
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSample preparation considerations for surface and crystalline properties and ecotoxicity of bare and silica-coated magnetite nanoparticles†
Lyubov Bondarenko *a,
Vera Terekhovab,
Anne Kahruc,
Gulzhian Dzhardimalievaad,
Elena Kelbyshevae,
Natalya Tropskayaaf and
Kamila Kydralievaa
*a,
Vera Terekhovab,
Anne Kahruc,
Gulzhian Dzhardimalievaad,
Elena Kelbyshevae,
Natalya Tropskayaaf and
Kamila Kydralievaa
aMoscow Aviation Institute (National Research University), Moscow, Russia. E-mail: l.s.bondarenko92@gmail.com
bLomonosov Moscow State University, 119991 Moscow, Russia
cNational Institute of Chemical Physics and Biophysics, 12618 Tallinn, Estonia. E-mail: anne.kahru@kbfi.ee
dInstitute of Problems of Chemical Physics, Chernogolovka, Moscow Region, Russia
eA. N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, Moscow, Russia
fSklifosovsky Institute for Emergency Medicine, Moscow, Russia
First published on 29th September 2021
Abstract
Magnetite (Fe3O4) nanoparticles (NPs) have widely used in various fields, including in medicine, due to their (super)paramagnetic properties. This requires a thorough evaluation of their possible hazardous effects. However, there is no standard procedure for the preparation of oxidation-prone NPs (such as magnetite) before subjecting them to biological assays. In this study we used Fe3O4 NPs (bare and silica-coated) as test samples to compare different preparation methods (ultrasound, centrifugation and filteration of NPs suspensions) based on X-ray and dynamic light scattering analysis and evaluation of microstructure and surface charge. After oxidation and functionalization, all samples retained their superparamagnetic behaviour. The toxicity of NP suspensions obtained by the methods described for Paramecium caudatum ciliates and Sinapis alba plants was evaluated.
1. Introduction
Different iron oxide compounds are abundant in nature, such as: hematite (a-Fe2O3), maghemite (γ-Fe2O3) and magnetite (Fe3O4).1,2 Iron oxide NPs can be easily synthesized in a broad range of sizes.3,4 Hematite is the most stable of these iron oxides, but only magnetite NPs have a large surface area (up to 120 m2 g−1)5 and possess superparamagnetic properties, i.e., unlike ferromagnetic materials, they do not retain magnetization when the external field is removed.6 These properties are highly useful in the development of various separation processes7,8 and several Fe3O4-based materials are already close to commercial use in this area.9 Iron oxides such as magnetite and maghemite demonstrated superparamagnetic behaviour at a size below 30 nm at room temperature and referred to as superparamagnetic iron oxide nanoparticles (SPIONs). Superparamagnetism can be defined as the ability of magnetic nanoparticles to exhibit a robust paramagnetic nature with high susceptibility and saturation magnetization under the influence of a magnetic field and the tendency to completely lose the same nature once the magnetic field is removed, resulting in zero magnetic remanence and zero coercivity.10 Thanks to their special magnetic properties, SPIONs are widely used in separation technology,11 protein immobilization,12 catalysis,13 medical science,14 and environmental applications.15 The medical science application of SPIONs is mainly focused on targeted drug/gene delivery,16 biosensors,17 magnetic resonance imaging (MRI),18 contrast enhancement19 and hyperthermia,20 biophotonics,21 cancer cell detection,22 magnetic field-assisted diagnosis and radiotherapy,23 and tissue engineering.24 Due to their widespread use in medicine, SPIONs must be stable and, above all, they must be thoroughly evaluated for their potential toxic effects, i.e., they must be tested biologically.However, upon exposure to ambient oxygen, the magnetite phase readily oxidizes into the maghemite phase with partial conversion of ferrous ions into ferric ions.25,26 In turn, a change in the magnetite phase leads to a change in its surface charge and magnetic properties. In addition, a change in environmental conditions can lead to the aggregation of magnetite nanoparticles, while an increase in size greater than 30 nm will result in the loss of their superparamagnetic properties.1
Currently, there are many approaches for the preparation of nanoparticle suspensions for biotesting.27–31 Literature is also available describing methodologies for a specific type of nanomaterials, e.g. “hard” nanoparticles such as metallic ZnO, TiO2 and gold nanoparticles,32 carbon nanotubes33 or polymeric nanoparticles.34 Not all of these methods cannot be applied to “soft” nanoparticles (such as liposomes, polymeric micelles, dendrimers)35 or oxygen-sensitive nanoparticles (NPs) such as iron oxides. Centrifugation or filtration can be used to separate nanoparticles of different sizes. In addition, changes in the functionalization of the NPs under the influence of external conditions will also lead to a change in the phase that elicits a biological response, which also needs to be evaluated. However, there is no special methodical approach or protocol for this type of preparation of oxygen-sensitive nanomaterials for biotesting. Available standard dispersion protocols have been developed for NPs of very slow transformation/dissolution rates, such as TiO2 and SiO2.36,37 As a general rule, NPs suspensions are subjected to ultrasound in the test medium prior to biological testing,38,39 but ultrasound energy levels suitable for TiO2 or SiO2 may not be optimal for other types of NPs, e.g., CuO NPs, and may result in changes in surface characteristics and dissolution properties40 and thus toxicity41 due to partial solubility of CuO and ion release, as illustrated in ref. 42. In addition to ultrasonication, NPs can be separated from agglomerates by centrifugation43 and/or filtration44,45 of the test suspensions. Among these, filtration represents a fast, cost-effective, and simple technique that can be performed at atmospheric pressure. Moreover, it can also be useful for other purposes, such as for the concentrating of colloidal nanomedicines or the reducing of their polydispersity. For example, Roy et al.46 produced Au and CdS nanoparticles and passed them through multiwall carbon nanotubes, used as a filter to remove larger particle sizes and thus reduce the polydispersity of the particle suspension.34,46,47
Centrifugation is another useful technique to separate larger agglomerates from nanomaterials suspensions, as the centrifugal force enhances the precipitation of nanomaterials due to the increased gravitational pull.48 Centrifugation is more efficient than filtration as it is fast, easy, and more cost-effective and can be applied to different types of nanoparticles. However, the centrifugation of large sample volumes requires special equipment and, in some cases, is complicated by the resuspension of settled agglomerates occurs, especially in case of organic nanomaterials which are already more difficult to separate by centrifugation than metallic ones.48,49 Moreover, the toxicity of metallic nanoparticles often depends on the speciation of the released metal species.50
In this study, we report how the properties of oxidation-prone iron oxide nanosuspensions change when using different standard sample preparation protocols to separate nano-sized (colloidal) species from larger agglomerates. More specifically, we evaluated how physico-chemical properties (crystal properties, surface charge, hydrodynamic size) affected the toxicity of these nanosuspensions to aquatic ciliates and terrestrial plant seeds. Fe3O4 NPs (bare and silica-coated under ambient and inert conditions) were used as sample magnetic NPs (MNPs).
2. Materials and methods
2.1 Synthesis of Fe3O4 NPs
Bare Fe3O4 NPs were prepared by the coprecipitation method described in ref. 6. Briefly, 7.56 g of FeCl3·6H2O and 2.78 g of FeCl2·4H2O were dissolved in 70 mL H2O, and 40 mL of 25% ammonium hydroxide solution was added at 50 °C under vigorous stirring either under argon flow or ambient aerobic conditions. The obtained Fe3O4 (Ar) and Fe3O4 (air) nanoparticles were washed five times with ultrapure water to remove the synthesis residues and then dried at 70 °C under vacuum.2.2 Synthesis of Fe3O4 NPs modified by 3-aminopropyltriethoxysilane (Fe3O4-APTES NPs)
The synthesis of silica-coated magnetite nanoparticles was carried out as described by Stöber & Fink.51 Briefly, the synthesized magnetite nanoparticles Fe3O4 (Ar) and Fe3O4 (air) described above were used as cores to be functionalized with the NH2-silica. 3-Aminopropyltriethoxysilane (APTES, 98% purity, Sigma-Aldrich) was used as NH2-silica precursor. The obtained NH2-silica functionalized magnetite nanoparticles are further designated as Fe3O4-APTES (Ar) and Fe3O4-APTES (air). As in the case of Fe3O4 (Ar) and Fe3O4 (air) synthesis, the silica coating was performed in argon and air atmosphere, respectively.According to ref. 52, 3.21 g of Fe3O4 NPs were dispersed in 150 mL of 98% ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water solution (1:1; volume ratio). Then, 13.6 g of APTES was added to the solution under argon or air atmosphere, respectively, at 40 °C for 2 h. A molar ratio of 4:1 (APTES:Fe3O4) was used. In the case of argon atmosphere, all manipulations were carried out in a glove box. After cooling the synthesized Fe3O4-APTES (Ar) and Fe3O4-APTES (air) NPs to room temperature, the MNPs were separated using a magnet (Nd, 0.3 Ts). To avoid the presence of silane non-specifically bound to particle surfaces, all samples were washed with water and ethanol prior to analysis or further use. Finally, the Fe3O4-APTES (Ar) and Fe3O4-APTES (air) samples were dried under vacuum at 70 °C for 2 hours.
:water solution (1:1; volume ratio). Then, 13.6 g of APTES was added to the solution under argon or air atmosphere, respectively, at 40 °C for 2 h. A molar ratio of 4:1 (APTES:Fe3O4) was used. In the case of argon atmosphere, all manipulations were carried out in a glove box. After cooling the synthesized Fe3O4-APTES (Ar) and Fe3O4-APTES (air) NPs to room temperature, the MNPs were separated using a magnet (Nd, 0.3 Ts). To avoid the presence of silane non-specifically bound to particle surfaces, all samples were washed with water and ethanol prior to analysis or further use. Finally, the Fe3O4-APTES (Ar) and Fe3O4-APTES (air) samples were dried under vacuum at 70 °C for 2 hours.
2.3 Separation of the MNP suspensions
The obtained NP suspensions were dispersed by ultrasound for 10 min at 30 Hz according to a dispersion protocol based on probe sonication commonly used in several EU projects studying NPs53,54 and were further subjected to sequential fractionation: starting with (1) centrifugation (5 min, 3000 rpm equal to 1660 g). After centrifugation, the supernatant and sediment were separated, and the surface charge, hydrodynamic size and toxicity of Fe3O4 particles in the supernatant were examined. The supernatant was then filtered through a “White ribbon” cellulose filter with a pore size of 8–12 μm and the parameters described above were analysed again (Fig. 1).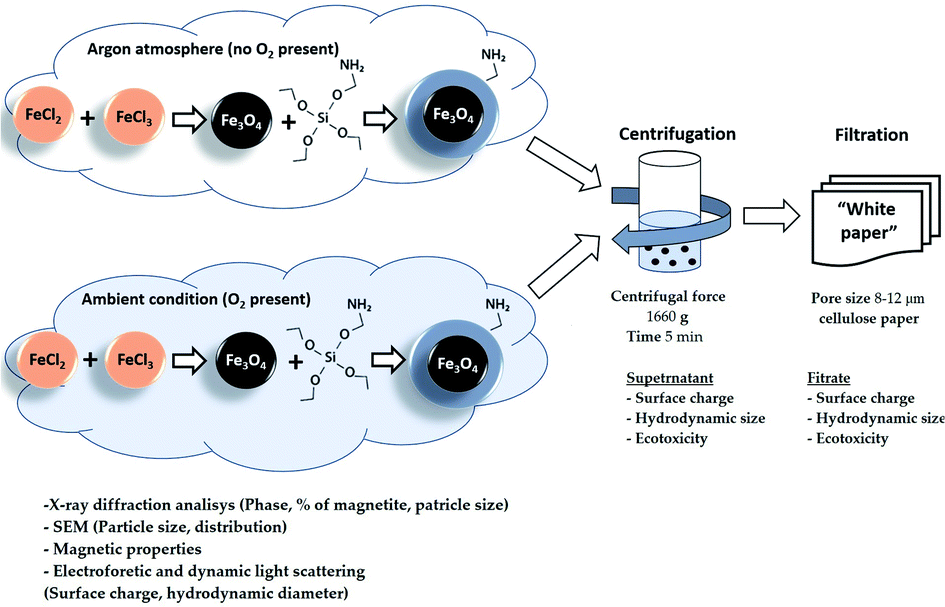 | ||
| Fig. 1 Formulation and separation scheme of MNPs. | ||
2.4 Characterization of the microstructure and colloidal stability of magnetic NPs
The structure of the surface layer of the NPs can have a profound effect on the toxicity of NPs, as this layer is the first layer to come into contact with cells. Therefore, the phase composition and primary particle size of the samples were determined by X-ray diffraction (XRD) analysis in Bragg–Brentano geometry using a Philips X-pert diffractometer (Philips Analytical, Eindhoven, The Netherlands, Cr-Kα radiation, λ = 2.29106 Å) as described in ref. 55. The full width at a half maximum (FWHM) of all reflections was used to determine particle size with the Scherrer equation. To quantify the oxidation progress, the reflection (440) was fitted with five different functions in Origin 2019 Pro.The lattice parameters determined for all samples formulated in this study were smaller than those previously reported for magnetite 8.396–8.400 Å (ICDD-PDF 19-629), but larger than those for maghemite 8.33–8.34 Å (ICDD-PDF 39-1346). This phenomenon can be explained by the partial oxidation of Fe2+ during formulation, which result in the non-stoichiometric formation of Fe3−δO4 where δ can range from zero (stoichiometric magnetite) to 1/3 (completely oxidized).56 The magnetic properties of the dried MNP powders were characterised using the Lake Shore Vibrating sample magnetometer (Lake Shore Cryotronics, Westerville, OH, USA) at 300 K.
The hydrodynamic size and surface charge of the particles were analysed by Dynamic Light Scattering (DLS) on a Brookhaven apparatus with a wave length of 633 nm and a solid-state He–Ne laser at a scattering angle of 173° at 25 °C. For analysis, each sample was diluted in distilled water to 0.1 g L−1. Prior to analysis, the samples were subjected to ultrasound (ultrasonic bath, 100 W, 40 kHz) for 10 s ultrasound followed by 100 s stop. The experiments were carried out in a disposable zeta cell (DTS 1070). Samples were analysed at their original pH (as synthesized, ranging from 5.4 to 7.5). The pH values were measured before and after the study.
2.5 Ecotoxicity test of magnetic NPs
The toxicity of aqueous suspensions of magnetic NPs to ciliates and Sinapis alba plants was tested in the concentration range from 0.1 to 1000 mg L−1.The exact concentrations/dilutions tested depended on the assay and the MNPs and are indicated in the figures and tables. Toxicity values (EC50) of the MNPs are presented as mg compound per L (nominal concentrations). Prior to testing, the prepared NP suspensions in distilled water were ultrasonically dispersed using the sonication bath (100 W, 40 kHz) (Heb Biotechnology, Shaanxi, China) for 10 min and centrifuged and filtered under the same conditions as for zeta potential measurements (see Section 2.3 – Separation of the MNP suspensions).
2.6 Paramecium caudatum acute toxicity test
The ecotoxicity of magnetic NPs (MNPs) to ciliates was determined by Paramecium caudatum Ehrenberg acute toxicity test performed following the protocol described in ref. 57. Briefly, the assay is based on measuring the mortality of Paramecium caudatum when exposed to toxic substances compared to the control. The assay was performed in multiwell polystyrene plates (8 × 12 wells; well size 1 mL; Eppendorf). P. caudatum stock cultures were maintained in Lozin-Lozinskiy mineral nutrient medium of the following composition, mg L−1: NaCl – 100.0, KCl – 10.0, CaCl2·2H2O – 10.0, MgCl2·6H2O – 10.0, NaHCO3 – 20.0 (Sigma-Aldrich Chemie GmbH, Steinheim, Germany). The stock cultures were maintained at room temperature (22 ± 2 °C), pH 7.5–8.0, and without the addition of any organic compounds. To initiate the test culture, approximately 1/3 of the stock culture was transferred into a Petri dish containing fresh nutrient medium and incubated at 22 ± 2 °C in the dark for 24 h.Using a stereoscopic microscope (Model MC-1, Micromed, Shanghai, China), 10–15 ciliates were transferred using a capillary pipette into each of 3–4 test wells containing fresh nutrient medium.
The volume of liquid when transferring the ciliates to the wells did not exceed 0.02 mL. In general, each set of wells (control and test wells) contained at least 30 ciliates. 0.6 mL of incubation medium was added to the control wells and 0.6 mL of the test sample was added to the test wells. The plates with samples and ciliates were incubated at 22 ± 2 °C in the dark. During the exposure period, no food or other supplements were added. After 24 hours of incubation, the viability of individuals in each well were checked using a stereoscopic microscope. Freely moving ciliates were considered viable and immobile individuals were considered dead. Mean values of live or dead organisms were calculated and compared with control values.
2.7 Sinapis alba L. acute toxicity test
The toxicity of MNPs to plants was measured using root growth inhibition assay of white mustard Sinapis alba L. (ISO 18763:2016 (ref. 58)) in Phytotoxkit format.59 Certified, high-quality, commercially available seeds were used for all experiments. Following the Phytotoxkit test format (plate assay), the 10 mL of previously shaken NP suspension in distilled water was poured onto transparent test plates (21 × 15.5 × 0.8 cm) covered by white filter paper and ten Sinapis alba seeds were placed on the paper. The test plates were closed with a transparent lid and incubated first horizontally at 20 ± 2 °C in the dark for 24 h, and then for 72 h vertically at 24 ± 2 °C, with an illumination period of 16 h per day at light intensity of 4000–7000 lx (light wavelength 400–700 nm, universal white). At the end of incubation, the length of the main root of the mustard seedlings was measured. Mean values were calculated and compared with those of the control. The test was performed in three replicates.2.8 Statistical analysis
The inhibitory effect of the tested compounds/dilutions compared to the control was calculated as a percentage. From dose–response curves, EC50 values (mg L−1 or mg Fe per L, depending on the compound) were calculated using the probit method (GraphPad Prism 9), fitted (OriginPro 2019) and expressed as mean value ± standard deviation (SD). ANOVA was used for analysis of statistically significant variances within and between test groups. The degree of statistical significance of the results was calculated in the GraphPad Prism 9 application (statistical significance between groups was fixed at p ≤ 0.05, the Tukey's and Šídák's criteria were used).3. Results
3.1 Microstructure of Fe3O4/APTES NPs
| Fe3O4 + OH− + H2O = γ-Fe2O3 + Fe(OH)3 |
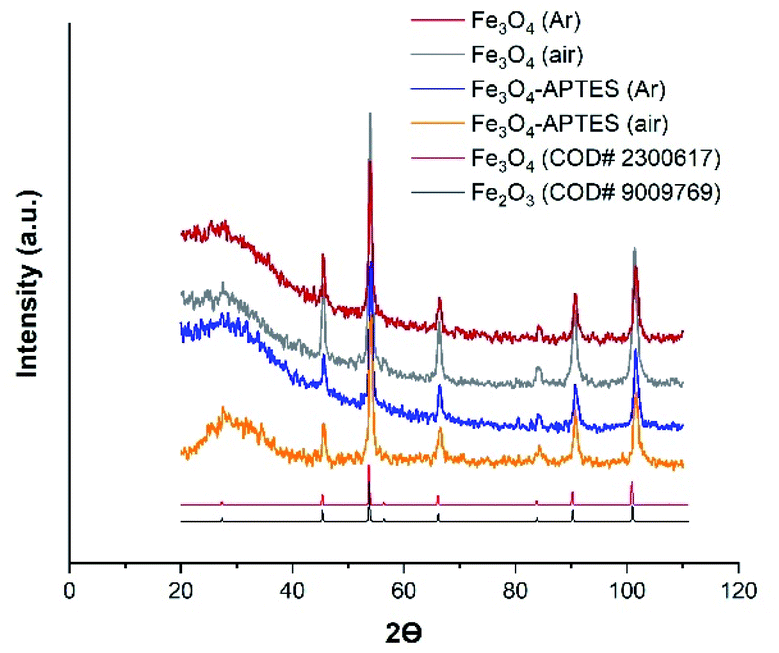 | ||
| Fig. 2 Powder X-ray diffraction (XRD) of MNPs products with the Crystallography Open Database (COD) magnetite and maghemite patterns Fe3O4 (Ar), Fe3O4-APTES (Ar) and Fe3O4-APTES (air) data have been reported in ref. 55. | ||
At the same time, coating the magnetite particles with silica prevented the dissolution and reconstructive transformation of magnetite particles. In fact, there was a small decrease in the proportion of magnetite from 78.8% to 75.8% under inert and from 50.7% to 42.7% under ambient conditions after APTES modification, respectively (Table 1). Alkoxysilanes can give a broad peak in the diffraction patterns, however, in this case, a broad peak can be attributed to the effect of sample preparation.
| Sample | Structure | % Fe3O4 | DXRD, nm | CV, % | DSEM, nm | CV, % |
|---|---|---|---|---|---|---|
| Fe3O4 (Ar) | Fe2.93O4 | 78.8 | 17.1 ± 2.3 | 13.5 | 32.1 ± 4.3 | 13.5 |
| Fe3O4 (air) | Fe2.84O4 | 50.7 | 14.9 ± 2.02 | 13.5 | 30.2 ± 2.3 | 12.1 |
| Fe3O4/APTES (Ar) | Fe2.92O4 | 75.8 | 20.5 ± 3.3 | 16.1 | 24.2 ± 2.8 | 11.6 |
| Fe3O4/APTES (air) | Fe2.81O4 | 42.4 | 16.5 ± 1.96 | 9.5 | 23.3 ± 3.1 | 10.5 |
Finally, the crystalline component composition of the samples can be assigned as follows: Fe2.93O4 and Fe2.92O4, Fe2.84O4 and Fe2.81O4 for Fe3O4 (Ar) and Fe3O4/APTES (Ar), Fe3O4 (air) and Fe3O4/APTES (air) samples, respectively (Table 1).
The size of the coherent-scattering region was derived from the powder XRD data by the Scherrer's method. The full width at half maximum (FWHM) of the reflections was used for particle size determination. Such magnitudes for the magnetic iron oxide nanoparticles are usual for superparamagnetic MNPS with high saturation magnetization and a high specific surface area.62,63 According to the particle diameter and standard deviation, there are no size difference between all MNPs. Although all samples are considered polydisperse according to ref. 64, the CV of 9.5% and SD = 1.96 indicate the homogeneity of the Fe3O4/APTES (air) NPs which is crucial for the ultimate performance of the surface-activated material. Thus, the synthesis atmosphere (argon or ambient) only moderately affected the particle size, but there was a noticeable impact on the lattice parameters of magnetite, causing deeper crystalline defects in it.
The particle size, studied by SEM, has been somewhat reduced. The diameter of bare Fe3O4 under argon condition changes after modification from 32.1 nm to 24.2 nm.55 The same effect was observed for MNPs under ambient conditions: functionalization by APTES leads to size reduction from 30.2 nm to 23.3 nm. There sizes for bare and modified magnetic NPs are typical for superparamagnetic MNPs that correlate with the value of saturation magnetization and other magnetic properties.
One of the most important properties of magnetic nanoparticles, which justifies their widespread use, is superparamagnetism. The magnetic properties of modified and bare MNPs were investigated (Table 2S, ESI†). The shape of the loops indicates the ferromagnetic nature of the material desired for separation application (Fig. 1S, ESI†). Modification by APTES did not produce significant changes in magnetic properties (saturation magnetization changed from 81.2 to 68.7 emu g−1 which correlates with XRD data, Table 1). In this case, saturation magnetization of Fe3O4 (Ar) decreased to 49.9 emu g−1 for Fe3O4 (air) and 30.8 emu g−1 for Fe3O4-APTES (air). However, the saturation magnetization for the MNP samples indicates that silane-stabilized magnetite nanoparticles oxidized in air still exhibit superparamagnetic properties at room temperature, which correlates with their sizes (Table 1).
3.2 Surface charge-bioactivity relationship for MNPs
Fe3O4 (Ar) has +20 mV at the maximum concentration of 1000 mg L−1 and pH 5.4, which correlates with other zeta potentials of MNPs studied65,66 and demonstrates it phase (Fig. 3). The preparation of Fe3O4 at ambient conditions leads to a decrease in the positive charge at native pH and 1000 mg L−1 for Fe3O4 (air) compared to Fe3O4 (Ar) due to the increase in the amount of OH-group on the surface after oxidation.67 The functionalization of APTES lead to an increase in surface charge close to +43 mV (1000 mg L−1, native pH) for both Fe3O4 (Ar) and Fe3O4 (air) due to the presence of positively charged amino groups on the surface.68 Dilution in water leads to charge changes of bare and modified MNPs from positive at 1000 mg L−1 to negative at 50 mg L−1: there is no effect of the subsequent dilution from 50 mg L−1 to 1 mg L−1 for all samples except Fe3O4/APTES (air) according to ANOVA. At 1 and 10 mg L−1 all samples have the same negative charge ∼−15 mV due to complete hydrolysis. The isoelectric point (IEP, charge is equal to zero, the suspension is totally unstable) of Fe3O4 (Ar) and Fe3O4 (air) are close to 500 mg L−1 and for both modified by APTES under argon and ambient conditions – close to 500 and 800 mg L−1 respectively. There data correlate with the hydrodynamic sizes of the MNPs: at IEP concentration the particles have the maximum sizes: 744 nm for Fe3O4 (Ar), 890 nm for Fe3O4 (air), 957 nm for Fe3O4-APTES (Ar) and 737 nm for Fe3O4-APTES (air). The use of suspensions at these IEP concentrations in ecotoxicity experiments can lead to an uneven distribution of nanoparticles in the medium of infusoria and higher plants.
 | ||
| Fig. 3 (a) Zeta-potential and (b) hydrodynamic diameter of aqueous suspensions of MNPs: a dose-effect study (ultrasonication 10 min, dilution in water). | ||
Bare and silica-coated MNPs were tested for toxicity at concentrations up to 1000 mg L−1 (ciliates test) and up to 1000 mg L−1 (plant test) (Fig. 4).
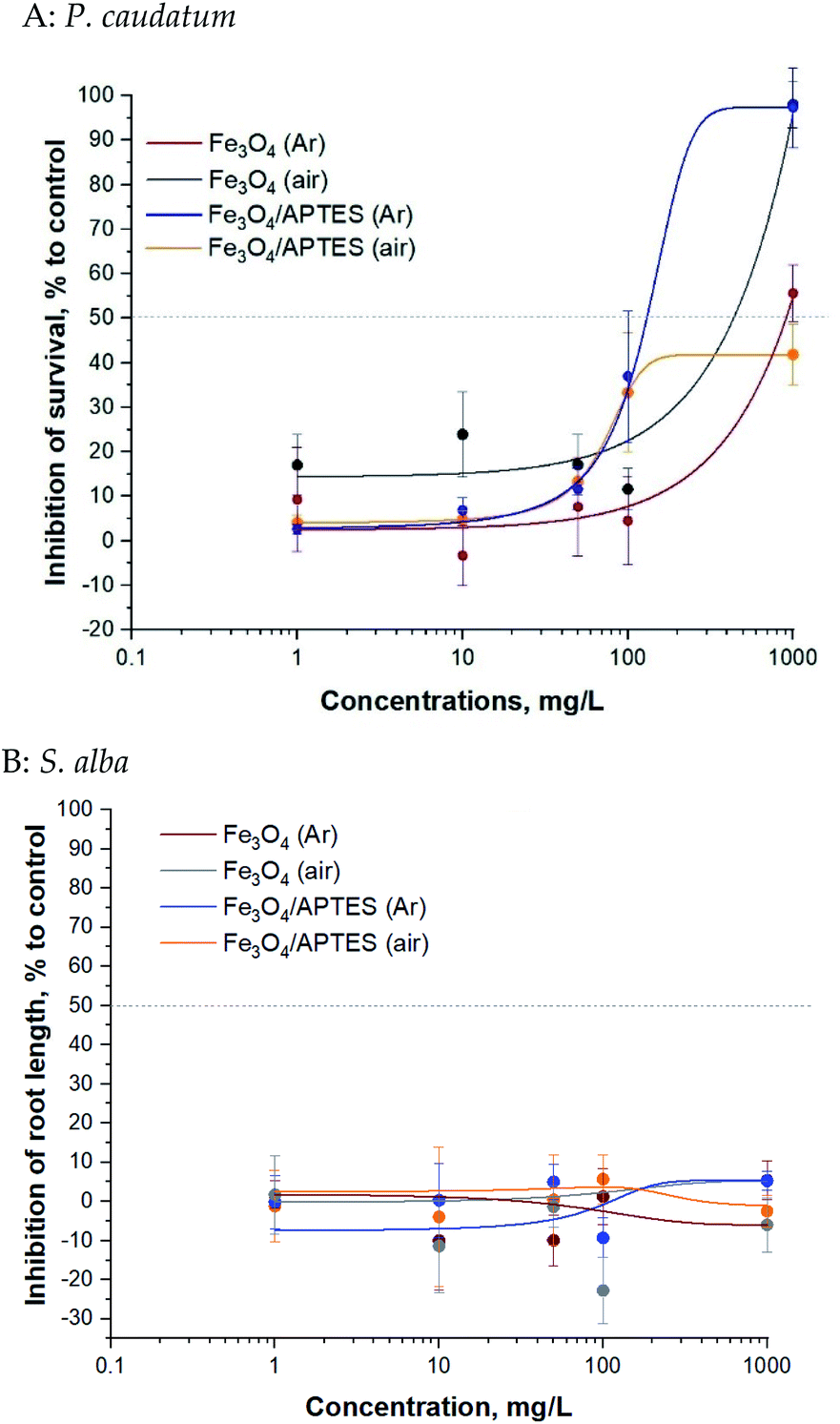 | ||
| Fig. 4 Toxicity of aqueous suspensions of MNPs to P. caudatum ciliates in the 24 h immobilization assay (A) and to higher S. alba plants in the 96 h root growth inhibition assay (B): a dose-effect study. All concentrations are nominal. Mean values ± SD of triplicates. | ||
Magnetic NPs were not toxic to P. caudatum ciliates: the EC50 value was >100 mg L−1 for all samples (Table 2). The EC50 value of Fe3O4/APTES (air) for ciliates was not reached at the highest concentration tested (1000 mg L−1) and could also be considered non harmful. Synthesis under ambient conditions (e.g. oxygen atmosphere) increased to some extent the toxicity of the bare MNPs69 and decreased that of the modified ones for ciliates only for the highest concentration of 1000 mg L−1: there is no difference between the bare MNPs at the rest of the concentration according to ANOVA (Table 2S†).
| Sample | 24 h EC50a for P. caudatum, mg L−1 | 96 h EC50a for S. alba, mg L−1 |
|---|---|---|
a The EC50 is the concentration of a sample that reduces root length or survival of ciliates by 50%.b Highest concentration that was tested. Color code: ≤1 mg L−1 ( ) = very toxic; >1–10 mg L−1 ( ) = very toxic; >1–10 mg L−1 ( ) = toxic; >10–100 mg L−1 ( ) = toxic; >10–100 mg L−1 ( ) = harmful; >100 mg L−1 ( ) = harmful; >100 mg L−1 ( ) = “not classified/not harmful”. EC50 data that do not allow classification are displayed on a white background. ) = “not classified/not harmful”. EC50 data that do not allow classification are displayed on a white background. |
||
| Fe3O4 (Ar) | 910 ( ) ) |
>1000b mg L−1 |
| Fe3O4 (air) | 230 ( ) ) |
|
| Fe3O4/APTES (Ar) | 130 ( ) ) |
|
| Fe3O4/APTES (air) | >1000b mg L−1 | |
Generally, regarding the effects of silica coating of MNPs by APTES for ciliates, functionalization in argon medium leads to an increase in toxicity, although functionalization under ambient conditions leads to a decrease in toxicity. As expected, dilution from 10 mg L−1 to 1 mg L−1 led to a significant decrease in inhibition: no effect at subsequent dilution according to ANOVA for bare MNPs and APTES modified MNPs.
The bare and modified MNPs evaluated for toxic effects in the present study were not toxic to S. alba plants in the root length inhibition test: the EC50 value was not reached at the highest concentration tested (1000 mg L−1) (Table 2). There was non clear dose–effect relationship: neither oxidation nor functionalization of APTES led to an increase in toxicity according to Tukey's multiple comparisons test. Dilution also does not affect toxicity.
By relating the physico-chemical properties of the studied NPs (zeta potential, percentage of magnetite) and the ecotoxicity of MNPs (Table 2, Fig. 2 and 3) it can be concluded that a change in charge leads to a change in the toxicity of the nanoparticles relative to ciliates. The higher the negative zeta potential, the lower the toxicity for both bare and modified nanoparticles. There is no correlation for S. alba plants: higher plants are not sensitive to changes in nanoparticles surface charge.
 | ||
| Fig. 5 View of MNPs suspension samples: 1st column – Fe3O4 (Ar), 2nd column – Fe3O4 (air), 3rd column – Fe3O4-APTES (Ar), 4th column – Fe3O4-APTES (air) before fractionation (initial), after centrifugation and after filtration (initial concentration of MNPs is 1 mg L−1). | ||
The present study shows that centrifugation and filtration lead to a consistent decrease in surface charge for all samples. Thus, during centrifugation, the zeta potential decreases from ±43 mV to 29.6 mV and 38.9 mV for both modified MNPs under inert and ambient conditions, respectively (Fig. 6A). The same tendency of decreasing charge from 20.1 mV to 1.3 mV is observed for Fe3O4 (Ar). Filtration leads to a complete change of the sign of the charged surface from positive to negative by more than 40 mV in the case of modified MNPs and Fe3O4 (Ar). However, for Fe3O4 (air), centrifugation does not change the charge. For this sample after filtration, it was not possible to measure the zeta potential due to the presence of only ions in the solution (no NPs).
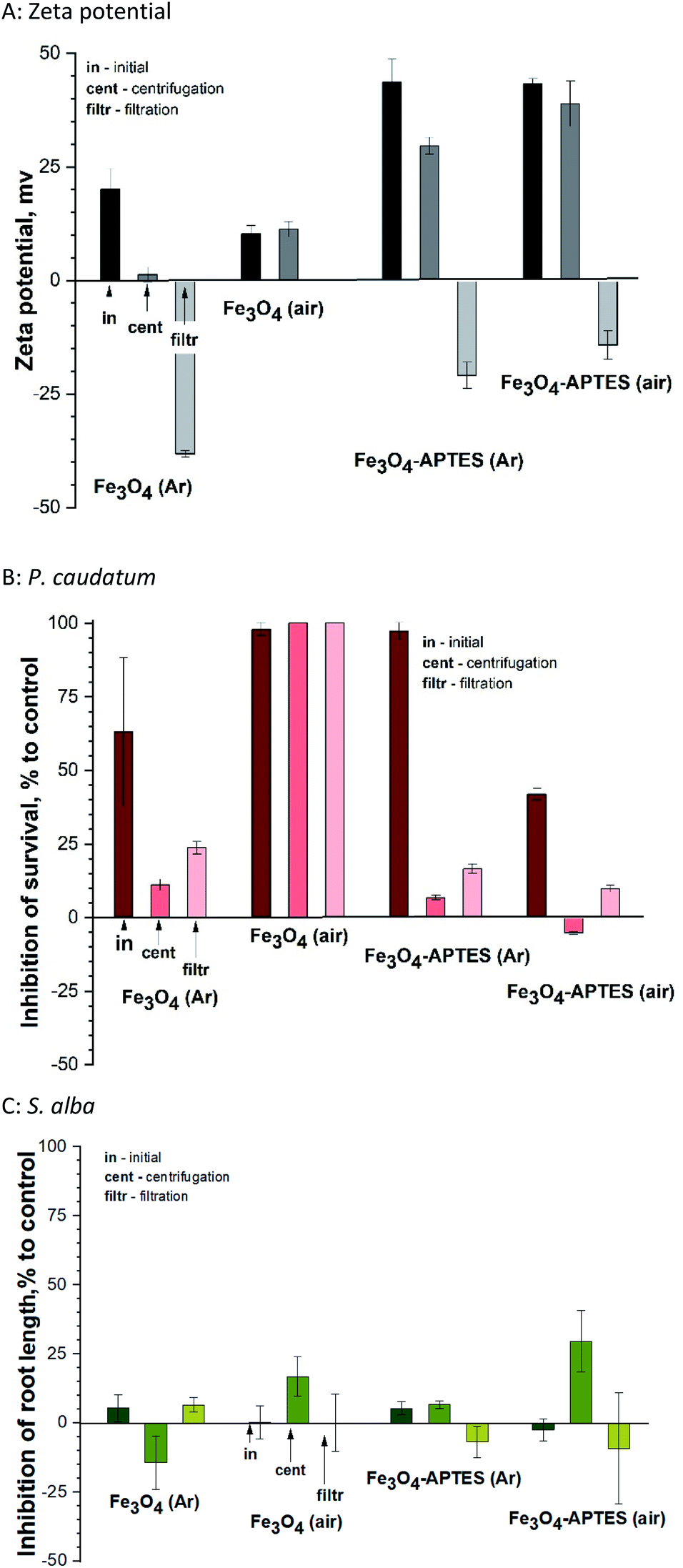 | ||
| Fig. 6 Dose–effect relationships and zeta potentials (A) of aqueous suspensions of MNPs after fractionation in the 24 h immobilization assay of P. caudatum ciliates (B) and higher S. alba plants (C): a dose–effect study. All concentrations are nominal. Mean ± SD values of triplicates. | ||
Fractionation affects the hydrodynamic diameter. After centrifugation, the diameter of nanoparticles decreases due to the removal of large particles from the suspension by centrifugal forces and subsequent decontamination. Filtration leads to an increase in hydrodynamic diameter due to aggregation of the remaining nanoparticles. From the stability point of view, fractionation does not lead to a decrease in stability for APTES-modified samples (surface charge is greater than ±20). At the same time, for Fe3O4 (Ar), centrifugation causes a loss of stability (charge of 1.3 mV). Subsequent filtration leads to in a further decrease in surface charge and an increase in stability. The sample is unstable both in the initial state and after centrifugation.
As for the effects of filtration on ciliates, centrifugation leads to a decrease in inhibition in the case of MNPs prepared and modified in an inert atmosphere (Fig. 6B). This correlates with a change in the zeta potential: the lower the charge modulus, the lower the toxicity, probably due to a decrease in concentration and the removal of large NPs, which have a mechanical effect on ciliates. The toxicity of Fe3O4 (air) and Fe3O4-APTES (air) does not change after centrifugation, which also correlates with the charge of the system. Subsequent filtration only increases the nanoparticles toxicity of all nanoparticles: for example, the inhibition increases from 11 to 23% for Fe3O4-APTES (Ar), which is a significant difference according to ANOVA. This is due to the higher toxicity of iron ions, which probably prevail in the suspension after filtration, compared to NPs, and this correlates well with our previous studies.70
A similar study in relation to plants showed no unique trend (Fig. 6C). Centrifugation leads to increased toxicity for air-derived and air-modified nanoparticles, growth stimulation due to the influence of Fe3O4 (Ar), and unchanged toxicity value for Fe3O4-APTES (Ar).
Filtration decreases in toxicity, except for Fe3O4 (Ar), where toxicity slightly rises. In general, plant toxicity remains within biosafety margins (−20% < inhibition of root length < 20%). There is no correlation with zeta potential due to the lower sensitivity of this test sample.
4. Conclusions
This work show how sample preparation methods – centrifugation and filtration – affect the crystalline properties and surface charge in the process of preparing particlulate suspensions, as well as the toxicity of the resulting suspensions. However, the unique self-assembly silylation processes inevitably lead to increased heterogeneity of silica-based MPNs, thus becoming the main barrier to obtaining homogenous samples. Here we have compared three types of samples that have been subjected to various techniques to separate MNPs to stable colloidal conditions. Standard centrifugation procedures do not guarantee a sufficiently good solution separation of MNP particles and require additional-separation step. Depending on the type of nanoparticles used, the optimal separation protocol to obtain homogenous stable suspensions may combine two or more of the techniques. Once the nanoparticle solutions have been fractionated, each fraction can be quantified. Thus, these techniques not only allow fractionation of samples extract nanoparticles with the desired physical properties, but also enable evaluation of various nanoparticle assembly techniques to determine which technique produces nanoparticles with the desired physical and biological properties.The sample preparation technique affects the surface charge and bioactivity of silica MNPs. Therefore, when comparing the toxicity effects of specific metal-containing systems, it is important to take into account the sample preparation procedure is used, as it greatly affects both the physicochemical properties of the nanoparticles and the toxicity responses. In other words, the toxicity response depends on the method of sample preparation.
The production of magnetite nanoparticles under ambient conditions leads to a change in the phase composition and oxidation of Fe3O4, while the APTES modification insignificantly oxidizes magnetite phase. Oxidation of MNPs leads to a recharge of the surface from positive to negative according to zeta-potential data. At the same time, the toxicity in relation to ciliates decreases. Regardless of the MNPs type, all MNPs suspensions demonstrate no toxicity to plants.
By exploring fractionation procedures, we have demonstrated that it is possible to obtain a homogenous preparation of NPs with modifiable physical and toxic properties. This work will help to achieve more consistent results in biological experiments and may drive the development of commercial applications of NPs in a number of fields.
Author contributions
Research, software, Lyubov Bondarenko; research, methodology, original draft-writing, resources, Vera Terekhova; methodology, writing—revising and editing, Anne Kahru; research, resources, supervision, Gulzhian Dzhardimalieva; research, Elena Kelbysheva; research, Natalya Tropskaya; conceptualization, original draft-writing, writing—revising and editing, supervision, Kamila Kydralieva. All authors have read and accepted the published version of the manuscript. The authors thank Pavel Uchanov and Tatiana Marochko for their help with biotesting and Andrei Tchourakov for improving English.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors are grateful to the Russian Foundation for Fundamental Research for financial support (No. 20-54-26012). Gulzhian Dzhardimalieva performed this study in accordance with the state tasks, state registration AAAA-A19-119032690060-9. Vera Terekhova performed the research of nanotoxicity according to the Development program of the Interdisciplinary Scientific and Educational School of M. V. Lomonosov Moscow State University The future of the planet and global environmental change.References
- A. G. Leonel, A. A. P. Mansur and H. S. Mansur, Water Res., 2020, 190, 116693 CrossRef PubMed
.
- A. V. Samrot, C. S. Sahithya, A. J. Selvarani, S. K. Purayil and P. Ponnaiah, Curr. Res. Green Sustain. Chem., 2020, 191, 100042 Search PubMed
- R. T. Reza, C. M. Pérez, M. Martínez and P. García-Casillas, NSTI-Nanotech 2010, 2010, vol. 1, pp. 534–538 Search PubMed
- S. Margel, T. Lublin-Tennenbaum, S. Gura, M. Tsubery, U. Akiva, N. Shpaisman and O. Ziv, Magnetic Cell Separation, 2007, vol. 32, pp. 119–1622 Search PubMed
- G. I. Dzhardimalieva, V. I. Irzhak, S. Y. Bratskaya, V. Y. Mayorov, Y. O. Privar, E. D. Kasymova, L. S. Kulyabko, S. Zhorobekova and K. A. Kydralieva, Colloid J., 2020, 82, 1–7 CrossRef CAS
- K. A. Kydralieva, A. A. Yurishcheva and G. I. Dzhardimalieva, et al., J. Inorg. Organomet. Polym. Mater., 2016, 26, 1212 CrossRef CAS
- G. Xianghai, Q. Wang and T. Chen, Authorea, 2020, DOI:10.22541/au.159884105.56132261.
- M. G. Santos, D. T. de Carvalho, L. B. Caminitti, B. B. A. de Lima, M. H. d. S. Cavalcanti, D. F. R. dos Santos, L. S. Virtuoso, D. B. Hirata and E. C. Figueiredo, Food Chem., 2021, 353(2), 129442 CrossRef CAS PubMed
- F. Piccinno, F. Gottschalk, S. Seeger and B. Nowack, J. Nanopart. Res., 2012, 14(9), 1–11 CrossRef
- G. Kandasamy and D. Maity, Int. J. Pharm., 2015, 496(2), 191–218 CrossRef CAS PubMed
- A. Samanta and B. J. Ravoo, Angew. Chem., Int. Ed., 2014, 53, 12946–12950 CrossRef CAS PubMed
- L. Xu, M. J. Kim, K. D. Kim, Y. H. Choa and H. T. Kim, Colloids Surf., A, 2009, 350, 8–12 CrossRef CAS
- S. Liu, H. Chen, X. Lu, C. Deng, X. Zhang and P. v. Yang, Angew. Chem., Int. Ed., 2010, 49(41), 7557–7561 CrossRef CAS PubMed
- Y.-C. Lee and I.-J. Kang, Bull. Korean Chem. Soc., 2017, 38(3), 313–319 CrossRef CAS
- Z. Q. Guo, Y. Li, S. H. Pan and J. Z. Xu, J. Mol. Liq., 2015, 206, 272–277 CrossRef CAS
- L. Shen, B. Li and Y. Qiao, Materials, 2018, 11(2), 324 CrossRef PubMed
- T. Zhang, H. Chai, F. Y. Meng, Z. Z. Guo, Y. Jiang and P. Miao, ACS Appl. Mater. Interfaces, 2018, 10(43), 36796–36804 CrossRef CAS PubMed
- R. Van Roosbroeck, W. Van Roy, T. Stakenborg, J. Trekker, A. D'Hollander and T. Dresselaers, ACS Nano, 2014, 8(3), 2269–2278 CrossRef CAS PubMed
- P. Z. Li, P. Chevallier, P. Ramrup, D. Biswas, D. Vuckovich and M. A. Fortin, et al., Chem. Mater., 2015, 27(20), 7100–7109 CrossRef CAS
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10(2), 2436–2446 CrossRef CAS PubMed
- D. V. Voronin, O. A. Sindeeva, M. A. Kurochkin, O. Mayorova and I. V. Fedosov, ACS Appl. Mater. Interfaces, 2017, 9(8), 6885–6893 CrossRef CAS PubMed
- J. W. Shen, K. Y. Li, L. Cheng, Z. Liu, S. T. Lee and J. Liu, ACS Appl. Mater. Interfaces, 2014, 6(9), 6443–6452 CrossRef CAS PubMed
- L. Wang, S. J. Chen, Y. Zhu, M. X. Zhang, S. X. Tang and J. Y. Li, et al., ACS Appl. Mater. Interfaces, 2018, 10(49), 42102–42114 CrossRef CAS PubMed
- M. Hogan, Y. T. Chen, A. G. Kolhatkar, C. J. Candelari, S. Madala and T. R. Lee, et al., ACS Biomater. Sci. Eng., 2016, 2(9), 1619–1629 CrossRef CAS PubMed
- S. P. Schwaminger, D. Bauer, P. Fraga-García, F. E. Wagner and S. Berensmeier, CrystEngComm, 2016, 19, 246–255 RSC
- L. Bondarenko, A. Kahru, V. Terekhova, G. Dzhardimalieva, P. Uchanov and K. Kydralieva, Nanomaterials, 2020, 10(10), 1–18 CrossRef PubMed
- OECD, Guidance on sample preparation and dosimetry for the safety testing of manufactured nanomaterials. Series on the Safety of Manufactured Nanomaterials, No. 36, 2012 Search PubMed
- T. Xia, R. F. Hamilton, J. C. Bonner, E. D. Crandall, A. Elder, F. Fazlollahi, T. A. Girtsman, K. Kim, S. Mitra and S. A. Ntim, Environ. Health Perspect., 2013, 121, 683–690 CrossRef PubMed
- J. Bonner, R. M. Silva, A. J. Taylor, J. M. Brown, S. C. Hilderbrand, V. Castranova, D. Porter, A. Elder, G. Oberdorster and J. R. Harkema, Environ. Health Perspect., 2013, 121, 676–682 CrossRef PubMed
- J. M. Cohen, J. G. Teeguarden and P. Demokritou, Part. Fibre Toxicol., 2014, 11, 20–32 CrossRef PubMed
- OECD, Advancing Adverse Outcome Pathway (AOP) Development for Nanomaterial Risk Assessment and Categorisation Part 1: Final Project Report and Recommendations with Methodology to Prioritise Key Events (KEs) Relevant for Manufactured Nanomaterials Series on the Safety of Manufactured Nanomaterials, 2020, vol. 93, p. 19 Search PubMed
- X. Sun, et al., Angew. Chem., Int. Ed., 2009, 48, 939–942 CrossRef CAS PubMed
- G. S. Duesberg, M. Burghard, J. Muster, G. Philipp and S. Roth, Chem. Commun., 1998, 3, 435–436 RSC
- C. Fornaguera and C. Solans, Int. J. Polym. Sci., 2018, 1–10 Search PubMed
- J. Estelrich, M. Quesada-Pérez, J. Forcada and J. Callejas-Fernández, Soft Nanoparticles for Biomedical Applications, 2014, 1, 1–18 Search PubMed
- J. M. Cohen, G. M. DeLoid and P. Demokritou, Nanomedicine, 2015, 10, 3015–3032 CrossRef CAS PubMed
- N. B. Hartmann, K. A. Jensen, A. Baun, K. Rasmussen, H. Rauscher, R. Tantra, D. Cupi, D. Gilliland, F. Pianella and J. M. Riego Sintes, J. Toxicol. Environ. Health, Part B, 2015, 18, 1–28 Search PubMed
- C. Lei, L. Zhang, K. Yang, L. Zhu and D. Lin, Environ. Pollut., 2016, 218, 505–512 CrossRef CAS PubMed
- H. Dong, F. Zhao, G. Zeng, L. Tang, C. Fan, L. Zhang, Y. Zeng, Q. He, Y. Xie and Y. Wu, J. Hazard. Mater., 2016, 312, 234–242 CrossRef CAS PubMed
- A. Kirje Käkinen, A. Kahru, H. Nurmsoo, A. L. Kubo and O. Bondarenko, Toxicol. In Vitro, 2016, 36, 172–179 CrossRef PubMed
- H. L. Karlsson, P. Cronholm, Y. Hedberg, M. Tornberg, L. De Battice, S. Svedhem and I. Odnevall Wallinder, Toxicology, 2013, 313, 59–69 CrossRef CAS PubMed
- L. Skjolding, S. Sørensen, N. Hartmann, R. Hjorth, S. Hansen and A. Baun, Angew. Chem., 2016, 128(55), 15224–15239 CrossRef PubMed
- M. Diao and M. Yao, Water Res., 2009, 43(20), 5243–5251 CrossRef CAS PubMed
- H. Zamani, A. Moradshahi, H. D. Jahromi and M. H. Sheikhi, Aquat. Toxicol., 2014, 154, 176–183 CrossRef CAS PubMed
- X. He, C. Xie, Y. Ma, L. Wang, X. He, W. Shi, X. Liu, Y. Liu and Z. Zhang, Aquat. Toxicol., 2019, 209, 113–120 CrossRef CAS PubMed
- S. Roy, V. Jain, R. Bajpai, P. Ghosh, A. S. Pente, B. P. Singh and D. S. Misra, J. Phys. Chem. C, 2012, 116(35), 19025–19031 CrossRef CAS
- C. Vauthier, B. Cabane and D. Labarre, Eur. J. Pharm. Biopharm., 2008, 69, 466–475 CrossRef CAS PubMed
- R. Scopes, Protein Purification: Principles and Practice, Springer, New York, NY, USA, 1994 Search PubMed
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83(12), 4453–4488 CrossRef CAS PubMed
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41, 8484–8490 CrossRef CAS PubMed
- W. Stöber and A. Fink, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef
- M. Ozmen, K. Can, G. Arslan, A. Tor, Y. Cengeloglu and M. Ersoz, Desalination, 2010, 254(1–3), 162–169 CrossRef CAS
- K. Alstrup Jensen, Y. Kembouche, E. Christiansen, N. Jacobsen, H. Wallin, C. Guiot, O. Spalla and O. Witschger, Nanogenotox, 2011, pp. 1–33 Search PubMed
- J. S. Taurozzi, V. Hackley and M. Wiesner, NIST Spec. Publ., 2012, 2, 1200–1212 Search PubMed
- L. Bondarenko, E. Illés, E. Tombácz, G. Dzhardimalieva, N. Golubeva, O. Tushavina, Y. Adachi and K. Kydralieva, Nanomaterials, 2021, 11(6), 1418 CrossRef CAS PubMed
- C. A. Gorski and M. M. Scherer, Am. Mineral., 2010, 95, 1017–1026 CrossRef CAS
- A. A. Rakhleeva and V. A. Terekhova, FR. 1.39.2006.02506, 2006.
- ISO 18763:2016, Soil quality—Determination of the toxic effects of pollutants on germination and early growth of higher plants, 2016 Search PubMed.
- O. V. Lisovitskaya and V. A. Terekhova, Eurasian Soil Sci., 2017, 50(9), 1105–1114 CrossRef
- W. Kim, C.-Y. Suh, S.-W. Cho, K.-M. Roh, H. Kwon, K. Song and I.-J. Shon, Talanta, 2012, 94, 348–352 CrossRef CAS PubMed
- Y. T. He and S. J. Traina, Clay Miner., 2007, 42, 13–19 CrossRef CAS
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14(8), 15977–16009 CrossRef PubMed
- H.-C. Roth, S. P. Schwaminger, M. Schindler, F. E. Wagner and S. Berensmeier, J. Magn. Magn. Mater., 2015, 377, 81–89 CrossRef CAS
- F. L. Romano, G. M. B. Ambrosano, M. B. Magnani and D. F. Nouer, J. Appl. Oral Sci., 2005, 13(3), 243–246 CrossRef PubMed
- E. Illés and E. Tombácz, Colloids Surf., A, 2003, 230, 99–109 CrossRef
- J. Wang, S. Zheng and Y. Shao, et al., J. Colloid Interface Sci., 2010, 349(1), 293–299 CrossRef CAS PubMed
- E. Tombácz, E. Illés, A. Majzik and A. Hajdú, et al., Croat. Chem. Acta, 2007, 80(3–4), 503–515 Search PubMed
- R. A. Bini, R. F. C. Marques and F. J. Santos, et al., J. Magn. Magn. Mater., 2012, 324(4), 534–539 CrossRef CAS
- S. P. Schwaminger, C. Syhr and S. Berensmeier, Crystals, 2020, 10(3), 214–226 CrossRef CAS
- S. Pradhan, J. Hedberg and E. Blomberg, et al., J. Nanopart. Res., 2016, 18, 285–294 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05703k |
| This journal is © The Royal Society of Chemistry 2021 |