DOI:
10.1039/D1RA05892D
(Paper)
RSC Adv., 2021,
11, 33149-33159
MoS2-assisted Fe2+/peroxymonosulfate oxidation for the abatement of phenacetin: efficiency, mechanisms and toxicity evaluation†
Received
3rd August 2021
, Accepted 4th October 2021
First published on 8th October 2021
Abstract
In this study, molybdenum disulfide (MoS2) was chosen as a co-catalyst to enhance the removal efficiency of phenacetin (PNT) in water by a ferrous ion-activated peroxymonosulfate (Fe2+/PMS) process. Operating parameters, such as the initial solution pH and chemical dose on PNT degradation efficiency were investigated and optimized. Under an initial pH of 3, an Fe2+ dose of 25 μM, a PMS dose of 125 μM and a MoS2 dose of 0.1 g L−1, the degradation efficiency of PNT reached 94.3%, within 15 min. The presence of common water constituents including Cl−, HCO3−, SO42− and natural organic matter (NOM) will inhibit degradation of PNT in the MoS2/Fe2+/PMS system. Radical quenching tests combined with electron paramagnetic resonance (EPR) results indicated that in addition to free radical species (˙OH, SO4˙− and O2˙−), nonradical reactive species (1O2) were also crucial for PNT degradation. The variations in the composition and crystalline structure of the MoS2 before and after the reaction were characterized by XPS and XRD. Further, the degradation pathways of PNT were proposed according to the combined results of LC/TOF/MS and DFT calculations, and primarily included hydroxylation of the aromatic ring, cleavage of the C–N bond of the acetyl-amino group, and cleavage of the C–O bond of the ethoxy group. Finally, toxicity assessment of PNT and its products was predicted using the ECOSAR program.
1. Introduction
The use of pharmaceutical and personal care products (PPCPs), such as antibiotics, care products, and medical drugs, has increased gradually in recent decades. However, because the traditional water treatment process cannot effectively remove these organic contaminants, the residues of PPCPs have been frequently detected in natural water and may pose a potential threat to our ecosystem and human health.1 The sources of PPCPs in natural water mainly include hospitals, pharmaceutical industries, domestic wastewater, agricultural activities, etc.2 As a typical PPCP with anti-inflammatory and analgesic effects, phenacetin (PNT), also known as N-(4-ethoxyphenyl) acetamide, has been widely detected in natural water. Due to its high solubility, multiple pKa values and resistance to microbial degradation in traditional wastewater treatment, PNT was detected at μg L−1 concentrations in wastewater treatment plants (WWTPs) and even in surface waters with concentrations at the ng L−1 level.3,4 Previous studies indicated that long-term exposure to PNT may result in side effects in aquatic organisms and on human health, such as anemia, cyanosis, hypoxia, kidney damage, and even cancer.5 Thus, it is essential to develop a reasonable and effective way to remove PNT from water.
Currently, advanced oxidation processes, abbreviated AOPs, have attracted increasing attention due to their successful application to decompose PPCPs in water by generating highly reactive radical species, i.e., hydroxyl radical (˙OH).6 More recently, sulfate radical (SO4˙−)-based AOPs have gained popularity for the treatment of PPCPs in water owing to their comparable standard redox potential (E0 = 2.5–3.1 V) with that of ˙OH (E0 = 2.8 V).7,8 In addition, SO4˙− was more selective than ˙OH, making SO4˙−-based AOPs less impacted by the presence of common inorganic anions and natural organic matter (NOM).9 Persulfate (PS) and peroxymonosulfate (PMS) are the two common sources for generating SO4˙−. PS and PMS, which have redox potentials of 2.01 V and 1.82 V, respectively, are powerful oxidizers.10 Nonetheless, PS or PMS cannot directly oxidize organic contaminants efficiently owing to their low reaction rates with target compounds at ambient temperatures; therefore, proper activation methods, such as thermal activation, UV irradiation, and transition metal activation, are needed for PS and PMS.10
Ferrous ion (Fe2+)-activated peroxymonosulfate (Fe2+/PMS) is one of the most regularly used SO4˙−-based AOPs attributable to its simple operation, low cost, and nontoxicity. In the Fe2+/PMS oxidation process, the degradation of organic contaminants always takes place in two stages: a fast stage during the initial period of the reaction and then a slow stage during the following reaction. During the initial stage, Fe2+ is oxidized to Fe3+ extremely readily, whereas the poor recovery of Fe2+ during the reaction leads to the lack of Fe2+ available for PMS activation.11 Thus, the usage efficiency of Fe2+ in the Fe2+/PMS system is not sufficient.12 To facilitate the transformation of Fe3+ to Fe2+, many methods have been extensively studied in recent years. For example, to maintain a proper amount of Fe2+ in the Fe2+/PMS system, zero-valent iron (ZVI) was always selected as an alternative to Fe2+ because it could generate Fe2+ more slowly by corrosion and then reduce Fe3+ to Fe2+ on the ZVI surface.13 In addition, chelating agents such as ethylenediamine tetraacetic acid (EDTA),14 ethylenediamine-N,N′-disuccinic acid (EDDS),15 and nitrilotriacetic acid (NTA)16,17 can complex with Fe2+ and slow its release, thus slowly catalyzing the reaction and improving the usage efficiency of Fe2+. Recently, as a vital member of transition metal dichalcogenides, MoS2 has received great attention because of its excellent performance in environmental remediation.18 Notably, MoS2 has been proven to promote the conversion of Fe3+ to Fe2+ and directly activate PMS to produce a certain amount of SO4˙−, enhancing the degradation efficiency of contaminants.19 Simultaneously, previous studies have also proved that MoS2, as a co-catalyst, can be recycled and reused.20–22 Therefore, the MoS2-assisted Fe2+/PMS oxidation process is a promising method to apply to PPCP-polluted water.
The purpose of this study was to investigate the performance and mechanism of the degradation of PNT by the MoS2/Fe2+/PMS system. Various parameters, such as Fe2+ dose, MoS2 dose and PMS dose, were investigated to optimize the process. The effects of initial solution pH, common anions (Cl− and HCO3−), and NOM on the PNT degradation performance were also evaluated. The reactive radical species responsible for PNT degradation were identified and analyzed. The chemical state of MoS2 before and after the reaction was measured by using XRD and XPS. Further, the degradation pathways of PNT were proposed based on LC/TOF/MS technique and density functional theory (DFT) calculation. Finally, the toxicity of PNT and its degradation products were assessed by Ecological Structure–Activity Relationships (ECOSAR) program.
2. Materials and methods
2.1. Chemicals and reagents
Phenacetin (PNT, ≥98.0%), p-benzoquinone (p-BQ, ≥98.0%) and humic acid (HA) were purchased from Sigma-Aldrich (USA). Molybdenum sulfide (MoS2, 99.5%, <2 μm), PMS (KHSO5·0.5KHSO4·0.5K2SO4), 5,5-dimethyl-1-pyrroline (DMPO, >99.0%) and 2,2,6,6-tetramethylpiperidine were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). FeSO4·7H2O (≥99.0%), sulfuric acid (H2SO4, ≥95.0%), sodium hydroxide (NaOH, ≥96.0%), sodium chloride (NaCl, ≥99.5%), sodium bicarbonate (NaHCO3, ≥99.5%), tert-butanol (TBA, ≥98.0%), and ethanol (EtOH, ≥99.5%) were provided by Sinopharm Chemical Reagent Co., Ltd. (Shang, China). All solutions were freshly prepared using Milli-Q purified water.
2.2. Experimental procedures
Batch experiments were conducted in an amber wide-mouth bottle containing 250 mL of reaction solution with magnetic stirring at 300 rpm at room temperature (20 ± 1 °C). In a typical run, a certain amount of MoS2 was added to the mixed solution of PNT and FeSO4, followed by 30 s of ultrasonic treatment to form a dispersion, which increased the contact area between the MoS2 and the reaction solution. The reaction was initiated by adding a certain amount of PMS into the solution. The initial solution pH was adjusted by adding 0.1 M H2SO4 or NaOH. At different time intervals, 1 mL samples were taken and immediately mixed with ethanol to terminate the reaction, and then the sample was filtered through a 0.22 μm membrane before further analysis. All the tests were performed at least in triplicate, and the results were averaged.
2.3. Analytical method
The concentration of PNT was measured by using HPLC (Waters e2695, USA) equipped with a UV detector set at 240 nm and a C18 column (250 mm × 4.6 mm, 5 μm, Agela Technologies) maintained at 35 °C. The mobile phase consisted of acetonitrile and water (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60, v/v) at a flow rate of 0.8 mL min−1, and the injection volume was 20 μL.
:60, v/v) at a flow rate of 0.8 mL min−1, and the injection volume was 20 μL.
Electron paramagnetic resonance (EPR) measurements were obtained on an EPR spectrometer (Bruker A300, Germany). X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha, and X-ray powder diffraction (XRD) patterns were determined using an X-ray diffractometer (Rigaku Ultima IV).
The PNT degradation products were identified using LC/TOF/MS (Agilent 1290 UPLC/6550 Q-TOF) in positive ionization mode using an electrospray ionization (ESI) source. Separation was accomplished using a Waters BEH C18 column (2.1 × 100 mm, 1.7 μm) with gradient elution, and the flow rate was set at 0.3 mL min−1. The mobile phase consisted of ultrapure water (A) and acetonitrile (B), and the gradient mode was programmed as follows: (1) 90% of A was maintained in the first 1 min; (2) from 1 to 9 min, A was linearly decreased from 90% to 10%, then kept for 3 min; (3) from 12 to 12.1 min, A and B were returned to the initial status, and then maintained for 0.9 min. The spray voltage, sheath gas temperature, and sheath gas flow were set at 4000 V, 350 °C, and 12 L min−1, respectively.
2.4. DFT calculation and toxicity assessment
Density functional theory (DFT) calculations (Gaussian 09) were used to predict the degradation products and transformation pathways of PNT using the B3LYP/6-311G basis set. The acute and chronic toxicity of PNT and its degradation products to fish, daphnia, and green algae were predicted by using ECOSAR program.
3. Results and discussion
3.1. Acceleration of PNT degradation by Fe2+/PMS by MoS2
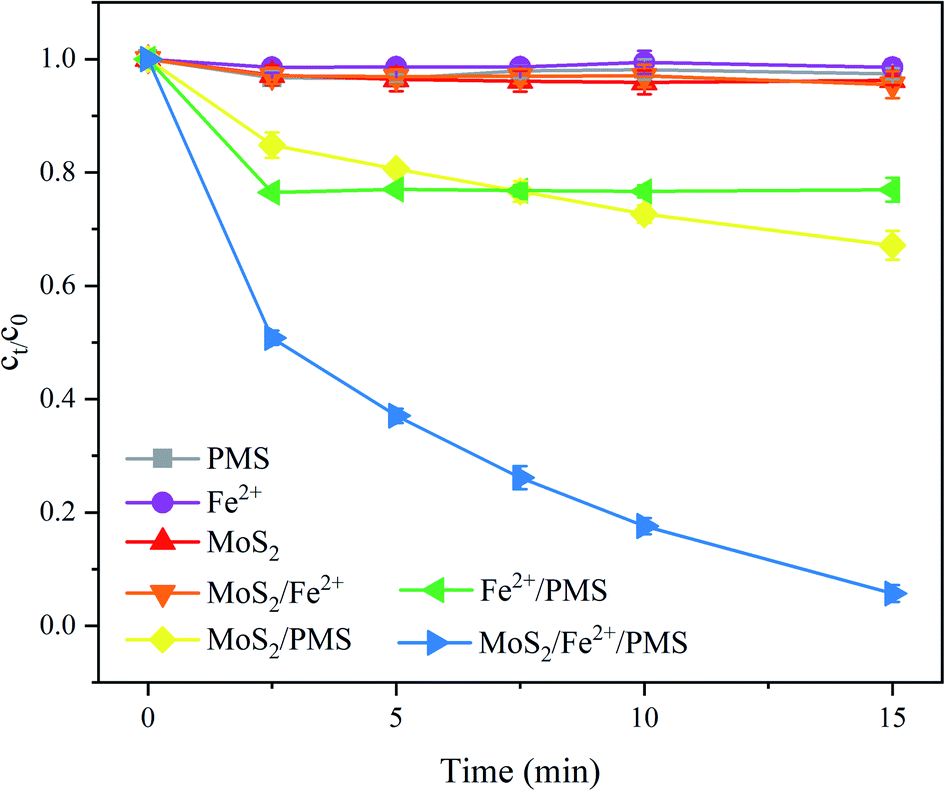
Fig. 1 compares the degradation of PNT in different systems, i.e., PMS alone, Fe2+ alone, MoS2 alone, MoS2/Fe2+, MoS2/PMS, Fe2+/PMS, and MoS2/Fe2+/PMS processes. After 15 min, the degradation of PNT by PMS or Fe2+ alone was negligible. The application of MoS2 and MoS2/Fe2+ also resulted in PNT removal within 5%, which indicated that PNT is resistant to the absorption of MoS2. Furthermore, the MoS2/PMS and Fe2+/PMS systems achieved PNT removal of 32.9% and 23.1%, respectively, showing that both Fe2+ and MoS2 could effectively activate PMS to produce SO4˙− and/or ˙OH, which account for PNT degradation (eqn (1)–(3)).12,19 During the Fe2+/PMS process, the PNT degradation slows suddenly after a period of reaction due to the fast consumption of Fe2+ and poor reduction of Fe3+ to Fe2+.11 Notably, the obvious enhancement of PNT degradation was observed by adding MoS2 to the Fe2+/PMS system, which resulted in a removal rate of 94.3% within 15 min, demonstrating the promoting effect of MoS2 in the Fe2+/PMS system. Additional experiments were also performed to estimate the role of MoS2 in the Fe3+/PMS system (Fig. S1†), and it was found that approximately 72.0% removal of PNT was also achieved in the MoS2/Fe3+/PMS system, which demonstrated the transformation of Fe3+ to Fe2+ in the presence of MoS2 (eqn (4)) since Fe3+ cannot serve as an efficient activator of PMS. A similar phenomenon was also reported for 2,4,6-trichlorophenol (TCP) and sulfamethoxazole (SMX) degradation in the Fe2+/PMS system by adding MoS2.20,22 It is generally believed that the unsaturated S atoms on the MoS2 surface can capture protons to form H2S and meanwhile expose Mo4+, which could accelerate the Fe3+/Fe2+ conversion (eqn (4)) and facilitate PMS conversion (eqn (3)), thus significantly improving the decomposition of PMS.23| | |
Fe2+ + HSO5− → SO4˙− + Fe3+ + ΟΗ−
| (1) |
| | |
Fe2+ + HSO5− →·OH + Fe3+ + SO42−
| (2) |
| | |
Interface: Mo4+ + HSO5− → SO4˙− + Mo6+ + OH−
| (3) |
| | |
Interface: Mo4+ + 2Fe3+ → Mo6+ + 2Fe2+
| (4) |
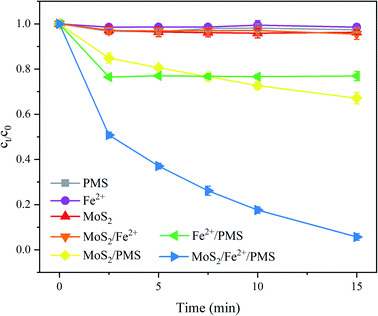 |
| | Fig. 1 Degradation of PNT in different systems. Experimental conditions: [PNT]0 = 25 μM, pH0 = 3.0, [Fe2+]0 = 25 μM, [PMS]0 = 125 μM, [MoS2]0 = 0.1 g L−1. | |
3.2. Optimal conditions
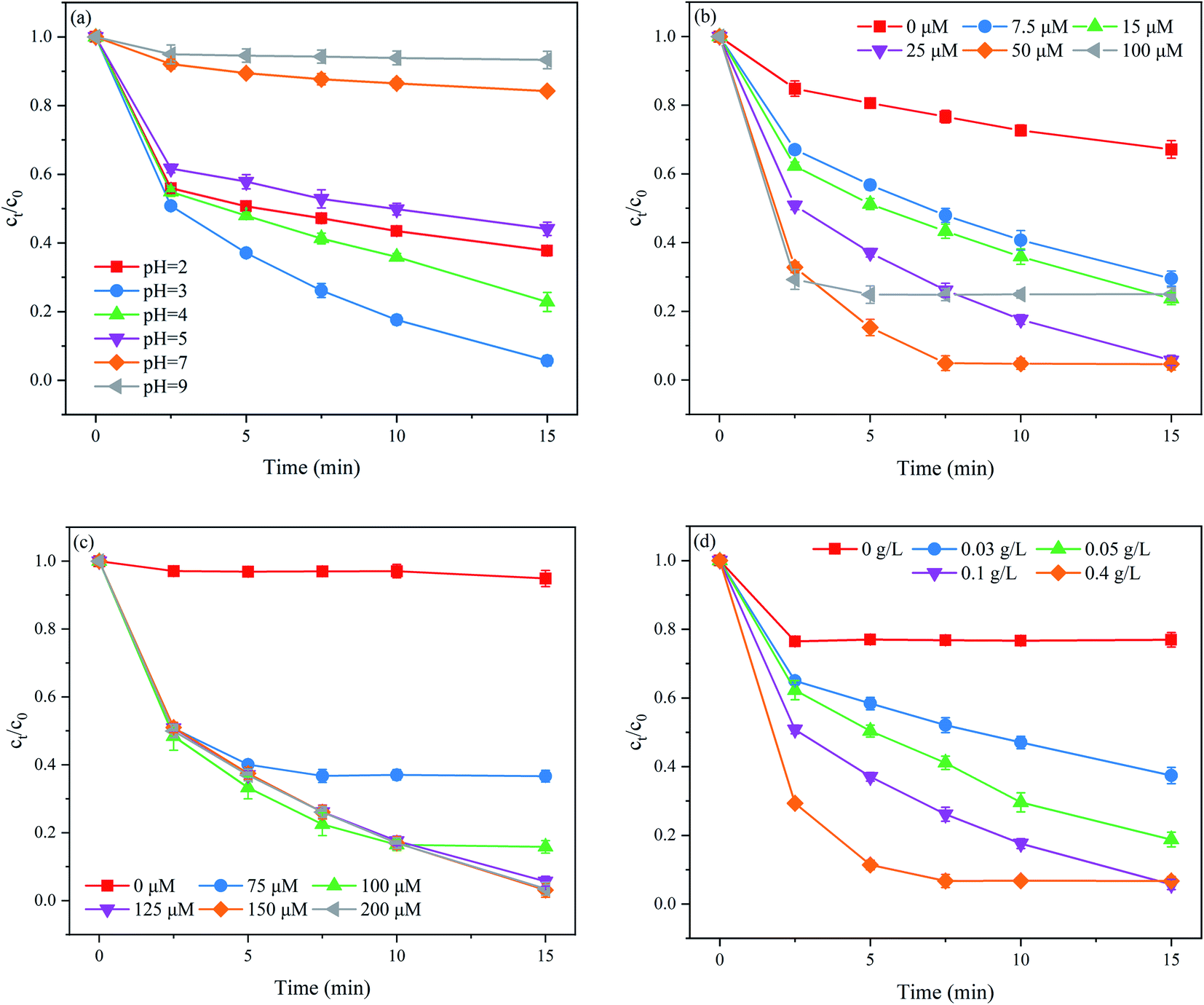
The solution pH has a complex role in the degradation of organic contaminants in SO4˙−-based AOPs, as it may affect the formation of reactive oxidizing species and the stability of PMS.24 Thus, it is necessary to evaluate the effect of the initial solution pH on the PNT degradation performance of the MoS2/Fe2+/PMS system. The effect of the initial solution pH on PNT degradation is illustrated in Fig. 2(a). As clearly seen, acidic conditions are more beneficial to PNT degradation than neutral or alkaline conditions. However, the inhibition of PNT degradation was observed at an extremely acidic pH (pH = 2). When the initial solution pH was decreased to 2, the generated radicals, such as SO4˙− and ˙OH, were consumed by excess hydrogen ions according to eqn (5) and (6).25 Furthermore, the formation of (Fe(H2O))2+ at an acidic pH negatively influenced PNT degradation because the free Fe2+ was reduced.26 As the initial solution pH increased from 3 to 9, the Fe3+ in the reaction system converted to the insoluble ferric hydroxide, which arrested the reduction of Fe3+ to Fe2+, thereby affecting PMS activation. Moreover, PMS dissociation increased through nonradical pathways at higher solution pH.27 In addition, under basic conditions, edge S would not dissociate, and as a result, S could not be captured, thus hindering the exposure of the Mo(IV) atom. Finally, MoS2 was negatively charged when the pH was higher, leading to strong repulsion between MoS2 and PMS, which could also hinder the PNT degradation.28| | |
SO4˙− + H+ + e− → HSO4˙−
| (5) |
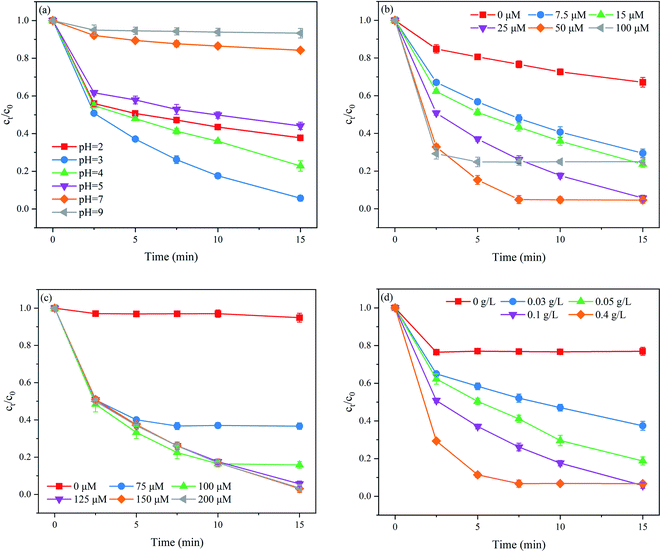 |
| | Fig. 2 Effects of (a) the initial solution pH, (b) Fe2+ dose, (c) PMS dose, and (d) MoS2 dose on PNT degradation in the MoS2/Fe2+/PMS process. Experimental conditions: [PNT]0 = 25 μM; (a) [Fe2+]0 = 25 μM, [PMS]0 = 125 μM, [MoS2]0 = 0.1 g L; (b) pH0 = 3.0, [PMS]0 = 125 μM, [MoS2]0 = 0.1 g L; (c) pH0 = 3.0, [Fe2+]0 = 25 μM, [MoS2]0 = 0.1 g L; (d) pH0 = 3.0, [Fe2+]0 = 25 μM, [PMS]0 = 125 μM. | |
The effect of the Fe2+ dose on PNT degradation is shown in Fig. 2(b). As shown, the removal rate of PNT increased markedly from 32.9% to 94.3% with increasing Fe2+ dose from 0 to 25 μM. As the Fe2+ dose further increased to 50 μM, the removal rate of PNT slightly increased to 95.4%. However, when the Fe2+ dose reached 100 μM, the removal rate of PNT decreased to 75.0%. Briefly, the removal rate of PNT gradually increased when the molar ratio of [Fe2+]0/[MoS2]0 ≤ 1:25, while further increased the molar ratio of [Fe2+]0/[MoS2]0 did not exhibited an obvious beneficial for PNT degradation or even decreased the removal rate of PNT. It is generally acknowledged that a large amount of Fe2+ corresponds to more PMS activator availability, which results in a faster reaction rate. However, excess Fe2+ also consumes reactive radicals (eqn (7) and (8)), slowing down the PNT degradation.29 Therefore, based on the activation efficiency along with the chemical cost, the optimum dose of Fe2+ was set to 25 μM ([Fe2+]0/[MoS2]0 = 1:25) in this study.
| | |
Fe2+ + SO4˙− → Fe3+ + SO42−
| (7) |
| | |
Fe2+ + ˙OH → Fe3+ + H2O
| (8) |
The PMS dose plays a vital role in removing organic contaminants in SO4˙−-based AOPs, as it serves as the primary source of reactive radicals. The effect of the PMS dose on PNT degradation is shown in Fig. 2(c). As shown, the removal rate of PNT increased from 5.1% to 94.3% as the PMS dose increased from 0 to 125 μM. However, the continuous increase in PMS dose did not further improve the removal rate of PNT. This phenomenon may be ascribed to the quenching effect of SO4˙− by excess PMS and the self-recombination of SO4˙− (eqn (9) and (10)).27 Similar findings have been reported for the removal of SMX at different PMS doses in the MoS2/Fe2+/PMS system.22
| | |
HSO5− + SO4˙− → SO5˙− + SO42− + H+
| (9) |
| | |
SO4˙− + SO4˙− → S2O82−
| (10) |
The effect of the MoS2 dose on PNT degradation is shown in Fig. 2(d). As shown, the removal rate of PNT increased monotonically from 23.1% to 94.3% as the MoS2 content increased from 0 to 0.1 g L−1. The number of exposed edge S atoms increased as the MoS2 dose increased, and then more active sites were available to accelerate the Fe3+/Fe2+ cycle, which facilitated the PNT degradation. Nevertheless, the removal rate of PNT slightly decreased when the MoS2 was increased to 0.4 g L−1 because Mo4+ acts as a scavenger of SO4˙−.21
3.3. Effects of common anions and NOM
Common anions (e.g., Cl−, HCO3−, and SO42−) and natural organic matter (NOM) are universal in natural water bodies and have been demonstrated to influence target organic contaminant degradation during SO4˙−-based AOPs. Therefore, it is necessary to evaluate the impact of these water components on the degradation performance of PNT once applying the MoS2/Fe2+/PMS process to the treatment of PNT in natural waters. The effects of different concentrations of Cl−, HCO3−, and HA on PNT degradation are shown in Fig. 3.
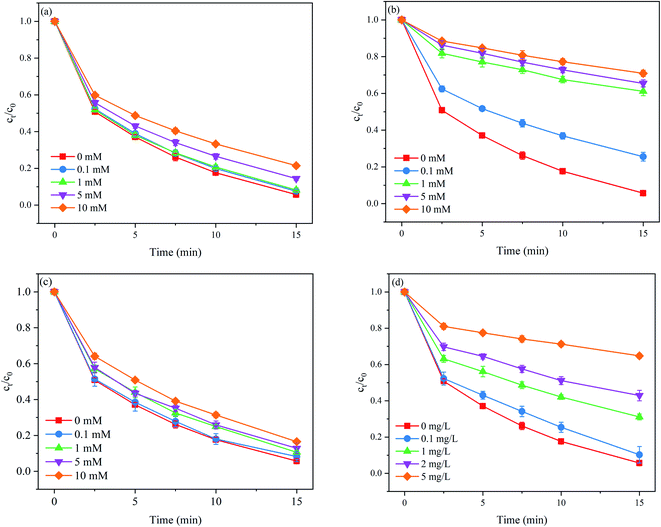 |
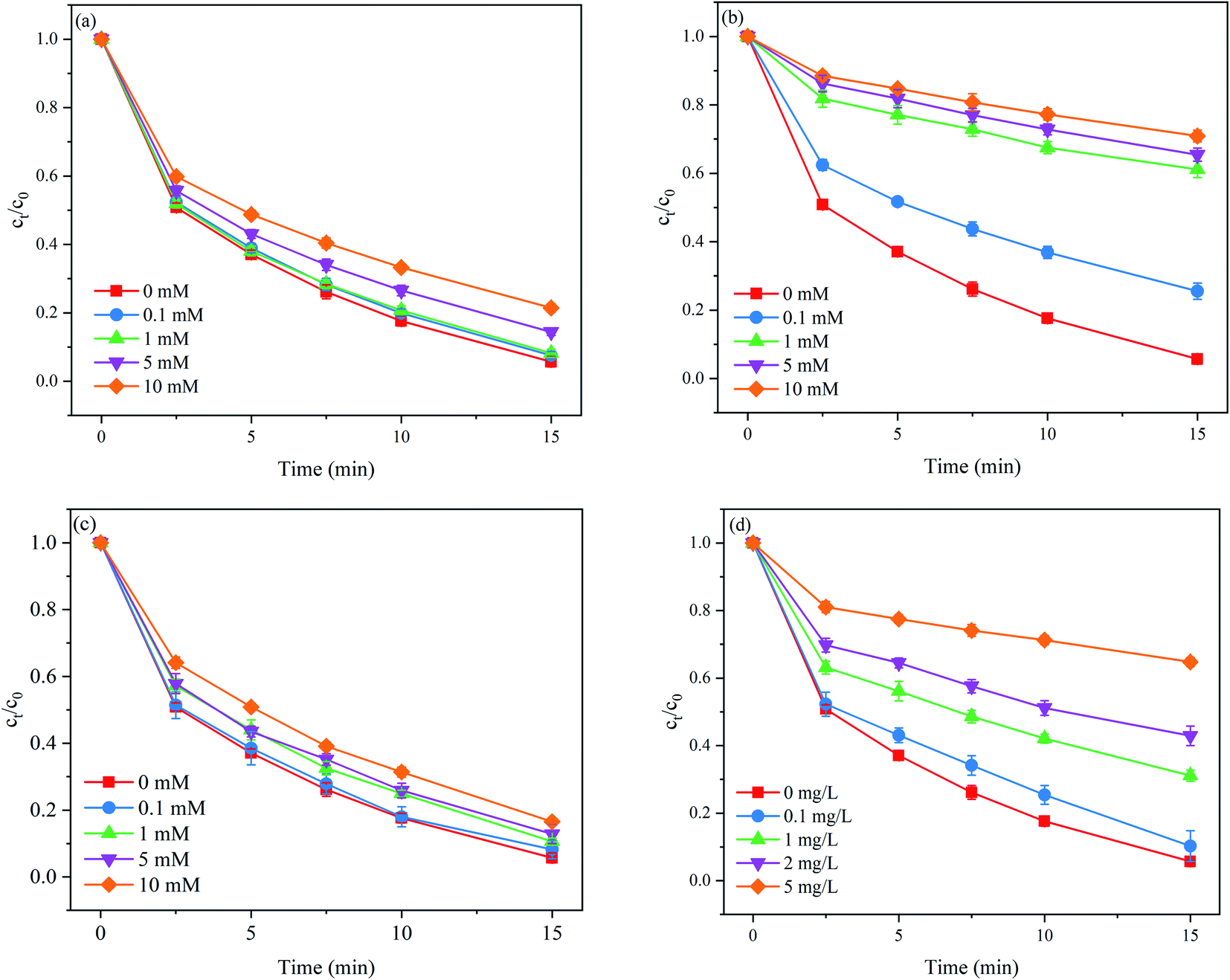
| | Fig. 3 Effects of (a) Cl−, (b) HCO3−, (c) SO42−, and (d) HA on PNT degradation in the MoS2/Fe2+/PMS process. Experimental conditions: [PNT]0 = 25 μM, pH0 = 3.0, [Fe2+]0 = 25 μM, [PMS]0 = 125 μM, [MoS2]0 = 0.1 g L−1. | |
As shown in Fig. 3(a), Cl− exerted an adverse effect on PNT degradation. The removal rate of PNT decreased from 93.0% to 78.6% as the concentration of Cl− increased from 0 to 10 mM. In the presence of Cl−, SO4˙− was rapidly scavenged at high reaction rate constants to form reactive chlorine species (eqn. (11) and (12)) such as Cl˙ and Cl2˙−, which exhibited lower redox potentials than SO4˙−.30 As seen in Fig. 3(b), HCO3− also exhibited adverse effects on PNT degradation. The removal rate of PNT decreased from 93.0% to 29.1% as the concentration of HCO3− increased from 0 to 10 mM. HCO3− is a well-known radical scavenger that can quench SO4˙− and ˙OH to produce less reactive radical species such as the carbonate radical (CO3˙−) (eqn. (13) and (14)).31 HCO3− can also directly consume PMS via eqn (15).32 In addition, the presence of HCO3− increased the solution pH, which was unfavorable to edge S atom capture. As seen in Fig. 3(c), the presence of SO42− also inhibited the PNT degradation. The removal rate of PNT decreased from 93.0% to 83.5% as the concentration of SO42− increased from 0 to 10 mM. The reason for this phenomenon lies in that the redox potential of SO4˙−/SO42− would decrease due to the presence of SO42−.33 As shown in Fig. 3(d), the removal rate of PNT decreased consistently from 93.0% to 35.2% as the concentration of HA increased from 0 to 5 mg L−1. NOM hinders the performance of MoS2/Fe2+/PMS by scavenging SO4˙− and ˙OH because the electron-rich moieties of NOM readily react with these electrophilic radicals, resulting in a decrease in the free radicals available for PNT degradation.34 These findings revealed that the impact of water characteristics should not be ignored once applying the MoS2/Fe2+/PMS process for practical use, and the operation parameters need to be carefully optimized to overcome the side effects caused by these water components in natural water.
| | |
Cl− + SO4˙− → Cl˙+ SO42−
| (11) |
| | |
HCO3− + SO4˙− → SO42− + H+ + CO3˙−
| (13) |
| | |
HCO3˙− + ·OH → CO3˙− + H2O
| (14) |
| | |
HCO3− +HSO5− → HSO42− + HCO4−
| (15) |
3.4. Identification of radical species
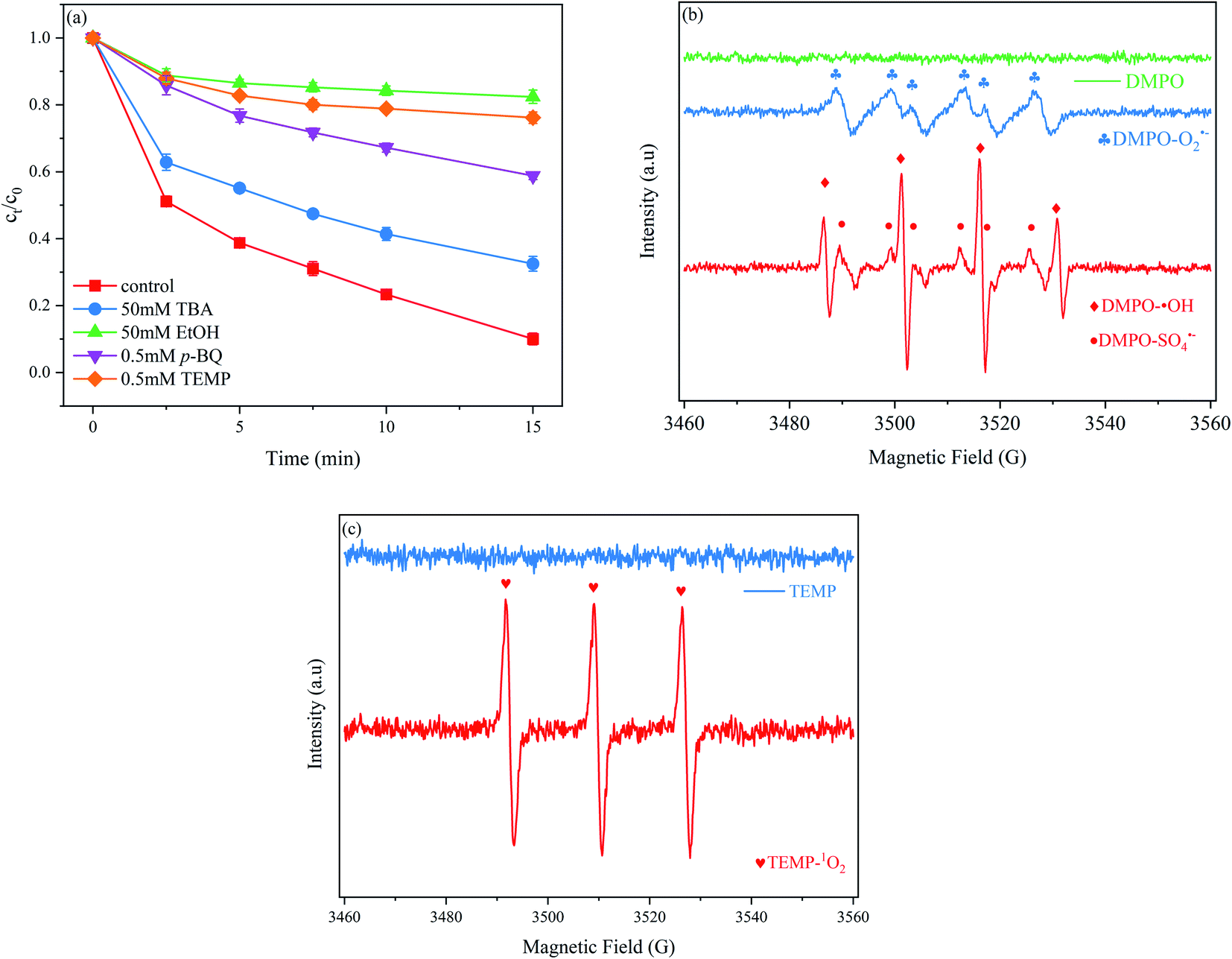
To investigate the role of radical species on PNT degradation, different quenchers, i.e. TBA, EtOH, p-BQ and TEMP were added to the MoS2/Fe2+/PMS system. TBA was selected as a ˙OH scavenger because its reaction with ˙OH had a rate constant that was approximately one thousand times faster ((3.8–7.6) × 108 M−1 s−1) than that of its reaction with SO4˙− ((4.0–9.1) × 105 M−1 s−1).35 As shown in Fig. 4(a), TBA slightly decreased the PNT removal rate. At 15 min, 67.5% was degraded with 50 mM TBA, which represents an efficiency that was 26.8% lower than that of the system without TBA. EtOH can significantly scavenge ˙OH and SO4˙− with second-order rate constants of (1.2–2.8) × 109 and (1.6–7.7) × 107 M−1 s−1, respectively. After adding 50 mM EtOH, the PNT removal rate markedly decreased to 17.6%, which was 76.7% lower than that of PNT without a scavenger. p-BQ was added to the reaction solution to further investigate the role of O2˙− because it can react with O2˙− with a second-order rate constant of (0.9–1.0) × 109 M−1 s−1.36 Our results show that the PNT removal rate decreased to 41.2% after the addition of 0.5 mM p-BQ, which indicated that O2˙− also participated in the degradation of PNT. Next, TEMP was added to the reaction solution to investigate the behavior of 1O2.37 The results show that PNT degradation was obviously hindered in the presence of 0.5 mM TEMP. These results indicated that in addition to the commonly-reported ˙OH and SO4˙−, a considerable amount of 1O2 and O2˙− is present in the MoS2/Fe2+/PMS system and simultaneously participates in the removal of PNT.
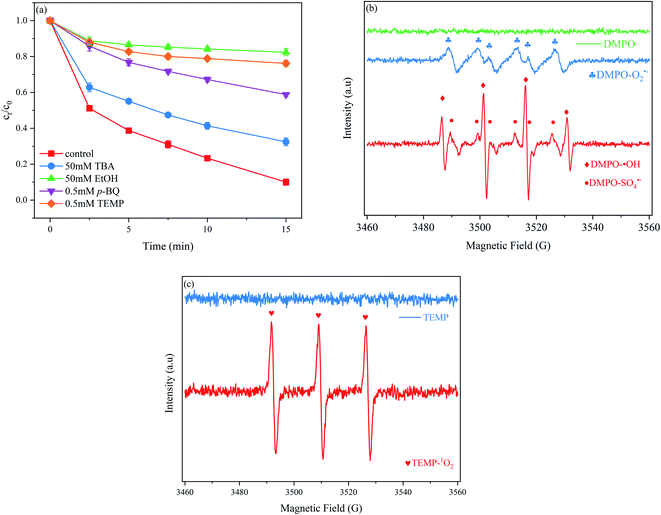 |
| | Fig. 4 (a) Effects of scavenger on PNT degradation; (b) EPR spectra of O2˙− ˙OH and SO4˙− using DMPO and (c) EPR spectra of 1O2 using TEMP. Experimental conditions: [PNT]0 = 25 μM, pH0 = 3.0, [Fe2+]0 = 25 μM, [PMS]0 = 125 μM, [MoS2]0 = 0.1 g L−1. | |
EPR was employed to directly detect the reactive species by using DMPO and TEMP as spin trap agents. DMPO could capture ˙OH, SO4˙−, and O2˙− to form different spin adducts with respective signals,38 while TEMP could capture 1O2.39 As shown in Fig. 4(b), no signal was detected in DMPO alone. In the MoS2/Fe2+/PMS system, four peaks with a 1:2:2:1 intensity ratio corresponding to the characteristic peak of DMPO–˙OH (αH = αN = 14.9 G) was observed, and six peaks with equal intensity corresponding to the characteristic peak of DMPO–SO4˙− (αN = 13.2 G, αH = 9.6 G, αH = 1.48 G, αN = 0.78 G) was also observed. The relatively weak signal of DMPO–SO4˙− was attributed to the lower sensitivity and shorter lifetime of the DMPO–SO4˙− peak.36 The peak of DMPO–O2˙− (αN = 17.4 G) adduct was also clearly detected. Fig. 4(c) exhibits strong triplet peaks with equal intensities; these signals are attributed to TEMP–1O2 and demonstrate the generation of 1O2 in the MoS2/Fe2+/PMS system. The EPR results were consistent with the results of the quenching experiments.
3.5. Characterization of MoS2 before and after reaction
The XRD patterns of MoS2 before and after reaction are given in Fig. S2.† The observed diffraction peaks were nearly unchanged after the reaction, and the peak positions were consistent with those reported by Xing et al.,18 reflecting the excellent reusability and stability of MoS2 in the MoS2/Fe2+/PMS system. The slight decrease in the peak intensity after the reaction was probably associated with the dissociation of Mo4+.
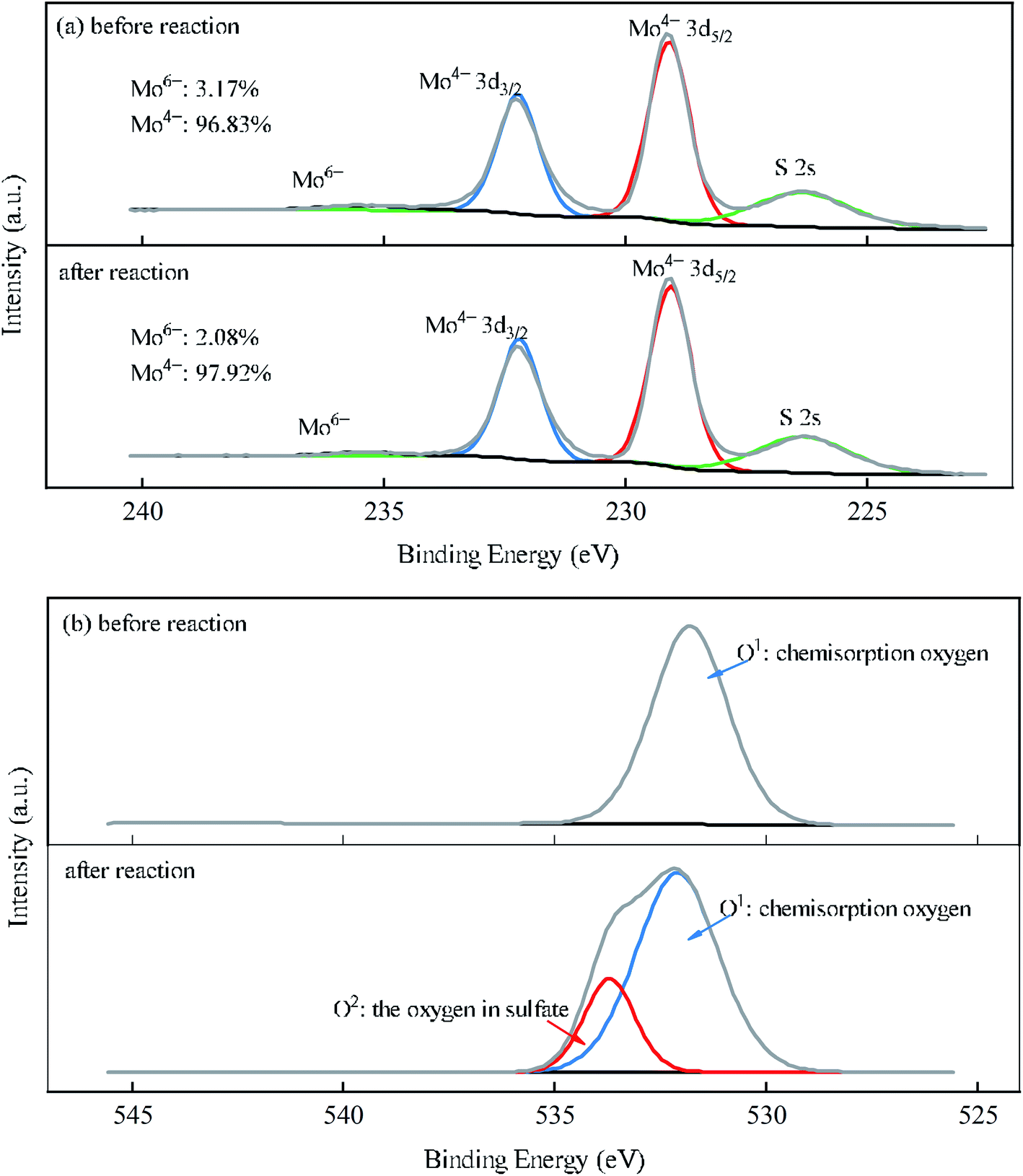
To better understand the roles of Mo and O species in the course of MoS2/Fe2+/PMS system, XPS analysis of MoS2 before and after reaction was performed (Fig. 5(a) and (b)). As shown in Fig. 5(a), four peaks at binding energies of 232.3 eV, 229.2 eV, 235.5 eV, and 226.5 eV correspond to the orbitals of Mo4+ 3d3/2, Mo4+ 3d5/2, Mo6+, and S 2s, respectively. After the reaction, the proportion of Mo6+ decreased, probably due to the dissolution of MoO3 in strong acid during the reaction or its reduction by HSO5− to regenerate Mo4+.40 As seen in Fig. 5(b), a peak emerged at 531.7 eV of MoS2 before the reaction, indicating the chemisorption of the oxygen of the raw MoS2, which has been proven to be closely related to the surface oxygen vacancies of the catalyst.41,42 Therefore, a small amount of oxygen may have been absorbed by oxygen vacancies, and electron transfer occurred through eqn (16).43 After the reaction, another peak formed at 533.3 eV, which was attributed to the oxygen in sulfate (SO42−).42
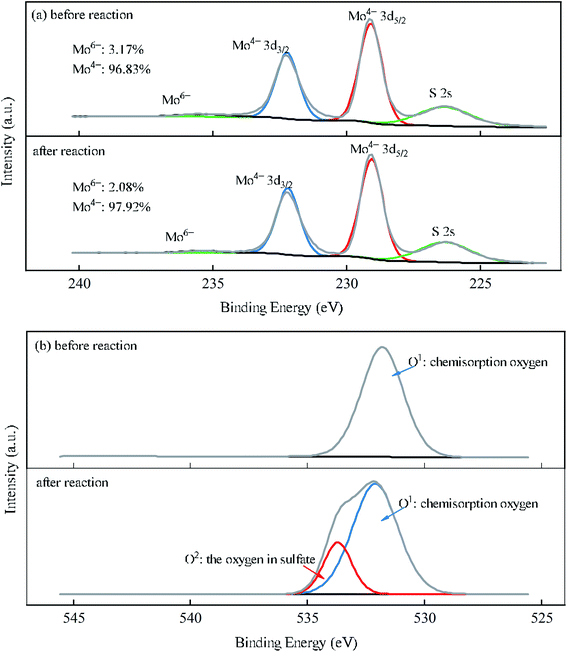 |
| | Fig. 5 XPS spectra of MoS2 before and after the reaction for the (a) Mo 3d and (b) O 1s orbital levels. | |
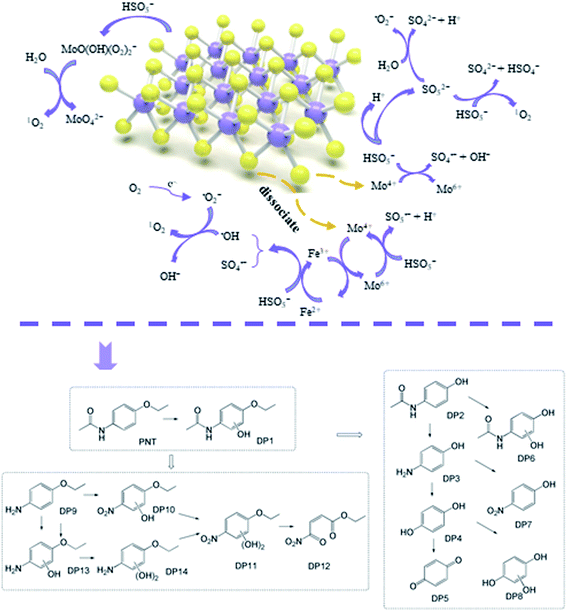
3.6. Proposed mechanisms of radical generation
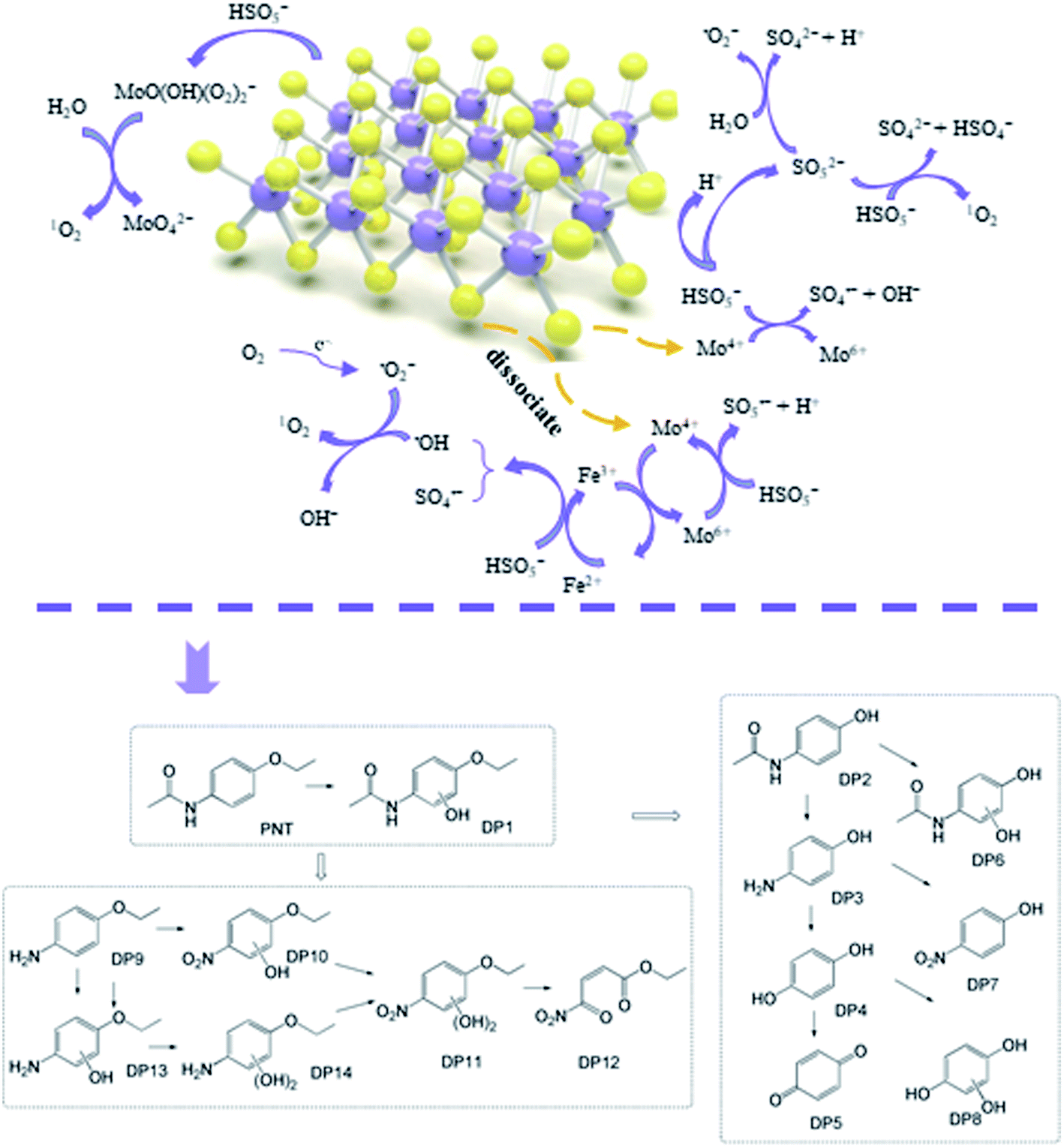
Based on the radical species and characterization of MoS2, the mechanisms of radical generation in the MoS2/Fe2+/PMS system can be elucidated as follows (Fig. 6). The initial activation of PMS by Fe2+ to generate SO4˙− and ˙OH has been widely acknowledged (eqn. (1) and (2)). The unsaturated S atoms on the surface of MoS2 can capture protons from the solution to form H2S and expose reductive Mo4+ active sites to accelerate the transformation of Fe3+ to Fe2+ (eqn (4)), which facilitates PMS activation. This was further supported by the variation of Fe2+ in the solution with and without MoS2 (Fig. S3†). As seen, the concentration of Fe2+ sharply decreased within 2.5 min and then gradually decreased to zero at 15 min in the Fe2+/PMS system. While, despite the concentration of Fe2+ also decreased significantly in the initial stage, and then it gradually increased along with the reaction time due to the transformation of Fe3+ to Fe2+ caused by the presence of MoS2. Similar result was also observed by other researchers.44–46 The reductive Mo4+ active sites are also directly involved in PMS activation (eqn (3)). In addition to the formation of ˙OH and SO4˙−, quenching experiments and EPR analysis also confirmed the formation of O2˙− and 1O2 in the MoS2/Fe2+/PMS system. The formation of O2˙− in the system may be attributed to PMS self-decomposition, in which electron transfer plays an important role (eqn (17) and (18)).47 On the other hand, the edge S of MoS2 dissociated under acidic conditions, resulting in the generation of S vacancies, which might absorb O2 to generate O2˙− (eqn (16)).43 To further confirm the main formation mechanism of O2˙− in the MoS2/Fe2+/PMS system, the degradation of PNT were carried out by sparging different gases (N2 and O2) (Fig. S4†). The degradation rate of PNT was only reduced by 7% when continuously sparging N2 into the solution. While, by sparging O2 inhibited the degradation of PNT probably due to the fast transformation of Fe2+ to Fe3+ in the presence of O2. Therefore, PMS self-decomposition play an important role on the formation of O2˙− in the MoS2/Fe2+/PMS system. Generally, the formation of 1O2 may originate from the reaction between the generated O2˙− and ˙OH or H2O (Eqs. (19) and (20)) as well as the self-decomposition of PMS (eqn (21)).43,47 In addition, previous study also indicated that the formed Mo6+ peroxo-complex may also serve as an activator of PMS to form 1O2 according to eqn (22).40| | |
SO52− + H2O → O2˙− + SO42− + H+
| (18) |
| | |
O2˙− +·OH → 1O2 + OH−
| (19) |
| | |
O2˙− + 2H2O → 1O2 + H2O2 + 2OH−
| (20) |
| | |
HSO5− + SO52− → HSO4− + SO42− + 1O2
| (21) |
| | |
MoO(OH)(O2)2− + H2O → MoO42− + 1O2 + H+
| (22) |
 |
| | Fig. 6 Proposed reaction mechanism and transformation pathways in the MoS2/Fe2+/PMS system. | |
3.7. DFT calculations and transformation pathways
Fig. S5† shows the chemical structure and Fukui function index (f0) of PNT. A higher f0 means that the PNT sites are attacked earlier by radicals, and when the f0 is higher, the PNT site is more vulnerable.48 Furthermore, LC/TOF/MS was performed to identify the degradation products of PNT in the MoS2/Fe2+/PMS system. After reaction for 5 min, new peaks appeared in the total ion chromatogram (TIC) (Fig. S6†). According to the results, approximately nine degradation products were identified, and all these products are listed in Table S1.† Based on the above DFT results, the detected products and previous studies, we proposed the possible transformation pathways of PNT, as shown in Fig. 6.
The chemical structure of PNT consists of an acetyl-amino group, an ethoxy group, and an aromatic ring. The sites of 6(C) and 9(C) have high f0 values and are easily attacked by free radicals. SO4˙− and ˙OH can react with the aromatic ring of PNT through electron transfer and hydrogen absorption to generate hydroxylated PNT. DP2, which was identified as paracetamol, a common drug and the metabolite of PNT in humans, may arise from the cleavage of the bond between 3(O) (f0 = 0.0639) and 2(C) (f0 = 0.0138).49 Then, the cleavage of the C–N bond of DP2 leads to the formation of aminophenol (DP3), which could be further oxidized to hydroquinone (DP4) and benzoquinone (DP5).50 SO4˙− is a single-electron oxidant that readily reacts with organic matter containing electron-rich functional groups such as aromatic and aniline moieties via an electron transfer mechanism.51 The amino group of DP3 is a potential reactive site for SO4˙−. Thus, the presence of nitrophenol (DP7) indicates that the amino group (–NH2) in 4-aminophenol can be oxidized into a nitro group (–NO2) via the attack of SO4˙−. This formation mechanism of nitrophenol has been previously described (Fig. S7†).52,53 DP6 and P8 were the hydroxylated products of paracetamol and DP5, respectively. The cleavage of the bond between 11(C) (f0 = 0.0697) and 10(N) (f0 = 0.0498) of acetyl-amino groups leads to the formation of DP9. After the amino group of DP9 is oxidized to a nitro group, the hydroxylation reaction is initiated by the attack of SO4˙−/˙OH to form two hydroxylated products, i.e., DP10 and DP11, and one open-ring product, DP12. DP13 and DP14 correspond to the mono- and dihydroxylated products of DP9, which form via hydroxylation driven by ˙OH and/or SO4˙− in a similar way. Therefore, the primary transformation pathways of PNT in the MoS2/Fe2+/PMS system include hydroxylation of the aromatic ring, cleavage of the C–N bond of the acetyl-amino group, and cleavage of the C–O bond of the ethoxy group.
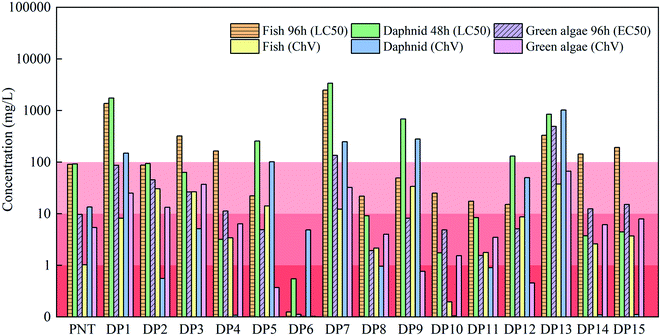
3.8. Toxicity assessment
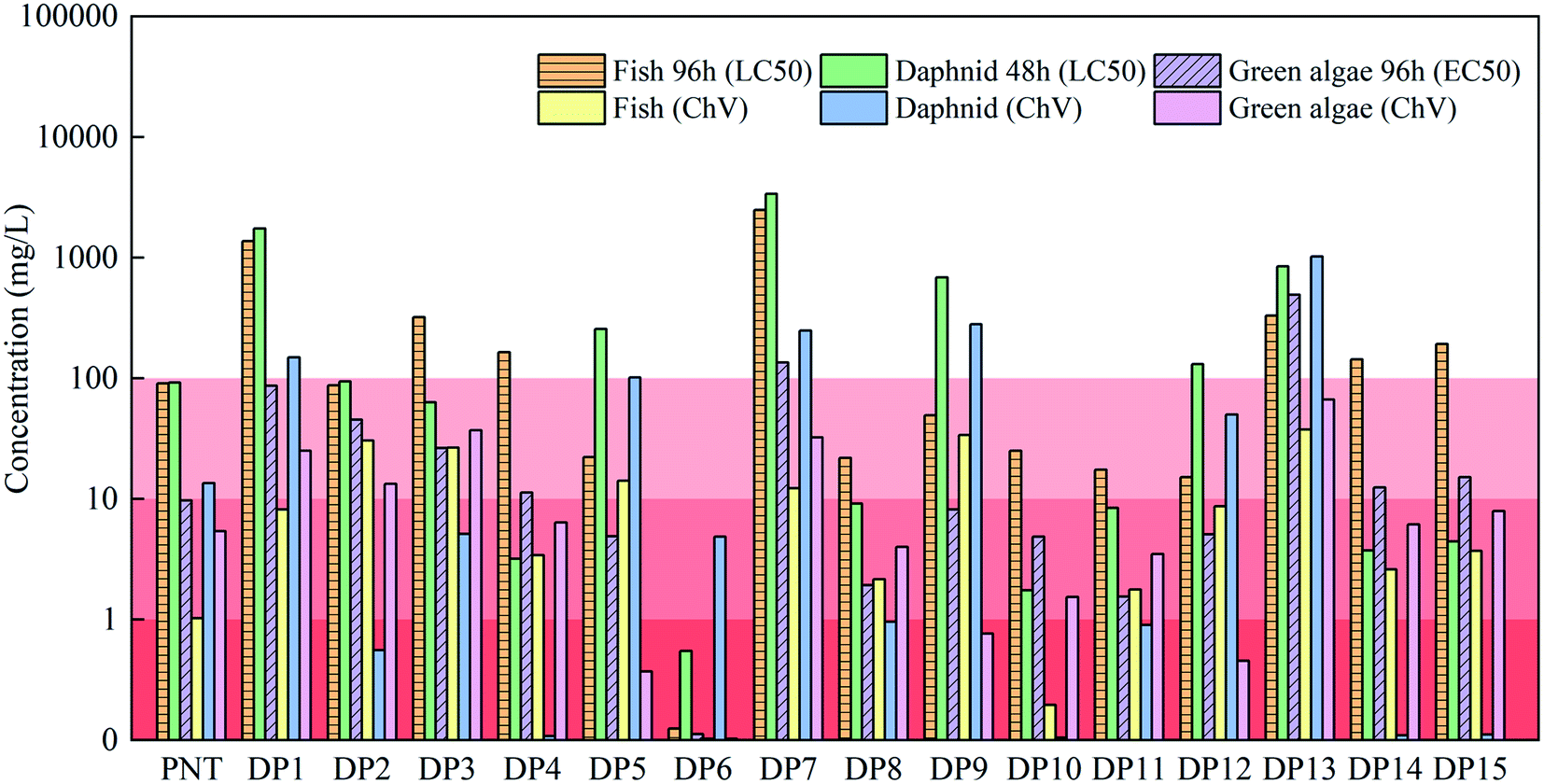
The TOC removal rate was only 2.8% at 15 min (Fig. S8†), indicating PNT may transform into intermediate products as discussed above rather complete mineralization. Thus, it is necessary to evaluate the toxicity of the degradation products since these products may be more toxic than their parent compound. In this study, the toxicity of PNT and its degradation products in the MoS2/Fe2+/PMS system were predicted by the ECOSAR program. According to the Globally Harmonized System of Classification and Labeling of Chemicals (GHS), the toxicity of substances can be divided into four levels, including not harmful (LC50/EC50/ChV > 100 mg L−1), harmful (10 mg L−1 < LC50/EC50/ChV ≤ 100 mg L−1), toxic (1 mg L−1 < LC50/EC50/ChV ≤ 10 mg L−1) and very toxic (LC50/EC50/ChV ≤ 1 mg L−1), where LC50, EC50 and ChV refer to half lethal concentration, half effective concentration and chronic toxicity, respectively.54 As shown in Fig. 7, green algae were more sensitive to acute toxicity, whereas fish and daphnid were more sensitive to chronic toxicity. Also, chronic toxicity was more significant to three species than that of acute toxicity. As for the acute toxicity, DP6 showed ‘very toxic’ and PNT, DP5, DP8, DP9, DP10, DP11 and DP12 exhibited ‘toxic’ to green algae. As for the chronic toxicity, DP6 and DP10 were more toxic than PNT for fish. Except for DP1, DP5, DP7, DP9, DP12 and DP13, all other degradation products showed more toxic than PNT for daphnid. The characteristics of degradation products' structure exert great impact on the toxicity. Therefore, the incomplete mineralization of PNT may lead to the formation of degradation products with higher toxicity, particularly hydroxylated products or products containing amino group, which should be carefully considered in practical application.55
 |
| | Fig. 7 Toxicity prediction of PNT and its degradation products. | |
4. Conclusion
In this study, we investigated the performance and mechanism of the MoS2-assisted Fe2+/PMS process of removing phenacetin. Compared to the Fe2+/PMS process, the MoS2/Fe2+/PMS process obviously enhanced PNT degradation. The best initial solution pH for the MoS2/Fe2+/PMS system for degrading PNT was 3. The optimum doses of Fe2+, PMS and MoS2 in terms of PNT degradation efficiency were determined. The presence of Cl−, HCO3−, SO42− and HA all exhibited inhibitory effects on the degradation of PNT. Quenching experiments and EPR analysis indicated that four types of reactive species, including ˙OH, SO4˙−, O2˙−, and 1O2, were detected in the MoS2/Fe2+/PMS system. The degradation pathways were proposed by LC/TOF/MS results and supported by the calculated Fukui index, and the primary transformation pathways of PNT were proposed. This mechanism can be briefly summarized as the hydroxylation of aromatic rings, cleavage of the C–N bonds of the acetyl-amino groups, and cleavage of the C–O bonds of the ethoxy groups. Toxicity analysis showed that most of the PNT degradation products are less toxic, but there are also generated products with higher toxic than that of PNT, which should be carefully considered in practical application. This study provides a deep understanding of the promoting effect of MoS2 in the Fe2+/PMS system and evaluates the characteristics and mechanism of PNT degradation in the SO4˙−-based oxidation process.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (51708348), the Scientific and Innovative Action Plan of Shanghai, China (19DZ1208204) and State Key Laboratory of Pollution Control and Resource Reuse Foundation (PCRRF18005).
References
- C. Su, Y. Cui, D. Liu, H. Zhang and Y. Baninla, Sci. Total Environ., 2020, 720, 137652 CrossRef CAS PubMed.
- D. Wu, Q. Sui, X. Yu, W. Zhao, Q. Li, D. Fatta-Kassinos and S. Lyu, Sci. Total Environ., 2021, 753, 141653 CrossRef CAS PubMed.
- L. Bo, L. Feng, J. Fu, X. Li, P. Li and Y. Zhang, J. Environ. Chem. Eng., 2015, 3, 2203–2211 CrossRef CAS.
- Y. Gao, J. Zhou, J. Zhang, C. Li, N. Gao and D. Yin, Sep. Purif. Technol., 2021, 256, 117819 CrossRef CAS.
- C. G. Daughton and I. S. Ruhoy, Sci. Total Environ., 2013, 443, 324–337 CrossRef CAS PubMed.
- N. Taoufik, W. Boumya, M. Achak, M. Sillanpää and N. Barka, J. Environ. Manage., 2021, 288, 112404 CrossRef CAS PubMed.
- Y. Q. Gao, N. Y. Gao, D. Q. Yin, F. X. Tian and Q. F. Zheng, Chemosphere, 2018, 201, 50–58 CrossRef CAS PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- X. He, A. A. De La Cruz and D. D. Dionysiou, J. Photochem. Photobiol., A, 2013, 251, 160–166 CrossRef CAS.
- P. Devi, U. Das and A. K. Dalai, Sci. Total Environ., 2016, 571, 643–657 CrossRef CAS PubMed.
- C. Tan, Y. Dong, L. Shi, Q. Chen, S. Yang, X. Liu, J. Ling, X. He and D. Fu, J. Taiwan Inst. Chem. Eng., 2018, 83, 74–81 CrossRef CAS.
- G. Liu, X. Li, B. Han, L. Chen, L. Zhu and L. C. Campos, J. Hazard. Mater., 2017, 322, 461–468 CrossRef CAS PubMed.
- Y. Q. Gao, J. Zhang, J. Q. Zhou, C. Li, N. Y. Gao and D. Q. Yin, RSC Adv., 2020, 10, 20991–20999 RSC.
- A. De Luca, R. F. Dantas and S. Esplugas, Water Res., 2014, 61, 232–242 CrossRef CAS PubMed.
- S. Miralles-Cuevas, I. Oller, J. A. S. Pérez and S. Malato, Water Res., 2014, 64, 23–31 CrossRef CAS PubMed.
- A. De Luca, R. F. Dantas and S. Esplugas, Appl. Catal., B, 2015, 179, 372–379 CrossRef CAS.
- W. Dong, Y. Jin, K. Zhou, S. P. Sun, Y. Li and X. D. Chen, Sci. Total Environ., 2019, 688, 513–520 CrossRef CAS PubMed.
- M. Xing, W. Xu, M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng and Y. Zhou, Chem, 2018, 4, 1359–1372 CAS.
- H. Zhou, L. Lai, Y. Wan, Y. He, G. Yao and B. Lai, Chem. Eng. J., 2020, 384, 123264 CrossRef CAS.
- B. Sheng, F. Yang, Y. Wang, Z. Wang, Q. Li, Y. Guo, X. Lou and J. Liu, Chem. Eng. J., 2019, 375, 121989 CrossRef CAS.
- D. He, Y. Cheng, Y. Zeng, H. Luo, K. Luo, J. Li, X. Pan, D. Barceló and J. C. Crittenden, Chemosphere, 2020, 240, 124979 CrossRef CAS PubMed.
- S. Wang, W. Xu, J. Wu, Q. Gong and P. Xie, Sep. Purif. Technol., 2020, 235, 116170 CrossRef CAS.
- M. Du, Q. Yi, J. Ji, Q. Zhu, H. Duan, M. Xing and J. Zhang, Chin. Chem. Lett., 2020, 31, 2803–2808 CrossRef CAS.
- M. I. Kanjal, M. Muneer, A. Abdelhaleem and W. Chu, Chin. J. Chem. Eng., 2020, 28, 2658–2667 CrossRef.
- A. Takdastan, B. Kakavandi, M. Azizi and M. Golshan, Chem. Eng. J., 2018, 331, 729–743 CrossRef CAS.
- Y. R. Wang and W. Chu, J. Hazard. Mater., 2011, 186, 1455–1461 CrossRef CAS PubMed.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171–179 CrossRef CAS.
- J. Yu, D. Ma, L. Mei, Q. Gao, W. Yin, X. Zhang, L. Yan, Z. Gu, X. Ma and Y. Zhao, J. Mater. Chem. B, 2018, 6, 487–498 RSC.
- H. Liu, T. A. Bruton, W. Li, J. Van Buren, C. Prasse, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2016, 50, 890–898 CrossRef CAS PubMed.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- C. Liang, Z. S. Wang and N. Mohanty, Sci. Total Environ., 2006, 370, 271–277 CrossRef CAS PubMed.
- M. Ahmadi and F. Ghanbari, Mater. Res. Bull., 2019, 111, 43–52 CrossRef CAS.
- X. Wu, X. Gu, S. Lu, Z. Qiu, Q. Sui, X. Zang, Z. Miao and M. Xu, Sep. Purif. Technol., 2015, 147, 186–193 CrossRef CAS.
- C. Luo, J. Jiang, J. Ma, S. Pang, Y. Liu, Y. Song, C. Guan, J. Li, Y. Jin and D. Wu, Water Res., 2016, 96, 12–21 CrossRef CAS PubMed.
- Y. Ji, Y. Fan, K. Liu, D. Kong and J. Lu, Water Res., 2015, 87, 1–9 CrossRef CAS PubMed.
- Z. Huang, P. Wu, J. Liu, S. Yang, M. Chen, Y. Li, W. Niu and Q. Ye, Chem. Eng. J., 2020, 395, 124936 CrossRef CAS.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- H. Luo, Y. Cheng, Y. Zeng, K. Luo, D. He and X. Pan, Sep. Purif. Technol., 2020, 248, 117023 CrossRef CAS.
- H. Sun, F. He and W. Choi, Environ. Sci. Technol., 2020, 54, 6427–6437 CrossRef CAS PubMed.
- Y. Zhang, J. Niu and J. Xu, Chem. Eng. J., 2020, 381, 122718 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- J. Li, J. Fang, L. Gao, J. Zhang, X. Ruan, A. Xu and X. Li, Appl. Surf. Sci., 2017, 402, 352–359 CrossRef CAS.
- S. Sun, Y. Gu, Y. Wang and Y. Liu, Chem. Phys. Lett., 2020, 752, 137557 CrossRef CAS.
- S. Qu, W. Wang, X. Pan and C. Li, J. Hazard. Mater., 2020, 384, 121494 CrossRef CAS PubMed.
- Y. Li, D. D. Cheng, Y. Luo and L. X. Yang, Rare Met., 2021, 40, 3543–3553 CrossRef CAS.
- X. X. Ji, H. F. Wang and P. J. Hu, Rare Met., 2019, 38, 783–792 CrossRef CAS.
- B. Liu, W. Song, H. Wu, Y. Xu, Y. Sun, Y. Yu and H. Zheng, Chem. Eng. J., 2020, 400, 125947 CrossRef CAS.
- X. Zhao, P. Du, Z. Cai, T. Wang, J. Fu and W. Liu, Environ. Pollut., 2018, 232, 580–590 CrossRef CAS PubMed.
- M. Xiao and Y. Zhang, Chemosphere, 2016, 152, 17–22 CrossRef CAS PubMed.
- M. Noorisepehr, B. Kakavandi, A. A. Isari, F. Ghanbari, E. Dehghanifard, N. Ghomi and F. Kamrani, Sep. Purif. Technol., 2020, 250, 116950 CrossRef CAS.
- L. Zhou, X. Yang, Y. Ji and J. Wei, Sci. Total Environ., 2019, 692, 201–208 CrossRef CAS PubMed.
- M. Mahdi Ahmed, S. Barbati, P. Doumenq and S. Chiron, Chem. Eng. J., 2012, 197, 440–447 CrossRef CAS.
- Y. Yang, X. Lu, J. Jiang, J. Ma, G. Liu, Y. Cao, W. Liu, J. Li, S. Pang, X. Kong and C. Luo, Water Res., 2017, 118, 196–207 CrossRef CAS PubMed.
- M. Xu, J. Deng, A. Cai, C. Ye, X. Ma, Q. Li, S. Zhou and X. Li, Chem. Eng. J., 2021, 413, 127533 CrossRef CAS.
- X. Zheng, Y. Li, J. Yang and S. Cui, Chem. Eng. J., 2021, 422, 130105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05892d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yan-yan Raoa,
Han Ninga,
Da-qiang Yinb and
Nai-yun Gaob
*a,
Yan-yan Raoa,
Han Ninga,
Da-qiang Yinb and
Nai-yun Gaob