
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-supporting carbon-rich SiOC ceramic electrodes for lithium-ion batteries and aqueous supercapacitors†‡
Shakir Bin Mujib a,
François Ribotb,
Christel Gervaisb and
Gurpreet Singh*a
a,
François Ribotb,
Christel Gervaisb and
Gurpreet Singh*a
aDepartment of Mechanical and Nuclear Engineering, Kansas State University, Manhattan, KS 66506, USA. E-mail: gurpreet@ksu.edu
bSorbonne Université, CNRS UMR 7574, Laboratoire de Chimie de la Matière Condensée de Paris, 75005 Paris, France
First published on 3rd November 2021
Abstract
Fabrication of precursor-derived ceramic fibers as electrodes for energy storage applications remains largely unexplored. Within this work, three little known polymer-derived ceramic (PDC)-based fibers are being studied systemically as potential high-capacity electrode materials for electrochemical energy devices. We report fabrication of precursor-derived SiOC fibermats via one-step spinning from various compositions of siloxane oligomers followed by stabilization and pyrolysis at 800 °C. Electron microscopy, Raman, FTIR, XPS, and NMR spectroscopies reveal transformation from polymer to ceramic stages of the various SiOC ceramic fibers. The ceramic samples are a few microns in diameter with a free carbon phase embedded in the amorphous Si–O–C structure. The free carbon phase improves the electronic conductivity and provides major sites for ion storage, whereas the Si–O–C structure contributes to high efficiency. The self-standing electrodes in lithium-ion battery half-cells deliver a charge capacity of 866 mA h gelectrode−1 with a high initial coulombic efficiency of 72%. As supercapacitor electrode, SiOC fibers maintain 100% capacitance over 5000 cycles at a current density of 3 A g−1.
1. Introduction
Despite the exponential rise in research activity in the design and development of micro-/nano-structured electrode materials for electrochemical energy storage devices,1–4 graphite or carbon-coated metal foil remain the electrode of choice for most capacitors and Li-ion battery (LIB) technologies. Materials with higher charge storage than traditional carbons such as silicon [almost 10× lithium (Li) storage capacity (∼4.2 A h g−1)] emerged as potential replacements for graphite owing to silicon's large abundance in the earth's crust, low toxicity, and well-established manufacturing technology for its large scale production.5–7 However, drastic volume variation and pulverization during charge/discharge, excessive formation of solid electrolyte interfaces (SEIs), and stress-induced cracks in Si electrodes has rendered bulk silicon unusable as an electrode for LIBs.6,8Efforts related to the development and use of nanostructured silicon-based electrodes (utilizing the size effect to achieve fast Li-ion transport, and prevent fracture propagation in electrodes), in the form of nanoparticles,;9–12 nanotubes and nanowires,13–17 and composite nanoparticles with carbon18–23 are now well-documented. Nevertheless, capacity decay over a long period of charge/discharge, high first cycle loss, high cost of material synthesis, and challenges associated with scalability limit their applications as electrodes for commercial applications.
To circumvent the issues associated with silicon electrodes, silicon oxycarbide (SiOC) derived from pyrolysis of ceramic precursor polymers has renewed attention in recent years from electrochemical storage point of view.24–33 Potential of polymer-derived ceramic (PDC) (e.g.; SiOC, SiCN) materials in energy storage, initially proposed by Dahn et al. in 1990s, largely depends on the chemical structures of the material.34,35 SiOC is an amorphous ceramic mainly consists of a Si–O–C glass phase with a free carbon region, where the mixed bonds of Si with O and C enable high Li storage.36,37 According to Graczyk-Zajac et al., mixed bonds in SiOC allows more disordered carbon phase for higher Li capacity (325 mA h g−1) as well as reversible storage sites at the interface of the carbon phase and amorphous network.38 Kasper et al. showed that the electrical conductivity of SiOC samples improved (0.07 to 2.2 S m−1) with the increasing amount of free carbon phase, in addition to enabling more active sites for Li.39 The edges of the free carbon phase and the interstitial spaces are major electrochemically active sites for Li ions, whereas micro-/nano-voids and amorphous Si–O–C network contribute to minor Li storage.25,40,41 Lee et al. reported that controlling the nanovoids in the Si–O–C domain derived from PSS-octakis(dimethylsilyloxy)silsesquioxane (POSS) provided efficient ion pathway and withstood structural degradation by buffering during lithiation/delithiation.33 As a result, the authors were able to achieve a capacity of 412 mA h g−1 at a high current density of 3600 mA g−1. Furthermore, pyrolysis parameters are always crucial in controlling the microstructure (e.g.; free/disordered carbon phase and nanovoids) and thus maintaining the electrochemical properties of SiOC. For example, pyrolysis temperature above 900 °C results in lower capacity and unstable cycling behavior of the SiOC ceramics compared to lower temperature.29,42 With increasing temperature disordered carbon phase arranges into ordered structure and amorphous Si–O–C phase decomposes to electrochemically inactive SiC crystals. In addition, metal foil-based electrode, conventionally prepared using a doctor blade, carries the inactive weight of polymeric binders, conducting agents (usually carbon black), and the foil itself, which do not contribute towards electrode capacity.31
Since the development of electrospinning in 1930s by Formhlas,43,44 the technique has been explored to fabricate a variety of artificial fibers from homogenous polymer solution using high potential electric field. Nanofibers produced by electrospinning can provide large surface area, exceptional tolerance against mechanical deformation with improved electrical conductivity and enhanced electrochemical performance. For example, electrospinning of PAN nanofibers with Si nanoparticles delivered a flexible electrode after carbonization that showed a high capacity of 1600 mA h g−1 in LIBs.45 Xiao et al. reported a stretchable PVDF nanofiber membrane which was coated with Ni and Si to form a Si/Ni/PVDF core–shell structure.46 This soft silicon electrode showed 56.9% capacity retention after 1000 cycles with 20% stretching capability.
Herein, we report on the fabrication of freestanding, binder-free SiOC electrodes by an electrospinning method using liquid precursor polymer, followed by thermal treatment and pyrolysis. Three new types of SiOC precursors, that are readily available, and easy to draw into low-cost, high-yield ceramic fibers compared to commercial ceramic fibers,47,48 are utilized in this study both for LIBs and supercapacitors. To the best of our knowledge such siloxanes have not been reported before for the fabrication of SiOC ceramic fibermats, that are investigated in electrochemical devices. Scanning- and transmission-electron microscopy reveals uniform, defect-free amorphous structure of the SiOC fibers. Fourier transform infrared spectroscopy (FTIR) and nuclear magnetic resonance (NMR) are utilized to study the polymeric precursor to ceramic transformation of the siloxanes, whereas Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) confirms the presence of “free carbon” content. Amorphous Si–O–C phase provides chemical stability and host sites for Li storage, and free carbon phase contributes to conductive network as well as the high Li capacity of the fibermat. As a result, as-prepared SiOC electrodes deliver specific capacity as high as 866 mA h gelectrode−1 (considering total weight of the electrode) with high first cycle coulombic efficiency of 72% in LIBs. Specific capacity of 800 mA h g−1 at a current density of 50 mA g−1 is achieved with a 100% capacity retention after 50 cycles for a particular SiOC electrode. Further, as supercapacitor electrode SiOC fibers have delivered a high gravimetric capacitance of 55 F g−1 at 100 mV s−1 and maintained 100% capacitance over 5000 cycles at 3 A g−1.
2. Experimental
2.1 Materials
Three types of commercially available oligomers precursors to silicon oxycarbide ceramic were sourced from Gelest, Inc. (Pennsylvania, USA). These SiOC precursors: (1) 1,5-divinyl-3,3-diphenyl-1,1,5,5-tetramethyltrisiloxane (cited as DDTS), (2) 1,3-divinyltetramethyldisiloxane (cited as DTDS), and (3) 1,3,5-trivinyl-1,1,3,5,5-pentamethyltrisiloxane (cited as TPTS) as shown in Fig. 1 have comparable chemical structures with vinyl groups which are potential crosslinking groups. Fig. 1 shows the compositions of the precursors where silicon (Si) to oxygen (O) atom ratio for DDTS and TPTS is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and for DTDS is 2:1.
:2 and for DTDS is 2:1.
 | ||
| Fig. 1 Molecular structure of three pre-ceramic silicon oligomers used in this study. | ||
To accomplish electrospinning of the preceramic oligomers 10 wt% of polyvinylpyrrolidone (PVP) with Mw ≈ 1300000 g mol−1 (Sigma Aldrich, Missouri, USA) was used as a spinning agent. Dicumyl peroxide (Sigma Aldrich, Missouri, USA) was used as a crosslinker for the oligomers and iso-propanol (Fisher Scientific, Massachusetts, USA) was used as a solvent. Ultra-high purity argon (Ar) gas was used for inert environment during pyrolysis which was supplied by Matheson (Kansas, USA).
2.2 Electrospinning and synthesis of fibermat
Various SiOC fibermat samples were fabricated via electrospinning of polymer solutions, followed by stabilization and pyrolysis at elevated temperatures. The electrospinning setup that was designed and built in the lab consisted of four major parts:47 (1) a syringe which is controlled with a stepper motor works as a feeder for polymer solution; (2) a voltage source (up to 30 kV) that controls the amount of electrical charge applied to the solution; (3) an aluminum roller that works as a fibermat collector; and (4) a microcontroller made of Arduino® that controls the motors' speed and the sensors. The major parts of the setup are illustrated in Fig. 2. | ||
| Fig. 2 Fabrication of SiOC ceramic fibermats via electrospinning process. (a) Electrospinning set-up, (b) as-spun fibermat collected to from the roller, (c) fibermat after crosslinking at ∼300 °C in air, (d) fiber mat after pyrolysis at ∼800 °C in Ar environment, and (e) a 6.35 mm dia. electrode punched out from the ceramic fibermat. | ||
The electrospinning solution was prepared by dissolving 10 wt% of PVP in 7500 mg of iso-propanol; after that preceramic oligomer was added into the solution with a weight ratio of 3:1 to PVP. DCP as a crosslinking catalyst was already dissolved into the preceramic oligomer, which was 1 wt% of the siloxane oligomer. The solution was then stirred for about 2 h to get a homogenous mixture of PVP and preceramic oligomer. The solution was then poured into the feeder syringe. During electrospinning, the feed rate was set at 5 mL h−1 while the voltage supply was maintained at 15 kV. The positive output was connected to the syringe nozzle and the collector was grounded. The nozzle to collector distance was set to 25 cm. The collector was wrapped with an Al foil and a fibermat of 15 × 15 cm2 was collected onto the foil after the electrospinning. The electrospun raw or as-spun fibermat is shown in Fig. 2b.
2.3 Crosslinking and pyrolysis of the fibermat
The electrospun fibermats of the three siloxanes were dried at room temperature overnight and then crosslinked at ∼300 °C in an oven for 8 h in the presence of air. The crosslinked fibermats (Fig. 2c) were then pyrolyzed in an alumina tube furnace at approx. 800 °C for 30 min in flowing Ar gas. The heating rate was approx. 2 °C min−1 and Ar gas flow rate was 5 mL min−1 in the furnace. The crosslinked fibermats were cut into smaller rectangular shape (50 × 25 mm2) to fit in the aluminum ceramic boat that was used to hold the samples in the furnace during pyrolysis. The fibermat polymer-to-ceramic conversion was complete at 800 °C; a representative of SiOC ceramic fibermat is shown in Fig. 2d. The lateral shrinkage during polymer-to-ceramic conversion for DDTS-derived SiOC ceramic fibermat was 40%, whereas for DTDS- and TPTS-derived SiOC ceramic fibermats were 60% and 55% respectively.2.4 Characterization techniques
Several material characterization techniques were employed to determine the morphological, compositional, and chemical conversions at various stages of processing of the fibers-as-spun, crosslinked, and pyrolyzed fibermats. The images of surface features, shapes, and diameters of the fibers in the SiOC ceramic fibermats were investigated using a FEI Nova NanoSEM 450 scanning electron microscope (SEM). An FEI Tecnai Osiris 200 kV (scanning) transmission electron microscope (TEM) was used to obtain morphological and crystallographic information as well as energy dispersive X-ray (EDX) mapping analysis of the ceramic fibermats.To determine the molecular structure and chemical interactions of the ceramic fibermats Raman spectroscopy and Fourier-transform infrared spectroscopy (FTIR) were used. Raman analysis in the range of 800–2000 cm−1 was performed to determine mainly the carbon vibrational modes using a confocal micro-Raman microscope (Horiba Jobin Yvon LabRam ARAMIS) equipped with a HeNe laser source (632.8 nm). The presence and evolution of various chemical functional groups of the siloxane precursors in the fibermats were investigated using a PerkinElmer Spectrum 400 FTIR spectrometer in the range of 500–3500 cm−1.
To further investigate the chemical bonds in each sample, nuclear magnetic resonance (NMR) spectroscopy was used. A Bruker AVANCE 300 spectrometer was used to record solid-state 13C CP MAS and 29Si MAS NMR spectra using following specifications (B0 = 7.0 T, υ0(1H) = 300.29 MHz, υ0(13C) = 75.51 MHz, υ0(29Si) = 59.66 MHz), and a 4 mm Bruker probe and spinning frequency of 10 kHz. 13C CP MAS experiments were recorded with ramped amplitude cross-polarization in the 1H channel to transfer magnetization from 1H to 13C (recycle delay = 3 s, CP contact time = 1 ms, optimized 1H spinal-64 decoupling). Single-pulse 29Si NMR MAS spectra were recorded with recycle delays of 60 s. Liquid-state NMR spectra were recorded on a Bruker AVANCE 300 spectrometer (B0 = 7.0 T, υ0(1H) = 300.13 MHz, υ0(13C) = 75.47 MHz, υ0(29Si) = 59.63 MHz). Chemical shift values were referenced to tetramethyl silane for 13C and 29Si.
X-ray photoelectron spectroscopy (XPS) was carried out to analyze the surface composition using a Thermo Scientific Al Kα+ ion beam (beam energy = 1486.6 eV and spot size = 400 μm) XPS. To remove the surface contamination of the ceramic fiber mats, initial sputtering of the surface with Ar+ at 3.0 keV for 2 min was performed.
2.5 Electrode preparation and electrochemical measurement
Ceramic fibermats were tested electrochemically both as organic electrolyte-based lithium-ion batteries (LIBs) and aqueous supercapacitors electrodes. Electrospun SiOC ceramic fibermats were used as freestanding electrodes in LIBs half-cells. A disk electrode was punched out from the pyrolyzed fibermat (Fig. 2e) with diameter of about 6.35 mm (1/4 inch), which was used as the working electrode. A glass fiber membrane (φ ≈ 19 mm, t ≈ 25 μm) (GE, USA) as separator, pure Li metal (φ ≈ 14.3 mm, t ≈ 75 μm) (Alfa Aesar, USA) as the counter electrode, and approximately 6 drops of 1 M lithium hexafluorophosphate (LiPF6) in (1:1 v/v) dimethyl carbonate (DMC):ethylene carbonate (EC) (Sigma Aldrich, USA) as the electrolyte were used. The cells were assembled in LIR 2032 coin cells in a glove box, and the assembled cells were tested using a multichannel BT2000 Arbin test system (Texas, USA) between 10 mV to 2.5 V vs. Li/Li+. The cells were subjected to symmetric cycling at current densities of 50, 100, 200, 400, 800, 400, 200, 100, 50 mA g−1 for 5 cycles each.
For supercapacitor testing, a three-electrode setup was used. Ceramic fibermats (as an active material) were mixed with 10 wt% carbon black (as a conducting agent) (Alfa Aesar, Massachusetts, USA) and 5 wt% polyvinylidene fluoride (PVDF) (as a binder) (Sigma Aldrich, Missouri, USA) thoroughly; approximately 4–6 drops of N-methyl 2 pyrrolidone (NMP) (Alfa Aesar, Massachusetts, USA) was also added to form a slurry of uniform consistency. The slurry was then pasted on stainless steel (SS) mesh (1 × 1 cm2) using a flat paint brush, followed by drying at 80 °C overnight in an oven. The ceramic coated SS was then used as working electrode in the three-electrode setup, where Pt wire and Ag/AgCl were used as the counter and reference electrode, respectively. 1 M NaCl was used as an electrolyte. A CHI 660 electrochemical workstation (CH Instruments, Inc., Texas, USA) was used to perform cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) of the electrodes. CV and GCD were performed in the potential window of 0–1 V at various scan rates and current densities, respectively. Electrochemical impedance spectroscopy (EIS) was also carried out from 0.01 Hz to 100 kHz at an amplitude of 5 mV. For CV and GCD, the specific capacitance values were calculated using eqn (1) and (2), respectively.49
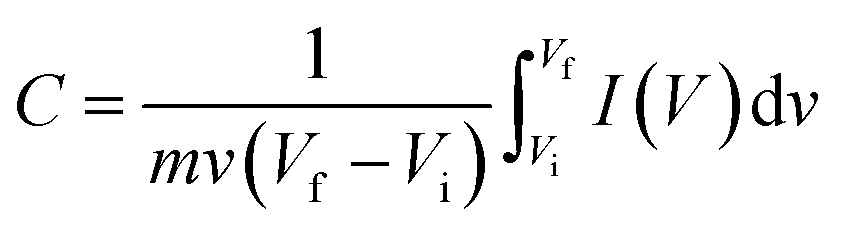 | (1) |
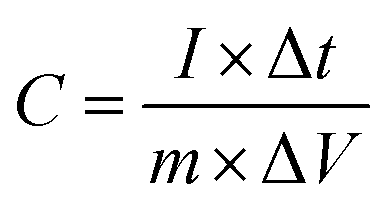 | (2) |
3. Results and discussions
3.1 Structure and morphology of fibermats
SEM and TEM images of the pyrolyzed samples revealed the surface and internal features of the SiOC fibers. Fig. 3 shows that SiOC fibermats fabricated from the three siloxane oligomer samples were largely uniform in diameter along the fiber length. For DDTS-derived SiOC fibers the average fiber diameter was between 1–3 μm, whereas for DTDS- and TPTS-derived SiOC fibers the average dimeter stayed between 0.2–1.5 μm. SiOC fibers derived from the siloxane oligomer with high molecular weight resulted in large diameter fibers compared to low molecular weight precursors. Furthermore, high molecular weight precursor produced thick and dense fibers as can be seen from the inset of the high-resolution SEM images (Fig. 3a). The thinner and hollow fibers derived from the DTDS and TPTS may have resulted from the steric hindrance, and molecular weight difference with the PVP (spinning agent) during the spinning process.47 As a result, in our case of single-nozzle electrospinning, the PVP molecules moved to the inner layers and preceramic polymer stayed in the outer layers, and the separation likely occurred during the crosslinking and pyrolysis processes.50 The core–shell structure of the hollow TPTS-derived SiOC fibers was also confirmed by TEM. In addition, SEM and TEM revealed a smoother surface for DDTS- and DTDS-SiOC compared to TPTS-SiOC. The hollow fibers with bead-on-string structure of the TPTS-SiOC was attributed to the low molecular weight as well as the concentration and viscosity of the spinning solution.51–53 High-resolution TEM images of the pyrolyzed fibermats, illustrated in Fig. S1,‡ suggested the amorphous or featureless structure of the three fiber types. The results of the EDX elemental analyses, also shown in Fig. S1,‡ confirmed the homogenous distribution of the Si, O, and C elements in the ceramic fibers. | ||
| Fig. 3 SEM (top and middle) and corresponding TEM (bottom) images of the electrospun SiOC fibermats. (a–c) The images reveal the formation of thick, smooth, and dense fibers for the SiOC samples produced from preceramic Si oligomers with high molecular weight compared to low molecular weights. | ||
3.2 Compositional analysis
Raman spectra (Fig. 4) show the presence of carbon domains which allows to evaluate corresponding microstructures in the ceramic materials. The D and G vibrational bands at 1342 and ∼1600 cm−1 for the DDTS- and DTDS-derived SiOC, suggested the presence of “free carbon” structures i.e., carbon bonded to carbon and not any other elements in the ceramic. The intense D bands suggested the existence of disordered aromatic rings, whereas G bands were associated with the sp2-hybridized carbon atoms.54 Additionally, under the fitted curves the T (1424 cm−1) and D′′ (∼1490 cm−1) bands indicated the likely presence of disordered sp2–sp3 bonds and amorphous carbons in the SiOC fibers, respectively. However, for TPTS-derived SiOC sample, no pronounced D and G peaks were observed as a strong fluorescence background was produced under the visible laser source (HeNe 632.8 nm). The presence of carbon domains in the TPTS-SiOC fibers were later confirmed using XPS analysis. | ||
| Fig. 4 Comparison of Raman spectra for the SiOC samples. (a) DDTS- and (b) DTDS-derived SiOC samples show integrated carbon D and G bonds, whereas (c) TPTS-derived SiOC show strong fluorescence. | ||
FTIR spectra, plotted in Fig. 5, shows the characteristic absorption bands of the PVP and polymer-to-ceramic conversion of preceramic Si precursors from the as-spun to pyrolyzed stages. For the PVP, as shown in Fig. 5a, as-spun and crosslinked samples exhibited the main peaks at 1660, 1427, 1291, and 570 cm−1, that were assigned to the stretching of C–O, C–H, C–H2, and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.55 The presence of these peaks in both as-spun and crosslinked PVP fibers suggested no noticeable crosslinking happened during heat treatment at ∼300 °C, while after pyrolysis at ∼800 °C these peaks disappeared. DDTS fibers electrospun with PVP (as spinning agent) showed (Fig. 5b) Si–CH3 (∼1260 cm−1), and Si–O–Si (∼1060 cm−1) stretching bonds in the as-spun fibers indicating the obvious presence of Si precursors in these samples.47,56 Si–CHCH2 (∼1660 cm−1) are superimposed with the C–O vibration. Reduction of Si–CH3 and Si–CHCH2 peak intensities in the crosslinked samples suggested crosslinking reactions occurred at approx. 300 °C. After pyrolysis, the two obvious peaks at ∼1060 and ∼800 cm−1 confirmed the presence of Si–O and Si–C bonds, respectively, in the pyrolyzed SiOC fibers.57 DTDS- and TPTS-derived SiOC fibers also showed similar characteristic peaks from as-spun to pyrolyzed stages, which are presented in Fig. 5c and d. However, Si–O bonds were more intense than Si–C bonds, especially in DTDS-derived SiOC fibers (Fig. 5c), indicating the presence of more Si–O bonds as compared to Si–C bonds in the ceramic samples. Note the peaks around 3000–3100 cm−1 for DDTS, which can be attributed to C–H stretching of the aromatic phenyl group.58
O bonds, respectively.55 The presence of these peaks in both as-spun and crosslinked PVP fibers suggested no noticeable crosslinking happened during heat treatment at ∼300 °C, while after pyrolysis at ∼800 °C these peaks disappeared. DDTS fibers electrospun with PVP (as spinning agent) showed (Fig. 5b) Si–CH3 (∼1260 cm−1), and Si–O–Si (∼1060 cm−1) stretching bonds in the as-spun fibers indicating the obvious presence of Si precursors in these samples.47,56 Si–CHCH2 (∼1660 cm−1) are superimposed with the C–O vibration. Reduction of Si–CH3 and Si–CHCH2 peak intensities in the crosslinked samples suggested crosslinking reactions occurred at approx. 300 °C. After pyrolysis, the two obvious peaks at ∼1060 and ∼800 cm−1 confirmed the presence of Si–O and Si–C bonds, respectively, in the pyrolyzed SiOC fibers.57 DTDS- and TPTS-derived SiOC fibers also showed similar characteristic peaks from as-spun to pyrolyzed stages, which are presented in Fig. 5c and d. However, Si–O bonds were more intense than Si–C bonds, especially in DTDS-derived SiOC fibers (Fig. 5c), indicating the presence of more Si–O bonds as compared to Si–C bonds in the ceramic samples. Note the peaks around 3000–3100 cm−1 for DDTS, which can be attributed to C–H stretching of the aromatic phenyl group.58
 | ||
| Fig. 5 FTIR spectra of electrospun (a) PVP, (b) DDTS-, (c) DTDS-, and (d) TPTS-derived samples show the evolution of chemical bonds from the as-spun to pyrolyzed stages. | ||
XPS survey spectra of the electrospun SiOC fibers, presented in Fig. S2,‡ showed similar Si 2p, C 1s, and O 1s peaks, irrespective of the source of the preceramic polymer. The surface composition of the elements was determined by integrating the area under the respective peaks and presented in Table 1. As expected, pyrolyzed PVP fibers mostly showed the presence of carbon (93.06 at%) after heat treatment. DDTS- and TPTS-derived ceramic fibers shows increased level of oxygen in the pyrolyzed samples indicating the inclusion of oxygen during crosslinking of the fibers. All specimens showed a significant amount of carbon, mainly free carbon, also proposed by Raman analysis. However, pyrolyzed samples presented a little amount of nitrogen, which was suspected to have come from PVP. High-resolution XPS spectra, plotted in Fig. 6, shows the bonding of the pyrolyzed SiOC samples derived from various siloxane precursors. Curve fittings were done to the Si 2p, C 1s, and O 1s peaks. The spectra under Si 2p band indicated the presence of SiCO3 (102.1 eV) and SiO4 (103.5 eV) peaks.31 Low intensity of SiCO3 compared to SiO4 for pyrolyzed-DDTS ceramic fibers were due to the low carbon content in the elemental composition. In addition, C–Si (284.0 eV), C–C (284.9 eV), and CO (287.1 eV) were observed in C 1s band.47 The higher amount of carbon in the pyrolyzed-DTDS and pyrolyzed-TPTS fibers lead to higher intensity of C–C and C–Si peaks. The intense C–C peaks also pointed to increased possibility of free carbon phase in the ceramic fibers. O 1s band were fitted with 3 peaks at 532.4, 532.9, and 534.4 eV corresponding to SiO2, O–Si, and C–O, respectively.
| Pyrolyzed samples | Elements (at%) | |||
|---|---|---|---|---|
| C | O | Si | N | |
| Pyrolyzed PVP | 93.06 | 4.84 | — | 2.10 |
| DDTS-derived SiOC | 32.35 | 37.58 | 29.70 | 0.37 |
| DTDS-derived SiOC | 73.80 | 9.42 | 16.09 | 0.68 |
| TPTS-derived SiOC | 58.00 | 19.28 | 21.74 | 0.98 |
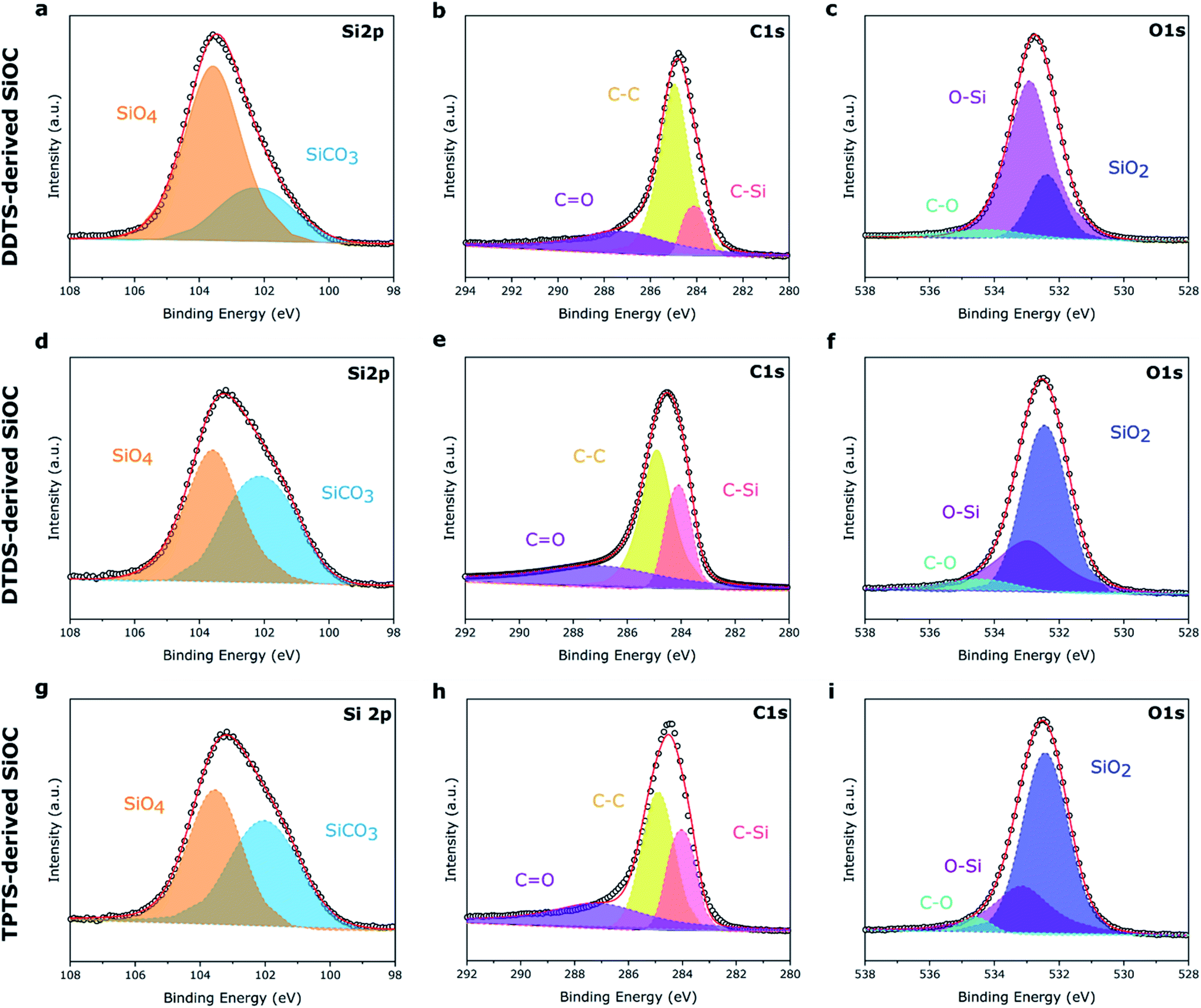 | ||
| Fig. 6 High-res XPS spectra of the three preceramic polymer-derived SiOC fiber samples. (a–c) DDTS-derived SiOC; (d–f) DTDS-derived SiOC; (g–i) TPTS-derived SiOC. | ||
The initial composition of the electrospinning solution was investigated by liquid state NMR. 29Si spectra (Fig. 7a) show for all systems a signal around −3 ppm corresponding to ViMe2SiO environments. In addition, signals are observed at −46.8 ppm and −34.0 ppm in DDTS and TPTS based solutions that can respectively be assigned to Ph2SiO2 and ViMeSiO2 groups. This assignment was confirmed by 2D experiments. For example, 1H–29Si HMBC map of DDTS solution (Fig. S3a‡) shows that the 29Si signal at −47 ppm shows cross-peaks with 1H phenyl signals between 7.5 and 8 ppm while the 29Si signal at −34 ppm shows cross-peaks with 1H vinyl signals between 6 and 5.5 ppm. The evolution of the various preceramic siloxane precursor structures during heat treatment and the resulting SiOC samples were also investigated using solid-state NMR. Spectra were recorded on bulk powders instead of fibers to improve the signal/noise ratio.
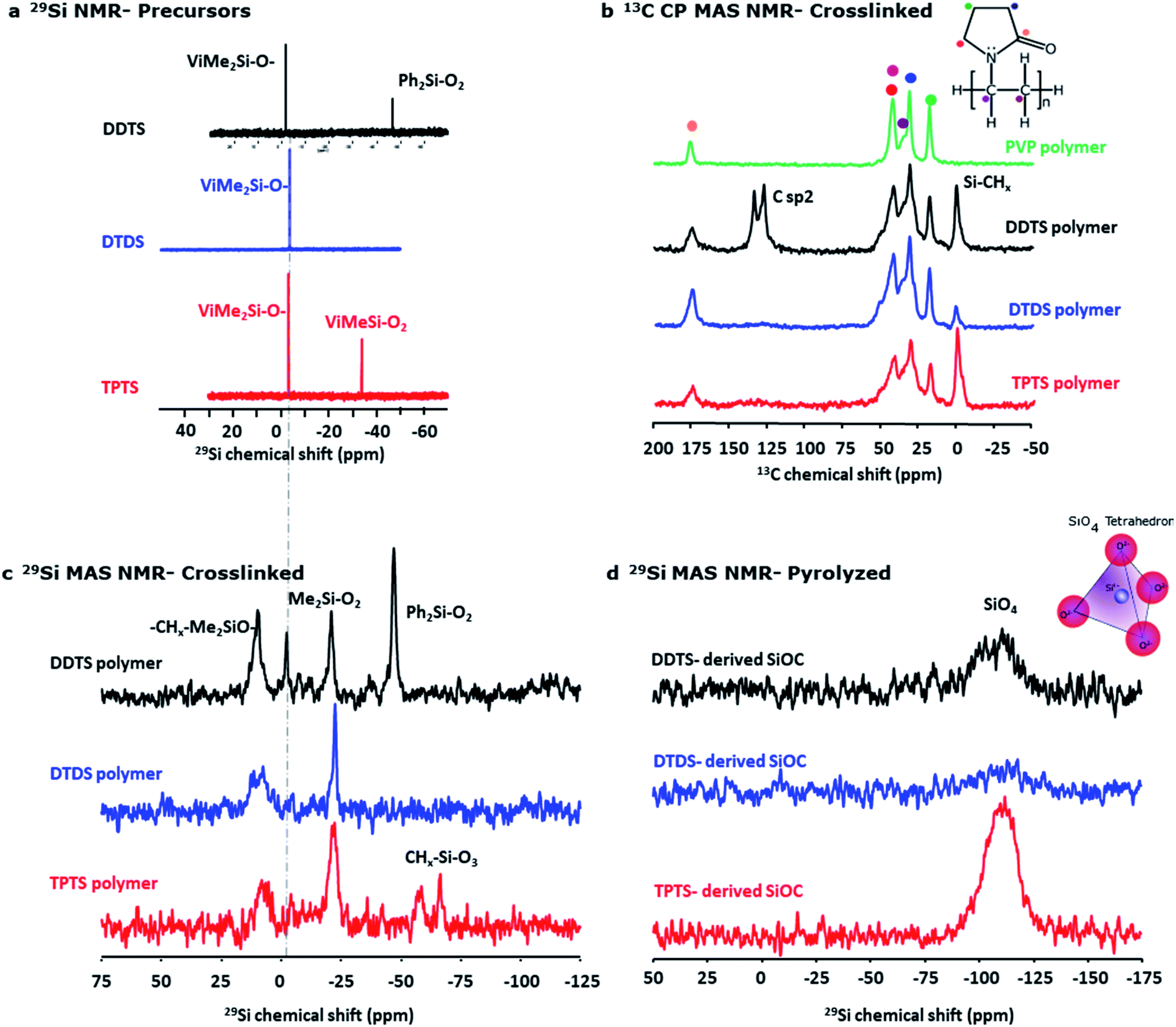 | ||
| Fig. 7 NMR spectra of precursors, crosslinked and pyrolyzed samples. (a) 29Si liquid state NMR of the electrospinning solution, (b) comparison of solid state 29Si MAS NMR spectra of crosslinked polymers of various preceramic Si precursors, (c) 13C CP MAS NMR spectra of crosslinked (at ∼300 °C) polymers in comparison with PVP, (d) comparison of 29Si MAS NMR spectra of pyrolyzed (at ∼800 °C) ceramic powders. | ||
Fig. 7b shows 29Si MAS NMR spectra for crosslinked (at ∼300 °C) polymers and compares the key features of the DDTS, DTDS, and TPTS polymers. Appearance of –CHx–Me2SiO (∼10 ppm) in the polymers indicated effective crosslinking of the vinyl groups occurred in presence of DCP.59 On the other hand, a signal appeared at −21 ppm which was characteristic of Me2SiO2 units in polydimethylsiloxane (PDMS) chains.60,61 This would suggest that part of the Si–CHCH2 groups were replaced by Si–O–Si bonds during heat-treatment as confirmed by the total disappearance of ViMeSiO2 units at −3 ppm in DTDS and TPTS cross-linked systems. Moreover, in the case of TPTS, ViMeSiO2 units observed at −34 ppm in the initial solution disappeared in the cross-linked system while new signals were present around −70 ppm, a chemical shift range characteristic of MeSiO3 environments. Here again, this suggested the replacement of Si–CHCH2 groups by Si–O–Si bonds. In the case of DDTS, Me2SiO2 units were observed but ViMeSiO2 were also present in small quantity in accordance with iR experiments showing remaining vinyl groups after crosslinking (Fig. 5b). The signal corresponding to Ph2SiO2 units (−48 ppm) was still intense after crosslinking suggesting that Si–Ph bonds were preserved. The 29Si MAS spectra of the samples pyrolyzed at 800 °C showed SiO4 bonds (−110.0 ppm) (Fig. 7d), while no Si–C bonds were found.62 This might be due to the fact mostly free C phase had formed during pyrolysis, and very low Si–C bonds were present in the ceramic samples.47 Nevertheless, high-res XPS of the SiOC fibers indicated the presence of Si–C and C–C bonds in the ceramic fibers.
13C cross polarization (CP) MAS NMR spectra in Fig. 7c show strong peaks of Si–CHx indicating the crosslinking of the Si precursor polymers. In the DTDS polymer, sp2 carbons were observed around 130 ppm that corresponded mainly to the phenyl groups. Indeed, from 1H–13C HSQC map of DDTS solution (Fig. S3b‡) cross-peaks with 1H phenyl signals (between 7.5 and 8 ppm) were observed for carbon signals at 127.6, 129.7 and 134.4 ppm, while the cross-peaks with 1H vinyl signals (between 6 and 5.5 ppm) were observed with carbon signals at 132.1 and 139.0 ppm. The absence of a clear 13C peak at 139 ppm in the DTDS cross-linked sample suggested that the proportion of vinyl groups was small compared to phenyl groups. No evidence of crosslinking behavior between PVP molecules and siloxane was observed in all the crosslinked samples when compared with PVP polymer. 13C CP MAS NMR spectrum of the TPTS-derived SiOC confirmed the presence of typical free carbon as shown in Fig. S4.‡ 13C spectra of the remaining SiOC samples were not acquired as it was obvious from the other characterization techniques that all the systems were full of graphite.
3.3 Electrochemical performance
The electrochemical energy storage capability of the precursor derived SiOC fibers were analyzed in Li half-cells. The as-prepared ceramic fibermats were used as electrodes. Fig. 8a–c shows the potential vs. capacity plots for various SiOC fiber electrodes. A closer look at the charge–discharge profiles of the samples at a current density of 50 mA g−1 showed that first cycle had experienced irreversible capacity decay. Among the SiOC electrodes, the DDTS-derived SiOC delivered initial discharge capacity of 1188 and charge capacities of 866 mA h g−1, corresponding to a high coulombic efficiency of over 72%. DTDS-derived SiOC electrode delivered 1332 and 800 mA h g−1, respectively for discharge and charge capacities in the first cycle with 59% efficiency. TPTS-derived SiOC provided 1150 and 636 mA h g−1 for discharge and charge capacities, respectively, with 52% coulombic efficiency. DDTS- and DTDS-SiOC electrodes showed stable response with coulombic efficiency of ∼100% in the second and third cycles, whereas TPTS-SiOC displayed poor reversibility with only ∼83% efficiency. Differential capacity plots, presented in Fig. S5,‡ showed two distinct regions for the SiOC electrodes: a sharp peak around 0.1 V, which corresponded to the Li insertion into the carbons; and a broad region in between 0.2–0.6 V, that was related to Li and Si–O–C phase.27Accordingly, when tested for cycling stability TPTS-SiOC electrode showed poor performance and the charge capacity dropped sharply to 100 mA h g−1 after 50 cycles at 50 mA g−1. For DDTS- and DTDS-SiOC electrodes, the capacities after 50 cycles were 580 and 800 mA h g−1, showing 100% capacity retention for the DTDS-derived SiOC fiber electrode. The distinct fiber morphology and the “free C“ content in the DTDS-SiOC particles provided additional Li-ion storing sights and improved the electrochemical properties of SiOC fibers.63 The rate capability performance of the various SiOC electrodes is presented in Fig. S5.‡ DTDS-SiOC delivered a high capacity of 450 mA h g−1 at a high current density of 800 mA g−1. However, thin diameter and hollow structure of the TPTS-SiOC fibers might have been destroyed during Li-ion insertion/extraction process, which resulted in poor cycleability and rate capability.64 The SEM and TEM images of the SiOC electrodes after cycling in LIBs are shown in Fig. S7,‡ where the broken fibers of the TPTS-SiOC can be seen post-cycling. Whereas poor rate capability performance of the DDTS-SiOC can be attributed to the low C content as well as dense structure of the fibers which limits the ion diffusion. To show the consistent performance of the cells, three Li half-cells of each type were assembled and tested and the performance of all the cells are presented in Fig. S5.‡ Fig. S5(a–c)‡ shows the differential capacity curves of the SiOC electrodes for the initial 3 cycles. The sharp cathodic peak at 0.005 V and the broad anodic peak can be attributed to the lithiation and delithiation of Li+ ions in the amorphous SiOC structure.54,65 The cathodic peaks in the range of 0.05–0.16 V can be attributed to the irreversible reaction in hard carbon present in the SiOC structure.25,65 However, cathodic peaks due to formation of SEI (typically at ∼0.5 V) are less pronounced in the dQ/dV plots. A comparison of the electrochemical performance of the polymer-derived SiOC anodes with reported SiOC anodes for LIBs is presented in Table S1.‡
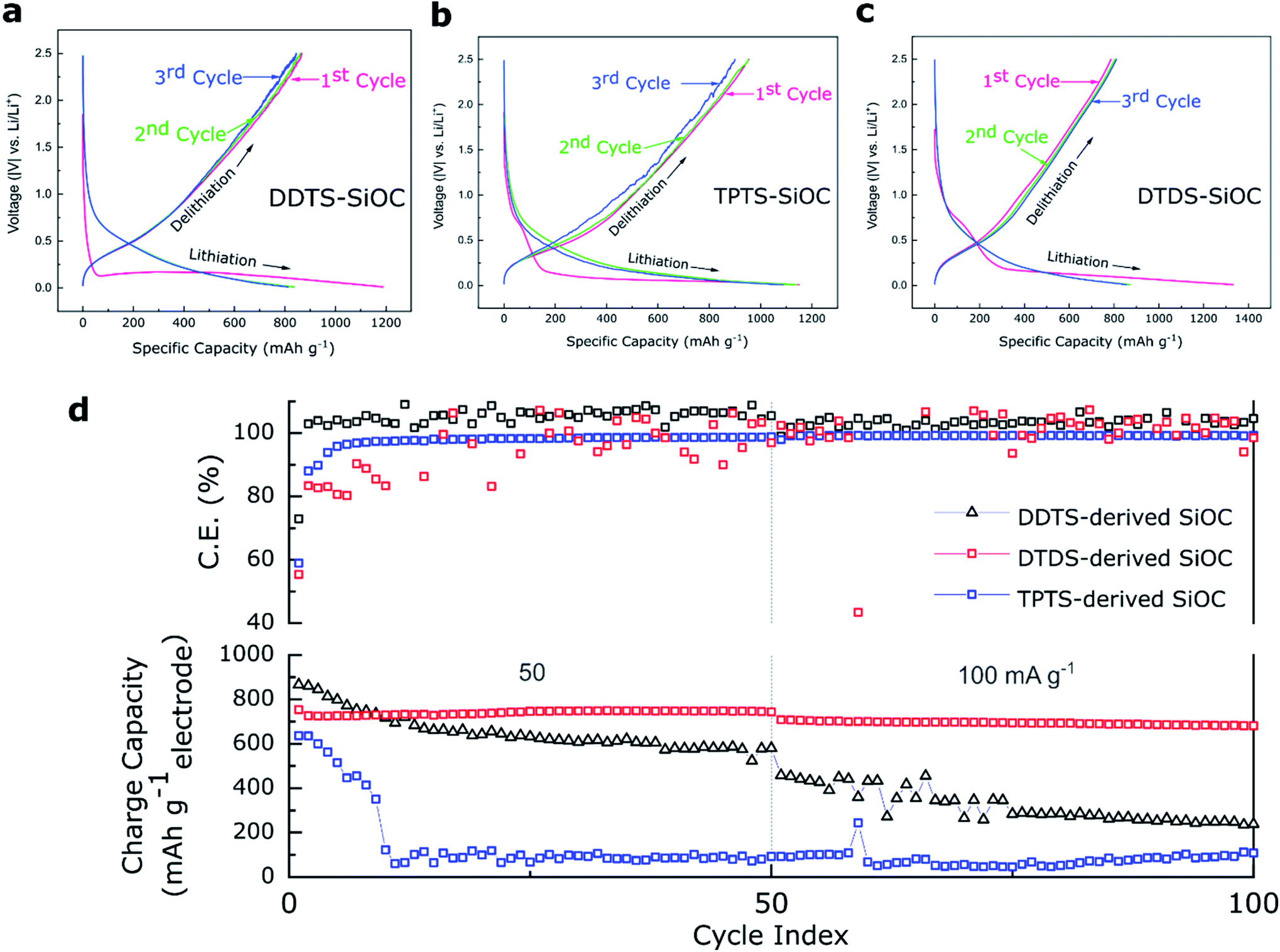 | ||
| Fig. 8 Charge-storage performances of the various precursor-derived SiOC freestanding fibermats in the Li half-cells. (a–c) Voltage profile of the SiOC electrodes show the charge–discharge profiles for the first three cycles, (d) cycling stability data of the samples for 100 cycles. DTDS-SiOC shows stable performance delivering 680 mA h g−1 at 100 mA g−1, whereas DDTS-SiOC and TPTS-SiOC deliver 240 and 110 mA h g−1, respectively after 100 cycles. | ||
The electrochemical storage capabilities of SiOC fibers in supercapacitors were also investigated using three electrode system and 1 M H2SO4 as electrolyte. CV plots obtained for TPTS-derived SiOC electrode at various scan rates of 2 to 500 mV s−1 from 0 to 1 V are shown in Fig. 9a. The almost rectangular shape of the voltammogram profiles indicated mostly the electrochemical double layer capacitive behavior (Type A) of the SiOC electrode during charge storage.66 However, at a low scan rate of 2 mV s−1, a broad redox peak was observed for TPTS-SiOC electrode in between 0.2 and 0.4 V (Fig. S6d‡). The charge storage mechanism at lower scan rate can be contributed to the combination of double-layer capacitance of C in SiOC and pseudocapacitance from Si–O and Si–Si components. The quasi-rectangular shape of the CV plot retained even at higher scan rate of 500 mV s−1, indicating higher ionic diffusivity and charge transfer of the TPTS-SiOC electrode. As a result, the electrode delivered high specific capacitances of 78, 69, 55, 47, and 38 F g−1 at the scan rates of 2, 10, 100, 200, and 500 mV s−1. The areal capacitance of the TPTS-SiOC electrode was also calculated and a high capacitance of 474 mF cm−2 was achieved at 2 mV s−1. The cyclic voltammograms of DTDS- and DDTS-derived SiOC supercapacitor electrodes are presented in Fig. S6.‡ DTDS-SiOC delivered 107 F g−1 at a scan rate of 2 mV s−1 and decreased to 31 F g−1 at 100 mV s−1, where DDTS-SiOC delivered 25 and 3 F g−1 at 2 and 100 mV s−1, respectively. The highest performance of TPTS-SiOC among the three SiOC electrodes was correlated to the lowest diameter and hollow core of the fibers, contributing to a higher electrochemically active area for double-layer capacitor. The BET analysis and avg. pore diameter of the SiOC samples are presented in ESI Section 7.‡ As-anticipated the TPTS-SiOC had a high specific surface area of 235 m2 g−1. In addition, the presence of “free C” contributed to the enhanced electronic conductivity. Fig. 9b demonstrates the charge/discharge profiles of the best performing TPTS-SiOC electrodes at various current densities from 0 to 1 V. The quasi-triangular shape of the GCD curves without any obvious plateau suggested the dominating double-layer capacitive behavior of the electrode, which was in accord with earlier results.67 The triangular shape of the GCD is a typical behavior of highly reversible supercapacitor with constant charge/discharge.68 As a result, TPTS-SiOC delivered 30 F g−1 even at a high current density of 10 A g−1. When tested for cycling stability, the electrode demonstrated ∼100% capacitance retention over 5000 cycles at 3 A g−1. DDTS- and DTDS-derived SiOC electrodes also showed stable cycling ability over 5000 cycles as shown in Fig. S6.‡
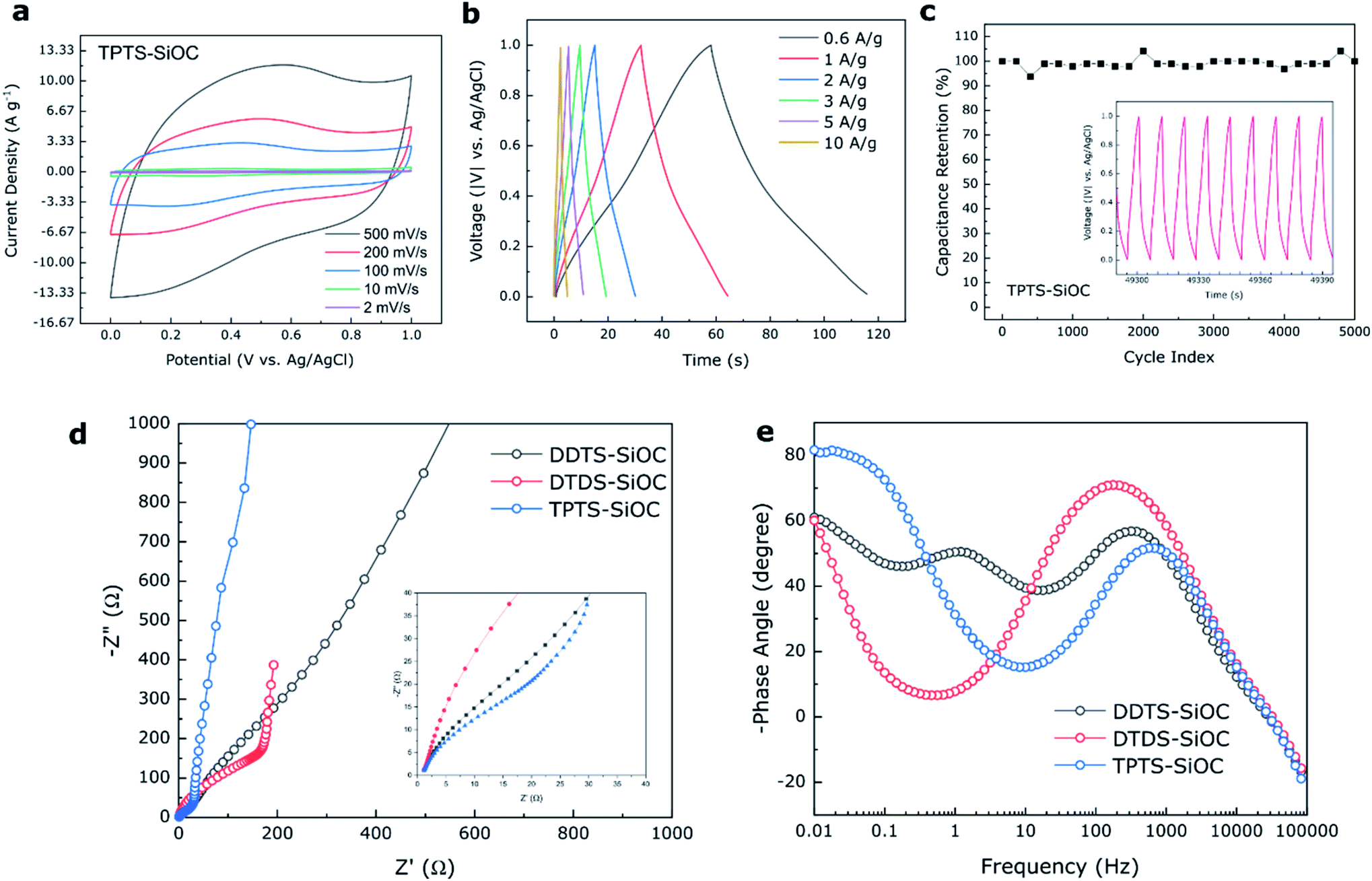 | ||
| Fig. 9 Electrochemical performance of TPTS-derived SiOC supercapacitor electrode in 1 M H2SO4 aqueous electrolyte. (a) CV profile of the electrode at various scan rate; (b) GCD curve of the electrode at various current density; (c) cycling performance of the TPTS-derived SiOC electrode over 5000 cycles shows 100% capacitance retention; (d) comparison of impedance spectra among the various SiOC electrodes; (e) comparison of Bode plots indicating variation of phase angles among the electrodes. | ||
EIS was done for the various SiOC electrodes to further understand the reaction kinetics of the supercapacitor samples. In Fig. 9d, the ohmic resistance, Rs between the aqueous electrolyte and the electrode was measured in the high frequency region where the curves intercept the Z′ axis. The semicircle in medium frequency region was attributed to the charge transfer resistance (RCT) of the electrolyte–electrode interface, and the inclined lines in the low frequency region corresponded to the ion diffusion into the SiOC electrode materials.54,69 An equivalent circuit was obtained for the electrodes, as shown in Fig. S6,‡ from which the calculated RCT for TPTS-SiOC electrode (6 Ω) was much lower than the DDTS-SiOC (15 Ω) and DTDS-SiOC (50 Ω) electrodes. In the low frequency region, the near vertical line to the real axis of the TPTS-SiOC electrode corresponded to the ideal capacitive behavior.70 Furthermore, the Bode plot (Fig. 9e) showed that TPTS-SiOC had the nearest phase angle (80.5°) to 90°, indicating the best capacitive behavior among the SiOC electrodes.71 At phase angle of 45°, the relaxation time constant, τo (τo = 1/fo) were measured to be 2.61, 6.81, and 56.18 ms for the TPTS-SiOC, DDTS-SiOC, and DTDS-SiOC electrodes, respectively. The significantly lower time constant of the TPTS-SiOC electrode confirmed the fast ion diffusion and transport characteristic as a supercapacitor electrode.
4. Conclusion
In summary, we have presented a method to fabricate scalable, self-supporting electrodes from precursor-based SiOC fibers utilizing three unique siloxane precursors. The electrospinning of the short-chain siloxane oligomers was not achievable without the addition of a spinning agent (such as PVP). In addition, the precursor-to-ceramic yield of the fibers was also observed to be lower than some of the previously reported polymer-derived ceramic materials. This phenomenon suggested limited thermal crosslinking of the fibers in the presence of 1 wt% DCP. The electron microscopy of the SiOC fibers featured rigid surface structures and small diameters (0.2–3 μm). Raman, XPS, FTIR, and NMR characterization techniques outlined the polymer to ceramic conversion stages. 29Si MAS NMR spectra of the SiOC fibers showed only SiO4 bonds, indicating mostly free C phase had formed during pyrolysis with a low amount of Si–C bonds in the ceramic samples. The amorphous SiOC structures, comprised of free carbon phase and Si–O–C mixed bonds of DTDS-derived SiOC contributed to high reversibility of Li storage. The free carbon phase served as electron conductor as well as the major electrochemically active sites for Li ions, while amorphous Si–O–C network contributed to minor Li storage. In terms of electrochemical properties, the SiOC electrodes displayed excellent capacity with a high coulombic efficiency in LIBs. As supercapacitor electrodes, superior cycleability of 100% capacitance retention over 5000 cycles was achieved. The as-prepared SiOC fiber mats can provide highly efficient, high-energy, and high-power electrodes and reduce the total weight of the electrochemical energy storage devices.Author contributions
S. M. carried out the preparation of all samples, perform characterization, data analysis and drafted the manuscript; F. R. and C. G. contributed to perform NMR characterizations, data analysis and drafting of the manuscript; G. S. conceived the idea, designed the project and helped with drafting the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
Financial support from National Science Foundation (NSF) Partnerships for International Research and Education (PIRE) grant number 1743701 is gratefully acknowledged. The authors thank Davi Marcelo Soares for his help in developing the electrospinning setup. The research was performed in part in the Nebraska Nanoscale Facility: National Nanotechnology Coordinated Infrastructure and the Nebraska Center for Materials and Nanoscience (and/or NERCF), which are supported by the National Science Foundation under Award ECCS: 2025298, and the Nebraska Research Initiative.References
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, A review of advanced and practical lithium battery materials, J. Mater. Chem., 2011, 21(27), 9938–9954 RSC
.
- M.-S. Balogun, W. Qiu, Y. Luo, H. Meng, W. Mai, A. Onasanya, T. K. Olaniyi and Y. Tong, A review of the development of full cell lithium-ion batteries: the impact of nanostructured anode materials, Nano Res., 2016, 9(10), 2823–2851 CrossRef CAS
- Poonam, K. Sharma, A. Arora and S. K. Tripathi, Review of supercapacitors: materials and devices, J. Electrochem. Energy Convers. Storage, 2019, 21, 801–825 CrossRef
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Review on supercapacitors: technologies and materials, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef
- T. H. Hwang, Y. M. Lee, B.-S. Kong, J.-S. Seo and J. W. Choi, Electrospun Core–Shell Fibers for Robust Silicon Nanoparticle-Based Lithium Ion Battery Anodes, Nano Lett., 2012, 12(2), 802–807 CrossRef CAS PubMed
- M. A. Rahman, G. Song, A. I. Bhatt, Y. C. Wong and C. Wen, Nanostructured Silicon Anodes for High-Performance Lithium-Ion Batteries, Adv. Funct. Mater., 2016, 26(5), 647–678 CrossRef CAS
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review, Adv. Energy Mater., 2014, 4(1), 1300882 CrossRef
- W. Tao, P. Wang, Y. You, K. Park, C.-Y. Wang, Y.-K. Li, F.-F. Cao and S. Xin, Strategies for improving the storage performance of silicon-based anodes in lithium-ion batteries, Nano Res., 2019, 12(8), 1739–1749 CrossRef CAS
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, Size-Dependent Fracture of Silicon Nanoparticles During Lithiation, ACS Nano, 2012, 6(2), 1522–1531 CrossRef CAS PubMed
- T. M. Higgins, S.-H. Park, P. J. King, C. Zhang, N. McEvoy, N. C. Berner, D. Daly, A. Shmeliov, U. Khan, G. Duesberg, V. Nicolosi and J. N. Coleman, A Commercial Conducting Polymer as Both Binder and Conductive Additive for Silicon Nanoparticle-Based Lithium-Ion Battery Negative Electrodes, ACS Nano, 2016, 10(3), 3702–3713 CrossRef CAS PubMed
- J. Park, S. Suh, S. Jeong and H.-J. Kim, New approach for the high electrochemical performance of silicon anode in lithium-ion battery: a rapid and large surface treatment using a high-energy pulsed laser, J. Power Sources, 2021, 491, 229573 CrossRef CAS
- D. Lin, Z. Lu, P.-C. Hsu, H. R. Lee, N. Liu, J. Zhao, H. Wang, C. Liu and Y. Cui, A high tap density secondary silicon particle anode fabricated by scalable mechanical pressing for lithium-ion batteries, Energy Environ. Sci., 2015, 8(8), 2371–2376 RSC
- X. H. Liu, L. Q. Zhang, L. Zhong, Y. Liu, H. Zheng, J. W. Wang, J.-H. Cho, S. A. Dayeh, S. T. Picraux, J. P. Sullivan, S. X. Mao, Z. Z. Ye and J. Y. Huang, Ultrafast Electrochemical Lithiation of Individual Si Nanowire Anodes, Nano Lett., 2011, 11(6), 2251–2258 CrossRef CAS PubMed
- B. Wang, J. Ryu, S. Choi, X. Zhang, D. Pribat, X. Li, L. Zhi, S. Park and R. S. Ruoff, Ultrafast-Charging Silicon-Based Coral-Like Network Anodes for Lithium-Ion Batteries with High Energy and Power Densities, ACS Nano, 2019, 13(2), 2307–2315 CrossRef CAS PubMed
- M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Silicon Nanotube Battery Anodes, Nano Lett., 2009, 9(11), 3844–3847 CrossRef CAS PubMed
- X. H. Liu, H. Zheng, L. Zhong, S. Huang, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J.-H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. Wang, S. X. Mao, Z. Z. Ye, S. Zhang and J. Y. Huang, Anisotropic Swelling and Fracture of Silicon Nanowires during Lithiation, Nano Lett., 2011, 11(8), 3312–3318 CrossRef CAS PubMed
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, High-performance lithium battery anodes using silicon nanowires, Nat. Nanotechnol., 2008, 3(1), 31–35 CrossRef CAS PubMed
- S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo and H.-K. Liu, Highly Reversible Lithium Storage in Spheroidal Carbon-Coated Silicon Nanocomposites as Anodes for Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2006, 45(41), 6896–6899 CrossRef CAS PubMed
- M. Ko, S. Chae, J. Ma, N. Kim, H.-W. Lee, Y. Cui and J. Cho, Scalable synthesis of silicon-nanolayer-embedded graphite for high-energy lithium-ion batteries, Nat. Energy, 2016, 1(9), 16113 CrossRef CAS
- G. Liang, X. Qin, J. Zou, L. Luo, Y. Wang, M. Wu, H. Zhu, G. Chen, F. Kang and B. Li, Electrosprayed silicon-embedded porous carbon microspheres as lithium-ion battery anodes with exceptional rate capacities, Carbon, 2018, 127, 424–431 CrossRef CAS
- K. Wang, S. Pei, Z. He, L.-a. Huang, S. Zhu, J. Guo, H. Shao and J. Wang, Synthesis of a novel porous silicon microsphere@carbon core–shell composite via in situ MOF coating for lithium ion battery anodes, Chem. Eng. J., 2019, 356, 272–281 CrossRef CAS
- P. Li, J.-Y. Hwang and Y.-K. Sun, Nano/Microstructured Silicon–Graphite Composite Anode for High-Energy-Density Li-Ion Battery, ACS Nano, 2019, 13(2), 2624–2633 CAS
- L. Zhang, C. Wang, Y. Dou, N. Cheng, D. Cui, Y. Du, P. Liu, M. Al-Mamun, S. Zhang and H. Zhao, A Yolk–Shell Structured Silicon Anode with Superior Conductivity and High Tap Density for Full Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2019, 58(26), 8824–8828 CrossRef CAS PubMed
- F. Hiroshi, O. Hisashi, H. Takakazu and K. Kiyoshi, Preparation of Microporous Si–O–C Composite Material and Its Lithium Storage Capability, Chem. Lett., 2009, 38(1), 86–87 CrossRef
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, A Si–O–C Composite Anode: High Capability and Proposed Mechanism of Lithium Storage Associated with Microstructural Characteristics, ACS Appl. Mater. Interfaces, 2010, 2(4), 998–1008 CrossRef CAS PubMed
- J. Shen, D. Ahn and R. Raj, C-rate performance of silicon oxycarbide anodes for Li+ batteries enhanced by carbon nanotubes, J. Power Sources, 2011, 196(5), 2875–2878 CrossRef CAS
- X. Liu, M.-C. Zheng and K. Xie, Mechanism of lithium storage in
Si–O–C composite anodes, J. Power Sources, 2011, 196(24), 10667–10672 CrossRef CAS
- S. Sen, S. J. Widgeon, A. Navrotsky, G. Mera, A. Tavakoli, E. Ionescu and R. Riedel, Carbon substitution for oxygen in silicates in planetary interiors, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(40), 15904–15907 CrossRef CAS PubMed
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Lithium insertion into carbon-rich SiOC ceramics: influence of pyrolysis temperature on electrochemical properties, J. Power Sources, 2013, 244, 450–455 CrossRef CAS
- P. Lv, H. Zhao, C. Gao, Z. Du, J. Wang and X. Liu, SiOx–C dual-phase glass for lithium ion battery anode with high capacity and stable cycling performance, J. Power Sources, 2015, 274, 542–550 CrossRef CAS
- L. David, R. Bhandavat, U. Barrera and G. Singh, Silicon oxycarbide glass-graphene composite paper electrode for long-cycle lithium-ion batteries, Nat. Commun., 2016, 7(1), 10998 CrossRef CAS PubMed
- M. Halim, C. Hudaya, A. Y. Kim and J. K. Lee, Phenyl-rich silicone oil as a precursor for SiOC anode materials for long-cycle and high-rate lithium ion batteries, J. Mater. Chem. A, 2016, 4(7), 2651–2656 RSC
- S. H. Lee, C. Park, K. Do and H. Ahn, Maximizing the utilization of active sites through the formation of native nanovoids of silicon oxycarbide as anode materials in lithium-ion batteries, Energy Storage Materials, 2021, 35, 130–141 CrossRef
- J. R. Dahn, A. M. Wilson, W. Xing and G. A. Zank, Electrodes for lithium ion batteries using polysilazanes ceramic with lithium, US Pat., US5631106A, 1997 Search PubMed
- J. R. Dahn, K. Eguchi, A. M. Wilson, W. Xing and G. A. Zank, Electrodes for lithium ion batteries using polysiloxanes, US Pat., US5824280A, 1998 Search PubMed
- P. E. Sanchez-Jimenez and R. Raj, Lithium Insertion in Polymer-Derived Silicon Oxycarbide Ceramics, J. Am. Ceram. Soc., 2010, 93(4), 1127–1135 CrossRef CAS
- D. Ahn and R. Raj, Cyclic stability and C-rate performance of amorphous silicon and carbon based anodes for electrochemical storage of lithium, J. Power Sources, 2011, 196(4), 2179–2186 CrossRef CAS
- M. Graczyk-Zajac, D. Vrankovic, P. Waleska, C. Hess, P. V. Sasikumar, S. Lauterbach, H.-J. Kleebe and G. D. Sorarù, The Li-storage capacity of SiOC glasses with and without mixed silicon oxycarbide bonds, J. Mater. Chem. A, 2018, 6(1), 93–103 RSC
- J. Kaspar, M. Graczyk-Zajac, S. Choudhury and R. Riedel, Impact of the electrical conductivity on the lithium capacity of polymer-derived silicon oxycarbide (SiOC) ceramics, Electrochim. Acta, 2016, 216, 196–202 CrossRef CAS
- M. Graczyk-Zajac, L. M. Reinold, J. Kaspar, P. V. W. Sasikumar, G.-D. Soraru and R. Riedel, New Insights into Understanding Irreversible and Reversible Lithium Storage within SiOC and SiCN Ceramics, Nanomaterials, 2015, 5(1), 233–245 CrossRef CAS PubMed
- S.-H. Baek, L. M. Reinold, M. Graczyk-Zajac, R. Riedel, F. Hammerath, B. Büchner and H.-J. Grafe, Lithium dynamics in carbon-rich polymer-derived SiCN ceramics probed by nuclear magnetic resonance, J. Power Sources, 2014, 253, 342–348 CrossRef CAS
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Carbon-rich SiOC anodes for lithium-ion batteries: Part II. Role of thermal cross-linking, Solid State Ionics, 2012, 225, 527–531 CrossRef CAS
- F. Anton, Process and apparatus for preparing artificial threads, US Pat., US1975504A, 1934 Search PubMed
- F. Anton, Method and apparatus for spinning, US Pat., US2160962A, 1939 Search PubMed
- Y. Xu, Y. Zhu, F. Han, C. Luo and C. Wang, 3D Si/C Fiber Paper Electrodes Fabricated Using a Combined
Electrospray/Electrospinning Technique for Li-Ion Batteries, Adv. Energy Mater., 2015, 5(1), 1400753 CrossRef
- Q. Xiao, Q. Zhang, Y. Fan, X. Wang and R. A. Susantyoko, Soft silicon anodes for lithium ion batteries, Energy Environ. Sci., 2014, 7(7), 2261–2268 RSC
- Z. Ren, C. Gervais and G. Singh, Fabrication and characterization of silicon oxycarbide fibre-mats via electrospinning for high temperature applications, RSC Adv., 2020, 10(63), 38446–38455 RSC
- Z. Ren and G. Singh, Nonoxide polymer-derived CMCs for “super” turbines, Am. Ceram. Soc. Bull., 2019, 98(3), 2–7 Search PubMed
- N. L. Torad, R. R. Salunkhe, Y. Li, H. Hamoudi, M. Imura, Y. Sakka, C.-C. Hu and Y. Yamauchi, Electric Double-Layer Capacitors Based on Highly Graphitized Nanoporous Carbons Derived from ZIF-67, Chem.–Eur. J., 2014, 20(26), 7895–7900 CrossRef CAS PubMed
- S. Homaeigohar, Y. Davoudpour, Y. Habibi and M. Elbahri, The Electrospun Ceramic Hollow Nanofibers, Nanomaterials, 2017, 7(11), 383 CrossRef PubMed
- J. F. Kim, J. H. Kim, Y. M. Lee and E. Drioli, Thermally induced phase separation and electrospinning methods for emerging membrane applications: a review, AIChE J., 2016, 62(2), 461–490 CrossRef CAS
- Y. Y. Zhao, Q. B. Yang, X. F. Lu, C. Wang and Y. Wei, Study on correlation of morphology of electrospun products of polyacrylamide with ultrahigh molecular weight, J. Polym. Sci., Part B: Polym. Phys., 2005, 43(16), 2190–2195 CrossRef CAS
- A. Koski, K. Yim and S. Shivkumar, Effect of molecular weight on fibrous PVA produced by electrospinning, Mater. Lett., 2004, 58(3), 493–497 CrossRef CAS
- B. Dong, Y. Han, T. Wang, Z. Lei, Y. Chen, F. Wang, H. Abadikhah, S. A. Khan, L. Hao, X. Xu, R. Cao, L. Yin and S. Agathopoulos, Hard SiOC Microbeads as a High-Performance Lithium-Ion Battery Anode, ACS Appl. Energy Mater., 2020, 3(10), 10183–10191 CrossRef CAS
- Kamaruddin, D. Edikresnha, I. Sriyanti, M. M. Munir and Khairurrijal, Synthesis of Polyvinylpyrrolidone (PVP)-Green Tea Extract Composite Nanostructures using Electrohydrodynamic Spraying Technique, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 202, 012043 CrossRef
- Y. Zhang, Y. Li, J. Shao and C. Zou, Fabrication of superhydrophobic fluorine-free films on cotton fabrics through plasma-induced grafting polymerization of 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane, Surf. Coat. Technol., 2015, 276, 16–22 CrossRef CAS
- M. Naviroj, S. M. Miller, P. Colombo and K. T. Faber, Directionally aligned macroporous SiOC via freeze casting of preceramic polymers, J. Eur. Ceram. Soc., 2015, 35(8), 2225–2232 CrossRef CAS
- J. Wang, C. He, Y. Lin and T. S. Chung, Studies on the thermal stability of F- and non-F-containing ladder polyepoxysilsesquioxanes by TGA-FTIR, Thermochim. Acta, 2002, 381(1), 83–92 CrossRef CAS
- S. Roualdes, R. Berjoan and J. Durand, 29Si NMR and Si 2p XPS correlation in polysiloxane membranes prepared by plasma enhanced chemical vapor deposition, Sep. Purif. Technol., 2001, 25(1), 391–397 CrossRef CAS
- B. Julián, C. Gervais, E. Cordoncillo, P. Escribano, F. Babonneau and C. Sanchez, Synthesis and Characterization of Transparent PDMS–Metal–Oxo Based Organic–Inorganic Nanocomposites, Chem. Mater., 2003, 15(15), 3026–3034 CrossRef
- R. K. Harris and M. L. Robins, 29Si nuclear magnetic resonance studies of oligomeric and polymeric siloxanes: 4. Chemical shift effects of end-groups, Polymer, 1978, 19(10), 1123–1132 CrossRef CAS
- D.-S. Ruan, Y.-L. Li, L. Wang, D. Su and F. Hou, Fabrication of silicon oxycarbide fibers from alkoxide solutions along the sol–gel process, J. Sol-Gel Sci. Technol., 2010, 56(2), 184–190 CrossRef CAS
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H.-J. Kleebe and R. Riedel, Stable SiOC/Sn Nanocomposite Anodes for Lithium-Ion Batteries with Outstanding Cycling Stability, Adv. Funct. Mater., 2014, 24(26), 4097–4104 CrossRef CAS
- M. Ma, H. Wang, X. Li, K. Peng, L. Xiong and X. Du, Free-standing SiOC/nitrogen-doped carbon fibers with highly capacitive Li storage, J. Eur. Ceram. Soc., 2020, 40(15), 5238–5246 CrossRef CAS
- H. Fukui, N. Nakata, K. Dokko, B. Takemura, H. Ohsuka, T. Hino and K. Kanamura, Lithiation and Delithiation of Silicon Oxycarbide Single Particles with a Unique Microstructure, ACS Appl. Mater. Interfaces, 2011, 3(7), 2318–2322 CrossRef CAS PubMed
- E. Raymundo-Piñero, M. Cadek and F. Béguin, Tuning Carbon Materials for Supercapacitors by Direct Pyrolysis of Seaweeds, Adv. Funct. Mater., 2009, 19(7), 1032–1039 CrossRef
- P. Pazhamalai, K. Krishnamoorthy, S. Sahoo, V. K. Mariappan and S.-J. Kim, Carbothermal conversion of siloxene sheets into silicon-oxy-carbide lamellae for high-performance supercapacitors, Chem. Eng. J., 2020, 387, 123886 CrossRef CAS
- J.-S. M. Lee, T.-H. Wu, B. M. Alston, M. E. Briggs, T. Hasell, C.-C. Hu and A. I. Cooper, Porosity-engineered carbons for supercapacitive energy storage using conjugated microporous polymer precursors, J. Mater. Chem. A, 2016, 4(20), 7665–7673 RSC
- Z. Sang, X. Yan, L. Wen, D. Su, Z. Zhao, Y. Liu, H. Ji, J. Liang and S. X. Dou, A graphene-modified flexible SiOC ceramic cloth for high-performance lithium storage, Energy Storage Materials, 2020, 25, 876–884 CrossRef
- L. Niu, Q. Li, X. Chen, F. Wei, H. Wang and H. Zhang, Study on the Impedance Characteristic and Electrocatalytic Activity of Platinum-Modified Polyaniline Film, J. Chin. Chem. Soc., 2003, 50(6), 1165–1170 CrossRef CAS
- Y. Shao, J. Li, Y. Li, H. Wang, Q. Zhang and R. B. Kaner, Flexible quasi-solid-state planar micro-supercapacitor based on cellular graphene films, Mater. Horiz., 2017, 4(6), 1145–1150 RSC
Footnotes |
| † In Memoriam Prof. Masaki Narisawa who passed away on 27 January 2021. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05968h |
| This journal is © The Royal Society of Chemistry 2021 |