar), 133.78 (CC), 135.64 (C-Ar), 136.26 (CC), 166.69 (COOH), 189.28 (CO); MS(ES+) m/z 254.1 [M(79Br) + H]+ and 256.1 [M(81Br) + H]+; HRMS calcd for C10H879BrO3 [M + H]+ 254.9585, found 254.9652.
O acid), 1418s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.69 (1H, d, J = 15.6 Hz, CHCH), 7.78–7.81 (2H, m, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.96–7.98 (2H, m, CH-Ar), 13.09 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 128.65 (CBr-Ar), 131.21 (C-Ar), 132.55 (C-Ar), 133.78 (CC), 135.64 (C-Ar), 136.26 (CC), 166.69 (COOH), 189.28 (CO); MS(ES+) m/z 254.1 [M(79Br) + H]+ and 256.1 [M(81Br) + H]+; HRMS calcd for C10H879BrO3 [M + H]+ 254.9585, found 254.9652.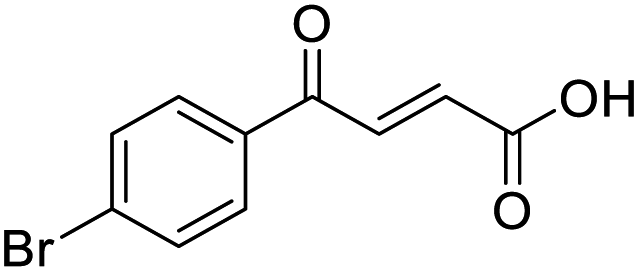 O acid), 1416s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.70 (1H, d, J = 15.6 Hz, CHCH), 7.55 (1H, t, J = 7.8 Hz, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.91–7.92 (1H, m, CH-Ar), 8.04–8.05 (1H, m, CH-Ar), 8.15–8.16 (1H, m, CH-Ar), 13.08 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 122.81 (CBr-Ar), 128.32 (C-Ar), 131.57 (C-Ar), 131.68 (C-Ar), 134.03 (CC), 136.24 (CC), 136.95 (C-Ar), 138.66 (C-Ar), 166.66 (COOH), 189.05 (CO); MS(ES+) m/z 254.1 [M(79Br) + H]+ and 256.1 [M(81Br) + H]+; HRMS calcd for C10H679BrO3 [M–H]− 252.9585, found 252.9503.
O acid), 1416s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 6.70 (1H, d, J = 15.6 Hz, CHCH), 7.55 (1H, t, J = 7.8 Hz, CH-Ar), 7.85 (1H, d, J = 15.6 Hz, CHCH), 7.91–7.92 (1H, m, CH-Ar), 8.04–8.05 (1H, m, CH-Ar), 8.15–8.16 (1H, m, CH-Ar), 13.08 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 122.81 (CBr-Ar), 128.32 (C-Ar), 131.57 (C-Ar), 131.68 (C-Ar), 134.03 (CC), 136.24 (CC), 136.95 (C-Ar), 138.66 (C-Ar), 166.66 (COOH), 189.05 (CO); MS(ES+) m/z 254.1 [M(79Br) + H]+ and 256.1 [M(81Br) + H]+; HRMS calcd for C10H679BrO3 [M–H]− 252.9585, found 252.9503.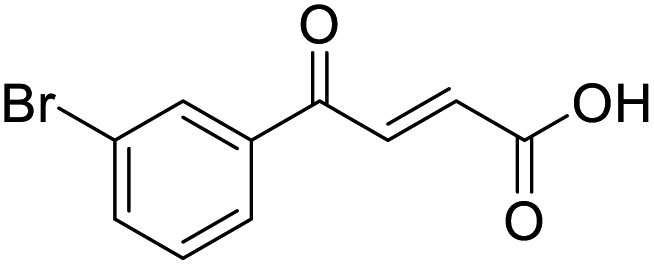
General procedure B for the synthesis of 3, 12–14 and 16–17
In a Biotage microwave vial, methyl ketone-containing compound (1 eq.), glyoxylic acid monohydrate (3 eq.) and acetic acid (1 eq.) were dissolved in MeOH (2.5 mL mmol−1) then pyrrolidine (1 eq.) was added. The vial was sealed with a 20 mm aluminium crimp cap with PTFE/silicon septa and the mixture stirred for 5 min using a PTFE stirring bar. The mixture was irradiated using the MW at 60 °C for 8 h while stirred at 600 rpm. The solvent was removed under vacuum. A typical scale was 2.4 mmol but the reaction was successfully scaled up and down.O acid), 1427s (O–H acid); 1H NMR (500 MHz; MeOD; Me4Si) δH 1.05–1.21 (5H, m, Cy), 1.51–1.67 (5H, m, Cy), 2.50 (1H, tt, J = 10.7, 3.4 Hz, Cy), 6.42 (1H, d, J = 15.9 Hz, CC), 6.91 (1H, d, J = 15.9 Hz, CC); 13C NMR (126 MHz; MeOD; Me4Si) δC 26.49 (Cyc), 29.31 (Cyc), 50.35 (Cyc), 132.21 (CC), 139.56 (CC), 168.50 (COOH), 204.38 (CO); MS(ES+) m/z 183.1 [M + H]+; HRMS calcd for C10H14O3 [M–H]− 181.0870, found 181.0870.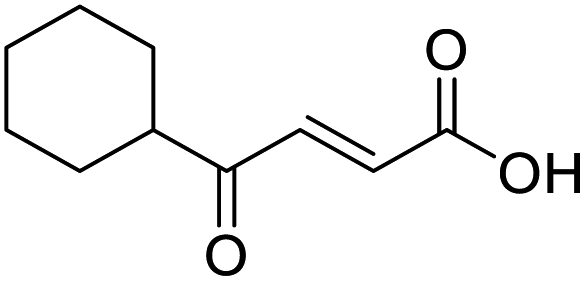 O acid), 1392s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 0.96–0.98 (2H, m, CHH-cyclopropane), 0.99–1.03 (2H, m, CHH-cyclopropane), 2.47–2.51 (1H, m, CH-cyclopropane), 6.70 (1H, d, J = 16.0 Hz, CHCH), 7.03 (1H, d, J = 16.0 Hz, CHCH), 13.06 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 12.07 (CH2), 19.82 (CH), 131.92 (CC), 139.56 (CC), 167.00 (COOH), 200.20 (CO); MS(ES+) m/z 141.1 [M + H]+; HRMS calcd for C7H8O3 [M–H]− 139.0473, found 139.0400.
O acid), 1392s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 0.96–0.98 (2H, m, CHH-cyclopropane), 0.99–1.03 (2H, m, CHH-cyclopropane), 2.47–2.51 (1H, m, CH-cyclopropane), 6.70 (1H, d, J = 16.0 Hz, CHCH), 7.03 (1H, d, J = 16.0 Hz, CHCH), 13.06 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 12.07 (CH2), 19.82 (CH), 131.92 (CC), 139.56 (CC), 167.00 (COOH), 200.20 (CO); MS(ES+) m/z 141.1 [M + H]+; HRMS calcd for C7H8O3 [M–H]− 139.0473, found 139.0400.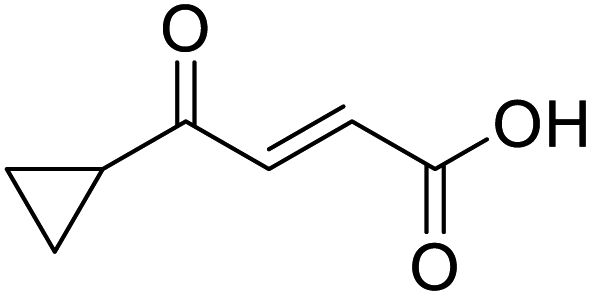 O acid), 1428s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 1.54–1.58 (4H, m, CH2CH2-cyclopentane), 1.64–1.85 (4H, m, CH2CH2-cyclopentane), 3.31 (1H, m, CH-cyclopenpane), 6.62 (1H, d, J = 15.6 Hz, CHCH), 6.99 (1H, d, J = 15.6 Hz, CHCH), 13.09 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 26.09 (CH2CH3-cyclopentane), 28.63 (CH2CH3-cyclopentane), 49.34 (CH), 132.00 (CC), 139.26 (CC), 166.97 (COOH), 202.25 (CO); MS(ES+) m/z 169.1 [M + H]+; HRMS calcd for C9H12O3 [M–H]− 167.0798, found 167.0725.
O acid), 1428s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 1.54–1.58 (4H, m, CH2CH2-cyclopentane), 1.64–1.85 (4H, m, CH2CH2-cyclopentane), 3.31 (1H, m, CH-cyclopenpane), 6.62 (1H, d, J = 15.6 Hz, CHCH), 6.99 (1H, d, J = 15.6 Hz, CHCH), 13.09 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 26.09 (CH2CH3-cyclopentane), 28.63 (CH2CH3-cyclopentane), 49.34 (CH), 132.00 (CC), 139.26 (CC), 166.97 (COOH), 202.25 (CO); MS(ES+) m/z 169.1 [M + H]+; HRMS calcd for C9H12O3 [M–H]− 167.0798, found 167.0725.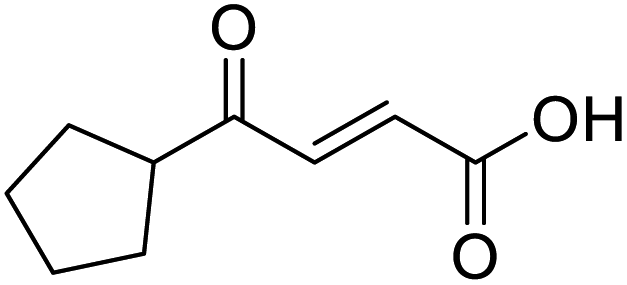 O acid), 1424s (O–H acid), 1117s (C–O ether); 1H NMR (500 MHz; MeOD; Me4Si) δH 1.53 (2H, dtd, J = 13.4, 11.4, 4.2 Hz, Pyr), 1.71 (2H, ddd, J = 13.4, 4.2, 2.1 Hz, Pyr), 2.93 (1H, tt, J = 11.4, 3.8 Hz, Pyr), 3.42 (2H, td, J = 11.4, 2.1 Hz, Pyr), 3.87 (2H, ddd, J = 11.4, 4.2, 2.1 Hz, Pyr), 6.62 (1H, d, J = 15.9 Hz, CC), 7.07 (1H, d, J = 15.9 Hz, CC); 13C NMR (126 MHz; MeOD; Me4Si) δC 29.06 (Pyr), 47.01 (Pyr), 68.07 (Pyr), 132.75 (CC), 139.07 (CC), 168.44 (COOH), 202.59 (CO); MS(ES+) m/z 185.1 [M + H]+; HRMS calcd for C9H12O4 [M + H]+ 183.0663, found 183.0644.
O acid), 1424s (O–H acid), 1117s (C–O ether); 1H NMR (500 MHz; MeOD; Me4Si) δH 1.53 (2H, dtd, J = 13.4, 11.4, 4.2 Hz, Pyr), 1.71 (2H, ddd, J = 13.4, 4.2, 2.1 Hz, Pyr), 2.93 (1H, tt, J = 11.4, 3.8 Hz, Pyr), 3.42 (2H, td, J = 11.4, 2.1 Hz, Pyr), 3.87 (2H, ddd, J = 11.4, 4.2, 2.1 Hz, Pyr), 6.62 (1H, d, J = 15.9 Hz, CC), 7.07 (1H, d, J = 15.9 Hz, CC); 13C NMR (126 MHz; MeOD; Me4Si) δC 29.06 (Pyr), 47.01 (Pyr), 68.07 (Pyr), 132.75 (CC), 139.07 (CC), 168.44 (COOH), 202.59 (CO); MS(ES+) m/z 185.1 [M + H]+; HRMS calcd for C9H12O4 [M + H]+ 183.0663, found 183.0644.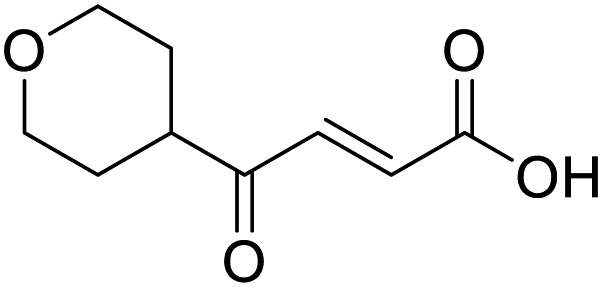 O acid), 1638s (CO ketone), 1466 (O–H acid); 1H NMR (500 MHz; MeOD; Me4Si) δH 0.99 (6H, d, J = 6.8 Hz, Me), 2.83 (1H, hept, J = 6.8 Hz, CH), 6.54 (1H, d, J = 15.9 Hz, CC), 7.01 (1H, d, J = 15.9 Hz, CC); 13C NMR (126 MHz; MeOD; Me4Si) δ 18.24 (Me), 40.57 (CMe2), 132.44 (CC), 139.36 (CC), 168.56 (COOH), 205.15 (CO); MS(ES+) m/z 143.1 [M + H]+; HRMS calcd for C7H10O3 [M–H]− 141.0557, found 141.0536.
O acid), 1638s (CO ketone), 1466 (O–H acid); 1H NMR (500 MHz; MeOD; Me4Si) δH 0.99 (6H, d, J = 6.8 Hz, Me), 2.83 (1H, hept, J = 6.8 Hz, CH), 6.54 (1H, d, J = 15.9 Hz, CC), 7.01 (1H, d, J = 15.9 Hz, CC); 13C NMR (126 MHz; MeOD; Me4Si) δ 18.24 (Me), 40.57 (CMe2), 132.44 (CC), 139.36 (CC), 168.56 (COOH), 205.15 (CO); MS(ES+) m/z 143.1 [M + H]+; HRMS calcd for C7H10O3 [M–H]− 141.0557, found 141.0536.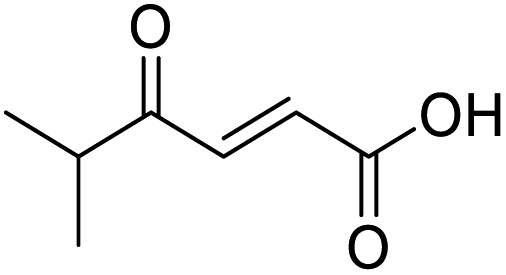 O acid), 1454s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 0.82 (3H, t, J = 7.2 Hz, CH2CH3), 1.02 (3H, d, J = 6.6 Hz, CH3), 1.37 (1H, dq, J = 6.6 Hz and 7.2 Hz, CHH), 1.37 (1H, dq, J = 6.6 Hz and 7.2 Hz, CHH), 2.89 (1H, sext, J = 6.6 Hz, CH), 6.62 (1H, d, J = 15.6 Hz, CHCH), 7.05 (1H, d, J = 15.6 Hz, CHCH), 13.10 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 11.61 (CH2CH3), 15.59 (CH3), 25.53 (CH2CH3), 45.52 (CH), 131.93 (CC), 138.73 (CC), 166.92 (COOH), 203.66 (CO); MS(ES+) m/z 157.1 [M + H]+; HRMS calcd for C8H12O3 [M–H]− 155.0786, found 155.0713.
O acid), 1454s (O–H acid); 1H NMR (600 MHz; DMSO-d6; Me4Si) δH 0.82 (3H, t, J = 7.2 Hz, CH2CH3), 1.02 (3H, d, J = 6.6 Hz, CH3), 1.37 (1H, dq, J = 6.6 Hz and 7.2 Hz, CHH), 1.37 (1H, dq, J = 6.6 Hz and 7.2 Hz, CHH), 2.89 (1H, sext, J = 6.6 Hz, CH), 6.62 (1H, d, J = 15.6 Hz, CHCH), 7.05 (1H, d, J = 15.6 Hz, CHCH), 13.10 (1H, br s, COOH); 13C NMR (150 MHz; DMSO-d6; Me4Si) δC 11.61 (CH2CH3), 15.59 (CH3), 25.53 (CH2CH3), 45.52 (CH), 131.93 (CC), 138.73 (CC), 166.92 (COOH), 203.66 (CO); MS(ES+) m/z 157.1 [M + H]+; HRMS calcd for C8H12O3 [M–H]− 155.0786, found 155.0713.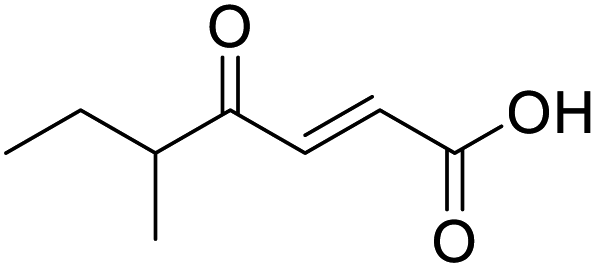 O acid), 1362 (O–H acid); 1H NMR (500 MHz; MeOD; Me4Si) δH 0.99 (3H, t, J = 7.5 Hz, Me), 2.37 (3H, s, COMe), 2.71 (2H, q, J = 7.5 Hz, CH2), 6.64 (1H, s, CC); 13C NMR (126 MHz; MeOD; Me4Si) δC 14.06 (Me), 20.87 (CH2), 26.56 (COMe), 128.17 (CC), 156.25 (CC), 169.07 (COOH), 201.87 (CO); MS(ES+) m/z 143.1 [M + H]+; HRMS calcd for C7H10O3 [M–H]− 141.0557, found 141.0560.
O acid), 1362 (O–H acid); 1H NMR (500 MHz; MeOD; Me4Si) δH 0.99 (3H, t, J = 7.5 Hz, Me), 2.37 (3H, s, COMe), 2.71 (2H, q, J = 7.5 Hz, CH2), 6.64 (1H, s, CC); 13C NMR (126 MHz; MeOD; Me4Si) δC 14.06 (Me), 20.87 (CH2), 26.56 (COMe), 128.17 (CC), 156.25 (CC), 169.07 (COOH), 201.87 (CO); MS(ES+) m/z 143.1 [M + H]+; HRMS calcd for C7H10O3 [M–H]− 141.0557, found 141.0560.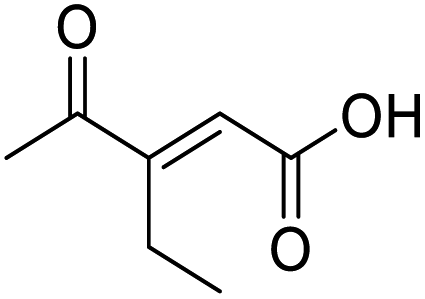
Author contributions
The investigation was performed by M. U., C. G., L. J. S. and H. L. under the supervision of M. J. W. A. G. L. carried out the molecular orbital calculations. The manuscript was written by M. U. and reviewed by M. J. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Newcastle University (SAgE doctoral training programme studentship award to MU), Cancer Research UK (C2115/A21421, Newcastle Drug Discovery Unit Programme Grant) and the Erasmus+ programme of the European Union (studentship award to LJS) for funding.Notes and references
- B. J. Drakulić, T. P. Stanojković, Ž. S. Žižak and M. M. Dabović, Eur. J. Med. Chem., 2011, 46, 3265–3273 CrossRef PubMed.
- M. D. Vitorović-Todorović, A. Erić-Nikolić, B. Kolundžija, E. Hamel, S. Ristić, I. O. Juranić and B. J. Drakulić, Eur. J. Med. Chem., 2013, 62, 40–50 CrossRef PubMed.
- A. Giordani, P. Pevarello, M. Cini, R. Bormetti, F. Greco, S. Toma, C. Speciale and M. Varasi, Bioorg. Med. Chem. Lett., 1998, 8, 2907–2912 CrossRef CAS PubMed.
- L. A. Arnold, A. Kosinski, E. Estébanez-Perpiñá and R. K. Guy, J. Med. Chem., 2007, 50, 5269–5280 CrossRef CAS PubMed.
- M. Bianchi, A. Butti, Y. Christidis, J. Perronnet, F. Barzaghi, R. Cesana and A. Nencioni, Eur. J. Med. Chem., 1988, 23, 45–52 CrossRef CAS.
- A. Dal Pozzo, M. Acquasaliente, G. Donzelli, P. De Maria and M. C. Nicoli, J. Med. Chem., 1987, 30, 1674–1677 CrossRef CAS PubMed.
- C. Xu, X. Bai, J. Xu, J. Ren, Y. Xing, Z. Li, J. Wang, J. Shi, L. Yu and Y. Wang, RSC Adv., 2017, 7, 4763–4775 RSC.
- S. P. Annangudi, M. Sun and R. G. Salomon, Synlett, 2005, 2005, 1468–1470 Search PubMed.
- B. G. Sundar, T. Bailey, E. Bacon, L. Aimone, Z. Huang, J. Lyons, R. Raddatz and R. Hudkins, Bioorg. Med. Chem. Lett., 2011, 21, 5543–5546 CrossRef CAS PubMed.
- X. Li, N. Liu, H. Zhang, S. E. Knudson, H. J. Li, C. T. Lai, C. Simmerling, R. A. Slayden and P. J. Tonge, ACS Med. Chem. Lett., 2011, 2, 818–823 CrossRef CAS PubMed.
- J. Sun, L. Chen, C. Liu, Z. Wang, D. Zuo, J. Pan, H. Qi, K. Bao, Y. Wu and W. Zhang, Chem. Biol. Drug Des., 2015, 86, 1541–1547 CrossRef CAS PubMed.
- J. Ren, J. Xu, G. Zhang, C. Xu, L. L. Zhao, X. F. You, Y. Wang, Y. Lu, L. Yu and J. Wang, Bioorg. Med. Chem. Lett., 2019, 29, 539–543 CrossRef CAS PubMed.
- P. Jakubec, D. Berkeš, A. Kolarovič and F. Považanec, Synthesis, 2006, 4032–4040 CAS.
- N. Chakor, S. Dallavalle, L. Musso and M. Moretti, Tetrahedron Lett., 2008, 49, 5056–5058 CrossRef CAS.
- W. L. Wan, J. B. Wu, F. Lei, X. L. Li, L. Hai and Y. Wu, Chin. Chem. Lett., 2012, 23, 1343–1346 CrossRef CAS.
- X. Wang, Y. Yan, B. Gong, X. Tang, Q. Li, Y. Meng, H. E. Xu and W. Yi, Bull. Korean Chem. Soc., 2013, 34, 3143–3146 CrossRef CAS.
- A. S. Amarasekara and U. Ha, Synth. Commun., 2018, 48, 2533–2538 CrossRef CAS.
- R. Scheffold and P. Dubs, Helv. Chim. Acta, 1967, 50, 798–808 CrossRef CAS.
- R. Messer, A. Schmitz, L. Moesch and R. Häner, J. Org. Chem., 2004, 69, 8558–8560 CrossRef CAS PubMed.
- V. P. Ghidu, M. A. Ilies, T. Cullen, R. Pollet and M. Abou-Gharbia, Bioorg. Med. Chem. Lett., 2011, 21, 259–261 CrossRef CAS PubMed.
- A. Bolognese and G. Scherillo, J. Org. Chem., 1977, 42, 3867–3869 CrossRef CAS.
- K. Kaczanowska, H. Eickhoff, K. Albert, K. H. Wiesmüller and A. P. Schaffner, J. Heterocycl. Chem., 2011, 48, 792–798 CrossRef CAS.
- C. Desel, K. Werninghaus, M. Ritter, K. Jozefowski, J. Wenzel, N. Russkamp, U. Schleicher, D. Christensen, S. Wirtz, C. Kirschning, E. M. Agger, C. P. da Costa and R. Lang, PLoS One, 2013, 8, 1–9 CrossRef PubMed.
- S. Zhao, J. B. Lin, Y. Y. Zhao, Y. M. Liang and P. F. Xu, Org. Lett., 2014, 16, 1802–1805 CrossRef CAS PubMed.
- M. A. I. Salem, M. I. Marzouk and A. M. El-Kazak, Molecules, 2016, 21, 249 CrossRef PubMed.
- S. K. Wang, M. T. Chen, D. Y. Zhao, X. You and Q. L. Luo, Adv. Synth. Catal., 2016, 358, 4093–4099 CrossRef CAS.
- H. Chen, S. Zhao, S. Cheng, X. Dai, X. Xu, W. Yuan and X. Zhang, J. Heterocycl. Chem., 2019, 56, 1672–1683 CrossRef CAS.
- M. Lemurell, J. Ulander, H. Emtenäs, S. Winiwarter, J. Broddefalk, M. Swanson, M. A. Hayes, L. Prieto Garcia, A. Westin Eriksson, J. Meuller, J. Cassel, G. Saarinen, Z. Q. Yuan, C. Löfberg, S. Karlsson, M. Sundqvist and C. Whatling, J. Med. Chem., 2019, 62, 4325–4349 CrossRef CAS PubMed.
- M. Yamaguchi, N. Maruyama, T. Koga, T. Kuroki, K. Kamei, M. Hamana, M. Akima and N. Ohi, Chem. Pharm. Bull., 1994, 42, 1850–1853 CrossRef CAS PubMed.
- M. Krasavin, A. Shetnev, S. Baykov, S. Kalinin, A. Nocentini, V. Sharoyko, G. Poli, T. Tuccinardi, M. Korsakov, T. B. Tennikova and C. T. Supuran, Eur. J. Med. Chem., 2019, 168, 301–314 CrossRef CAS PubMed.
- J. Krall, C. H. Jensen, F. Bavo, C. B. Falk-Petersen, A. S. Haugaard, S. B. Vogensen, Y. Tian, M. Nittegaard-Nielsen, S. B. Sigurdardóttir, J. Kehler, K. T. Kongstad, D. E. Gloriam, R. P. Clausen, K. Harpsøe, P. Wellendorph and B. Frølund, J. Med. Chem., 2017, 60, 9022–9039 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian09, Revision D.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05539a |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2021 |