
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinylene-bridged donor–acceptor type porous organic polymers for enhanced photocatalysis of amine oxidative coupling reactions under visible light†
Bang Wua,
Xinyue Jianga,
Yang Liua,
Qiu-Yan Li *a,
Xinsheng Zhaob and
Xiao-Jun Wang*a
*a,
Xinsheng Zhaob and
Xiao-Jun Wang*a
aJiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, P. R. China. E-mail: xjwang@jsnu.edu.cn; qyli@jsnu.edu.cn
bSchool of Physics and Electronic Engineering, Jiangsu Normal University, Xuzhou 221116, P. R. China
First published on 14th October 2021
Abstract
Porous organic polymers (POPs), owing to their abundant porosity, high stability and well-tunable properties, are promising candidates as heterogeneous photocatalysts for organic transformations. Here we report two vinylene-bridged donor–acceptor (D–A) structural POPs (TpTc-POP and TbTc-POP) that are facilely constructed by the electron-rich triarylamine and electron-deficient tricyanomesitylene as key building blocks by the organic base catalyzed Knoevenagel condensation. Both TpTc-POP and TbTc-POP possess hierarchical meso- and micro-pores with a high surface area. Furthermore, the unsubstituted vinylene linkages of D–A moieties in their polymer backbones extend their π-conjugation and render their broad absorption range in the visible-light region. Thus, these DA-POPs exhibited highly effective photocatalytic activities for aerobic oxidative coupling of amines to imines under visible light irradiation. This study shows the great potential of conjugated POPs with a D–A structural feature in designing highly efficient and active heterogeneous photocatalytic systems.
Introduction
Visible light photoredox catalysis has gained remarkably extensive research attention in the recent decades, because it provides a sustainable and environmentally friendly method for a wide range of organic transformations.1 Since a large proportion of organic substrates cannot absorb photons in the visible region, a variety of photocatalysts that absorb strongly in the visible spectrum are often employed in these photochemical processes, including noble metal Ru2+ or Ir3+ complexes,2 as well as organic dyes.3 However, these homogeneous catalytic systems would suffer from several inherent drawbacks to some extent, such as the high price and scarcity of precious metals, the photobleaching of organic dyes, and the low recyclability and reusability of photocatalysts, which impede their practical applications in scale-up synthesis. The hybridization of these photocatalysts with solid-state materials, such as metal–organic frameworks and dye doped metal oxides, only partially alleviates the difficulty of recyclability.4 Consequently, the development of efficient heterogeneous photocatalysts that are low cost and recyclable, have outstanding catalytic activity and are free of precious metals is highly desirable.Porous organic polymers (POPs) as a novel class of porous materials are constructed by robust covalent bonds between various organic building blocks, featuring high porosity, structural tunability and diversity, together with highly physicochemical stability.5 They have been widely investigated in a plethora of applications, such as gas sorption/separation, chemical sensing, heterogeneous catalysis and water treatment.5,6 Among them, conjugated POPs with extended π-conjugated skeletons have emerged as a versatile platform for efficient photocatalytic organic transformations.7 In particular, the simultaneous introduction of electron donor and acceptor (D–A) building units into their conjugated polymer backbones can greatly enhance the visible-light harvesting capability and charge separation efficiency of photogenerated carriers, thereby resulting in an improvement of photocatalytic performance.7,8 Moreover, these properties also are closely related to the intrinsic chemical linkage of D–A building units in conjugated POPs.
Among the various linkages in POPs, sp2 carbon vinylene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linkage that can enhance both of the chemical stability and π-electron delocalization of conjugated polymer skeletons has witnessed a rapidly increasing attention since the first report in 2016.9,10 These unique features make vinylene-linked POPs emerging as promising heterogeneous photocatalysts in a diverse array of photocatalytic applications along with improved photocatalytic performance in comparison to their analogues with other dynamic linkages.11 For example, Zhang and co-workers employed electron-deficient tricyanomesitylene moiety as a key building block to obtain various olefin-bridged conjugated porous polymers or covalent organic frameworks, which exhibited highly photocatalytic activities for water-splitting reactions or aerobic oxidation of arylboronic acids.12 Wang's group utilized electron-accepting benzothiadiazole and electron-donating pyrene to construct a donor–acceptor sp2-carbon-linked covalent organic framework for efficient photocatalytic thioamide cyclization and oxidative amine coupling.13
C linkage that can enhance both of the chemical stability and π-electron delocalization of conjugated polymer skeletons has witnessed a rapidly increasing attention since the first report in 2016.9,10 These unique features make vinylene-linked POPs emerging as promising heterogeneous photocatalysts in a diverse array of photocatalytic applications along with improved photocatalytic performance in comparison to their analogues with other dynamic linkages.11 For example, Zhang and co-workers employed electron-deficient tricyanomesitylene moiety as a key building block to obtain various olefin-bridged conjugated porous polymers or covalent organic frameworks, which exhibited highly photocatalytic activities for water-splitting reactions or aerobic oxidation of arylboronic acids.12 Wang's group utilized electron-accepting benzothiadiazole and electron-donating pyrene to construct a donor–acceptor sp2-carbon-linked covalent organic framework for efficient photocatalytic thioamide cyclization and oxidative amine coupling.13
However, in this regard, there are still only limited reports regarding vinylene-bridged donor–acceptor structural POPs to date. Thus, it is in high demand to couple various polyarene building blocks into these POPs in order to finely tune their photophysical and electronic properties. Here, we report two vinylene-linked D–A structural POPs composed of triarylamine donor and tricyanomesitylene acceptor (termed as TpTc-POP and TbTc-POP). Due to the extended π-conjugation of D–A units in polymer backbones, they exhibited highly photocatalytic activities for visible-light-driven aerobic oxidative coupling of amines to imines.
Results and discussion
Triphenylamine (TPA) and its derivatives are well-known photoactive molecules along with excellent electron-donating abilities, and they have been widely used in various optoelectronic materials.14 Thus, electron-rich TPA motif based aldehydes [i.e. tris(4-formylphenyl)amine (TFPA) or tris[1-(1′-formyl-4,4′-biphenyl)]amine (TFBA)] was used to readily react with electron-deficient tricyanomesitylene (TCM) by organic base piperidine catalyzed Knoevenagel condensation at 150 °C for 72 h in DMF, resulting in the formation of two vinylene-bridged D–A structural POPs TpTc-POP and TbTc-POP as depicted in Scheme 1. Besides, in order for comparison, the model compound (termed as DpTc) and control POP (termed as TfTc-POP) were prepared by reacting TCM with p-(diphenylamino)benzaldehyde and 1,3,5-tris(p-formylphenyl)benzene under the similar reaction conditions, respectively (Schemes S1 and S2 in ESI†).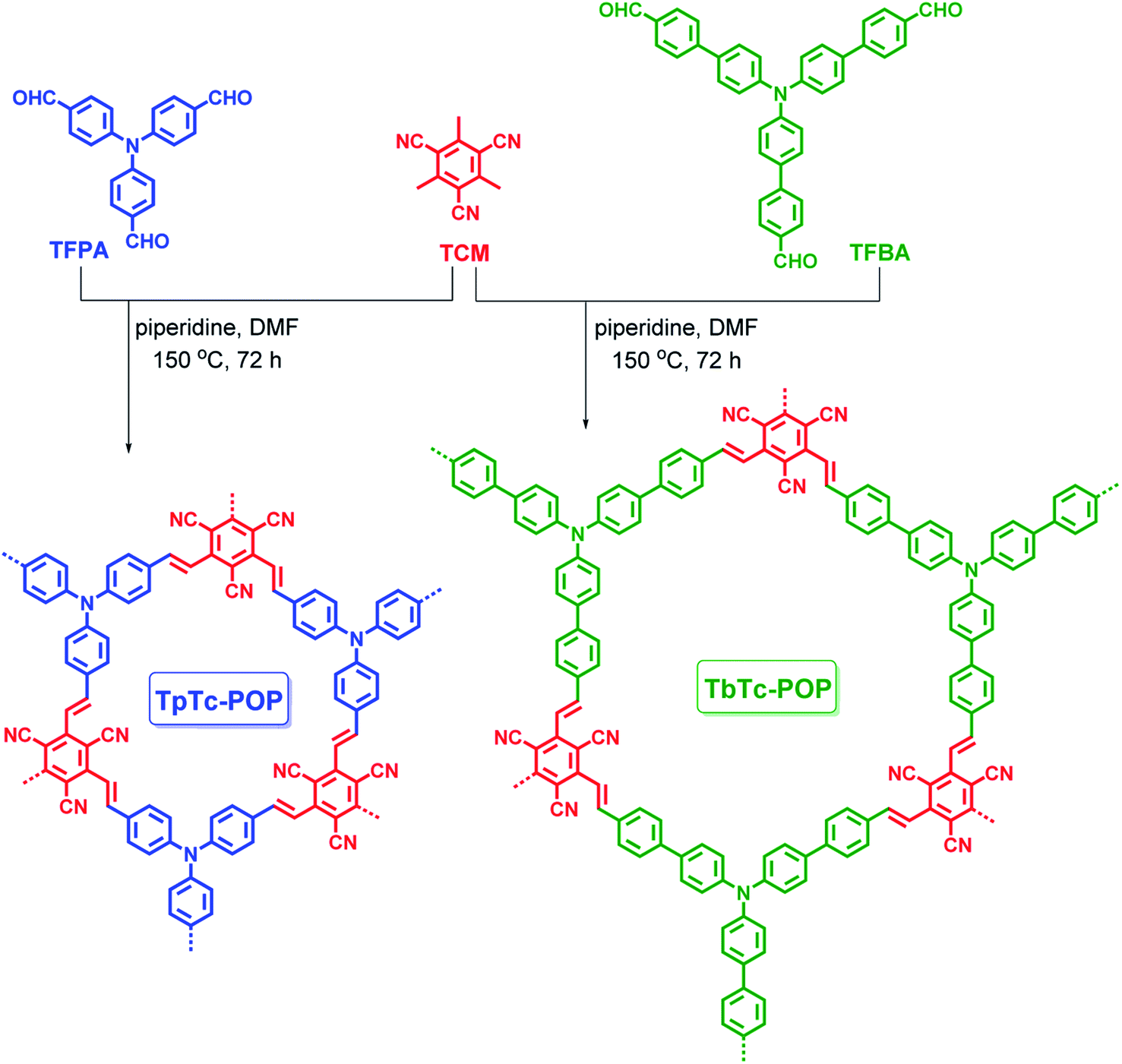 | ||
| Scheme 1 Synthetic routes of vinylene-bridged D–A porous organic polymers TpTc-POP and TbTc-POP. | ||
The chemical structures of TpTc-POP and TbTc-POP were confirmed by solid-state 13C cross-polarization/magic angle spinning (CP/MAS) nuclear magnetic resonance (NMR) and Fourier transform infrared (FT-IR) spectral analyses. As shown in Fig. 1a, the 13C CP/MAS NMR of TpTc-POP and TbTc-POP are combined and compared with 13C NMR spectra of model compound DpTc in CDCl3. The peaks at around 115 ppm were assigned to cyano groups (c). The most intense signals at 128 ppm were ascribed to the newly formed olefinic carbon (a) overlapped with aromatic carbons (h). The broad peaks at around 148 ppm were attributed to a combination of the carbon atoms (b, d, f and g) in POPs. Furthermore, FT-IR spectra of TpTc-POP and TbTc-POP revealed the stretching vibration signals of newly formed CC moieties at 1626 and 970 cm−1 in both POPs, verifying the presence of the trans-formed vinylene linkages in the as-prepared POP skeletons (Fig. 1b). The peak at 2226 cm−1 was attributed to the stretching vibration of cyano group (CN). Moreover, the peaks at ca. 1700 cm−1 corresponding to CO stretching vibration of aldehyde monomers disappeared in POP samples, indicative of their high polymerization degrees (Fig. S1 in ESI†). The morphologies of POPs examined by scanning electron microscopy (SEM) revealed their nanoscale particles in a relatively tight texture (Fig. S2 in ESI†). Powder X-ray diffraction (PXRD) profiles of TpTc-POP and TbTc-POP indicated their amorphous nature with the broad peaks at around 20°, suggesting the partial π–π stacked structure in polymer skeletons (Fig. S3 in ESI†). On the other hand, we have tried to optimize the reaction conditions in order to obtain the crystalline frameworks for these two POPs. However, no desirable results were achieved at present stage. This should be ascribed to the conformation of TPA unit, in which three phenyl rings are not in the same plane. Thermogravimetric analysis (TGA) revealed that they possessed favorable stability up to 400 °C with only 5–10% weight loss under a nitrogen atmosphere (Fig. S4 in ESI†). These results strongly supported the facile formation of target vinylene-linked D–A conjugated polymers by efficient polycondensation of TPA aldehydes and tricyanomesitylene.
 | ||
| Fig. 1 Solid-state 13C CP/MAS NMR (a) and FT-IR (b) spectra of TpTc-POP, TbTc-POP and the model compound DpTc. | ||
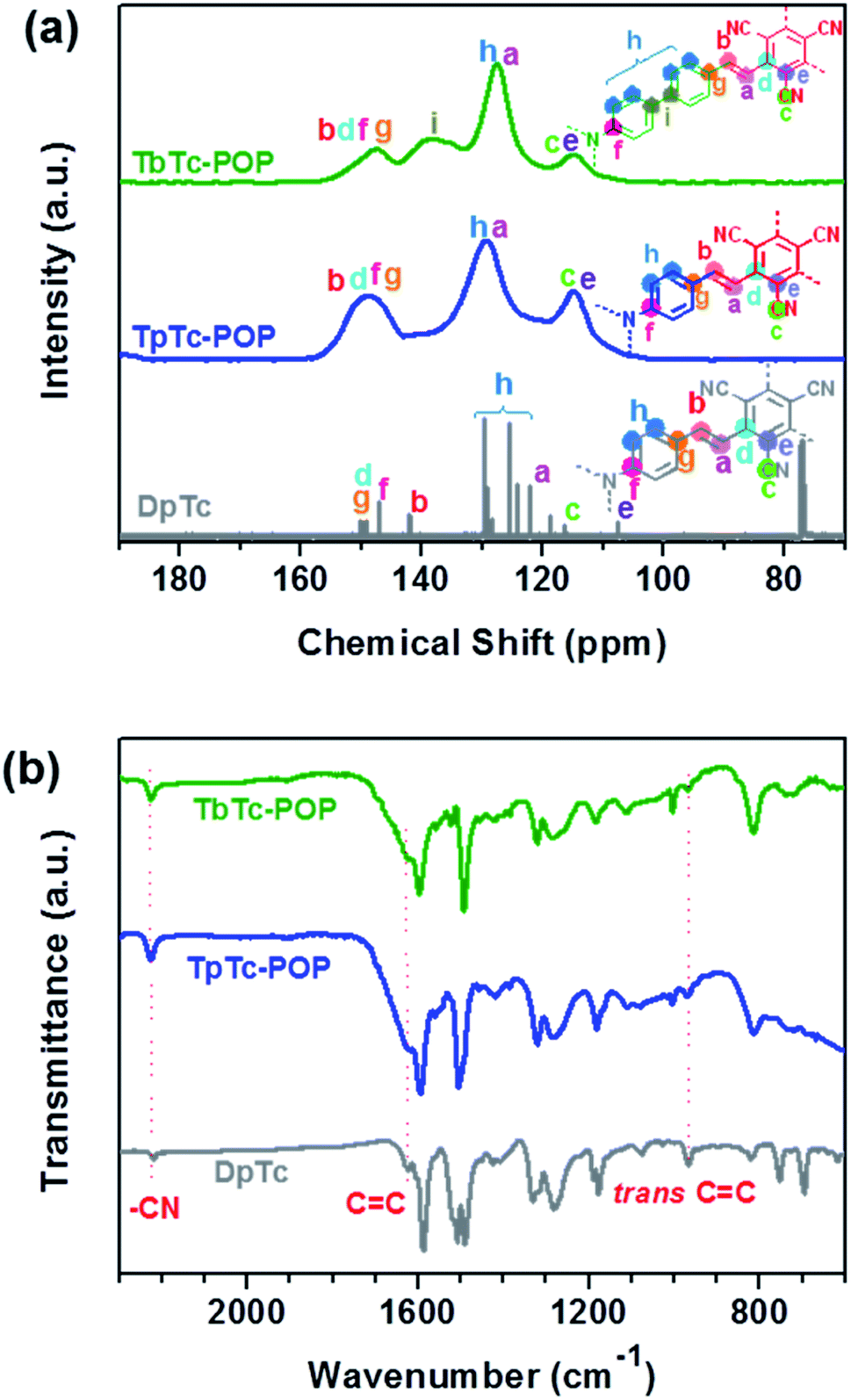
As shown in Fig. 2, the porosities and specific surface areas of TpTc-POP and TbTc-POP were characterized by nitrogen sorption isotherms measured at 77 K. Both of them displayed a combination of type I and IV nitrogen sorption isotherms according to the IUPAC classification,15 suggesting the co-existence of micropores and mesopores in two POPs. The obtained curves showed a steep increase at low pressure of P/P0 < 0.1, corresponding to their microporous nature. And the desorption hysteresis at high pressure was attributed to the extensive mesopores. Besides, the Brunauer–Emmett–Teller (BET) surface area was calculated to be 966 and 538 m2 g−1 for TpTc-POP and TbTc-POP, respectively. The nonlocal density functional theory (NLDFT) was utilized to calculate their pore-size distributions, which revealed two type of micro- and meso-porous distribution of 0.78, 1.69, 5.25 nm for TpTc-POP and 0.84, 1.73, 5.26 nm for TbTc-POP, respectively. Their hierarchical porous structures are beneficial for the diffusion and transformation of substrates in the photocatalytic process.
 | ||
| Fig. 2 Nitrogen adsorption and desorption isotherms of TpTc-POP (a) and TbTc-POP (b). Insets: the pore size distributions derived from NLDFT. | ||
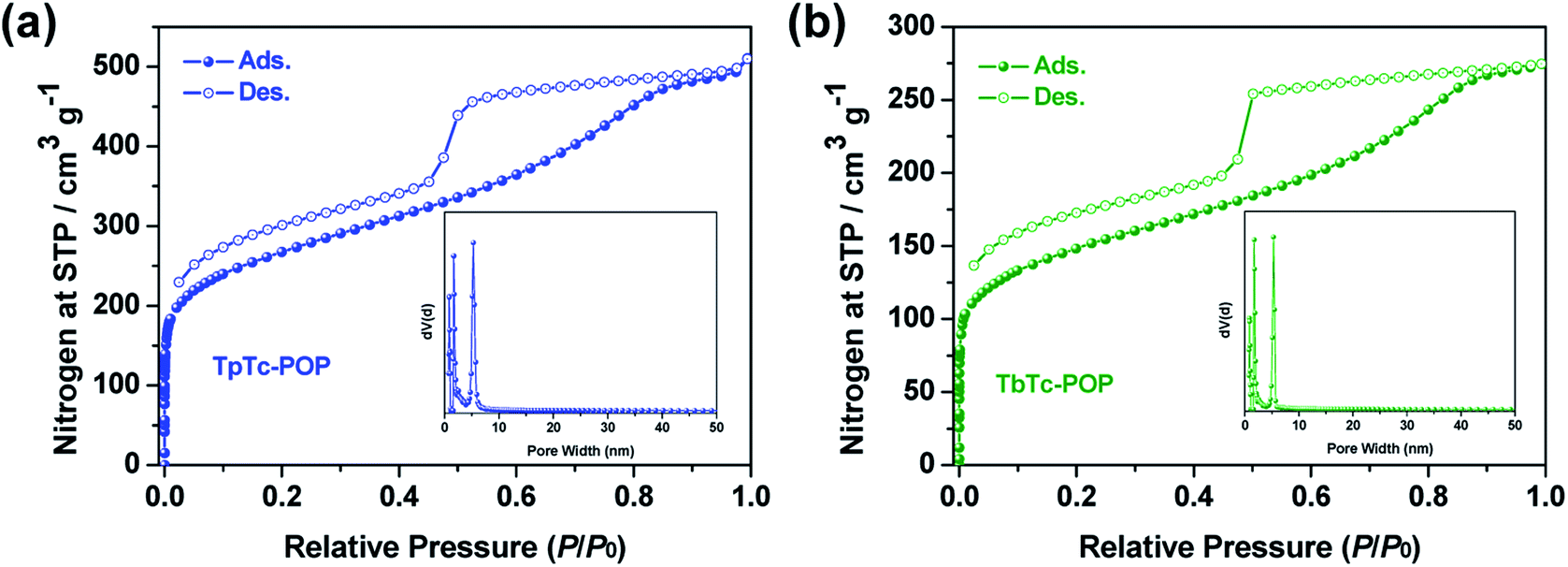
The electronic structures of TpTc-POP, TbTc-POP and the control TfTc-POP were studied by ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) and ultraviolet photoelectron spectroscopy (UPS). As shown in Fig. 3a, their UV-vis DRS showed a similar absorption range with a maximum of 300–600 nm and a gradual decrease until even near infrared around 900 nm. Accordingly, their optical band gaps were calculated from Kubelka–Munk function as 1.93, 1.95, and 1.98 eV for TpTc-POP, TbTc-POP and TfTc-POP, respectively (Fig. 3b). These results confirmed their strong visible-light harvesting abilities and low band gap characters caused by the extended sp2-carbon π-conjugation of D–A units in polymer backbones.7,8 Besides, the solid samples of POPs exhibited a photoluminescence (PL) band with a maximum peak centered at 585 nm for TfTc-POP, 638 nm for TbTc-POP and 650 nm for TpTc-POP, respectively (Fig. 3c). Their PL decay curves are fitted with the average lifetimes of 1.7 ns for TfTc-POP, 2.9 ns for TbTc-POP and 3.2 ns for TpTc-POP (Fig. S5 in ESI†). In comparison to TfTc-POP, the distinct PL redshifted by over 50 nm and the elongated lifetimes of TpTc-POP and TbTc-POP should be attributed to the stronger electron-donating ability of TPA units in polymers, thus, benefitting the charge separation of D–A components.
 | ||
| Fig. 3 (a) UV-vis diffuse reflectance spectra of TpTc-POP, TbTc-POP and control TfTc-POP. (b) The corresponding band gaps determined from the Kubelka–Munk-transformed reflectance spectra. (c) Photoluminescence spectra of TpTc-POP, TbTc-POP and control TfTc-POP measured in the solid state. (d) Band structures and the thermodynamic equilibrium redox potential for O2 in vacuum scale. | ||
Furthermore, the energy of valence band maximum (EVB in relative to the vacuum level) of TpTc-POP, TbTc-POP and TfTc-POP were evaluated by UPS analyses to be −5.6, −5.5, and −6.0 eV, respectively (Fig. 3d and S6 in ESI†). Thereby, the energy of the conduction band minimum (ECB) of TpTc-POP, TbTc-POP and TfTc-POP were calculated as −3.67, −3.55 and −4.02 eV, respectively. Since the ECB of all three POPs are higher than the potential of O2/O2˙− (−4.16 eV), they allowed for the reduction of oxygen to a superoxide radical anion upon photoexcitation.1,16
Encouraged by these POPs' photoelectronic properties, we thus evaluated and compared their photocatalytic activities by using visible light-driven aerobic oxidative coupling of benzylamine to imine as a model reaction. As shown in Fig. 4, the photocatalytic reactions with 1 mol% POPs loading in acetonitrile were performed in an open air atmosphere under white LEDs irradiation at room temperature and the progresses were monitored by 1H NMR. As expected, all three POPs can smoothly photocatalyze this transformation of benzylamine to imine and afford good conversions. The yields of N-benzylidenebenzylamine after 6 h irradiation are 95%, 85% and 62% for TpTc-POP, TbTc-POP and TfTc-POP, respectively. It is clear that TpTc-POP and TbTc-POP containing TPA moieties exhibited higher photocatalytic efficiency than their control POP TfTc-POP. This should be attributed to the fact that TPA units possess the strong electron-donating abilities. Besides, the recyclability and reusability of TpTc-POP and TbTc-POP were also examined for this photocatalytic model reaction. After they were separated by simple centrifugation from the reaction mixture, both of them remained highly active for at least five cycles (Fig. S7 in ESI†). On the other hand, nitrogen sorption isotherms of TpTc-POP and TbTc-POP after five successive photocatalysis demonstrated a few decrease in gas uptake and surface area (Fig. S8 in ESI†). However, their pore size distributions are almost consistent before and after photocatalysis. These results verified their moderate to good photochemical stabilities.
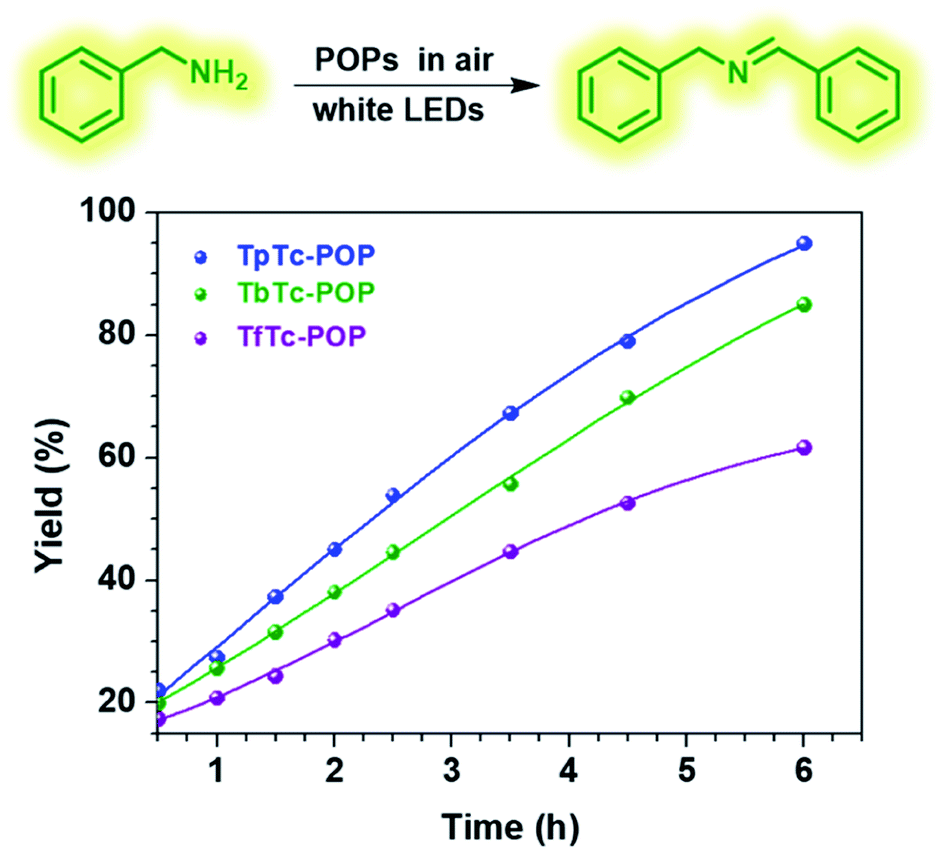 | ||
| Fig. 4 Oxidative coupling of benzylamine to imine over time with different POP photocatalysts under the irradiation of white-LEDs in an open air atmosphere. | ||
To gain insights into the reaction mechanism and the key active species involved in the photochemical conversion, several trapping experiments using different scavengers were performed as shown in Table 1. The control experiments revealed that photocatalyst TpTc, light and oxygen are all indispensable in this photo-induced reaction; otherwise, only trace conversion can be observed (Table 1, entries 2–4). Upon adding KI as the hole scavenger, a low yield of 11% was obtained (Table 1, entry 5). NaN3 as a singlet oxygen scavenger was added into the reaction mixture, and the transformation was suppressed to some extent by NaN3 with a yield of 47% (Table 1, entry 6). After adding p-benzoquinone, a typical superoxide radical anion O2˙− scavenger, the reaction yield was greatly reduced from 95% to 20% (Table 1, entry 7). Thus, these quenching experiments imply that the hole and O2˙− are determined as the major reactive species for this photocatalytic transformation. And the singlet oxygen can also promote the proceeding of this reaction as well. Then, electron paramagnetic resonance (EPR) measurements were performed to trap the involved reactive species of O2˙− and 1O2 by using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP).17 As expected, both O2˙− and 1O2 characteristic signals of adducts with DMPO and TEMP were observed in EPR spectra upon the irradiation of TpTc-POP in CH3CN solution, respectively (Fig. S9 in ESI†). In addition, N,N,N′,N′-tetramethyl-phenylenediamine (TMPD) was used to detect the generation of O2˙− from the photochemical reaction mixture.18 As shown in Fig. 5a, a deep blue-colored solution with two strong absorption bands at 564 and 613 nm indicated the formation of the cationic radical TMPD˙+ and the superoxide radical anion O2˙− after light irradiation of TMPD solution in the presence of TpTc-POP.
| Entry | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4c | 5d | 6e | 7f | |
| a Reaction conditions: benzylamine (0.5 mmol), TpTc-POP (5 mg), solvent acetonitrile (1.5 mL), 6 h, white LEDs (3 W, ∼100 mW cm−2), room temperature.b Determined by 1H-NMR analysis.c Under nitrogen atmosphere.d KI as the hole scavenger.e NaN3 as the single oxygen 1O2 scavenger.f p-Benzoquinone as the superoxide scavenger. | |||||||
| TpTc-POP | + | + | — | + | + | + | + |
| hv | + | — | + | + | + | + | + |
| Air | + | + | + | — | + | + | + |
| Yieldb (%) | 95 | Trace | Trace | Trace | 11 | 47 | 20 |
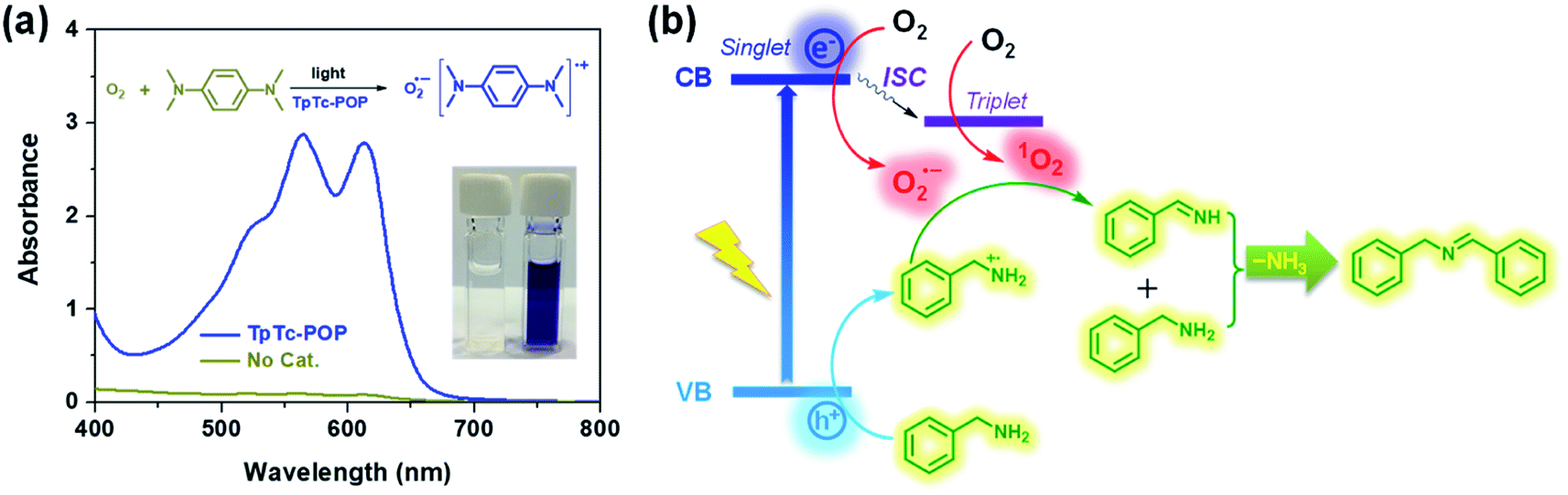 | ||
| Fig. 5 (a) UV-vis spectra and photograph of the cationic radical of TMPD produced by TpTc-POP in the presence of light and oxygen. (b) The corresponding proposed mechanism for the oxidative coupling of benzylamine. | ||
Therefore, based on these above experimental results and literature reports,18 we proposed a plausible mechanism for the photocatalytic oxidative coupling of benzylamine by POPs, as illustrated in Fig. 5b. Under the light irradiation, the polymer photocatalyst is excited to produce excitons of bound electron–hole pairs. The photogenerated electrons can activate oxygen via a single electron transfer (SET) process or energy transfer, resulting in a superoxide radical O2˙− and singlet oxygen 1O2. Meanwhile, the holes are transferred to amine, producing amine radical cations. A subsequent reaction between the amine radical cation with O2˙− and 1O2 gives the intermediate imine, which can be further reacted with another amine molecule to form the desired product after removal of ammonia.
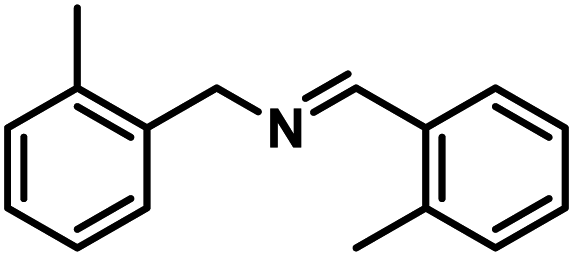
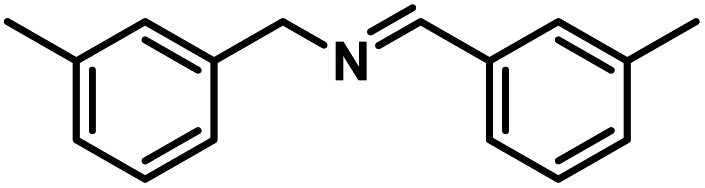
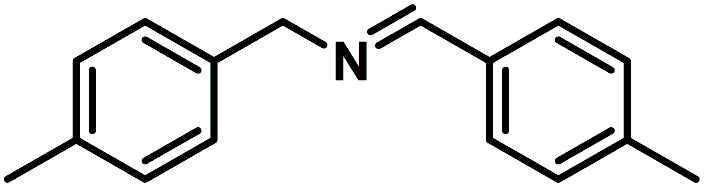
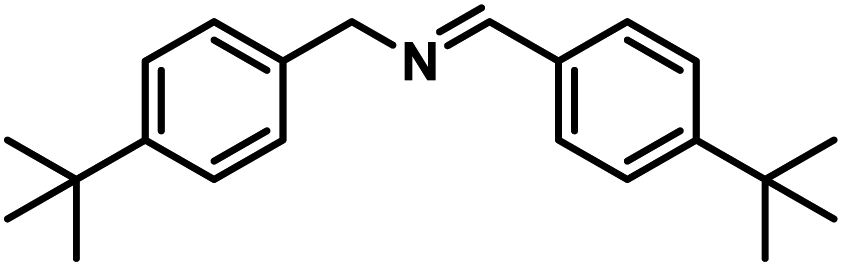
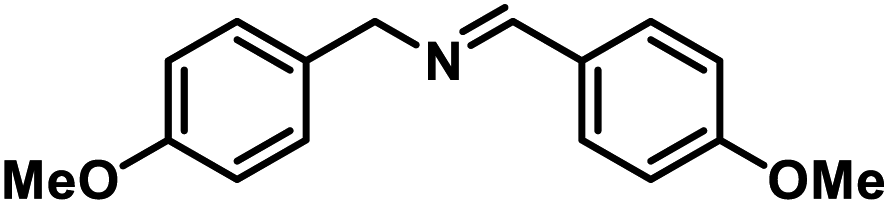
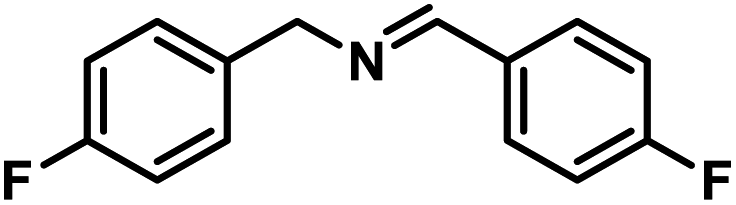
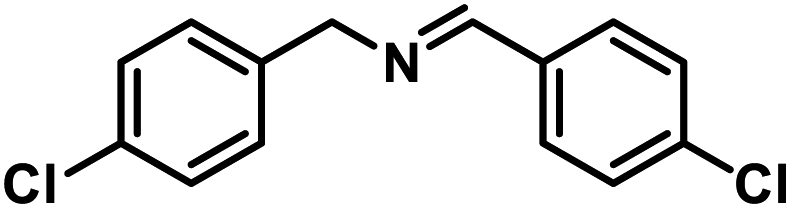
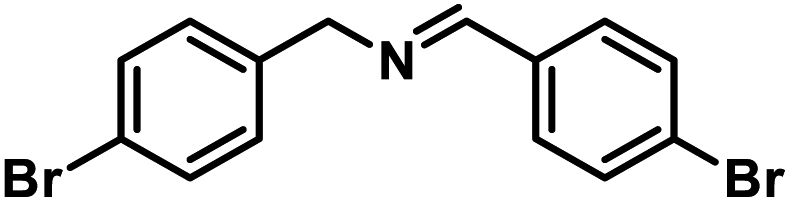
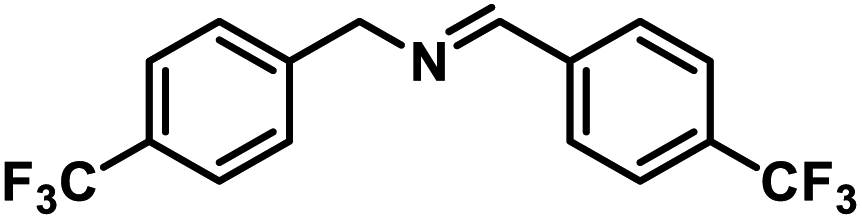
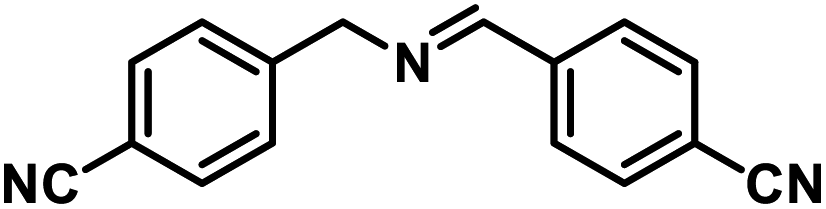
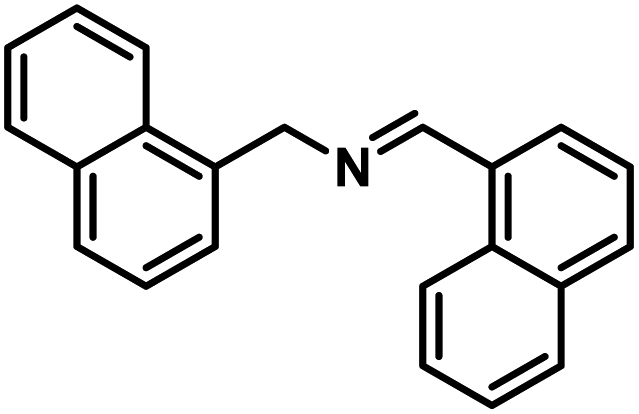
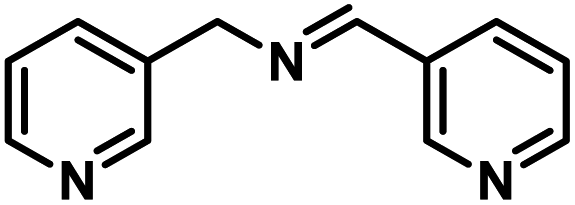
Next, the substrate scopes of oxidative coupling reactions photocatalyzed by TpTc-POP and TbTc-POP were expanded to various benzylamines with different functional groups (Tables 2 and S1 in ESI†). Various benzylamines bearing electron-donating or -withdrawing groups can be readily converted into their corresponding imine products in high yields by these two POPs photocatalysis with different reaction time. However, the electronic properties of the substituents have some effects on the efficiency of the photocatalyzed reactions. The benzylamines with strong electron-withdrawing groups (such as –CF3 and –CN, entries 10 and 11) slowed down the reaction rate and prolonged the reaction time in comparison to those with electron donating groups. Moreover, polycyclic aromatic and heterocyclic amines, such as 1-naphthalenemethylamine, 3-picolylamine and 2-thenylamine, can also be transformed into the corresponding imine with high yields (entries 12–14).
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: benzylamines (0.5 mmol), TpTc-POP (5 mg), CH3CN (1 mL), irradiation with white LEDs (3 W, ∼100 mW cm−2).b Determined by 1H NMR analysis. | ||||
| 1 |  |
 |
6 | 95 |
| 2 | 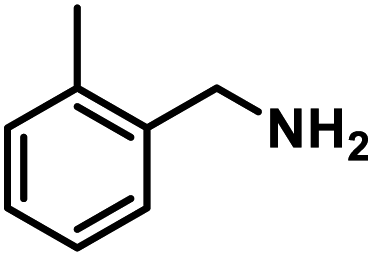 |
 |
6 | 89 |
| 3 |  |
 |
6 | 90 |
| 4 | 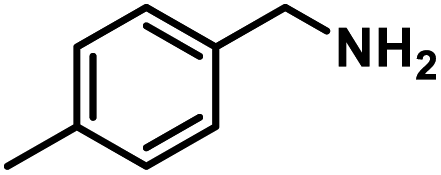 |
 |
6 | 99 |
| 5 |  |
 |
6.5 | 91 |
| 6 |  |
 |
6 | 99 |
| 7 | 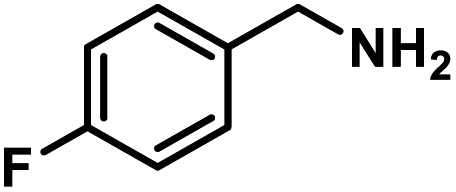 |
 |
6 | 99 |
| 8 | 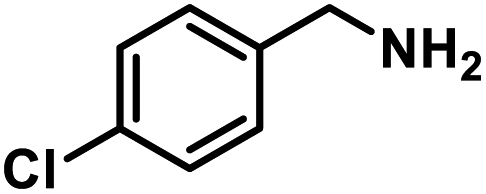 |
 |
6 | 99 |
| 9 |  |
 |
6 | 95 |
| 10 | 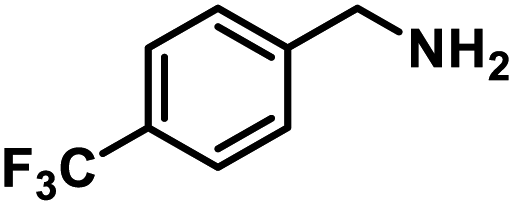 |
 |
8 | 99 |
| 11 | 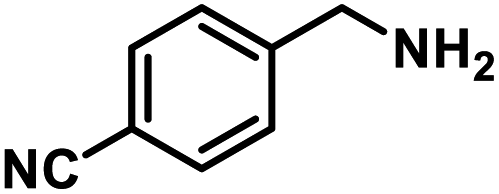 |
 |
12 | 93 |
| 12 | 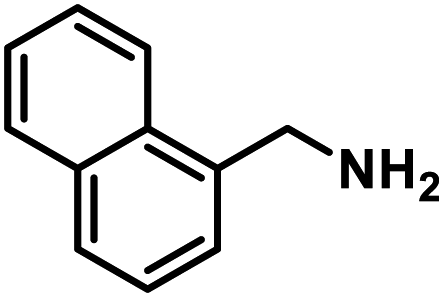 |
 |
8 | 98 |
| 13 | 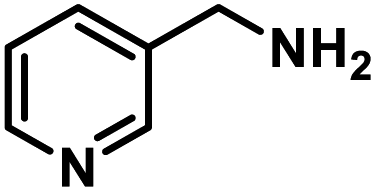 |
 |
8 | 97 |
| 14 |  |
 |
8 | 93 |
Conclusions
In summary, we have constructed two vinylene-bridged D–A structural POPs by using the electron-rich TPA and electron-deficient TCM as key building blocks and further explored their applications in photocatalytic aerobic oxidative coupling of amines to imines. The fully sp2-carbon-connected polymer skeletons allowed for their intensive light-harvesting capabilities and tunable band structures. More importantly, due to the strong electron-donating ability of TPA units in polymers, the two POPs exhibited more impressive semiconducting properties and enhanced photocatalytic activity in comparison to the benzene-centered analogous POP. This work revealed that it is highly promising for the rational design of various D–A structural POPs with the enhanced photocatalytic performance by tuning donor and acceptor moieties at a molecular level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported from Natural Science Foundation of Jiangsu Province (BK20181001), National Natural Science Foundation of China (22001100).References
- (a) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (c) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed; (d) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (e) A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401–4423 RSC; (f) X.-B. Li, Z.-K. Xin, S.-G. Xia, X.-Y. Gao, C.-H. Tung and L.-Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC; (g) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- (a) W. M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (b) F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405 Search PubMed; (c) X.-L. Yang, J.-D. Guo, H. Xiao, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 5365–5370 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed; (c) C. Zhou, T. Lei, X.-Z. Wei, C. Ye, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142, 16805–16813 CrossRef CAS PubMed; (d) A. Srivastava, P. K. Singh, A. Ali, P. P. Singh and V. Srivastava, RSC Adv., 2020, 10, 39495–39508 RSC; (e) T. Ju, Y.-Q. Zhou, K.-G. Cao, Q. Fu, J.-H. Ye, G.-Q. Sun, X.-F. Liu, L. Chen, L.-L. Liao and D.-G. Yu, Nat. Catal., 2021, 4, 304–311 CrossRef CAS.
- (a) X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC; (b) J. Chen, J. Cen, X. Xu and X. Li, Catal. Sci. Technol., 2016, 6, 349–362 RSC; (c) C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed; (d) Y.-Y. Zhu, G. Lan, Y. Fan, S. S. Veroneau, Y. Song, D. Micheroni and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 CrossRef CAS PubMed; (e) X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed; (f) H.-P. Liang, Q. Chen and B.-H. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS; (g) L. E. Oi, M.-Y. Choo, H. V. Lee, H. C. Ong, S. B. A. Hamid and J. C. Juan, RSC Adv., 2016, 6, 108741–108754 RSC; (h) X. Yang, T. Huang, S. Gao and R. Cao, J. Catal., 2019, 378, 248–255 CrossRef CAS.
- (a) A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed; (b) S. Das, P. Heasman, T. Ben and S. L. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed; (c) Y. Yuan and G. S. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS PubMed; (d) K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed; (e) D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- (a) L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29 Search PubMed; (b) W. Ji, T.-X. Wang, X. Ding, S. Lei and B.-H. Han, Coord. Chem. Rev., 2021, 439 Search PubMed; (c) Q. Sun, B. Aguila, Y. Song and S. Ma, Acc. Chem. Res., 2020, 53, 812–821 CrossRef CAS PubMed; (d) Q. Sun, Z. F. Dai, X. J. Meng and F. S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC; (e) P. Zhang, S. Wang, S. Ma, F.-S. Xiao and Q. Sun, Chem. Commun., 2020, 56, 10631–10641 RSC; (f) D. Chen, C. Liu, J. Tang, L. Luo and G. Yu, Polym. Chem., 2019, 10, 1168–1181 RSC.
- (a) Y. H. Xu, S. B. Jin, H. Xu, A. Nagai and D. L. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC; (b) T. Zhang, G. Xing, W. Chen and L. Chen, Mater. Chem. Front., 2020, 4, 332–353 RSC; (c) D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC; (d) R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS; (e) J. Byun and K. A. I. Zhang, Mater. Horiz., 2020, 7, 15–31 RSC; (f) T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC; (g) C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef CAS PubMed; (h) C. Xu, W. Zhang, J. Tang, C. Pan and G. Yu, Front. Chem., 2018, 6, 592 CrossRef CAS PubMed; (i) J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS.
- (a) H. Liu, X. Yan, W. Chen, Z. Xie, S. Li, W. Chen, T. Zhang, G. Xing and L. Chen, Sci. China: Chem., 2021, 64, 827–833 CrossRef CAS; (b) W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y.-B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed; (c) Y. Zhi, S. Ma, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, Appl. Catal., B, 2019, 244, 36–44 CrossRef CAS; (d) J. Luo, J. Lu and J. Zhang, J. Mater. Chem. A, 2018, 6, 15154–15161 RSC.
- (a) S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS; (b) X. Li, Mater. Chem. Front., 2021, 5, 2931–2949 RSC; (c) T. He, K. Geng and D. Jiang, Trends Chem., 2021, 3, 431–444 CrossRef CAS.
- (a) X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC; (b) E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- (a) R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed; (b) Y. Zhao, H. Liu, C. Wu, Z. Zhang, Q. Pan, F. Hu, R. Wang, P. Li, X. Huang and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 5376–5381 CrossRef CAS PubMed; (c) J.-L. Shi, R. Chen, H. Hao, C. Wang and X. Lang, Angew. Chem., Int. Ed., 2020, 59, 9088–9093 CrossRef CAS PubMed; (d) E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CrossRef CAS; (e) S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed.
- (a) S. Bi, Z.-A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef; (b) S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS PubMed; (c) J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- (a) P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC; (b) Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed; (c) A. Mahmood, Sol. Energy, 2016, 123, 127–144 CrossRef CAS; (d) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. R. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- (a) Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS; (b) S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- (a) S. Bandyopadhyay, S. Kundu, A. Giri and A. Patra, Chem. Commun., 2018, 54, 9123–9126 RSC; (b) S. Kundu, B. Behera, A. Giri, N. Saha and A. Patra, Chem. Commun., 2021, 57, 6875–6878 RSC.
- (a) Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS PubMed; (b) Y. Zhi, K. Li, H. Xia, M. Xue, Y. Mu and X. Liu, J. Mater. Chem. A, 2017, 5, 8697–8704 RSC; (c) Z. Li, S. Han, C. Li, P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06118f |
| This journal is © The Royal Society of Chemistry 2021 |