
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxazolomycins produced by Streptomyces glaucus and their cytotoxic activity†
Yu Mu‡
a,
Yi Jiang‡*b,
Xiaodan Qua,
Bo Liu a,
Junfeng Tana,
Guiding Lib,
Mingguo Jiang*c,
Liya Lia,
Li Hana and
Xueshi Huang*a
a,
Junfeng Tana,
Guiding Lib,
Mingguo Jiang*c,
Liya Lia,
Li Hana and
Xueshi Huang*a
aInstitute of Microbial Pharmaceuticals, College of Life and Health Sciences, Northeastern University, Shenyang 110819, China. E-mail: huangxs@mail.neu.edu.cn; Fax: +86-24-83656106; Tel: +86-24-83656106
bYunnan Institute of Microbiology, Yunnan University, Kunming 650091, China. E-mail: jiangyi@ynu.edu.cn; Fax: +86-871-65173878; Tel: +86-871-65034073
cGuangxi Key Laboratory for Polysaccharide Materials and Modifications, School of Marine Sciences and Biotechnology, Guangxi University for Nationalities, Nanning 530008, China. E-mail: mzxyjiang@gxun.edu.cn; huangxs@mail.neu.edu.cn; Fax: +86-771-3267023; Tel: +86-771-3267023
First published on 28th October 2021
Abstract
Six oxazolomycins (1–6) were isolated from the fermentation broth of a soil-borne bacterial strain, Streptomyces glaucus. The structures of the new compounds, oxazolomycins D–F (1–3) and glaucumycins A, B (6a/6b), were elucidated by detailed spectroscopic data analysis. Oxazolomycins 1, 2, 4, and 5 demonstrated weak or modest cytotoxic activities against four human cancer cell lines, with IC50 values ranging from 10.6 ± 1.7 to 89.5 ± 6.6 μM (or >100 μM). Further study showed that 4 caused S phase cell cycle arrest in SMMC7721 cells through down-regulating the protein expression of cyclin A2, CDK2. Meanwhile, 4 induced apoptosis in SMMC7721 cells through down-regulating the protein levels of Bcl-2, up-regulating the levels of Bax, and activating the cleavage of caspase-3.
1. Introduction
Oxazolomycin and its congeners are a relatively rare family of natural products produced by streptomycetes.1,2 The common structural characteristics of this class of compounds contain a unique spiro fused β-lactone-γ-lactam core and a terminal oxazole ring, and these two structural moieties are linked by a triene and an (E,E)-diene aliphatic chain.1,2 Natural sources of oxazolomycins are extremely rare, and only fifteen members, including neooxazolomycin,3 oxazolomycins A, B, C,4,5 curromycins A, B (also known as triedimycins A, B),6,7 16-methyl oxazolomycin,8,9 KSM-2690 B, KSM-2690 C,10 lajollamycins,11,12 bisoxazolomycin A, and oxazolomycin A2 (ref. 13) have been found up to this point, and all of them were isolated from Streptomyces spp. Furthermore, several oxazolomycin related compounds, inthomycins and phthoxazolins, which only possess the oxazole ring and a triene fragment were also reported.14,15 The oxazolomycins showed a wide range of biological activities, e.g. oxazolomycin A, neooxazolomycin, curromycin A, 16-methyl oxazolomycin, KSM-2690 B, KSM-2690 C, lajollamycin, bisoxazolomycin A, and oxazolomycin A2 displayed cytotoxic activity;2 oxazolomycin A, curromycin A, 16-methyl oxazolomycin, KSM-2690 B, lajollamycin, bisoxazolomycin A, and oxazolomycin A2 exhibited antibacterial activity mainly against Gram-positive bacteria;2 oxazolomycin A showed antiviral activity against influenza A, herpes simplex, and vaccinia;1,2 oxazolomycins A, B, C and curromycins A, B inhibited crown gall formation on potato tubers.16–18 The unique structure and multiple biological activities of oxazolomycins has attracted much attention from organic chemists. The total synthesis of neooxazolomycin,19,20 oxazolomycin A,21 and lajollamycin22 has been accomplished. In addition, several synthesis method to establish the different structural segments of oxazolomycins have been reported.23–27In our continuous search for bioactive natural compounds from microbial sources, the secondary metabolites of a soil-borne Streptomyces glaucus (YIM 33872) were investigated. Streptomyces glaucus was isolated from the soil sample collected from the rainforest of Xishuangbanna, Yunnan, China. A small scale (100 mL) fermentation broth of S. glaucus showed diversiform chemical constituents illustrated through HPTLC and HPLC-MS analysis. Chromatographic separation of an EtOAc extract of a large scale fermentation broth of S. glaucus led to the isolation of six oxazolomycins, including four new compounds, oxazolomycins D–F (1–3), and a pair of isomers, named as glaucumycins A and B (6a/6b), as well as two known congeners KSM-2690 B and C (4–5) (Fig. 1).9 The structures of new compounds oxazolomycins D–F (1–3) and glaucumycins A, B (6a/6b) were elucidated by extensive spectroscopic data analysis as well as UV, IR, and MS spectra. The cytotoxic effects of 1, 2, 4, 5 were evaluated in four human carcinoma cell lines. The apoptosis and cell cycle arrest of 4 in SMMC7721 cell lines were also investigated. Herein, we report the fermentation, isolation, structural elucidation, and cytotoxic activity of oxazolomycins 1–6.
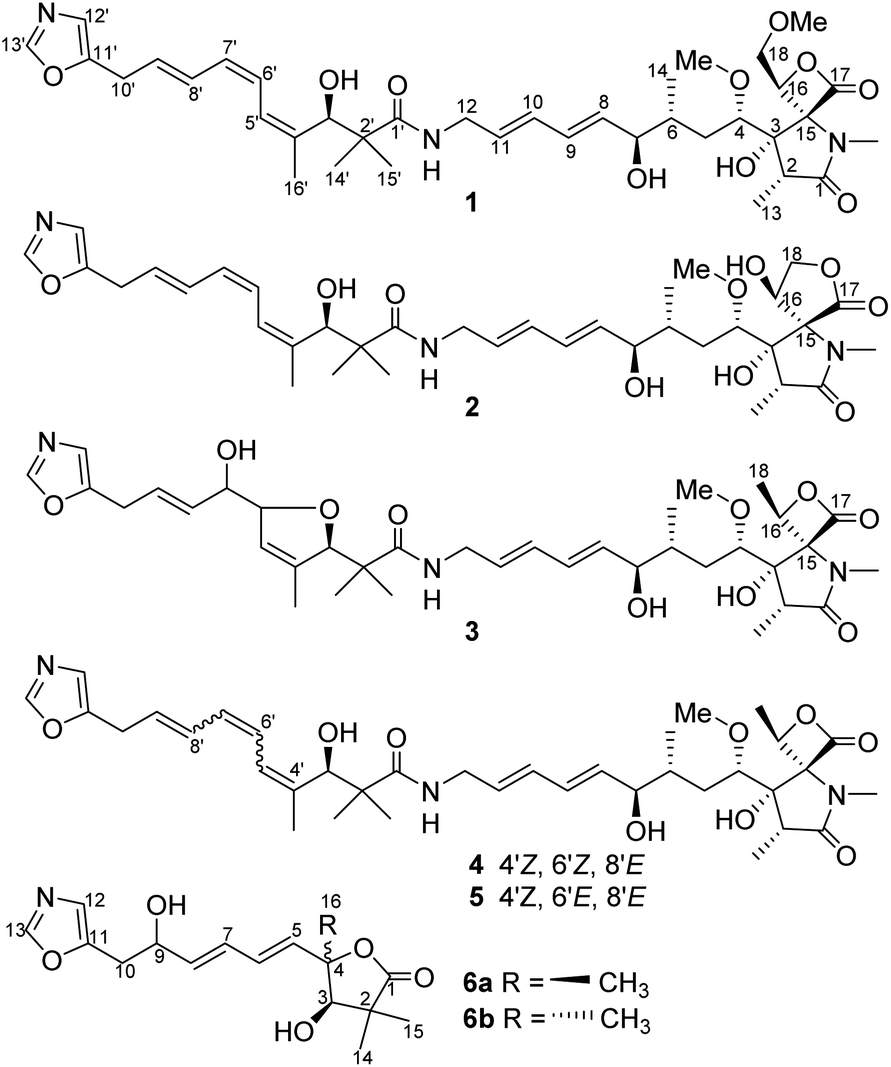 | ||
| Fig. 1 Structures of compounds 1–6. | ||
2. Materials and methods
2.1. General experimental procedures
Optical rotation were determined using an Anton Paar MCP200 automatic polarimeter (Graz, Austria). Ultraviolet spectra were obtained with a Beckman Coulter DU 730 nucleic acid/protein analyzer. IR spectra were recorded with a Bruker Tensor 27 FT-IR spectrometer. ESI-MS were recorded on an Agilent 1290–6420 Triple Quadrupole LC-MS spectrometer (Santa Clara, CA, USA). HRESI-MS experiments were performed using a Bruker Micro TOF-Q mass spectrometer (Bruker Daltonics, Billerica, MA). NMR spectra were recorded on a Bruker Advance III-600 MHz spectrometer (Bruker, Rheinstetten, Germany). Flow cytometric analysis were conducted on an LSR-Fortessa flow cytometer (BD, Franklin Lakes, NJ, USA). MTT and protein quantification were analyzed by a microplate reader (BioTek Synergy H1, BioTek Instruments, Inc., Vermont, USA). Silica gel (100–200 mesh, 200–300 mesh, Qingdao Marine Chemical, Ltd., Qingdao, China), MCI gel (CHP-20P, Mitsubishi Chemical Corp., Tokyo, Japan), and YMC*GEL ODS-A (S-50 μm, 12 nm) (YMC Co., Ltd., Kyoto, Japan) were used for column chromatography. The primary antibodies for cyclin A2, CDK2, p21, Bax, Bcl-2, caspase-3, GAPDH, and horse-radish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling Technology (Dancers, MA, USA).2.2. Microbial material
The producing organism was isolated from a rainforest soil sample collected in Xishuangbanna, Yunnan Province, China. The strain was assigned to be Streptomyces glaucus based on morphological characteristics and 16S rRNA gene sequences. The Blast result showed that the sequence was identical (100%) to the sequence of S. glaucus LMG 19902 (T) (GenBank accession no. AJ787332). The strain (No. YIM 33872) was deposited in Yunnan Institute of Microbiology, Yunnan University, China.2.3. Fermentation, extraction, and isolation
A slant culture of S. glaucus was inoculated into 500 mL Erlenmeyer flasks each containing 100 mL of seed medium composed of 4 g L−1 yeast extract, 4 g L−1 glucose, 5 g L−1 malt extract, 3.75 mg L−1 multiple vitamins solution (thiamine 0.5 mg, riboflavin 0.5 mg, niacin 0.5 mg, pyridoxine 0.5 mg, inositol 0.5 mg, calcium pantothenate 0.5 mg, p-aminobenzoic acid 0.5 mg, biotin 0.25 mg), and 1 mL L−1 trace element solution (2 g L−1 FeSO4·7H2O, 1 g L−1 MnCl2·4H2O, 1 g L−1 ZnSO4·7H2O). The pH was 7.2 with no adjustment, and the flasks were incubated for 2 days at 28 °C on a rotary shaker at 180 rpm. The seed culture was used to inoculate the fermentation medium with 10% volume. The large-scale fermentation was carried out in 1000 mL/2000 mL Erlenmeyer flasks with 200 mL/400 mL of fermentation medium containing soybean meal 10 g L−1, peptone 2 g L−1, glucose 20 g L−1, soluble starch 5 g L−1, yeast extract 2 g L−1, NaCl 4 g L−1, K2HPO4 0.5 g L−1, MgSO4·7H2O 0.5 g L−1, CaCO3 2 g L−1, with a pH of 7.8 with no adjustment, and the flasks were incubated for 7 days at 28 °C on a rotary shaker at 180 rpm.The 120 L fermentation broth was collected and clarified with a centrifuge to obtain a culture supernatant. Then, the supernatant was extracted with EtOAc three times. The combined EtOAc extracts were concentrated in vacuo to yield 32.7 g dried extract. The total extract was subjected to open silica gel (100–200 mesh) column chromatography with a CH2Cl2–MeOH solvent system (from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 50:1, 20:1, 5:1, and 1:1 at last) to yield seven fractions. Fraction 3 was subjected to silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to yield three subfractions (Fr.3.1–Fr.3.3). Fr.3.3 was further separated by ODS column chromatography and eluted with MeOH–H2O (60:40) to afford compound 4 (30.6 mg). Fraction 4 was subjected to an ODS column chromatography eluting with MeOH–H2O (60:40) to afford four subfractions (Fr.4.1–Fr.4.4). Fr.4.2 was further separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compounds 2 (6.7 mg) and 6 (3.9 mg). Fr.4.3 was separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 3 (6.5 mg). Fr.4.4 was separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 1 (7.1 mg). Fr.5 was subjected to a MCI gel CHP-20P column and eluted with MeOH–H2O (from 0:100–100:0) to yield five subfractions (Fr.5.1–Fr.5.5). Fr.5.5 was separated by ODS column chromatography eluting with MeOH–H2O (60:40) and further purified by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 5 (8.2 mg).
:1, 50:1, 20:1, 5:1, and 1:1 at last) to yield seven fractions. Fraction 3 was subjected to silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to yield three subfractions (Fr.3.1–Fr.3.3). Fr.3.3 was further separated by ODS column chromatography and eluted with MeOH–H2O (60:40) to afford compound 4 (30.6 mg). Fraction 4 was subjected to an ODS column chromatography eluting with MeOH–H2O (60:40) to afford four subfractions (Fr.4.1–Fr.4.4). Fr.4.2 was further separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compounds 2 (6.7 mg) and 6 (3.9 mg). Fr.4.3 was separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 3 (6.5 mg). Fr.4.4 was separated by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 1 (7.1 mg). Fr.5 was subjected to a MCI gel CHP-20P column and eluted with MeOH–H2O (from 0:100–100:0) to yield five subfractions (Fr.5.1–Fr.5.5). Fr.5.5 was separated by ODS column chromatography eluting with MeOH–H2O (60:40) and further purified by silica gel (200–300 mesh) column chromatography (petroleum ether–EtOAc 1:1) to give compound 5 (8.2 mg).
Oxazolomycin D (1): yellow oil; [α]20D = 97.3 (c 0.37, MeOH); UV (MeOH) λmax (logε) 232 (4.47), 274 (4.31) nm; IR (film) νmax 3344, 2929, 1820, 1677, 1553, 1458, 1385, 1262, 1093, 1051, 1026, 997 cm−1; 1H and 13C NMR see Table 1; HRESIMS m/z: 700.3738 [M + H]+ (calcd for C37H54N3O10+, 700.3804).
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | |
| 1 | 174.6, C | 175.9, C | 174.8, C | |||
| 2 | 43.7, CH | 2.37, q (7.3) | 42.9, CH | 2.42, q (7.5) | 44.0, CH | 2.36, q (7.3) |
| 3 | 81.3, C | 86.1, C | 81.1, C | |||
| 4 | 83.1, CH | 3.33, overlapped | 81.8, CH | 3.15, overlapped | 83.0, CH | 3.35, t (4.6) |
| 5 | 32.3, CH2 | 1.96, dt (16.0, 6.5) | 32.8, CH2 | 1.96, dt (16.6, 4.6) | 32.4, CH2 | 1.94, dt (15.2, 5.2) |
| 1.12, m | 1.10, m | 1.13, m | ||||
| 6 | 37.2, CH | 1.59, m | 38.2, CH | 1.52, m | 37.1, CH | 1.60, m |
| 7 | 75.7, CH | 3.83, m | 75.6, CH | 3.79, q (5.8) | 75.5, CH | 3.82, q (5.6) |
| 8 | 134.9, CH | 5.60, m | 135.1, CH | 5.60, m | 134.8, CH | 5.59, dd (14.3, 4.7) |
| 9 | 130.3, CH | 6.11, m | 130.2, CH | 6.12, m | 130.2, CH | 6.11, m |
| 10 | 130.7, CH | 6.12, m | 130.7, CH | 6.12, m | 130.6, CH | 6.11, m |
| 11 | 130.4, CH | 5.61, m | 130.4, CH | 5.61, m | 130.3, CH | 5.61, m |
| 12 | 40.9, CH2 | 3.71, m | 40.5, CH2 | 3.72, m | 40.7, CH2 | 3.68, dt (15.8, 5.6) |
| 3.73, dt (15.8, 5.6) | ||||||
| 13 | 10.2, CH3 | 1.03, d (7.5) | 11.1, CH3 | 1.06, d (7.5) | 10.2, CH3 | 1.03, d (7.5) |
| 14 | 16.5, CH3 | 0.86, d (6.6) | 16.8, CH3 | 0.86, d (6.6) | 16.4, CH3 | 0.86, d (6.8) |
| 15 | 85.6, C | 71.6, C | 83.9, C | |||
| 16 | 79.4, CH | 4.95, dd (9.1, 2.4) | 69.9, CH | 4.83, m | 77.7, CH | 4.95, q (6.6) |
| 17 | 170.7, C | 173.7, C | 170.5, C | |||
| 18 | 72.0, CH2 | 4.13, dd (12.0, 9.1) | 69.1, CH2 | 4.40, t (7.9) | 17.2 | 1.70, d (6.5) |
| 3.99, dd (12.0, 2.4) | 4.17, t (7.9) | |||||
| 1′ | 176.5, C | 176.5, C | 175.2, C | |||
| 2′ | 46.3, C | 46.4, C | 46.4, C | |||
| 3′ | 73.5, CH | 4.64, d (4.6) | 73.5, CH | 4.63, d (4.8) | 91.7, CH | 4.78, d (5.7) |
| 4′ | 140.4, C | 140.4, C | 138.4, C | |||
| 5′ | 123.9, CH | 6.41, d (12.0) | 123.9, CH | 6.41, d (11.9) | 124.1, CH | 5.47, brs |
| 6′ | 124.9, CH | 6.32, t (11.3) | 124.9, CH | 6.32, t (11.9) | 88.5, CH | 4.51, brs |
| 7′ | 127.6, CH | 5.93, t (11.3) | 127.6, CH | 5.93, t (11.9) | 73.7, CH | 3.89, q (5.3) |
| 8′ | 128.5, CH | 6.75, dd (14.5, 11.3) | 128.5, CH | 6.75, dd (14.6, 11.9) | 133.5, CH | 5.53, dd (15.4, 5.3) |
| 9′ | 129.4, CH | 5.79, dt (14.5, 7.0) | 129.5, CH | 5.79, dt (14.6, 7.0) | 125.5, CH | 5.68, dt (15.4, 6.4) |
| 10′ | 28.7, CH2 | 3.55, d (7.0) | 28.7, CH2 | 3.55, d (7.0) | 28.1, CH2 | 3.40, d (6.4) |
| 11′ | 150.9, C | 151.0, C | 151.3, C | |||
| 12′ | 122.5, CH | 6.90, s | 122.5, CH | 6.90, s | 122.3, CH | 6.83 s |
| 13′ | 151.8, CH | 8.24, s | 151.8, CH | 8.25, s | 151.6, CH | 8.21, s |
| 14′ | 25.2, CH3 | 1.09, s | 25.2, CH3 | 1.10, s | 24.5, CH3 | 1.11, s |
| 15′ | 22.0, CH3 | 0.97, s | 22.0, CH3 | 0.97, s | 18.7, CH3 | 0.93, s |
| 16′ | 20.4, CH3 | 1.75, s | 20.5, CH3 | 1.73, s | 14.0, CH3 | 1.55, s |
| 4-OCH3 | 56.2, CH3 | 3.14, s | 56.2, CH3 | 3.11, s | 56.3, CH3 | 3.15, s |
| 18-OCH3 | 58.9, CH3 | 3.28, s | ||||
| N-CH3 | 26.5, CH3 | 2.82, s | 26.4, CH3 | 2.63, s | 26.3, CH3 | 2.78, s |
| 3-OH | 5.61, brs | 6.06, brs | 5.32, brs | |||
| 7-OH | 4.86, d (4.0) | 4.85, d (5.5) | 4.81, d (4.1) | |||
| 12-NH | 7.67, t (5.7) | 7.67, t (5.5) | 7.67, t (5.7) | |||
| 16-OH | 7.14, d (5.5) | |||||
| 3′-OH | 5.49, d (4.6) | 5.47, d (4.8) | ||||
| 7′-OH | 4.90, d (5.3) | |||||
Oxazolomycin E (2): yellow oil; [α]20D = 59.3 (c 0.27, MeOH); UV (MeOH) λmax (logε) 232 (4.22), 274 (4.22) nm; IR (film) νmax 3367, 2928, 1765, 1680, 1532, 1455, 1396, 1287, 1205, 1099, 1049, 1026, 999 cm−1; 1H and 13C NMR see Table 1; HRESIMS m/z: 684.3500 [M − H]− (calcd for C36H50N3O10−, 684.3502).
Oxazolomycin F (3): yellow oil; [α]20D = 36.5 (c 0.15, MeOH); UV (MeOH) λmax (logε) 236 (4.06) nm; 1H and 13C NMR see Table 1; HRESIMS m/z: 708.3462 [M + Na]+ (calcd for C36H51N3NaO10+, 708.3467).
Glaucumycins A and B (6a/6b): colorless oil; UV (MeOH) λmax (logε) 232 (4.07) nm; IR (film) νmax 3367, 2979, 1756, 1511, 1384, 1278, 1223, 1089, 1025 cm−1; 1H and 13C NMR see Table 2; HRESIMS m/z: 330.1315 [M + Na]+ (calcd for C16H21NNaO5+, 330.1312).
| Position | 6a | 6b | ||
|---|---|---|---|---|
| δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | |
| 1 | 183.3, C | 182.6, C | ||
| 2 | 45.7, C | 44.7, C | ||
| 3 | 81.4, CH | 3.93, brs | 83.5, CH | 3.95, brs |
| 4 | 87.7, C | 86.0, C | ||
| 5 | 138.2, CH | 5.89, d (14.5) | 134.7, CH | 6.01, dd (14.8, 2.6) |
| 6 | 129.1, CH | 6.27, m | 129.7, CH | 6.27, m |
| 7 | 130.7, CH | 6.27, m | 131.1, CH | 6.27, m |
| 8 | 137.2, CH | 5.80, dd (14.6, 6.4) | 136.7, CH | 5.77, dd (14.5, 6.4) |
| 9 | 71.0, CH | 4.43, q (6.4) | 71.1, CH | 4.43, q (6.4) |
| 10 | 34.3, CH2 | 2.94, dd (15.6, 6.2) | 34.3, CH2 | 2.94, dd (15.6, 6.2) |
| 2.91, dd (15.6, 7.2) | 2.91, dd (15.6, 7.2) | |||
| 11 | 151.7, C | 151.7, C | ||
| 12 | 124.0, CH | 6.90, s | 124.0, CH | 6.90, s |
| 13 | 152.6, CH | 8.09, s | 152.6, CH | 8.08, s |
| 14 | 20.4, CH3 | 1.19, s | 19.7, CH3 | 1.04, s |
| 15 | 26.2, CH3 | 1.22, s | 25.3, CH3 | 1.21, s |
| 16 | 22.0, CH3 | 1.43, s | 27.4, CH3 | 1.49, s |
2.4. Cytotoxicity assay
The cytotoxicities of 1–2, 4–6 were evaluated against four human cancer cell lines including a gastric carcinoma cell line (BGC-823), a large-cell lung carcinoma cell line (H460), a breast adenocarcinoma cell line (MDA-MB-231), and a hepatocellular carcinoma cell line (SMMC-7721). The cell lines were cultured in RPMI-1640 or DMEM medium supplemented with 10% heat-inactivated FBS. After incubation for 12 h, the cells were treated with compounds 1–2, 4–6 at different concentrations (100, 33, 10, 3.3, 1.0, and 0.33 μM) for 48 h. The cytotoxic effects were assayed by the MTT method. Adriamycin was used as a positive control.2.5. Cell cycle assay
SMMC7721 cells were seeded in 6-well plates with a density of 5 × 105 cells per well and treated with KSM-2690 B (4) (0 or 12.5 μM) for 24 h. The cells were then collected and fixed overnight in 75% ethanol at −20 °C. The next day, the cells were washed twice with cold PBS and gently resuspended in 500 μL propidium iodide (PI) working fluid (Dingguo Changsheng Biotechnology Co. Ltd., Beijing, China) and then incubated for 15 min in the dark at room temperature. Cell cycle analysis was performed by using a LSR-Fortessa flow cytometer with a collection of 20000 cells. The population of cells in each phase was analyzed using ModFit LT 4.1 software (Verity Software House, Topsham, Me, USA). Each experiment was conducted three times.
2.6. Cell apoptosis assay
SMMC7721 cells were seeded in 6-well plates with a density of 5 × 105 cells per well and exposed to KSM-2690 B (4) (0 or 12.5 μM) for 24 h. Then, cells were collected, washed with cold PBS, and resuspended in 490 μL binding buffer and mixed with 5 μL of fluorescein isothiocyanate (FITC)-conjugated annexin-V reagent and 5 μL PI (BD, Franklin Lakes, NJ, USA). After 15 min incubation at room temperature in the dark, samples were analyzed by flow cytometry with the Annexin V-FITC/PI apoptosis method. Cytographs were presented by BD FACSDiva Software (BD, Franklin Lakes, NJ, USA). Each experiment was conducted three times.2.7. Western blot analysis
SMMC 7721 cells were cultured in 6-well plates at a density of 5 × 105 cells per well for 12 h, and then treated with compound 4 (0, 6.25, 12.5, and 25 μM) for 24 h. After that, the cells were collected and lyzed by RIPA buffer and then extracted by protein extraction kits (Beyotime, Shanghai, China). The concentration of protein was determined by a BCA protein assay kits (Beyotime, Shanghai, China). Western blot analysis procedures were carried out as our previously described.28 GAPDH expression was used as internal control. The GIS Gel Image System (Tanon, Shanghai, China) was used to quantitatively analyze the grayscale of bands.2.8. Data analysis
The results are presented as the mean ± standard deviation (SD) from at least three independent experiments. The differences of groups were analyzed using one-way analysis of variance with SPSS 13.0 (SPSS Inc., Chicago, IL, USA), followed by SNK (Student-Newman-Keuls) post hoc test. The threshold value for acceptance of difference was 5% (p ≤ 0.05). The statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., CA, USA).3. Results and discussion
3.1. Isolation and structural elucidation
The 120 L fermentation broth of S. glaucus was clarified with a centrifuge to obtain a culture supernatant. The supernatant was extracted by EtOAc three times. The combined EtOAc extract (32.7 g) were isolated by sequential column chromatography over silica gel, ODS, and MCI gel CHP-20P to yield oxazolomycins 1–6.Compound 1 was obtained as a yellow oil and its molecular formula was determined as C37H53N3O10 by HRESIMS at m/z [M + H]+ 700.3738 and 13C NMR data. The degrees of unsaturation of 1 was 13. The UV (λmax 232 nm for a conjugated diene and λmax 274 nm for a conjugated triene groups) and IR (νmax 1820 cm−1 for a β-lactone andνmax 1677 cm−1 for a γ-lactam) spectra showed the typical characteristics of oxazolomycins.1 The 1H NMR (Table 1) data of 1 displayed the presence of two singlet aromatic heterocyclic hydrogens at δH 8.24, 6.90, nine olefinic hydrogens at δH 6.75, 6.41, 6.32, 6.12, 6.11, 5.93, 5.79, 5.61, and 5.60, four oxygenated methines at δH 4.95 (dd, J = 9.1, 2.4 Hz), 4.64 (d, J = 4.6 Hz), 3.83 (1H, m), 3.33 (1H, m), four methylenes including two methylenes bonded with heteroatom at δH 4.13 (dd, J = 12.0, 9.1 Hz), 3.99 (dd, J = 12.0, 2.4 Hz) and δH 3.71 (2H, m), two aliphatic methylenes at δH 3.55 (2H, d, J = 7.0 Hz), δH 1.96 (dt, J = 16.0, 6.5 Hz) and δH 1.12 (1H, m), two methines at δH 2.37, 1.59, two methoxyls at δH 3.28, 3.14, and six methyl groups including four singlet methyls at δH 2.82, 1.75, 1.09, 0.97 and two doublet methyls at δH 1.03, 0.86. In addition, four active hydrogens were observed at δH 7.67, 5.61, 5.49, 4.86. The 13C NMR (Table 1) data of 1 exhibits 37 carbon signals, the type of these carbons were attributed to three carbonyls, three aromatic carbons, ten olefinic carbons, eight carbons bonded with heteroatom (four methines, two methylenes, and two carbons without directly bonded hydrogens), five aliphatic carbons (two methines, two methylenes, and one quaternary carbon), two methoxyls, and six methyls with the aid of HSQC experiment. The UV, IR, 1H, and 13C NMR data of 1 indicated it was a member of oxazolomycin family and was very similar to the known compound curromycin A.6,7 The only difference between these two compounds was no substituent on oxazole moiety in 1 which was explained by the two singlet aromatic hydrogens at δH 8.24, 6.90 belonging to the oxazole. The planar structure of 1 was further confirmed by HMBC and 1H–1H COSY correlations (Fig. 2). The conjugated triene fragment was determined as 4′Z,6′Z,8′E according to the coupling constants 3JH-6′/H-7′ = 11.3 Hz, 3JH-8′/H-9′ = 14.5 Hz and NOE correlations between H-5′ (δH 6.41) and H3-16′ (δH 1.75). Even though the geometry of conjugated diene could not confirmed by analysis the coupling constants of H-8/H-9 and H-10/H-11 because of the overlapped signals, the 8Z,10Z configurations could be determined by comparing the 1H and 13C NMR data with those of this family compounds.10 The stereochemistry of characteristic oxazolomycins had been determined through X-ray crystallography and total synthesis experiments.2,21–27 The stereochemistry of the spiro β-lactone-γ-lactam unit in natural oxazolomycins possessed 2R,3S,15S-configuration.21–27 The configurations of the chirality carbons on the aliphatic chain of compound 1 were deduced as 4S,6R,7R,3′R based on the almost same 13C NMR data of 1 with those of oxazolomycin A, lajollamycin, and synthesized oxazolomycins with the same side chain fragment.21–27 The configuration of C-16 was suggested as 16R through analyzing the NOE correlations observed between 3-OH (δH 5.61) and H2-18 (δH 4.13, 3.99), between H-16 (δH 4.95) and N-CH3 (δH 2.82) (Fig. 3). Thus, the structure of 1 was elucidated and named oxazolomycin D.
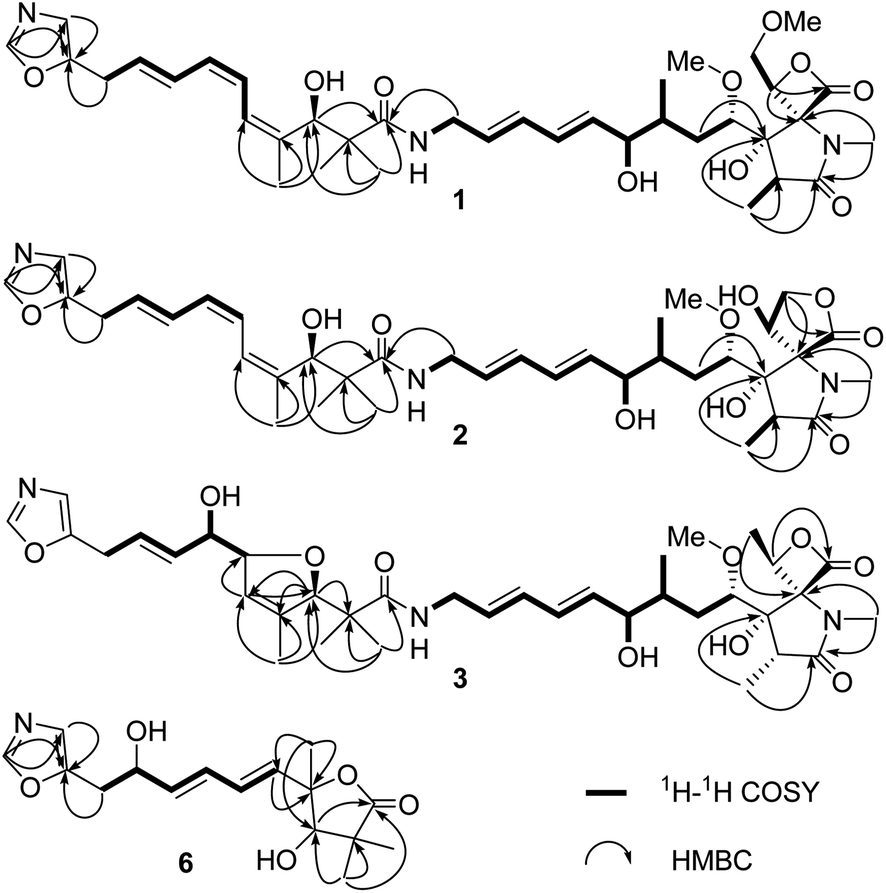 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of compounds 1–3, 6. | ||
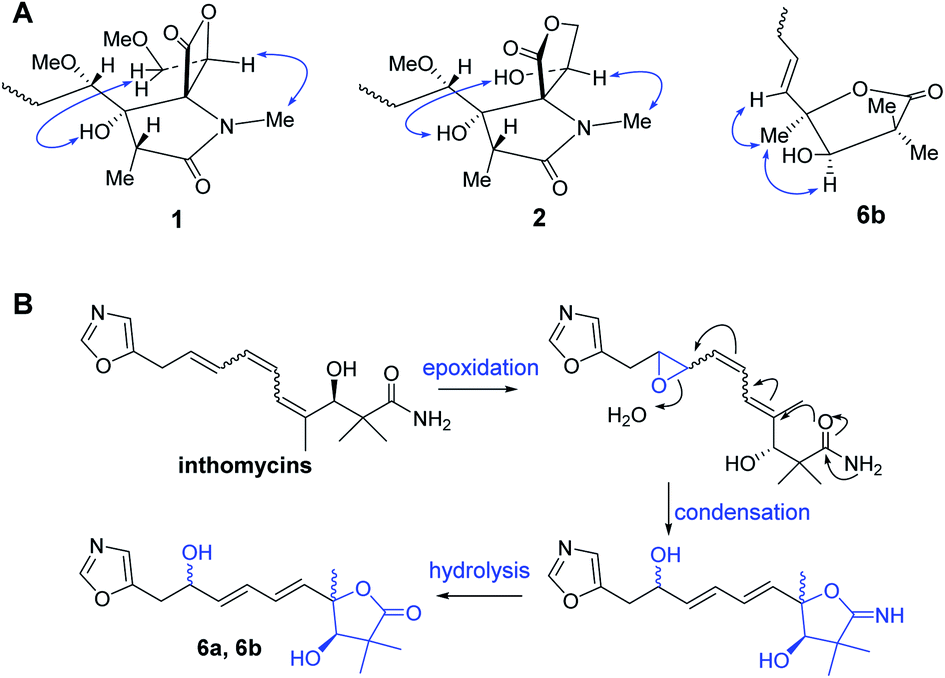 | ||
| Fig. 3 (A) Key NOE correlations of compounds 1–2, 6; (B) proposed biosynthetic pathway of 6a and 6b. | ||
Compound 2 was isolated as a yellow oil and possessed the molecular formula C36H51N3O10, as deduced by HRESIMS and 13C NMR data, with 13 degrees of unsaturation. The 1H and 13C NMR (Table 1) data of 2 showed the feature of oxazole, triene, diene, and amide fragments which indicated 2 was a member of oxazolomycins. The NMR data of 2 was very similar to those of 1 except the methoxymethyl-β-lactone ring. Compared with 1, the chemical shift of C-15, C-16, C-18 of 2 upshifted to δC 71.6, 69.9, 69.1, respectively and C-17 downshifted to δC 173.7. In addition, a methoxy was missing. These difference implied the methoxymethyl-β-lactone ring in 1 have changed. HMBC correlations between Ha-18 (δH 4.40) and C-15 (δC 71.6), C-17 (δC 173.7), and 1H–1H COSY correlations from H-16 (δH 4.83) to H-18 (δH 4.40, 4.17) indicated hydroxymethyl at C-16 and carbonyl at C-17 forming a γ-lactone (Fig. 2). Moreover, the peaks at 1765 cm−1 and 1680 cm−1 in IR spectrum showed typical features of moieties of γ-lactone and γ-lactam in molecule. The same double-bond geometries as those of 1 were determined on the basis of 1H–1H coupling constants and NOE correlations. The 2R,3S,15S configuration of spiro fused γ-lactone-γ-lactam were suggested basis on the same biosynthesis reaction according to the ref. 29. NOE correlations observed between 3-OH (δH 6.06) and 16-OH (δH 7.14), between H-16 (δH 4.83) and N-CH3 (δH 2.63) suggested the 16R configuration (Fig. 3). Identical 13C NMR of aliphatic chain part of 2 with those of 1 showed they possessing the same configuration of C-4, C-6, C-7, C-3′. Therefore, 2 was determined and named oxazolomycin E.
Compound 3 was purified as a yellow oil. The molecular formula of 3 was determined to be C36H51N3O10 on the basis of HRESI mass spectrometry as well as 1H and 13C NMR data (Table 1). Interpretation of the 1H, 13C NMR and UV spectra suggested that 3 was also a congener of oxazolomycin and related to KSM-2690 B.10 3 differs from KSM-2690 B in the conjugated triene fragment. Four olefinic carbons at δC 138.4, 133.5, 125.5, 124.1 and two additional oxygenated methines at δC 88.5, 73.7 observed in the 13C NMR spectrum of 3 suggested one double bond was oxygenated. The structure of 3 was partial changed during storage in the DMSO-d6, and the 2D NMR appeared many disturbed signals. The definite 1H–1H COSY correlations from H-10′ (δH 3.40) to H-9′ (δH 5.68), from H-9′ (δH 5.68) to H-8′ (δH 5.53), from H-8′ (δH 5.53) to H-7′ (δH 3.89), from H-7′ (δH 3.89) to H-6′ (δH 4.51) and 7′-OH (δH 4.90) could confirmed the C-6′ and C-7′ were oxygenated. Even though there was no HMBC correlation found between H-3′ to C-6′ or between H-6′ to C-3′, a furan ring was formed through analysis the molecular formula and 13 degrees of unsaturation. The 2R,3S,4S,6R,7R,15S,16S were suggested through comparing the 13C NMR data with those natural and synthesized congeners.21–27 3′R configuration was suggested on the basis of the biogenetic route and the configuration of C-6′ and C-7′ were undetermined. Thus, the structure of 3 was elucidated and named oxazolomycin F.
Compound 6a/6b were obtained as a mixture of two epimers in a nearly 1.2:1 ratio, according to the integration of the 1H NMR data. Their molecular formulas were established to be C16H21NO5 by HRESIMS and 13C NMR data. The signals of major isomer 6a revealed that the presence of a oxazole (δC 152.6, 151.7, 124.0, δH 8.09, 6.90), a diene moiety (δC 138.2, 137.2, 130.7, 129.1, δH 6.27, 6.27, 5.89, 5.80), a α,α-dimethyl-β-hydroxy carbonyl group (δC 183.3, 81.4, 45.7, 26.2, 20.4, δH 3.93), a –CH2CH(OH)- fragment (δC 71.0, 34.3, δH 4.43, 2.94, 2.91). The 1H NMR and 13C NMR data of 6a showed typical characteristic of the structure motif of ozazolomycins and were very similar to those of inthomycins and phthoxazolins.14,15 1H–1H COSY established the fragment from H-5 to H-10 (Fig. 2). The conjugated diene fragment was determined as 5E,7E according to the coupling constants 3JH-5/H-6 = 14.5 Hz, 3JH-7/H-8 = 14.6 Hz. HMBC correlations from CH3-14/15 (δH 1.22, 1.19) to C-1 (δC 183.3), C-2 (δC 45.7), C-3 (δC 81.4), from CH3-16 (δH 1.43) to C-3 (δC 81.4), C-4 (δC 87.7), C-5 (δC 138.2), from H-5 (δH 5.89) to C-4 (δC 87.7) established the linkage of α,α-dimethyl-β-hydroxy carbonyl group and C-4, diene fragment and C-4. Even though there was no direct correlation evidence, a γ-lactone ring formed between C-1 and C-4 was deduced from the molecular formula and 7 degrees of unsaturation. This conclusion was further supported by the absorption peak observed at 1756 cm−1 in IR spectrum which revealed the typical characteristic of moieties of γ-lactone. The C-3 was suggested as R according to the same biosynthesis way as oxazolomycins and inthomycins. The only difference between 6a/6b was the chemical shifts of Me-16 (δC 22.0 for 6a and δC 27.4 for 6b). Moreover, the different chemical shifts of C-3 (Δδ6a–6b −2.1), C-4 (Δδ6a–6b 1.7), C-5 (Δδ6a–6b 3.5) of 6a/6b implied different configuration of C-4. The NOE correlations observed between H3-16 (δH 1.49) and H-3 (δH 3.95), H-5 (δH 6.01) of 6b suggested the methyl at C-4 was α-oriented in 6b. Consequently, the methyl at C-4 was β in 6a. Therefore, 6a and 6b were determined and named glaucumycins A and B, respectively. They were possibly derived from inthomycins in microorganisms by the proposed biosynthetic pathway in Fig. 3B.
3.2. Cytotoxic activity
Many members of oxazolomycin family showed cytotoxicities according to the ref. 2. The cytotoxic activities of compounds 1–2, 4–6 against four human cancer cell lines were evaluated. The bioactive evaluation of 3 was not carried out because of the lack of material. As shown in Table 3, oxazolomycins 1, 2, 4, 5 exhibited weak or moderate cytotoxic activity against the tested cancer cell lines, while compounds 6 was inactive at 100 μM. Compared with compound 2, KSM-2690 B and C (4–5) showed stronger cytotoxicities. These results indicated the spiro fused β-lactone-γ-lactam core was quite favorable for the cytotoxic effect than γ-lactone-γ-lactam core (4, 5 vs. 2). The methoxymethyl group at C-16 of β-lactone core was not benefit for its cytotoxic activity (4 vs. 1).| Compound | 1 | 2 | 4 | 5 | 6 | Adriamycin |
|---|---|---|---|---|---|---|
| BGC-823 | 89.5 ± 6.6 | >100 | 36.7 ± 3.5 | 40.4 ± 5.2 | >100 | 1.5 ± 0.1 |
| H460 | 66.8 ± 3.4 | 75.6 ± 5.7 | 20.2 ± 1.6 | 34.8 ± 2.4 | >100 | 1.0 ± 0.2 |
| MDA-MB-231 | 80.2 ± 6.3 | >100 | 10.6 ± 1.7 | 28.3 ± 2.3 | >100 | 2.0 ± 0.4 |
| SMMC7721 | 62.3 ± 4.5 | 78.1 ± 10.2 | 27.3 ± 2.1 | 43.2 ± 4.7 | >100 | 2.2 ± 0.3 |
3.3. KSM-2690 B (4) caused SMMC 7721 cells S phase arrest
The ability to escape from controlled cell division is a hallmark of cancer. To investigate the possible molecular target resulting in the cytotoxicity, the effect of KSM-2690 B (4) on cell cycle distribution was analyzed by flow cytometry. As shown in Fig. 4A and B, the percentage of SMMC7721 cells in S phase obviously increased after KSM-2690 B treatment. These results indicated that 4 caused S phase cell cycle arrest. As well known, cells through the different phases of cell cycle is regulated by specific cyclins and cyclin-dependent kinases (CDKs). CDK2 is activated by the binding of cyclin A and plays a key role throughout the S-phase in dividing cells.30 At the same time, the up-regulation of the cyclin kinase inhibitor p21 have been reported to cause S phase cell-cycle arrest and its abnormal levels also influence the progress of DNA replication during cell-cycle progression at S phase. To further investigate the mechanism of 4 in S phase arrest, related S phase regulators cyclin A2, CDK2, and p21 were examined by western blot method. As depicted in Fig. 4C–F, KSM-2690 B (4) decreased the protein levels of cyclin A2 and CDK2, and increased that of p21 in a dose-dependent manner.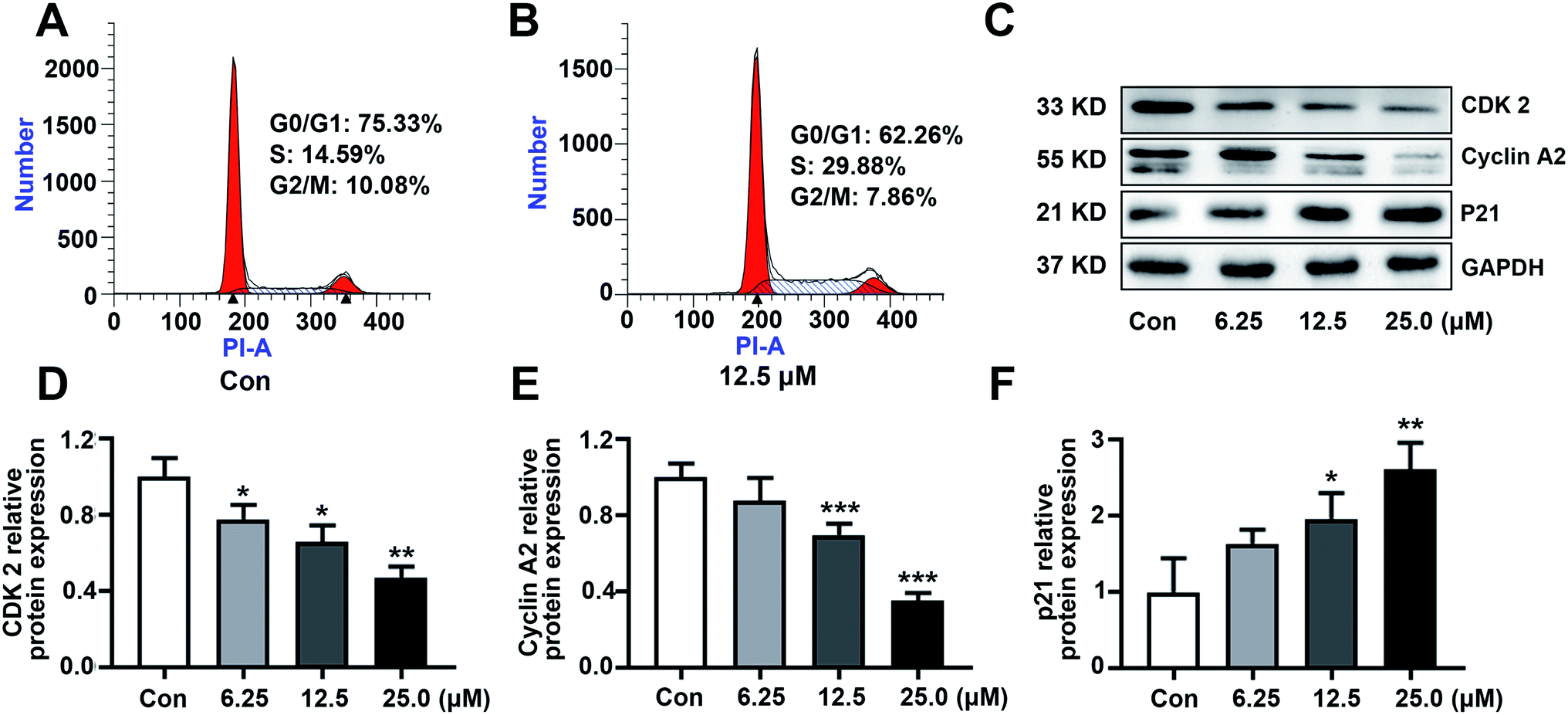 | ||
| Fig. 4 KSM-2690 B (4) caused S phase cell cycle arrest in SMMC 7721 cells. (A and B) Cell cycle distribution were analyzed by flow cytometry in SMMC7721 cells treated with 4 (0 or 12.5 μM). (C) The protein levels of cyclin A2, CDK2, and p21 in SMMC7721 cells treated with KSM-2690 B (4) (6.25, 12.5, and 25 μM) were evaluated by western blot. (D–F) The relative quantities analysis of cyclin A2, CDK2, and p21 in SMMC7721 cells. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. control. | ||
3.4. KSM-2690 B (4) induced apoptosis in SMMC-7721 cells
To explore the effect of KSM-2690 B on apoptosis of SMMC7721 cells, apoptosis detection was carried out by flow cytometric assay. As shown in Fig. 5A and B, the population of apoptotic cells increased in 4-treated SMMC7721 cells compared to the vehicle group. The results exhibited that 4 induced apoptosis in SMMC7721 cells. The balance of the pro-apoptotic factor Bax and anti-apoptotic factor Bcl-2 play an important role in progress of initiation of the apoptosis. The enhancement of Bax over Bcl-2 leads to the activation of caspases. The caspase-3 are crucial in the apoptosis cascade. Activation of executioner caspase-3, which results in cleaving structural proteins and causing the degradation of chromosomal DNA.31 To further investigate the underlying mechanism of KSM-2690 B on apoptosis, the expression of key factors associated with apoptosis was examined by western blot. As shown in Fig. 5C–F, KSM-2690 B up-regulated the protein levels of Bax, whereas down-regulated that of Bcl-2 in SMMC7721 cells in a dose-dependent manner. In addition, the expression levels of cleaved caspase-3 was obviously increased compared to those of the vehicle group.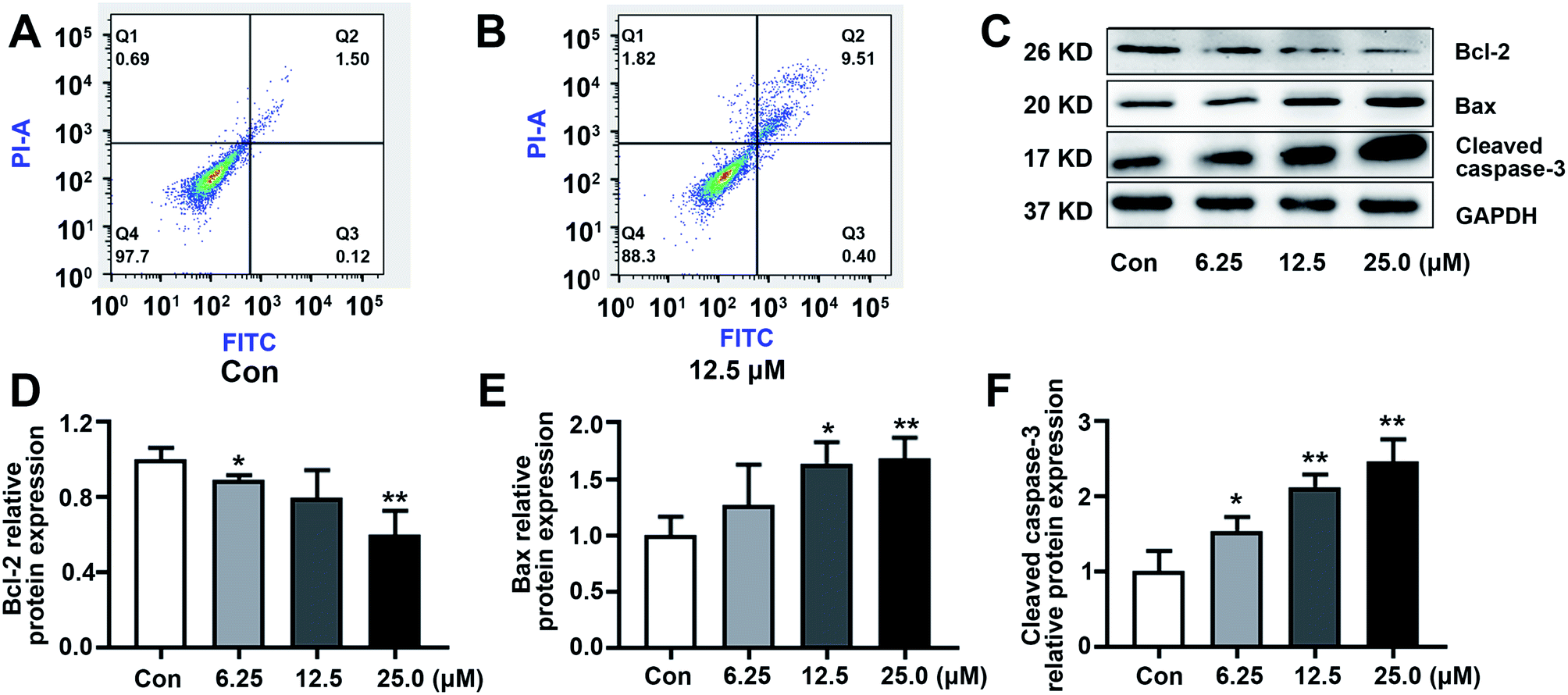 | ||
| Fig. 5 KSM-2690 B (4) induced apoptosis in SMMC7721 cells. (A and B) KSM-2690 B (4) induced apoptosis was analyzed by flow cytometry in SMMC7721 cells treated with 4 (0 or 12.5 μM). (C) The protein levels of Bax, Bcl-2, and cleaved caspase-3 in SMMC7721 treated with KSM-2690 B (4) (6.25, 12.5, and 25 μM) were evaluated by western blot. (D–F) The relative quantities analysis of Bax, Bcl-2, and cleaved caspase-3 in SMMC7721 cells. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. control. | ||
4. Conclusion
In summary, oxazolomycins D–F (1–3), a mixture of two isomers, glaucumycins A, B (6a/6b), as well as two known oxazolomycins (4–5) were isolated from Streptomyces glaucus. Oxazolomycins 1, 2, 4, 5 demonstrated weak or modest cytotoxic activity against four human cancer cell lines, with IC50 values ranging from 10.6 ± 1.7 to 89.5 ± 6.6 μM (or >100 μM). Further study exhibited that 4 caused S phase cell cycle arrest and induced apoptosis in SMMC7721 cells through regulation the expression of related protein cyclin A2, CDK2, p21, Bcl-2, Bax, and activation the cleavage of caspase-3.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Specific Research Project of Guangxi for Research Bases and Talents (grant no. AD18281066), P. R. China, and Science and Technology Major Project of Guangxi, P. R. China (grant no. AA18242026).References
- M. G. Moloney, P. C. Trippier, M. Yaqoob and Z. Wang, Curr. Drug Discovery Technol., 2004, 1, 181–199 CrossRef CAS PubMed.
- P. Oleksak, J. Gonda, E. Nepovimova, K. Kuca and K. Musilek, RSC Adv., 2020, 10, 40745–40794 RSC.
- K. Takahashi, M. Kawabata, D. Uemura, S. Iwadare, R. Mitomo, F. Nakano and A. Matsuzaki, Tetrahedron Lett., 1985, 26, 1077–1078 CrossRef CAS.
- T. Mori, K. Takahashi, M. Kashiwabara, D. Uemura, C. Katayama, S. Iwadare, Y. Shizuri, R. Mitomo, F. Nakano and A. Matsuzaki, Tetrahedron Lett., 1985, 26, 1073–1076 CrossRef CAS.
- H. Kanzaki, K. Wada, T. Nitoda and K. Kawazu, Biosci., Biotechnol., Biochem., 1998, 62, 438–442 CrossRef CAS PubMed.
- M. Ogura, H. Nakayama, K. Furihata, A. Shimuza, H. Seto and N. Otake, J. Antibiot., 1985, 38, 669–673 CrossRef CAS PubMed.
- Y. Ikeda, S. Kondo, H. Naganawa, S. Hattori and M. Hamada, J. Antibiot., 1991, 44, 453–455 CrossRef CAS PubMed.
- G. Ryu, S. Hwang and S. Kim, J. Antibiot., 1997, 50, 1064–1067 CrossRef CAS PubMed.
- G. Ryu and S. Kim, J. Antibiot., 1999, 52, 193–197 CrossRef CAS PubMed.
- T. Otani, K. Yoshida, H. Kubota, S. Kawai, S. Ito, H. Horic, T. Ishiyama and T. Oki, J. Antibiot., 2000, 53, 1397–1400 CrossRef CAS PubMed.
- R. R. Manam, S. Teisan, D. J. White, B. Nicholson, J. Grodberg, S. T. C. Neuteboom, K. S. Lam, D. A. Mosca, G. K. Lloyd and B. C. M. Potts, J. Nat. Prod., 2005, 68, 240–243 CrossRef CAS PubMed.
- K. Ko, S. H. Lee, S. H. Kim, E. H. Kim, K. B. Oh, J. Shin and D. C. Oh, J. Nat. Prod., 2014, 77, 2099–2104 CrossRef CAS PubMed.
- W. Koomsiri, Y. Inahashi, T. Kimura, K. Shiomi, Y. Takahashi, S. Omura, A. Thamchaipenet and T. Nakashima, J. Antibiot., 2017, 70, 1142–1145 CrossRef CAS PubMed.
- Y. Tanaka, I. Kanaya, Y. Takahashi, M. Shinose, H. Tanaka and S. Omura, J. Antibiot., 1993, 46, 1208–1213 CrossRef CAS PubMed.
- K. Shiomi, N. Arai, M. Shinose, Y. Takahashi, H. Yoshida, J. Iwabuchi, Y. Tanaka and S. Omura, J. Antibiot., 1995, 48, 714–719 CrossRef CAS PubMed.
- S. Kawai, G. Kawabata, A. Kobayashi and K. Kawazu, Agric. Biol. Chem., 1989, 53, 1127–1133 CAS.
- K. Kawazu, H. Kanzaki, G. Kawabata, S. Kawai and A. Kobayashi, Biosci., Biotechnol., Biochem., 1992, 56, 1382–1385 CrossRef CAS.
- H. Kanzaki, T. Ichioka, A. Kobayashi and K. Kawazu, Biosci., Biotechnol., Biochem., 1996, 60, 1535–1537 CrossRef CAS.
- E. O. Onyango, J. Tsurumoto, N. Imai, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., 2007, 119, 6823–6825 CrossRef.
- J. H. Kim, I. Kim, Y. Song, M. J. Kim and S. Kim, Angew. Chem., 2019, 131, 11134–11138 CrossRef.
- K. Eto, M. Yoshino, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Lett., 2011, 13, 5398–5401 CrossRef CAS PubMed.
- T. Nishimaru, K. Eto, K. Komine, J. Ishihara and S. Hatakeyama, Chem.–Eur. J., 2019, 25, 7927–7934 CrossRef CAS PubMed.
- M. Kumar, L. Bromhead, Z. Anderson, A. Overy and J. W. Burton, Chem.–Eur. J., 2018, 24, 1–5 CrossRef.
- M. Yoshino, K. Eto, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Biomol. Chem., 2012, 10, 8164–8174 RSC.
- J. P. N. Papillon and R. J. K. Taylor, Org. Lett., 2000, 2, 1987–1990 CrossRef CAS PubMed.
- S. Balcells, M. B. Haughey, J. C. L. Walker, L. Josa-Culleré, C. Towers and T. J. Donohoe, Org. Lett., 2018, 20, 3583–3586 CrossRef CAS PubMed.
- K. Eto, J. Ishihara and S. Hatakeyama, Tetrahedron, 2018, 74, 711–719 CrossRef CAS.
- X. Gao, H. Yao, Y. Mu, P. Guan, G. Li, B. Lin, Y. Jiang, L. Han, X. Huang and C. Jiang, Bioorg. Chem., 2019, 93, 103311 CrossRef CAS PubMed.
- C. Zhao, J. M. Coughlin, J. Ju, D. Zhu, E. Wendt-Pienkowski, X. Zhou, Z. Wang, B. Shen and Z. Deng, J. Biol. Chem., 2010, 285, 20097–20108 CrossRef CAS PubMed.
- S. Tadesse, E. C. Caldon, W. Tilley and S. Wang, J. Med. Chem., 2019, 62, 4233–4251 CrossRef CAS PubMed.
- F. Edlich, Biochem. Biophys. Res. Commun., 2018, 500, 26–34 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRMS spectra for compounds 1–3, 6 (PDF). See DOI: 10.1039/d1ra06182h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |