
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanded scope of Griesbaum co-ozonolysis for the preparation of structurally diverse sensors of ferrous iron†
Jun Chen,
Ryan L. Gonciarz and
Adam R. Renslo*
and
Adam R. Renslo*
Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94143, USA. E-mail: adam.renslo@ucsf.edu
First published on 22nd October 2021
Abstract
Sterically shielded 1,2,4-trioxolanes prepared by Griesbaum co-ozonolysis have been utilized as chemical sensors of ferrous iron in several recently described chemical probes of labile iron. Here we report optimized conditions for co-ozonolysis that proceed efficiently and with high diastereoselectivity across an expanded range of substrates, and should enable a new generation of labile iron probes with altered reaction kinetics and physicochemical properties.
In the mid-1990s,1 Griesbaum and co-workers reported the co-ozonolysis of ketone and ketoxime reactants for the preparation of unsymmetrically substituted 1,2,4-trioxolanes. When cyclic ketone and oxime co-reactants are employed in this process, the resulting trioxolane adducts can be remarkably stable due to shielding of the endoperoxide bond by proximal axial C–H bonds of the surrounding carbocyclic ring systems. Vennerstrom and co-workers exploited this reaction and the shielding effect of the rigid adamantane ring system to develop the antimalarial agents arterolane2 (Fig. 1) and artefenomel.3 The hindered endoperoxide embedded within their structures, like that in the 1,2,4-trioxane ring of artemisinin derivatives, confers an antimalarial effect via initial Fenton-type reaction with unbound, or “labile”, ferrous iron sources in the parasite.
 | ||
| Fig. 1 Structures of recently described probes of ferrous iron employing a 1,2,4-trioxolane-based sensor of ferrous iron, inspired by the synthetic antimalarial arterolane. | ||
Increasing appreciation for the importance of labile iron as the bioavailable pool of iron in cells has motivated the development of chemical probes capable of detecting iron with oxidation-state specificity.4 Detection of ferrous iron has been achieved largely through reactivity-based approaches4c,d in which ferrous iron promotes the reduction of N–O or O–O bonds to activate a fluorophore (e.g. SiRhoNox5 and related analogs6), separate a FRET pair (e.g., TRX-FRET,7 FIP-1 (ref. 8)), release a tethered reporter payload (e.g., TRX-PURO,7 ICL-1,9 HNG10), or covalently sequester a PET radionuclide in cells/tissues of animals (18F-TRX11,12). Trioxane and trioxolane-based reagents have also been employed for chemoproteomic studies of the malaria parasite13 and of mammalian cancer cells (FIPC-1 (ref. 14)). Whilst an arterolane-like pharmacophore has figured prominently in many of these first-generation probes, their further development and optimization is likely to require access to trioxolane systems exhibiting a broader range of iron reactivities and enhanced physiochemical properties for in vivo applications. We therefore reinvestigated the Griesbaum co-ozonolysis with the aim of enabling new structural architectures of potential utility for ferrous iron-reactive therapeutics and chemical probes.
The Griesbaum co-ozonolysis proceeds via [3 + 2]/retro-[3 + 2] reaction of the oxime reactant with ozone to afford a carbonyl oxide intermediate. This species then reacts with the ketone component in a final, stereochemistry determining [3 + 2] cycloaddition to afford 1,2,4-trioxolane adducts. The reaction of adamantane oximes with substituted cyclohexanones is known to proceed selectively via axial addition of carbonyl oxide to ketone, affording cis-4′ or trans-3′ adducts with useful (∼9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.) diastereoselectivity (Scheme 1).15 The seminal reports from Griesbaum described mostly undecorated alkyl and cycloalkyl substrates, whereas more recent work7,8,14,16 has focused on the adamantane oximes that were found to yield pharmacologically active products, though other groups have explored reactions of non-adamantane substrates.17–19 We previously reported15 an optimized protocol to access 3′-hydroxy adducts useful for drug and reporter payload delivery in an iron(II)-dependent fashion. This protocol involved use of the ketone component as limiting reagent, and proceeds in good yields (∼70%) with adamantane oxime at 0 °C in CCl4. Unfortunately, we have since found these conditions to be unsatisfactory when applied to substituted adamantanes, and particularly to non-adamantyl systems, with yields often in the range of 5–23% and in some cases failing altogether to afford the desired adducts.
:1 d.r.) diastereoselectivity (Scheme 1).15 The seminal reports from Griesbaum described mostly undecorated alkyl and cycloalkyl substrates, whereas more recent work7,8,14,16 has focused on the adamantane oximes that were found to yield pharmacologically active products, though other groups have explored reactions of non-adamantane substrates.17–19 We previously reported15 an optimized protocol to access 3′-hydroxy adducts useful for drug and reporter payload delivery in an iron(II)-dependent fashion. This protocol involved use of the ketone component as limiting reagent, and proceeds in good yields (∼70%) with adamantane oxime at 0 °C in CCl4. Unfortunately, we have since found these conditions to be unsatisfactory when applied to substituted adamantanes, and particularly to non-adamantyl systems, with yields often in the range of 5–23% and in some cases failing altogether to afford the desired adducts.
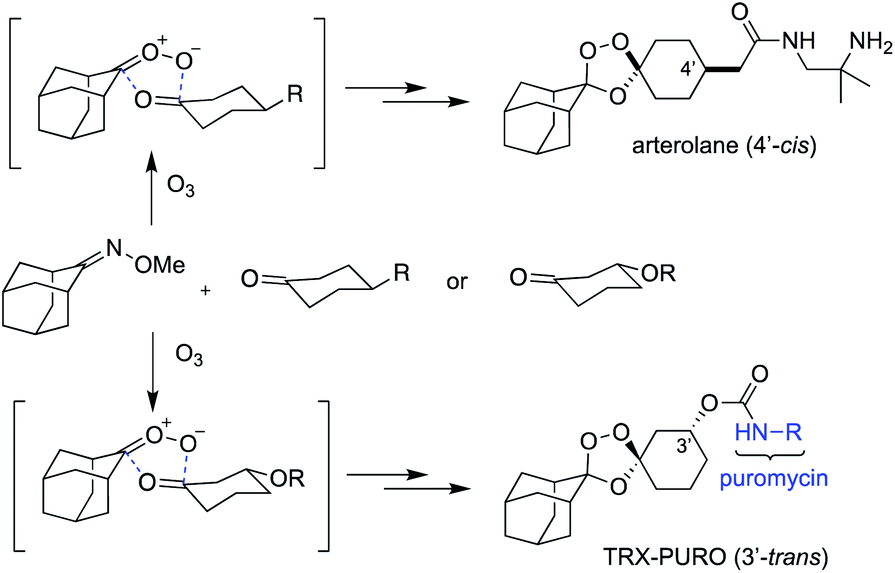 | ||
| Scheme 1 Griesbaum co-ozonolysis proceeds via conversion of the oxime to carbonyl oxide, followed by diastereoselective reaction with the ketone co-reactant, with axial addition favored, as shown. | ||
We hypothesized that side-reactions of ozone and/or the highly reactive carbonyl oxide intermediate20 may have contributed to poor yields with certain substrates. To explore this possibility, we evaluated the reaction of enantiopure ketone 2 (ref. 21) with a variety of substituted adamantanone oxime substrates under low temperature conditions reported previously19 for a different substrate (Table 1). We were pleased to find that reactions of 2 with various oximes (3 equiv.) at −78 °C in hexanes, using an oxygen flow rate = 1.1 liters per minute, or 6 g h−1 O3, afforded modest (50% for 3b) to excellent (77–94% for 3a, and 3c–3e) isolated yields of the desired adducts (Table 1). Our previous conditions afforded adduct 3a in variable yields ranging from 91% (ref. 21) to as low as 48%, while substituted adducts 3b–3e were obtained in only very poor yield (Table 1). Notably, the diastereofacial selectivity of the final [3 + 2] cycloaddition is further improved under the low temperature conditions. Thus, adducts 3a–3e were formed as a single trans diastereomer, as shown, whereas ∼10% of the cis diastereomer is formed under the original conditions (see ESI† for NMR spectra). As was expected, little diastereofacial selectivity is observed with respect to unsymmetrical carbonyl oxides during the [3 + 2] cycloaddition (note that only one of the two diastereomers of 3b–3e is shown Table 1).
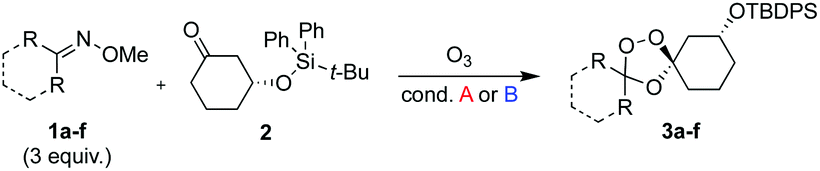

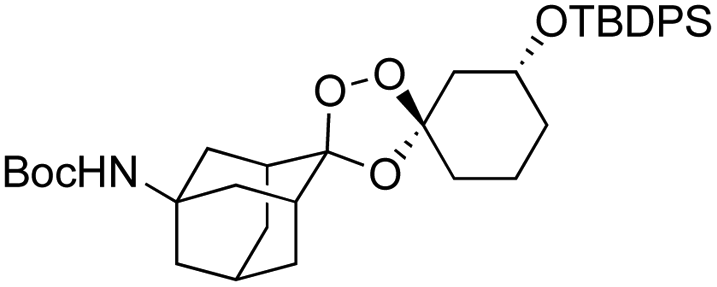
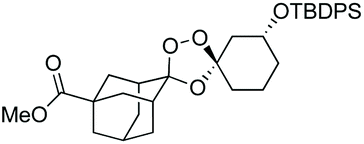

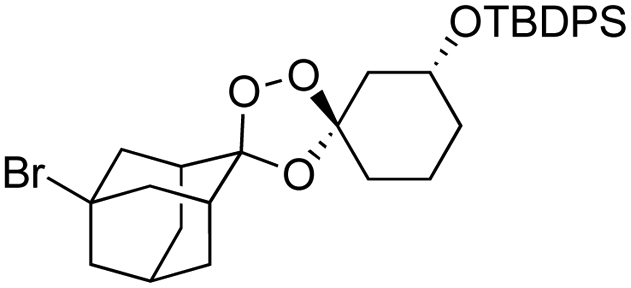
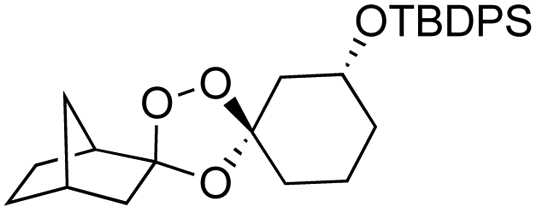
As noted above, the Fenton-type, iron(II)-specific reactivity of reporter-based probes like TRX-PURO and ICL-1 is modulated by the axial C–H bonds surrounding the endoperoxide function.22 To evaluate the potential of other aliphatic bicyclic ring systems to similarly shield the endoperoxide function, we computed minimized conformations of several potential adducts using MarvinSketch software (see ESI, Fig. S1†). From this analysis, we selected the bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, and bicyclo[3,3,1]nonane ring systems as likely to produce trioxolane adducts with desirable iron reactivity kinetics. Using low temperature reaction condition B, we were pleased to find that co-ozonolysis of 2 with bicyclo[2.2.1]heptan-2-one methyl oxime provided the desired adduct 3f in 81% isolated yield as a mixture of stereoisomers that were partially resolved by 1H NMR (three distinct resonances in a 13:8:1 ratio). The same reaction failed to afford any isolable amount of 3f under condition A (Table 1).
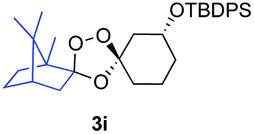
Buoyed by the successful generation of 3f, we explored the co-ozonolysis of additional oxime substrates under low temperature conditions (Table 2). Thus, reaction of 2 with bicyclo[2.2.2]octan-2-one methyl oxime or bicyclo[3.3.1]nonan-9-one methyl oxime afforded the desired adducts 3g and 3h in modest (26–44%) and excellent (80–87%) yields, respectively. The methyl oxime of camphor failed to afford adduct 3i, perhaps due to a more hindered steric environment around the oxime. Substituted cyclohexyl, cyclopentyl, and acyclic oximes were also investigated as substrates in the process. Cyclohexan-1-one oximes substituted at the 4-position afforded the desired adducts 3j (71%) and 3l (75%) in good yields, while 2-bromocyclohexan-1-one methyl oxime afforded 3k in 53% yield. Unexpectedly, cyclopentanone oxime substrates failed to yield isolable quantities of the expected adducts 3m, 3n, and 3o. It is possible that the failure of these reactions reflects instability of the trioxolane adducts, or competing reactions of the carbonyl oxide (e.g., dimerization). Similarly, the methyl oximes of acetophenone and 4-methoxyphenylacetone failed to afford useful yields of the expected adducts 3p and 3q.
|
|||
|---|---|---|---|
| Product (3f–k) | Isolated yield | Product (3l–q) | Isolated yield |
 |
70–82% |  |
75% |
 |
26–44% | 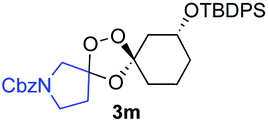 |
0% |
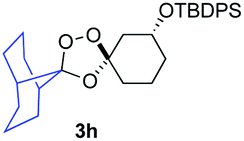 |
80–87% | 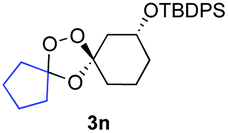 |
0% |
 |
0% | 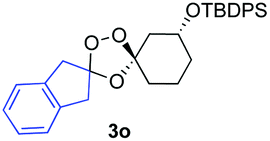 |
0% |
 |
71% | 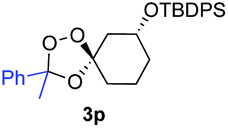 |
0% |
 |
53% |  |
4% |

Next, we sought to evaluate the Fe2+ reactivity of novel adducts like 3f, 3g, and 3h in the context of payload bearing trioxolane conjugates. Using conditions described previously by our group23 for the conversion of 3a to the mefloquine conjugate TRX-MFQ, 3f–3h were similarly converted to iron(II)-sensitive conjugates of mefloquine (MFQ) and morpholine (Scheme 2). Hence, intermediates 3f–h were treated with TBAF in THF to afford the alcohols 4f–h, which were then converted immediately to the para-nitrophenyl carbonate intermediates 5f–h before a final coupling with either mefloquine to afford 6f–h or morpholine to afford 7f–h (Scheme 2).
 | ||
| Scheme 2 Synthesis of mefloquine and morpholine conjugates 6f–h and 7f–h from intermediates 3f–h. MFQ = mefloquine, an antimalarial drug with a secondary amine function serving as the site of conjugation. | ||
As a surrogate measure of Fe2+ reactivity under physiological conditions, and to evaluate their ability to undergo iron(II)-dependent payload release, we evaluated 6f–h and 7f–h along with TRX-MFQ23 (8) as positive control, for activity against cultured P. falciparum parasites (W2 strain) using a standard protocol (Chart 1).24 Mefloquine bearing conjugates 6f–h exhibited potent IC50 values of 74 nM (for 6f) and 24 nM (6g and 6h), which were similar to that of the positive control 8 (IC50 = 17 nM). By contrast, morpholine-bearing conjugates 7f–h were markedly less potent, with IC50 values between 340 and 2700 nM (Chart 1), which is some 10–100 fold weaker than observed previously for congeneric adamantane-derived trioxolane comparators with a morpholine side chain.21 Accordingly, the potent anti-plasmodial activities of 6f–h can be inferred to result from iron(II)-dependent activation and release of the mefloquine payload by the canonical mechanism7,25 of payload release from “TRX” conjugates.
 | ||
| Chart 1 In vitro antiplasmodial activity of 6f–h, 7f–h and the known conjugate 8 against W2 P. falciparum parasites (IC50 ± SEM). Reported IC50 values are the means of at least three determinations. IC50 values for artefenomel and chloroquine controls are indicated at bottom left. The superior potency of conjugates 6 vs. 7 imply their activity is derived from mefloquine (MFQ) release. | ||
To further study the kinetics and regioselectivity of iron(II)-dependent activation, we followed the reaction of 6f and 8 with ferrous ammonium sulfate by UPLC/MS. As we have described previously for the progenitor TRX moiety,7,25 efficient activation and payload release requires regioselective activation of the endoperoxide bond by Fe2+ such that the ketone intermediate A is produced preferentially over the alternative adamantan-2-one product (Fig. 2). In adamantane-based systems this regioselectivity is conferred by the steric effect of the adamantane ring, as noted previously.22 We predicted based on modeling that the bicyclo[2.2.1]heptane moiety of 6f should similarly shield the proximal oxygen atom from inner-sphere coordination with Fe2+ leading to regioselective peroxide scission. In the event, exposure of either 6f or 8 to Fe2+ (as ferrous-ammonium sulfate with pH 7.4 Tris buffer) led within minutes to clean conversion to the common cyclohexanone intermediate A (Fig. 2). No detectable quantity of the alternate bicyclo[2.2.1]heptan-2-one product was detected in the reactions of 6f, thus confirming that the process is highly regioselective. Conversion of 6f to A was moderately faster than for comparator 8, with 6f fully consumed by the 11 minute time point. The kinetics of mefloquine release from intermediate A were comparable within experimental error. Taken together, the antiplasmodial and cell-free Fe2+ reactivity data indicate that efficient iron(II)-dependent uncaging and traceless release of payloads can be realized from non-adamantane based scaffolds such as 6f and likely as well from the other scaffolds described herein.
 | ||
| Fig. 2 In vitro iron fragmentation studies of the previously described, in vivo efficacious mefloquine conjugate 8 and the new congener 6f bearing a bicyclo[2.1.1]heptane ring in place of the adamantane. Fragmentation of the trioxolane with FAS is rapid for both conjugates, with β-elimination from common intermediate A being the rate limiting step in mefloquine (MFQ) release. | ||
Conclusions
In summary, we explored the scope of Griesbaum co-ozonolysis under optimized low temperature reaction conditions. Overall, these conditions afford improved yields, substrate scope, and diastereoselectivity as compared to our previously described conditions. Highly hindered ketoximes remain problematic substrates, while trioxolane adducts lacking a sufficiently shielded endoperoxide bond are likely unstable to isolation. However, with the appropriate selection of ketoxime and ketone reactants, a variety of new adducts could be prepared using the improved conditions. Three of these new adducts were conjugated to drug or control payloads and shown in antiplasmodial assays and chemical Fe2+ reactivity studies to be competent sensors of ferrous iron, much like the parental TRX system.These findings are significant insofar as they should enable iron(II)-activated chemistries to be applied to a broader range of drug or reporter payloads whilst maintaining physiochemical properties and iron(II)-dependent reactivity within a pharmacologically desirable range. As such, our findings have implications for antimalarial drug discovery, iron(II)-dependent drug delivery, and the continuing development of new chemical tools to study labile ferrous iron in biological settings.
Author contributions
All authors participated in the design of experiments, interpretation of data, and writing the manuscript. J. Chen and R. Gonciarz performed experiments. A. R. Renslo supervised research.Conflicts of interest
The authors declare the following competing financial interest(s): A. R. R. is an advisor and co-founder of Tatara Therapeutics.Acknowledgements
A. R. R. acknowledges funding from National Institutes of Health grant AI105106 and Congressionally Directed Medical Research Program, grants W81XWH1810763 and W81XWH1810754. We thank Jennifer Legac (UCSF) for antimalarial testing of analogues 6f–h and 7f–h.Notes and references
- (a) K. Griesbaum, X. Liu, A. Kassiaris and M. Scherer, Liebigs Ann., 1997, 1997, 1381 CrossRef; (b) K. Griesbaum, B. Övez, T. S. Huh and Y. Dong, Liebigs Ann., 1995, 1995, 1571 CrossRef.
- J. L. Vennerstrom, S. Arbe-Barnes, R. Brun, S. A. Charman, F. C. K. Chiu, J. Chollet, Y. Dong, A. Dorn, D. Hunziker, H. Matile, K. McIntosh, M. Padmanilayam, J. Santo Tomas, C. Scheurer, B. Scorneaux, Y. Tang, H. Urwyler, S. Wittlin and W. N. Charman, Nature, 2004, 430, 900 CrossRef CAS PubMed.
- S. A. Charman, S. Arbe-Barnes, I. C. Bathurst, R. Brun, M. Campbell, W. N. Charman, F. C. K. Chiu, J. Chollet, J. C. Craft, D. J. Creek, Y. Dong, H. Matile, M. Maurer, J. Morizzi, T. Nguyen, P. Papastogiannidis, C. Scheurer, D. M. Shackleford, K. Sriraghavan, L. Stingelin, Y. Tang, H. Urwyler, X. Wang, K. L. White, S. Wittlin, L. Zhou and J. L. Vennerstrom, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4400 CrossRef CAS PubMed.
- (a) A. T. Aron, A. G. Reeves and C. J. Chang, Curr. Opin. Chem. Biol., 2018, 43, 113 CrossRef CAS PubMed; (b) C. J. Chang, Nat. Chem. Biol., 2015, 11, 744 CrossRef CAS PubMed; (c) K. J. Bruemmer, S. W. M. Crossley and C. J. Chang, Angew. Chem., Int. Ed., 2020, 59, 13734 CrossRef CAS PubMed; (d) T. Hirayama, Free Radical Biol. Med., 2019, 133, 38 CrossRef CAS PubMed.
- T. Hirayama, H. Tsuboi, M. Niwa, A. Miki, S. Kadota, Y. Ikeshita, K. Okuda and H. Nagasawa, Chem. Sci., 2017, 8, 4858 RSC.
- T. Hirayama, A. Miki and H. Nagasawa, Metallomics, 2018, 11, 111 CrossRef PubMed.
- B. Spangler, C. W. Morgan, S. D. Fontaine, M. N. Vander Wal, C. J. Chang, J. A. Wells and A. R. Renslo, Nat. Chem. Biol., 2016, 12, 680 CrossRef CAS PubMed.
- A. T. Aron, M. O. Loehr, J. Bogena and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 14338 CrossRef CAS PubMed.
- A. T. Aron, M. C. Heffern, Z. R. Lonergan, M. N. Vander Wal, B. R. Blank, B. Spangler, Y. Zhang, H. M. Park, A. Stahl, A. R. Renslo, E. P. Skaar and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12669 CrossRef CAS PubMed.
- S. Xu, H.-W. Liu, L. Chen, J. Yuan, Y. Liu, L. Teng, S.-Y. Huan, L. Yuan, X.-B. Zhang and W. Tan, J. Am. Chem. Soc., 2020, 142, 2129 CrossRef CAS PubMed.
- N. Zhao, Y. Huang, Y. H. Wang, R. K. Muir, Y. C. Chen, N. Hooshdaran, J. Wei, P. Viswanath, Y. Seo, D. Ruggero, A. R. Renslo and M. J. Evans, J. Nucl. Med., 2021, 62, 949 CrossRef CAS PubMed.
- R. K. Muir, N. Zhao, J. Wei, Y.-h. Wang, A. Moroz, Y. Huang, Y.-C. Chen, R. Sriram, J. Kurhanewicz, D. Ruggero, A. R. Renslo and M. J. Evans, ACS Cent. Sci., 2019, 5, 727 CrossRef CAS PubMed.
- (a) H. M. Ismail, V. E. Barton, M. Panchana, S. Charoensutthivarakul, G. A. Biagini, S. A. Ward and P. M. O'Neill, Angew. Chem., Int. Ed., 2016, 55, 6401 CrossRef CAS PubMed; (b) C. Wei, C.-X. Zhao, S. Liu, J.-H. Zhao, Z. Ye, H. Wang, S.-S. Yu and C.-J. Zhang, Chem. Commun., 2019, 55, 9535 RSC.
- Y.-C. Chen, J. A. Oses-Prieto, L. E. Pope, A. L. Burlingame, S. J. Dixon and A. R. Renslo, J. Am. Chem. Soc., 2020, 142, 19085 CrossRef CAS PubMed.
- S. D. Fontaine, A. G. DiPasquale and A. R. Renslo, Org. Lett., 2014, 16, 5776 CrossRef CAS PubMed.
- J. Wu, X. Wang, F. C. K. Chiu, C. Häberli, D. M. Shackleford, E. Ryan, S. Kamaraj, V. J. Bulbule, A. I. Wallick, Y. Dong, K. L. White, P. H. Davis, S. A. Charman, J. Keiser and J. L. Vennerstrom, J. Med. Chem., 2020, 63, 3723 CrossRef CAS PubMed.
- E. Y. Yamansarov, O. B. Kazakova, N. I. Medvedeva, D. V. Kazakov, O. S. Kukovinets and G. A. Tolstikov, Russ. J. Org. Chem., 2014, 50, 1043 CrossRef CAS.
- Y. Dong, J. Chollet, H. Matile, S. A. Charman, F. C. K. Chiu, W. N. Charman, B. Scorneaux, H. Urwyler, J. Santo Tomas, C. Scheurer, C. Snyder, A. Dorn, X. Wang, J. M. Karle, Y. Tang, S. Wittlin, R. Brun and J. L. Vennerstrom, J. Med. Chem., 2005, 48, 4953 CrossRef CAS PubMed.
- O. B. Kazakova, E. F. Khusnutdinova, A. V. Petrova, E. Y. Yamansarov, A. N. Lobov, A. A. Fedorova and K. Y. Suponitsky, J. Nat. Prod., 2019, 82, 2550 CrossRef CAS PubMed.
- R. Chhantyal-Pun, M. A. H. Khan, C. A. Taatjes, C. J. Percival, A. J. Orr-Ewing and D. E. Shallcross, Int. Rev. Phys. Chem., 2020, 39, 385 Search PubMed.
- B. R. Blank, R. L. Gonciarz, P. Talukder, J. Gut, J. Legac, P. J. Rosenthal and A. R. Renslo, ACS Infect. Dis., 2020, 6, 1827 Search PubMed.
- S. A. Charman, C. S. Perry, F. C. Chiu, K. A. McIntosh, R. J. Prankerd and W. N. Charman, J. Pharm. Sci., 2006, 95, 256 CrossRef CAS PubMed.
- E. M. W. Lauterwasser, S. D. Fontaine, H. Li, J. Gut, K. Katneni, S. A. Charman, P. J. Rosenthal, M. Bogyo and A. R. Renslo, ACS Med. Chem. Lett., 2015, 6, 1145 CrossRef CAS PubMed.
- P. S. Sijwali and P. J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4384 CrossRef CAS PubMed.
- S. D. Fontaine, B. Spangler, J. Gut, E. M. Lauterwasser, P. J. Rosenthal and A. R. Renslo, ChemMedChem, 2015, 10, 47 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05932g |
| This journal is © The Royal Society of Chemistry 2021 |