
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtmosphere-sensitive photoluminescence of CoxFe3−xO4 metal oxide nanoparticles†
Julian Klein a,
Laura Kampermanna,
Sascha Saddelerb,
Jannik Kortea,
Oliver Kowollika,
Tim Smolaa,
Stephan Schulzb and
Gerd Bacher*a
a,
Laura Kampermanna,
Sascha Saddelerb,
Jannik Kortea,
Oliver Kowollika,
Tim Smolaa,
Stephan Schulzb and
Gerd Bacher*a
aWerkstoffe der Elektrotechnik and CENIDE, Universität Duisburg-Essen, Bismarckstraße 81, 47057 Duisburg, Germany. E-mail: gerd.bacher@uni-due.de
bInstitute of Inorganic Chemistry and CENIDE, University of Duisburg-Essen, Universitätsstraße 7, 45141 Essen, Germany
First published on 18th October 2021
Abstract
In this work the photoluminescence (PL) of CoxFe3−xO4 spinel oxide nanoparticles under pulsed UV laser irradiation (λexc = 270 nm) is investigated for varying Co/Fe ratios (x = 0.4⋯2.5). A broad emission in the green spectral range is observed, exhibiting two maxima at around 506 nm, which is dominant for Fe-rich nanoparticles (x = 0.4, 0.9), and at around 530 nm, that is more pronounced for Co-rich nanoparticles (x > 1.6). As examinations in different atmospheres show that the observed emission reacts sensitively to the presence of water, it is proposed that the emission is mainly caused by OH groups with terminal or bridging metal–O bonds on the CoxFe3−xO4 surface. Raman spectroscopy supports that the emission maximum at 506 nm corresponds to terminal OH groups bound to metal cations on tetrahedral sites (i.e., Fe3+), while the maximum around 530 nm corresponds to terminal OH groups bound to metal cations on octahedral sites (i.e., Co3+). Photoinduced dehydroxylation shows that OH groups can be removed on Fe-rich nanoparticles more easily, leading to a conversion process and the formation of new OH groups with different bonds to the surface. As such behavior is not observed for CoxFe3−xO4 with x > 1.6, we conclude that the OH groups are more stable against dehydroxylation on Co-rich nanoparticles. The higher OH stability is expected to lead to a higher catalytic activity of Co-rich cobalt ferrites in the electrochemical generation of oxygen.
Introduction
Metal oxide nanostructures are a promising class of materials for a variety of applications in catalysis, energy storage and sensor technology1–4 due to their unique chemical and physical properties, including large specific surface, high electronic conductivity, and low toxicity, as well as inexpensive production.1,2,5,6 In recent years, the suitability of metal oxide nanostructures for generating “green energy” has come to the fore. In photoelectrochemical cells (PECs), solar energy is stored by generating hydrogen using a catalytically active material.7–9 Due to their outstanding electrochemical activity and stability, spinel-type oxides with the empirical formula, AxB3−xO4 (with A, B = Mg, Ni, Co, Fe,…) are promising candidates for technical applications in PECs,7,10–12 in photocatalytic processes and electrochemical energy storage7,13,14 as well as in rechargeable metal–air batteries,15–18 respectively. The catalytic efficiency of spinel-type oxide nanocatalysts depends on their size, shape, composition and, in particular, on the nanoparticle surface with its defect structure and adsorbed molecular species.19–29 A common method to examine the surface of catalytically active metal oxides is photoluminescence (PL) spectroscopy. PL spectroscopy can be used to investigate surface-active sites on the spinel oxide catalysts in connection with adsorption or photocatalytic processes.30,31For a variety of catalytically active metal oxide materials, like (iron doped) TiO2 nanocrystal films,32 HfO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 or ZrO234 nanoparticles as well as ThO235 or Al2O336–38 bulk materials, it is quite typical to observe a broad emission band with a maximum in the green spectral range. Even if this emission band is well recognized and has been known for decades, the underlying mechanism is still controversially discussed. It is often assumed that the broad green emission is caused by the presence of oxygen defects in different oxidation states.33,39–41 Models have been postulated that assume optical transitions from the conduction band edge to oxygen defect states40 or transitions from oxygen defect states to the valence band.41 Other concepts do not assign the observed green emission to optical transitions between energy bands and (bulk) defect levels, but to emission centers on the metal oxide surface. On the one side, the PL of insulating oxides such as alkaline earth oxides is related to surface excitons and results from charge transfer processes involving metal and oxygen ions on the surface.31,42 This emission is sensitive to atmosphere and can usually only be detected after treatment at high temperatures in vacuum and with the exclusion of quenching gases such as oxygen.31 On the other side, the broad green emission of metal oxides is explained by molecular adsorbates bound to the metal oxide surface.38,43 The molecular species that are usually linked to the emission are OH groups that are bonded to surface metal cations.36,38 When exposed to UV irradiation, photoinduced OH radicals can form and be brought into an excited electronic state.44 The vibration properties and thus the subsequent radiative recombination process as well as the emission wavelength are very sensitive to the environment and the binding of the OH species.44–46 Another molecule that is always present on the surface of metal oxides in ambient conditions is molecular oxygen (O2). Molecular O2 can also be excited under UV irradiation, causing emission lines in the UV/blue spectral range and thus contribute to the broad metal oxide emission.47
33 or ZrO234 nanoparticles as well as ThO235 or Al2O336–38 bulk materials, it is quite typical to observe a broad emission band with a maximum in the green spectral range. Even if this emission band is well recognized and has been known for decades, the underlying mechanism is still controversially discussed. It is often assumed that the broad green emission is caused by the presence of oxygen defects in different oxidation states.33,39–41 Models have been postulated that assume optical transitions from the conduction band edge to oxygen defect states40 or transitions from oxygen defect states to the valence band.41 Other concepts do not assign the observed green emission to optical transitions between energy bands and (bulk) defect levels, but to emission centers on the metal oxide surface. On the one side, the PL of insulating oxides such as alkaline earth oxides is related to surface excitons and results from charge transfer processes involving metal and oxygen ions on the surface.31,42 This emission is sensitive to atmosphere and can usually only be detected after treatment at high temperatures in vacuum and with the exclusion of quenching gases such as oxygen.31 On the other side, the broad green emission of metal oxides is explained by molecular adsorbates bound to the metal oxide surface.38,43 The molecular species that are usually linked to the emission are OH groups that are bonded to surface metal cations.36,38 When exposed to UV irradiation, photoinduced OH radicals can form and be brought into an excited electronic state.44 The vibration properties and thus the subsequent radiative recombination process as well as the emission wavelength are very sensitive to the environment and the binding of the OH species.44–46 Another molecule that is always present on the surface of metal oxides in ambient conditions is molecular oxygen (O2). Molecular O2 can also be excited under UV irradiation, causing emission lines in the UV/blue spectral range and thus contribute to the broad metal oxide emission.47
The emission behavior of CoxFe3−xO4 (nanoparticles) under UV excitation is almost completely unknown. The limited literature on this topic describes emissions that differ greatly, especially in the energetic position of the emission bands, and no clear picture of the emission cause exists.48–51 A systematic investigation of the emission behavior of CoxFe3−xO4 and the cause of the emission is still missing. Due to the high sensitivity regarding smallest changes in the surface or bulk material, PL measurements could provide new information about the dynamics of catalytic processes, such as the migration of molecular adsorbates, surface charge transfer processes or the characterization and identification of active centers of CoxFe3−xO4.31,42
Here, we demonstrate that a broad green emission is detected for CoxFe3−xO4 (x = 0.4 to 2.5) nanoparticles under UV laser irradiation (λexc = 270 nm) as well. Although a change in the Co/Fe ratio should result in a change in the spinel-type structure and the band gap energy,52 no systematic variation of the emission wavelength with Co/Fe ratio is found. To gain further insights into a possible molecular character of the observed broad green emission, atmosphere-dependent investigations are carried out indicating terminal and bridging surface OH groups as emissive species. Using Raman spectroscopy, a link between individual emission lines and OH groups bound to specific metal cations is found. Finally, photoinduced dehydroxylation is used to investigate the influence of the Co/Fe ratio on the stability of surface OH groups against dehydroxylation. A higher OH stability could result in a better performance of Co-rich nanoparticles in the electrochemical splitting of water.
Experimental
Materials
Metal acetylacetonates (Co(acac)2, 99%; Fe(acac)3, 97%), poly(ethyleneimine) (PEI; branched, MW = 800 by LS) and triethylene glycol (TEG, 99%) were purchased from Sigma-Aldrich and used without further purification.General synthesis of CoxFe3−xO4 nanoparticles
Co(acac)2 and/or Fe(acac)3 (Table 1) were suspended in 150 mg of polyethylenimine (PEI) and 10 g of triethylene glycol (TEG), degassed at 120 °C for 1 h and then stirred at 250 °C for 1 h. After cooling down to ambient temperature, the particles were precipitated by adding a 1:1 mixture of acetone and ethyl acetate, and the precipitate was isolated by centrifugation (3000 rpm, 10 min). The powder was washed two times with acetone and dried at ambient temperature in vacuo. TEM investigations showed that the size of the resulting nanoparticles varied between 3.5 ± 0.7 nm and 7.6 ± 1.8 nm (Fig. S1 and Table S1†).
Sample preparation for Raman and PL spectroscopy
All CoxFe3−xO4 nanoparticles were suspended in ethanol (5.5 ± 2.5 mg mL−1) followed by drop-casting of multiple layers on silicon substrates (5 × 5 mm2). For PL measurements at low temperatures, GaAs substrates were used instead of silicon.Energy dispersive X-ray spectroscopy
To analyze the nanoparticles composition energy dispersive X-ray (EDX) spectroscopy studies on the nanoparticles were performed using a JEOL JSM6510 scanning electron microscope (SEM) equipped with an energy dispersive X-ray detector (Bruker Quantax 400). The obtained spectra were quantified by using the software Esprit 1.9 (Bruker). The determined compositions of the nanoparticles are summarized in Table S2 in the ESI.†Transmission electron microscopy
Size and morphology of the nanoparticles were analyzed by using a JEOL 2010 transmission electron microscope (TEM) operated at 200 kV accelerating voltage. All TEM samples were prepared by drop casting a powder dispersion in ethanol onto a carbon film supported copper grid. The determined particle size distribution is shown in Fig. S1 and Table S1 in the ESI.†X-ray diffraction analysis
X-ray diffraction (XRD) patterns of the nanoparticle powders were measured at room temperature by using a Bruker D8 Advance powder diffractometer in Bragg–Brentano geometry with CuKα radiation (λ = 0.15418 nm, U = 40 kV and I = 40 mA). The powder samples were investigated in the diffraction angle range 5° to 90°. The analysis of the crystal structure by XRD is shown in Fig. S2 in the ESI.†Raman spectroscopy
Raman spectroscopy was performed in ambient atmosphere with a NTEGRA Spectra system from NT-MDT with a laser wavelength of 532 nm, a spot size of 0.6 μm (100× objective), a laser power density of S = 0.3 × 103 kW cm−2, a 600 mm−1 grating and a spectral resolution of 6.5 cm−1. For the Raman spectra a 10 μm × 10 μm area was scanned and a total of 100 measurements were averaged. The analysis of the crystal structure by Raman spectroscopy is shown in Fig. S3 in the ESI.†Photoluminescence spectroscopy
Photoluminescence spectroscopy was performed with a pulsed frequency-tripled titanium-sapphire laser (Mira 900 from Coherent, frequency ∼76 MHz, pulse width of ∼130 fs) with a laser wavelength of 270 nm and a pulse fluence ρ = 2 μJ cm−2. The photoluminescence of the samples was collected with an iHR320 monochromator and a Symphony II CCD camera from Horiba Scientific. The spectral resolution Δλ is around 3 nm for a 150 mm−1 grating and a 150 μm slit width.Atmosphere dependent investigations were carried out in a cryostat (Janis ST-300) which was evacuated for vacuum investigations. For investigations in an oxygen-rich atmosphere the cryostat was flooded with synthetic air (20 ± 2% O2/N2). In order to carry out investigations in humid air, room air from the environment with a relative humidity (RH) of 45 ± 1% was used. During the PL measurements the cryostat was permanently open to these ambient conditions. Before a PL investigation was taken in humid air, the nanoparticle samples were stored in humid air for a few days. Before measurements were taken in vacuum atmosphere, a vacuum was drawn for over 16 hours with a final pressure of ∼10−6 mbar.
Time dependent photoluminescence measurements during which the atmosphere was changed were carried out without interruption and removing the sample. For this purpose, a vacuum was first generated again for 16 hours and PL measurements were carried out in vacuum atmosphere at constant time intervals. Without interrupting the continuously running PL measurements, the cryostat was then flooded with humid room air (45 ± 1%). PL measurements with a change in the atmospheric conditions from vacuum to a dry, oxygen-rich atmosphere (20 ± 2% O2/N2) were carried out in the same way without interruption or removal of the sample. For low temperature photoluminescence measurements, the cryostat was cooled with liquid helium.
Results and discussion
To identify possible contributions of surface adsorbates on the PL emission of CoxFe3−xO4 nanoparticles, we on purpose change both, atmosphere as well as UV irradiation time. Fig. 1a shows the PL spectrum of CoxFe3−xO4 nanoparticles with a Co content of x = 0.4 and 2, respectively, under pulsed UV excitation (λexc = 270 nm). The measurement of the nanoparticle's emission is carried out in humid air (RH of ∼45 ± 1%). A broad emission between 350 nm and 750 nm with two emission maxima at around 506 nm and 530 nm is detected for all nanoparticles. The spectral position of these two emission maxima is the same for all nanoparticles (Fig. S4†), only the respective intensity changes. For Fe-rich samples (x = 0.4 and 0.9), the emission maximum at around 506 nm is dominant, while for Co-rich samples (x > 1.6), the emission maximum at around 530 nm is more intense. According to the position of these two emission maxima, characteristic emissions at 506 nm (emission line S1) and 530 nm (emission line S2) are identified.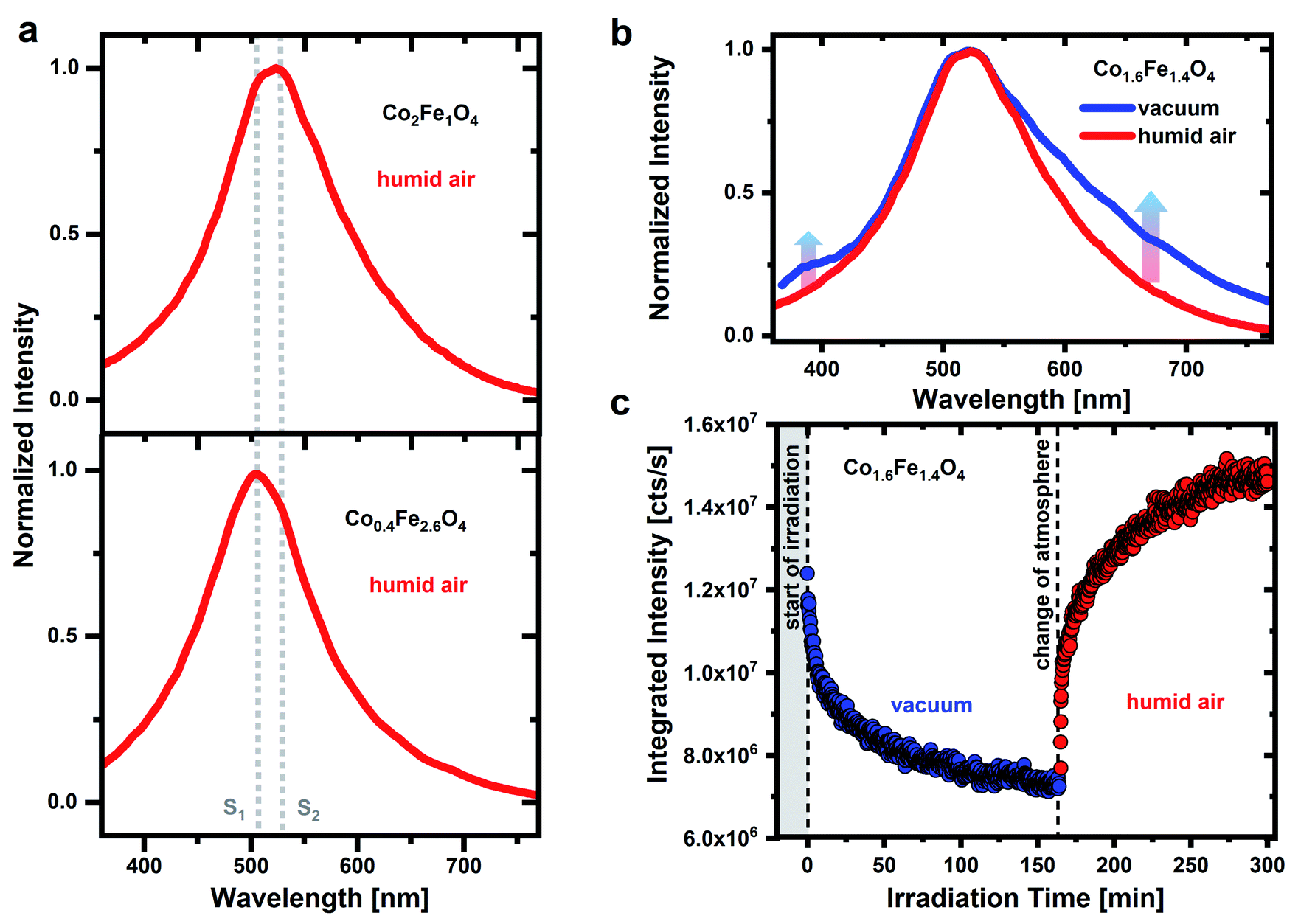 | ||
| Fig. 1 (a) Normalized PL spectra of CoxFe3−xO4 nanoparticles with x = 0.4 and 2, respectively, in humid air (RH of ∼45 ± 1%). (b) Comparison of the normalized PL spectra of Co1.6Fe1.4O4 nanoparticles in humid air and vacuum (∼10−6 mbar). (c) Time dependent behavior of the integrated PL intensity of the Co1.6Fe1.4O4 nanoparticle emission under UV irradiation. The nanoparticles are first examined in vacuum (0 to 160 min) and immediately afterwards in humid air. The PL measurements are detected under pulsed UV irradiation (λexc = 270 nm, ρ = 2 μJ cm−2). All spectra are recorded immediately after starting the UV irradiation. | ||
A broad green emission for metal oxides was previously addressed to optical transitions associated with oxygen defect states under the participation of the band edges or near band edge defects states.33,39–41 Such an emission process can occur via the recombination of an electron from the conduction band with a hole trapped by an oxygen vacancy.40,53 Alternatively, an electron can be trapped by an oxygen vacancy and then recombine with a hole in the valence band or a nearby defect state.41,54 In both cases either the conduction or the valence band edge is involved in the emission process. However, if the Co/Fe ratio for CoxFe3−xO4 changes from Fe-rich (x = 0.4 and 0.9) to Co-rich nanoparticles (x > 1.6), a change in the spinel crystal structure from a more inverse spinel phase to a more normal spinel phase occurred (Fig. S2 and S3†) and thus a change in the electronic band structure occurs as well.52 As a result, the detected emission would have to shift with Co/Fe ratio, which cannot be seen in Fig. 1 and S4.† The emission lines S1 and S2 remain unchanged for all CoxFe3−xO4 nanoparticles regarding their spectral position. Consequently, an optical transition via oxygen vacancies appears unlikely.
Charge transfer processes between surface metal and oxygen ions are often discussed as a cause of emissions for metal oxides as well. Under UV excitation, the metal–oxygen complex can be brought into the excited state and lead to the emission of photons in the green spectral range during the subsequent radiative recombination.31,55 However, if metal oxides are exposed to humid air, OH groups as well as (physisorbed) water molecules or oxygen species are present on the surface.46,56,57 As a result of this surface coverage, which is unavoidable in humid air, nearly no emission from surface metal–oxygen compounds can occur. Emissions related to such surface charge transfer processes are almost completely quenched by adsorbed molecules (i.e. O2) and can only be detected in vacuum after annealing at high outgassing temperatures.31 Therefore, an optical transition related to charge transfer processes as the cause of the detected emission under ambient conditions appears to be unlikely.
When exposed to UV light, surface adsorbates themselves can also cause an emission. The adsorbates that are primarily made responsible for the occurrence of a broad green emission are OH groups with absorption bands in an energy range > 3.4 eV and thus excitable by high-energy UV irradiation.35–38,58 OH groups present in the gas phase are usually assigned to narrow emission lines in the UV range.44 These emissions related to OH species are attributed to the photoinduced generation of OH radicals (OH*) and their subsequent radiative recombination.43,44,47,59 After UV excitation, optical transitions between the first excited electronic state A2Σ+ and the electronic ground state X2Π of OH* can occur.44,47 However, if the OH groups are bound to a (metal oxide) surface, broad emissions at lower energies are observed.36,38,45,46 Here, the OH groups can be excited either directly or indirectly via the metal oxide surface and a subsequent energy transfer to an OH group.46
By studying the emission of gradually annealed Al2O3, it was shown that dehydroxylation of the surface and the removal of OH groups significantly reduces the intensity of a broad green emission with a maximum at around 570 nm.38 Additional gravimetric investigations proved that this high temperature-dependent decrease in emission intensity follows the decrease in the density of OH groups on the metal oxide surface.36,38 By investigating hydroxylated MgO surfaces different types of surface OH groups were identified via photoluminescence measurements.46 A broad emission in the range from 300 nm to 600 nm was detected and emission maxima at 410 nm and 470 nm were assigned to different types of surface OH groups. Their electronic structure and vibration properties are very sensitive to the local structure and topology of the metal oxide surface and, in particular, to the coverage and environment of the OH groups.45,46 Adsorbates such as OH groups as the cause of the emission would also explain why similar broad emission bands in the green spectral range were observed in various metal oxides with completely different electronic structures.32,34,38 Beside OH groups, adsorbate emissions from oxygen species on the metal oxide surface have to be considered as well. If an excitation wavelength in a range between 250 nm and 300 nm is used, emission lines between 340 nm and 420 nm through A′3Δu → X3Σg− progressions of the O2 molecule can occur47,60 and thus also make a contribution to the broad emission observed.
In case the emission is caused by adsorbates like OH groups and/or O2 molecules on the nanoparticle surface, two assumptions can be made regarding the emission behavior. First, the emission should be sensitive to the atmosphere and a reduction in the coverage of the nanoparticle surface with adsorbates should result in a reduction in emission intensity. This is a crucial difference between an emission caused by adsorbates and an emission related to charge transfer processes of surface metal–oxygen complexes as the latter should increase with decreasing surface coverage. Second, this process should be reversible, and the initial emission intensity should be reached again when the initial coverage is restored.
Fig. 1b shows a comparison of the Co1.6Fe1.4O4 nanoparticle emission spectrum in humid air and in vacuum. Before the emission spectrum is recorded in humid air, the sample was stored in air for several days and before the measurement is taken in vacuum atmosphere, a vacuum was drawn for 16 hours. The emission spectrum widens in vacuum towards the low-energy tail and the formation of several weak emission shoulders is detected. In addition, another shoulder is formed on the high-energy side in a wavelength range of 350 nm to 430 nm. Besides these changes, the emission maximum does not shift. The detected emission in the UV spectral range can result from oxygen molecules. For the free O2 molecule in the gas phase, A′3Δu → X3Σg− transitions between the ground vibrational mode of the excited state (ν′ = 0) and the vibrational modes ν′′ = 3 to ν′′ = 7 of the ground state result in emission lines between 337 nm and 420 nm.47 Emission lines for the 0–5 to 0–7 transitions occur at wavelengths of around 375 nm, 396 nm and 420 nm.21,60 An assignment of the emission features observed here at around 350 nm to 430 nm to these transitions of the O2 molecule is confirmed by low-temperature studies at 10 K of the CoxFe3−xO4 nanoparticles (Fig. S5†). Here, emission lines at 376 nm, 393 nm and 418 nm are well resolved and in good agreement with the 0–5 to 0–7 transitions of the free O2 molecule. The O2 molecules that are responsible for this UV emission do not stem from the background atmosphere but are rather formed during dehydroxylation under UV irradiation.47 However, if the surface coverage with water or OH groups is too high, these adsorbates are expected to interact with the O2 molecules and weaken their emissions. For this reason, the emission can only be observed in a protective (vacuum) atmosphere. This contradiction at first sight, that water/OH groups are prerequisites for the O2 emission but prevent the same emission if the coverage is too high, is explained in more detail below.
Fig. 1c shows a temporally resolved PL study for Co1.6Fe1.4O4 nanoparticles in two different atmospheres. Under permanent UV irradiation, the nanoparticles are first examined for 160 min in a vacuum atmosphere and then for further 140 min in humid air. In vacuum, the integrated emission intensity drops rapidly after the start of irradiation with a time constant of about 3 min. After this pronounced initial drop, the emission intensity reaches ∼0.6 times the initial intensity after 160 min of irradiation. It must be noted that the samples were stored in vacuum (10−6 mbar) over a period of 16 hours prior to these measurements. By creating a vacuum, adsorbates are partially removed from a metal oxide surface. Physisorbed molecules, such as water or O2, weakly bound to a metal oxide surface through van der Waals forces, are expected to be removed from the surface in vacuum even at room temperature.61,62 In addition to physisorbed water, water molecules can also be bound to the surface through dissociative chemisorption. This second type of bond creates OH groups on the surface that desorb in a vacuum only at higher temperatures.61,62 Thus, it is not possible to completely remove all OH groups from the surface of metal oxides as even under ultra-high vacuum (UHV) conditions, some of the OH groups always remain on the surface.63
If metal oxides are now irradiated with high-energy UV photons, further OH groups can be removed from the surface by a photochemical dissociation process.44,64 In case the PL is caused by OH groups, this should lead to a drop in intensity as a result,38 in good agreement with our findings. The observed decrease in emission intensity in vacuum can therefore be explained by a dehydroxylation process and the removal of OH groups on the nanoparticles surface, triggered by UV irradiation. If such a dehydroxylated nanoparticle surface is exposed to humid air, it is again covered with OH groups by (photoinduced) dissociation of water molecules from the environment. The dissociation of water and formation of new OH groups under UV irradiation apparently predominates the photoinduced removal of OH groups in humid air after a vacuum treatment. Thus, the emission intensity increases (up to 1.2 times of the initial emission intensity), consistent with our assumption of a reversible removal of OH groups from the surface in vacuum and a (re-) coverage with OH groups in humid air.
The importance of a humid environment and water/OH groups on the surface for the observed emission is supported by studies in a dry, oxygen-rich environment (Fig. 2a). Physisorbed water was again removed by drawing a vacuum (for 16 hours) and subsequent UV irradiation (0 to 11 min) before the sample was exposed to synthetic air (20 ± 2% O2/N2). Fig. 2a shows a decrease of the integrated PL intensity in vacuum atmosphere due to the removal of residual surface OH groups as discussed above. After exposing the nanoparticles to synthetic air, the emission intensity drops even further, in strong contrast to what is observed after exposure to humid air. Hereby, a steady decrease in emission intensity would be expected. When changing the atmosphere, however, it cannot be ruled out that there will be a minimal displacement of the sample. This leads to the irradiation of a different sample area, resulting in a slight increase in emission intensity when the atmosphere is changed. The decrease in emission intensity in synthetic air with time clearly demonstrates that oxygen adsorbates cannot be responsible for the green emission and strengthens the importance of a humid environment. The experimental finding agrees well with the assignment of the broad green emission primarily to OH groups on the nanoparticles surface.
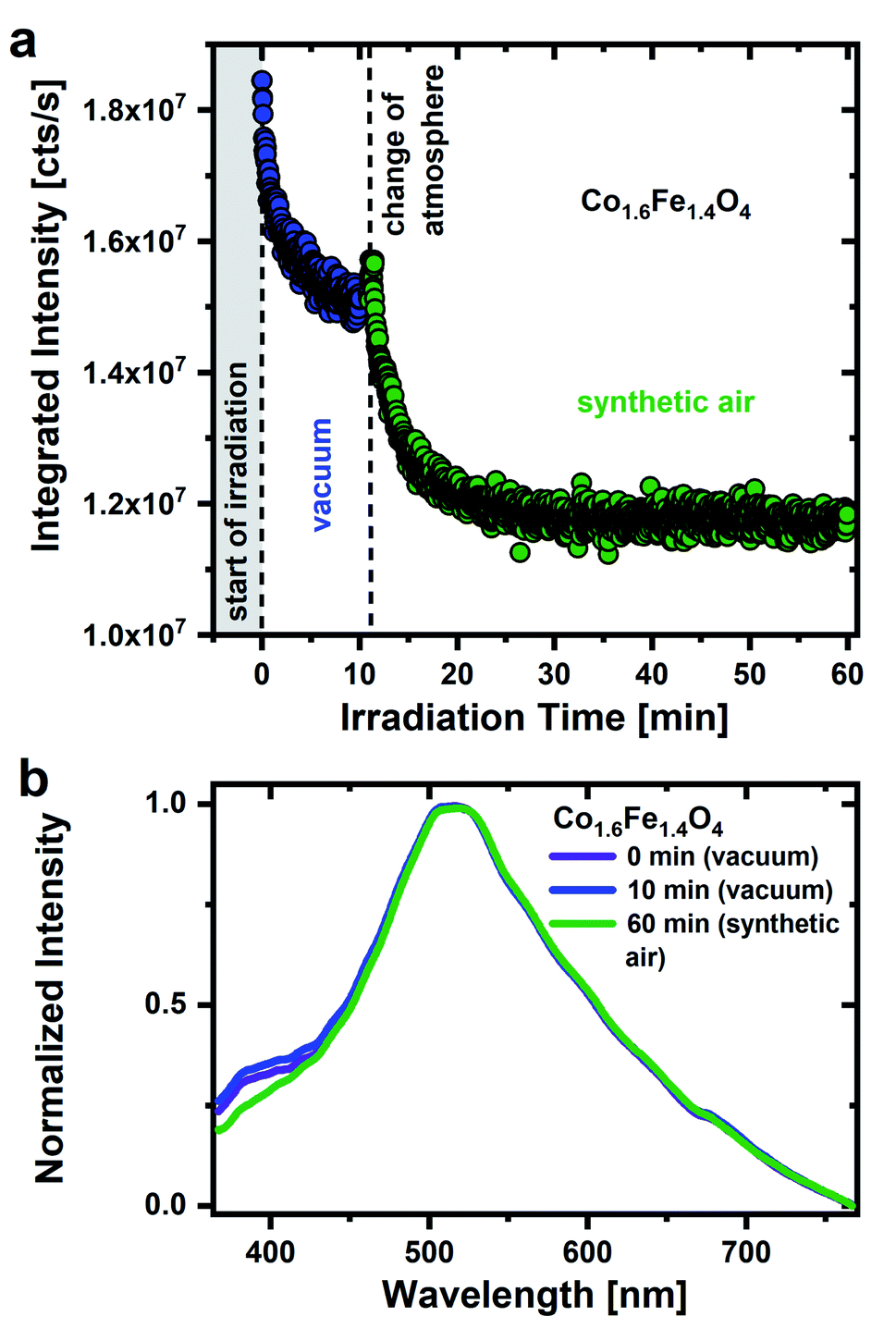 | ||
| Fig. 2 (a) Time dependent behavior of the integrated PL intensity of the Co1.6Fe1.4O4 nanoparticle emission under UV irradiation (λexc = 270 nm, ρ = 2 μJ cm−2). The sample was first examined for 11 min in vacuum atmosphere and immediately afterwards exposed to synthetic air (20 ± 2% O2/N2). (b) Associated emission spectra after an irradiation time of 0 and 10 min in vacuum and after 60 min in synthetic air. | ||
Fig. 2b shows the associated emission spectra after 0, 10 and 60 min. The main emission with the maximum in the green spectral range as well as the low-energy side remain unchanged over the whole time range. Surprisingly, a pronounced emission feature around 380–430 nm becomes stronger in vacuum with respect to a synthetic air environment as was previously reported for Al2O3 and SiO2 nanoparticles.47 Anjiki et al. detected three emission lines at 372 nm (374 nm), 392 nm (394 nm) and 414 nm (416 nm), which only occurred in vacuum atmosphere. As described before, these emission lines are in accordance with our low temperature measurements (Fig. S5†) and therefore assigned to optical transitions of O2 molecules that are formed under UV irradiation. According to Anjiki et al.,47 the oxygen species responsible for these emission lines do not originate from adsorbed O2 molecules but are photoinduced during surface dehydroxylation. As a result of the surface dehydroxylation, a M–O2 defect pair is formed, which split under UV irradiation resulting in a UV/blue emission from the excited O2 molecule.47 This is in agreement with our observations. The emission feature at 380–430 nm cannot be observed in an O2-rich atmosphere, instead the presence of water/OH appears to be a prerequisite. On the other hand, if the surface coverage with water is too high, however, it also seems to weaken the O2 related emission. In humid air, neighboring surface water molecules can interact with the M–O2 defect pair via hydrogen bonds.47 This affects the vibrational properties of the defect pair, the emission smears out and a weaker spectrally broad emission feature in the UV/blue range is observed in humid air.47 For this reason, the O2 emission resulting from photoinduced dehydroxylation is only observed in a protective atmosphere. Here, CoxFe3−xO4 nanoparticle do not necessarily have to be in a vacuum atmosphere. By storing CoxFe3−xO4 nanoparticles in a N2 protective atmosphere, the UV/blue emission can be detected as well (Fig. S6†).
As seen in Fig. 1 and 2, the temporal behavior of the OH groups-related green emission of CoxFe3−xO4 nanoparticles is very sensitive to both UV irradiation as well as atmospheric conditions, supporting the adsorbate character of the broad green emission. The Co/Fe ratio of the nanoparticles does not change the spectral position of the emission lines, whereas it plays a crucial role in the temporal behavior of the emission. The time dependent behavior of the broad green emission of Co0.4Fe2.6O4 nanoparticles with a low Co content is shown in Fig. 3 under permanent UV irradiation in humid air. For these Fe-rich samples (x = 0.4), initially an increase in emission intensity by a factor of 3.4 is detected. After an irradiation time of 12 minutes, the integrated emission intensity saturates and then decreases until it nearly reaches the original emission intensity (2 × 106 cts s−1) after 90 minutes. Interestingly, pronounced low-energy features in the PL spectra evolve under UV irradiation for Fe-rich nanoparticles in humid air (Fig. 3b). At the beginning of irradiation, an emission between 350 nm and 750 nm with a maximum at a wavelength of 506 nm (S1) is observed. After 10 minutes, the emission maximum has shifted to 530 nm (S2) and further secondary maxima begin to develop on the low-energy side. After 90 min of irradiation, four further maxima have finally formed at around 561 nm (S3), 602 nm (S4), 640 nm (S5) and 686 nm (S6). All nanoparticles with a low Co-content (x = 0.4 to x = 1.6) show a similar behavior of the emission intensity under UV irradiation in humid air (Fig. S7†).
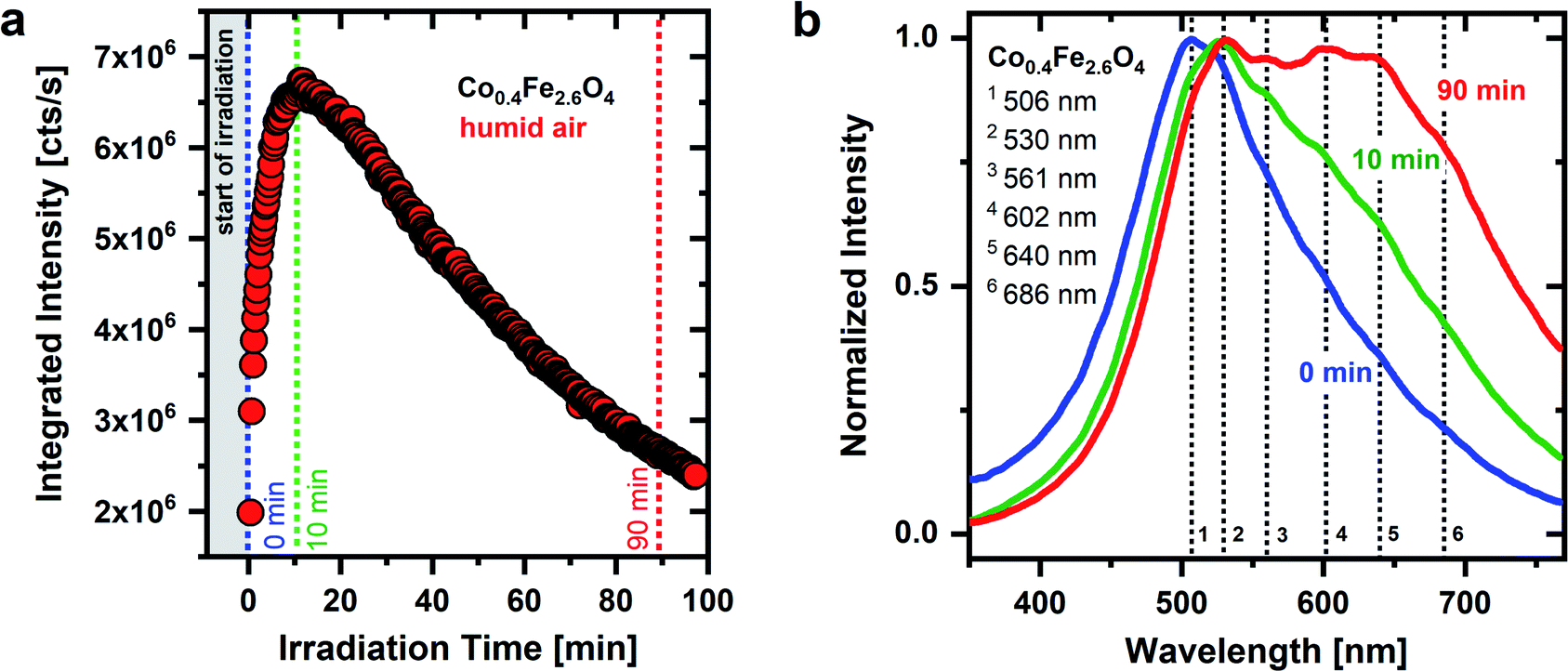 | ||
| Fig. 3 (a) Time dependent behavior of the integrated PL intensity of the Co0.4Fe2.6O4 nanoparticle emission under UV irradiation (λexc = 270 nm, ρ = 2 μJ cm−2) in humid air; (b) associated emission spectra at the beginning (0 min and 10 min) and at the end of UV irradiation (90 min) in humid air. | ||
As described above, a possible explanation for the observed time dependent behavior of the PL is the dehydroxylation and decrease of the surface coverage under UV irradiation. The emission of an adsorbate emission species, such as OH groups, is expected to be weakened and/or broadened by the presence of water. Charge transfers between emission species and water are discussed as the cause of the PL quenching of the emission species.65 Thus, if physisorbed water molecules are removed from the surface, an increase in intensity of the OH-related emission results. In addition, OH groups may form on the metal oxide surface because of water dissociation, which contributes to an increase in the emission intensity as well.
The drop in emission intensity for nanoparticles with x = 0.4 to 1.6 after about 10 min indicates that not only the amount of water molecules on the surface, but also the amount of OH groups successively decrease. Stoyanovskii et al.38 made similar observations investigating the emission of δ- and γ-Al2O3 oxides which had been gradually dehydroxylated by thermal treatment. They detected a broad green emission with a maximum at ∼570 nm, which was also assigned to surface OH groups. The intensity of the OH related emission increased at low annealing temperatures due to the removal of water. Afterwards, at higher temperatures, the intensity decreased due to the removal of OH groups.38 During surface dehydroxylation Stoyanovskii et al. observed, as in this work, additional emission lines formed on the low-energy side of the broad PL. This spectral change during annealing has been assigned to the formation of new types of OH groups because of surface dehydroxylation. The new OH group types generate new emission lines at wavelengths >570 nm and thus cause a broadening and shift of the emission to lower energies.38 This observation is in nice agreement with the generation of the discrete emission maxima S3–S6 found in our experiments after 90 min of UV irradiation in humid air.
There are various studies and models that describe the behavior of OH groups on metal oxide surfaces during such a dehydroxylation process.35,66–68 According to a model of Tsyganenko et al.,67 which was developed for spinel-type oxides using the example of γ-Al2O3, the surface of an ideal spinel oxide is covered with different types of OH groups. This coverage is unavoidable on a spinel oxide in humid air. OH groups can attach to the spinel oxide surface through a bond to one, two or three metal ions, whereby a distinction is made whether the metal ion is located on a tetrahedral (MT) or an octahedral site (MO). This results in a total of six different OH groups (OHT, OHO, OHTT, OHTO, OHTTT and OHTTO) with bonds to metal cations that can be found on a spinel oxides surface.67
When a dehydroxylation of the surface starts, for example by thermal treatment or UV laser irradiation, water and/or OH groups are removed from the surface.38 According to Breysse et al.35 the removal of water on a metal oxide occurs at an earlier stage than the removal of OH groups. If an OH group is removed, a metal cation with a reduced coordination number remains. If this metal cation is involved in the coordination sphere of another OH group, the bonding properties of this remaining OH group change and consequently a new OH type arises on the surface.67 Thus, through this coupling of two neighboring OH groups, i.e., the removal of one and the conversion of the other group, new kinds of OH can come into being during a dehydroxylation process. This expected behavior is in nice agreement with the spectral change that is detected in the emission spectra during the UV irradiation of Co0.4Fe2.6O4 nanoparticles.
The emission behavior over time, which is shown in Fig. 3, can therefore be divided into two steps. (i) At the beginning of the UV irradiation the emission intensity increases. This behavior can be explained by the removal of (physisorbed) water molecules from the surface and/or the conversion into OH groups, dominating over the removal of individual OH groups from the surface. (ii) The emission intensity reaches its maximum after 10 minutes of irradiation and starts to drop. This indicates that the weaker bound physisorbed water has been eliminated from the surface and OH groups desorb more and more. At the same time, after the model of Tsyganenko et al.,67 neighboring OH groups begin to couple with one another and new emission lines form on the low-energy side while the overall emission intensity decreases. This behavior is consistent with the hypothesis that neighboring OH groups couple with each other during dehydroxylation, thereby forming new OH groups with different bonds to surface metal cations. These new OH groups generate new emission lines (S3–S6) on the low energy side.
Surprisingly, a different time dependent PL behavior under UV irradiation is observed for CoxFe3−xO4 nanoparticles with a higher Co-content (x = 2) (see Fig. 4). Analogous to the Fe-rich sample, the emission intensity of Co2Fe1O4 nanoparticles initially increases and reaches 2.3 times the original emission intensity after about 10 minutes. However, in contrast to Co0.4Fe2.6O4, nanoparticles (see Fig. 3), there is no subsequent drop in emission intensity. Instead, the intensity continues to increase and reaches 3.8 times the original intensity after ∼50 minutes. Interestingly, in addition to this different development of the emission intensity over time, there is no spectral change in emission. Over the entire time range considered, the shape of the Co2Fe1O4 emission remains almost unchanged with a maximum at around 530 nm. Regarding its spectral position, this emission maximum agrees well with the emission line S2 of the Co0.4Fe2.6O4 nanoparticles. It appears that for Co-rich nanoparticles there is primarily a decrease in physisorbed water on the surface responsible for the rising PL intensity with time, and no decrease in the amount of OH groups bound to the surface. Consequently, no emission from the S3–S6 bands is observed, i.e., a coupling of neighboring OH groups during dehydroxylation and the related formation of new OH groups with different bonds to surface metal cations does not occur for Co-rich nanoparticles. If new OH groups are formed on Co-rich nanoparticles, these primarily have the same bonds to the surface that cause the S2 emission again. OH groups with different bonds to the surface (S3–S6) can only occur on dehydroxylated surfaces (see Fig. 3).
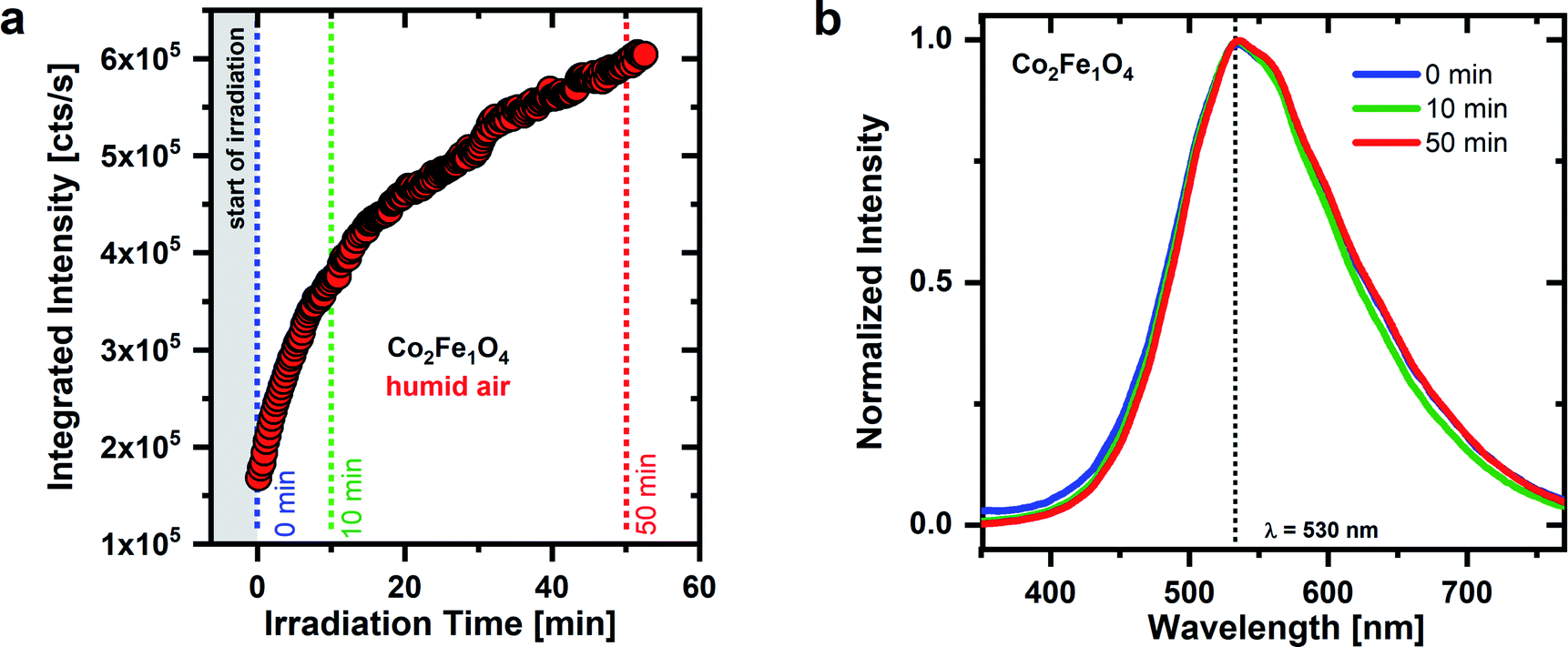 | ||
| Fig. 4 (a) Time dependent behavior of the integrated PL intensity of the Co2Fe1O4 nanoparticle emission under UV irradiation (λexc = 270 nm, ρ = 2 μJ cm−2) in humid air; (b) associated emission spectra at the beginning (0 min and 10 min) and at the end of UV irradiation (50 min) in humid air. | ||
In order to understand the differences in the temporal emission behavior of Fe-rich and Co-rich CoxFe3−xO4 nanoparticles in more detail, a connection between the nanoparticles surface structure and the OH-related emission must be established. When considering real spinel structures, vacancies, edges or corners must also be taken into account. It is expected that the latter will have no influence on the type of OH group (still only six OH types occur) but on their respective quantity.67 E.g., at edges, OH groups have a lower number of surrounding metal cations, which is why terminal OH groups, i.e. OHT or OHO, are primarily formed. This effect is particularly dominant in the case of nanoparticles and must be considered when interpreting the PL spectra.67 Under the simplified assumption that each OH group generates the same number of photons, we assign the two emission maxima at the beginning of the irradiation (S1, S2) to OHT and OHO.
Furthermore, the dehydroxylation of the surface of a spinel oxide and the reaction of neighboring OH groups with one another does not take place in a random process. Desorption occurs first in couples in which the first partner A is a basic group and has the highest negative charge (i.e. OHT) and partner B is the most acidic and thus has the lowest negative charge (i.e. OHTTO).66,67 We thus expect that due to their larger negative charge, OHT groups are converted into new OH groups earlier in time than OHO and are afterwards no longer present on the surface.67 Therefore, we assign the initial emission maximum for Fe-rich nanoparticles (x = 0.4, see Fig. 3) at 506 nm (S1) to OHT and the second emission maximum at 530 nm (S2), which is present until the end of the irradiation, to OHO groups. An exact assignment of the emission lines that occur on the low-energy side with increasing irradiation time is not possible. These occur through terminal and bridging OH groups that arise because of dehydroxylation.
In a second step, it is important to understand the change in the crystal structure from Co-rich (x = 2.5) to Fe-rich (x = 0.4) nanoparticles. For this purpose, the CoxFe3−xO4 nanoparticles are examined by XRD analysis and Raman spectroscopy (for a detailed analysis see Fig. S2 and S3†). Using these methods, it is determined that the crystal structure converts from a predominantly normal spinel for Co-rich nanoparticles to a more mixed inverse spinel for Fe-rich nanoparticles.69 This in turn affects which metal cations occupy the octahedral and tetrahedral sites, respectively. Raman spectroscopy indicates that in Co-rich nanoparticles, Co2+ ions are mainly present on the tetrahedral sites (Co2+tet) and Co3+ as well as Fe3+ ions occupy mainly octahedral sites (Co3+oct or Fe3+oct). As the Fe content increases, Co2+tet are exchanged by Fe3+ ions. The octahedral sites of Fe-rich nanoparticles are mainly occupied by Fe3+oct ions (as well as Co2+oct) and the spinel structure becomes more inverse.
By comparing the PL spectra of the Fe-rich and Co-rich nanoparticles (see Fig. 3 and 4) with these data, the emission lines are connected to OH groups on specific metal cations. The emission line at 506 nm (S1) is dominant for Fe-rich nanoparticles at the beginning of the UV irradiation, so it appears to be related to Fe cations and is assigned to terminal OH groups on tetrahedral sites as well. With increasing Co content, Fe3+tet ions are exchanged by Co2+tet ions and the S1 maximum no longer dominates the PL spectrum. The emission line at 530 nm (S2) is dominant for Co-rich nanoparticles, thus S2 is attributed to Co cations and as mentioned before, assigned to terminal OH groups on octahedral sides. With increasing Co content, the octahedral sites are mainly occupied by Co3+oct (as well as Fe3+oct) and the emission maximum S2 dominates the spectrum. Thus, it appears that the emission line S1 is dominant if Fe3+ is mainly present on tetrahedral sites and the emission line S2 if Co3+ is on the octahedral sites.
Such surface OH groups bound to metal cations are expected to be of importance for the (electro) catalytic properties of metal oxides.70,71 Especially OH groups bound to Co3+ are of significance for the splitting of water via cobalt oxide catalysts.72 In case of Co3O4 nanoparticles two active centers were identified. A slow site was assigned to CoIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O surface complexes and a fast site with higher catalytic efficiency to pairs of CoIII–OH surface centers linked by O atoms. The oxo-bridged CoIII centers are in the octahedral coordination of the Co3O4 crystal structure.72 A prerequisite for the high catalytic activity in the water oxidation of Co3O4 is therefore the presence of OH groups on CoIII complexes on octahedral sites. In addition to the catalysts' active sites, the stability of surface adsorbates and reaction products plays a significant role for the oxygen evolution reaction (OER).70,73 Our investigations show that Co-rich nanoparticles with a strong emission line S2 related to OH on Co3+oct appear to be more stable to dehydroxylation than their Fe-rich counterparts (see Fig. 3 and 4). A difference in the OH group stability affects the OER performance73 and can thus provide additional information about the overpotential of Co-rich CoxFe3−xO4. A four-step proton/electron mechanism is typically given for the OER reaction on the surface of (metal) oxide catalysts.73,74 Here, *OOH, *OH, and *O represent intermediate surface species bound to active sites (denoted by *).
O surface complexes and a fast site with higher catalytic efficiency to pairs of CoIII–OH surface centers linked by O atoms. The oxo-bridged CoIII centers are in the octahedral coordination of the Co3O4 crystal structure.72 A prerequisite for the high catalytic activity in the water oxidation of Co3O4 is therefore the presence of OH groups on CoIII complexes on octahedral sites. In addition to the catalysts' active sites, the stability of surface adsorbates and reaction products plays a significant role for the oxygen evolution reaction (OER).70,73 Our investigations show that Co-rich nanoparticles with a strong emission line S2 related to OH on Co3+oct appear to be more stable to dehydroxylation than their Fe-rich counterparts (see Fig. 3 and 4). A difference in the OH group stability affects the OER performance73 and can thus provide additional information about the overpotential of Co-rich CoxFe3−xO4. A four-step proton/electron mechanism is typically given for the OER reaction on the surface of (metal) oxide catalysts.73,74 Here, *OOH, *OH, and *O represent intermediate surface species bound to active sites (denoted by *).
| * + H2O → *OH + H+ + e− | (1) |
| *OH → *O + H+ + e− | (2) |
| *O + H2O → *OOH + H+ + e− | (3) |
| *OOH → * + O2 + H+ + e− | (4) |
The reaction step between two absorbed intermediate products with the maximum difference in the chemical Gibbs binding energy ΔGi–j is the rate-determining step and crucial for the OER overpotential.73,75,76 The rate of the individual steps is limited by the formation and stability of the intermediate products (O*, OH* and OOH*) bound to the active sites. In literature, there is no uniform picture regarding the rate-limiting step of OER on cobalt oxide systems. The crucial role for the OER overpotential is assigned to the first, second or third reaction step.71,77 Theoretical calculations by Bajdich et al.71 for Co3O4 and CoOOH showed that the first and third reaction step, depending on the crystal planes considered, and the formation of OH* and HOO* are of decisive importance. Chen et al.70 investigated cobalt oxides and the influence of Ni doping on water splitting via CoOOH. In agreement with Bajdich et al., the lower catalytic activity of undoped CoOOH was assigned to the limitation of CoOOH by the first reaction step. The formation of OH groups on undoped CoOOH is costly in terms of energy, but they observed a higher stability of the OH groups after doping with Ni. The higher OH binding energy on the doped system improves the OH formation and thus decreases the overpotential.70
Our optical investigations of CoxFe3−xO4 nanoparticles show an insightful observation regarding the stability of surface OH groups against dehydroxylation. A dominant emission line S2 can be observed on Co-rich nanoparticles, which is associated with OH on Co3+oct. Further emission lines S3–S6 (due to new OH groups formed during dehydroxylation) are not observed. In contrast to this, Fe-rich samples show a dominant emission line S1, which is supposed to be caused by OH groups bound to Fe3+tet. With increasing irradiation time of Fe-rich nanoparticles, emission line S1 becomes weaker and the emission lines S2–S6 dominate the spectrum. Thus, OH groups on Co-rich nanoparticles (with Co3+oct on octahedral sites) appear to have a higher stability regarding photoinduced dehydroxylation. According to this correlation, one might expect lower overpotentials of Co-rich CoxFe3−xO4 nanoparticles caused by the presence of OH bound to Co3+oct, their higher stability and the resulting improvement in the formation of OH in the OER. This observation for CoxFe3−xO4 nanoparticles agrees well with theoretical and experimental investigations of Co3O4 and CoOOH.71,73
Conclusion
In conclusion, we investigated the atmosphere dependent PL emission behavior of CoxFe3−xO4 nanoparticles (x = 0.4 to 2.5). All nanoparticles show a broad green emission under high-energy UV irradiation of 270 nm with an emission maximum consisting of two emission lines at around 506 nm and 530 nm. Using time-dependent PL investigations in humid air, vacuum and dry oxygen environment, the broad emission was attributed to different types of terminal and bridging OH groups on the nanoparticles surface. By additionally performing XRD and Raman spectroscopy, a connection between these emission lines and the surface structure was established. The dominant emission lines at around 506 nm and 530 nm are caused by OH groups terminally bound to metal cations on the tetrahedral (Fe3+tet) or octahedral site (Co3+oct), respectively. The surface coverage of Co-rich CoxFe3−xO4 nanoparticles (x = 2) was found to be significantly more stable with respect to photoinduced dehydroxylation. We hypothesize, that this enhanced stability of OH groups could be a possible cause of a better electrocatalytic performance of Co-rich nanoparticles compared to their Fe-rich counterparts.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. S. S. and S. S. contributed to the synthesis of CoxFe3−xO4 nanoparticles and XRD, EDX and TEM measurements. J. K., L. K., J. K., O. K., T. S., and G. B. contributed to Raman, absorption, and PL spectroscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 388390466-TRR 247 within the collaborative research center/transregio 247 “Heterogeneous Oxidation Catalysis in the Liquid Phase”, projects B3 and C3.References
- Y. Song, X. Li, C. Wei, J. Fu, F. Xu, H. Tan, J. Tang and L. Wang, Sci. Rep., 2015, 5, 8401 CrossRef PubMed.
- Q. Li, H. Li, Q. Xia, Z. Hu, Y. Zhu, S. Yan, C. Ge, Q. Zhang, X. Wang, X. Shang, S. Fan, Y. Long, L. Gu, G.-X. Miao, G. Yu and J. S. Moodera, Nat. Mater., 2021, 20, 76–83 CrossRef CAS PubMed.
- H. Ji, W. Zeng and Y. Li, Nanoscale, 2019, 11, 22664–22684 RSC.
- Y. Zhang, L. Li, H. Su, W. Huang and X. Dong, J. Mater. Chem. A, 2015, 3, 43–59 RSC.
- J. C. Védrine, ChemSusChem, 2019, 12, 577–588 CrossRef PubMed.
- K. Y. Kumar, H. B. Muralidhara, Y. A. Nayaka, J. Balasubramanyam and H. Hanumanthappa, Powder Technol., 2013, 246, 125–136 CrossRef CAS.
- D. H. Taffa, R. Dillert, A. C. Ulpe, K. C. L. Bauerfeind, T. Bredow, D. W. Bahnemann and M. Wark, J. Photonics Energy, 2017, 7, 12009 CrossRef.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- Y. J. Jang and J. S. Lee, ChemSusChem, 2019, 12, 1835–1845 CrossRef CAS PubMed.
- S. Chandrasekaran, C. Bowen, P. Zhang, Z. Li, Q. Yuan, X. Ren and L. Deng, J. Mater. Chem. A, 2018, 6, 11078–11104 RSC.
- N. Guijarro, P. Bornoz, M. Prévot, X. Yu, X. Zhu, M. Johnson, X. Jeanbourquin, F. Le Formal and K. Sivula, Sustainable Energy Fuels, 2018, 2, 103–117 RSC.
- H. M. Gobara, I. M. Nassar, A. M. El Naggar and G. Eshaq, Energy, 2017, 118, 1234–1242 CrossRef CAS.
- C. Yuan, H. B. Wu, Y. Xie and X. W. D. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- F. M. Ismail, M. Ramadan, A. M. Abdellah, I. Ismail and N. K. Allam, J. Electroanal. Chem., 2018, 817, 111–117 CrossRef CAS.
- H.-J. Qiu, L. Liu, Y.-P. Mu, H.-J. Zhang and Y. Wang, Nano Res., 2015, 8, 321–339 CrossRef CAS.
- T. Chen, S. Li, L. Ma, X. Zhao and G. Fang, Nanotechnology, 2019, 30, 395403 CrossRef CAS PubMed.
- S. Li, J. Xu, Z. Ma, S. Zhang, X. Wen, X. Yu, J. Yang, Z.-F. Ma and X. Yuan, Chem. Commun., 2017, 53, 8164–8167 RSC.
- Y. Dai, J. Yu, M. Ni and Z. Shao, Chem. Phys. Rev., 2020, 1, 11303 CrossRef.
- S. Cao, F. F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- L. Wang, Y. Yu, H. He, Y. Zhang, X. Qin and B. Wang, Sci. Rep., 2017, 7, 12845 CrossRef PubMed.
- J. Goodman and L. E. Brus, J. Chem. Phys., 1977, 67, 4398–4407 CrossRef CAS.
- H. Petitjean, H. Guesmi, H. Lauron-Pernot, G. Costentin, D. Loffreda, P. Sautet and F. Delbecq, ACS Catal., 2014, 4, 4004–4014 CrossRef CAS.
- A. K. Kharade and S. Chang, J. Phys. Chem. C, 2020, 124, 10981–10992 CrossRef CAS.
- A. S. Burange and C. S. Gopinath, in Catalytic and Noncatalytic Upgrading of Oils, ed. A. K. Dalai, D. B. Dadyburjor, Y. Zheng, A. Duan, W. L. Roberts and S. Nanda, American Chemical Society, Washington DC, 2021, pp. 291–318 Search PubMed.
- S. Saddeler, U. Hagemann and S. Schulz, Inorg. Chem., 2020, 59, 10013–10024 CrossRef CAS PubMed.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- C. Li, X. Han, F. Cheng, Y. Hu, C. Chen and J. Chen, Nat. Commun., 2015, 6, 7345 CrossRef CAS PubMed.
- M. Matsuoka, T. Kamegawa and M. Anpo, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2008, vol. 3, pp. 1065–1073 Search PubMed.
- M. Anpo and M. Che, in Advances in Catalysis, ed. W. Haag, B. Gates and H. Knoezinger, Elsevier, 1st edn, 1999, vol. 44, pp. 119–257 Search PubMed.
- J. Zhang, X. Chen, Y. Shen, Y. Li, Z. Hu and J. Chu, Phys. Chem. Chem. Phys., 2011, 13, 13096–13105 RSC.
- E. Rauwel, A. Galeckas and P. Rauwel, Mater. Res. Express, 2014, 1, 15035 CrossRef CAS.
- S. Mochizuki and T. Saito, Phys. B, 2012, 407, 2911–2914 CrossRef CAS.
- M. Breysse, B. Claudel, L. Faure and M. Guenin, J. Colloid Interface Sci., 1979, 70, 201–207 CrossRef CAS.
- H. Jeziorowski and H. Knözinger, Chem. Phys. Lett., 1977, 51, 519–522 CrossRef CAS.
- V. V. Mikho and A. P. Fedchuk, J. Appl. Spectrosc., 1974, 21, 1331–1333 CrossRef.
- V. O. Stoyanovskii and V. N. Snytnikov, Kinet. Catal., 2009, 50, 450–455 CrossRef CAS.
- M. Bhatnagar, V. Kaushik, A. Kaushal, M. Singh and B. R. Mehta, AIP Adv., 2016, 6, 95321 CrossRef.
- A. van Dijken, E. A. Meulenkamp, D. Vanmaekelbergh and A. Meijerink, J. Phys. Chem. B, 2000, 104, 1715–1723 CrossRef CAS.
- E. R. Viana, J. C. González, G. M. Ribeiro and A. G. de Oliveira, J. Phys. Chem. C, 2013, 117, 7844–7849 CrossRef CAS.
- G. Metra, E. Gianotti and S. Coluccia, in Metal Oxide Catalysis, ed. S. D. Jackson and J. S. Hargreaves, Wiley-VCH, Weinheim, 1st edn, 2008, vol. 2, pp. 51–94 Search PubMed.
- M. Anpo, T. Shima and Y. Kubokawa, Chem. Lett., 1985, 14, 1799–1802 CrossRef.
- H. J. Maria and S. P. Mc Glynn, J. Chem. Phys., 1970, 52, 3402 CrossRef CAS.
- C. Chizallet, M. Digne, C. Arrouvel, P. Raybaud, F. Delbecq, G. Costentin, M. Che, P. Sautet and H. Toulhoat, Top. Catal., 2009, 52, 1005–1016 CrossRef CAS.
- C. Chizallet, G. Costentin, H. Lauron-Pernot, J.-M. Krafft, M. Che, F. Delbecq and P. Sautet, J. Phys. Chem. C, 2008, 112, 19710–19717 CrossRef CAS.
- A. Anjiki and T. Uchino, J. Phys. Chem. C, 2012, 116, 15747–15755 CrossRef CAS.
- B. Ren, W. Shen, L. Li, S. Wu and W. Wang, Appl. Surf. Sci., 2018, 447, 711–723 CrossRef CAS.
- A. V. Ravindra, M. Chandrika, C. Rajesh, P. Kollu, S. Ju and S. D. Ramarao, Eur. Phys. J. Plus, 2019, 134, 296 CrossRef CAS.
- P. Annie Vinosha, L. Ansel Mely, J. Emima Jeronsia, K. Raja, D. Queen Sahaya Tamilarsi, A. C. Fernandez, S. Krishnan and S. Jerome Das, Optik, 2016, 127, 9917–9925 CrossRef.
- R. K. Singh, A. Narayan, K. Prasad, R. S. Yadav, A. C. Pandey, A. K. Singh, L. Verma and R. K. Verma, J. Therm. Anal. Calorim., 2012, 110, 573–580 CrossRef CAS.
- K. Dileep, B. Loukya, N. Pachauri, A. Gupta and R. Datta, J. Appl. Phys., 2014, 116, 103505 CrossRef.
- P. Felbier, J. Yang, J. Theis, R. W. Liptak, A. Wagner, A. Lorke, G. Bacher and U. Kortshagen, Adv. Funct. Mater., 2014, 24, 1988–1993 CrossRef CAS.
- E. Rauwel, A. Galeckas, M. Rosário Soares and P. Rauwel, J. Phys. Chem. C, 2017, 121, 14879–14887 CrossRef CAS.
- Q. Li, M. Anpo and X. Wang, Res. Chem. Intermed., 2020, 46, 4325–4344 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Grätzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1999, 99, 77–174 CrossRef CAS PubMed.
- M. Iwamoto, Y. Yoda, N. Yamazoe and T. Seiyama, J. Phys. Chem., 1978, 82, 2564–2570 CrossRef CAS.
- V. V. Mikho, J. Appl. Spectrosc., 1981, 35, 1170–1173 CrossRef.
- T. I. Quickenden, A. R. Hanlon and C. G. Freeman, J. Phys. Chem. A, 1997, 101, 4511–4516 CrossRef CAS.
- T. G. Slanger and P. C. Cosby, J. Phys. Chem., 1988, 92, 267–282 CrossRef CAS.
- K. Wandelt, Surface and Interface Science, Wiley-VCH, Weinheim, 2012 Search PubMed.
- M. Nagao, J. Phys. Chem., 1971, 75, 3822 CrossRef CAS.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737–1757 RSC.
- M. Wolf, S. Nettesheim, J. M. White, E. Hasselbrink and G. Ertl, J. Chem. Phys., 1990, 92, 1509–1510 CrossRef CAS.
- G. E. Dobretsov, T. I. Syrejschikova and N. V. Smolina, Biophysics, 2014, 59, 183–188 CrossRef CAS.
- H. Knözinger and P. Ratnasamy, Catal. Rev., 1978, 17, 31–70 CrossRef.
- A. A. Tsyganenko and P. P. Mardilovich, J. Chem. Soc., Faraday Trans., 1996, 92, 4843–4852 RSC.
- J. B. Peri, J. Phys. Chem., 1965, 69, 220–230 CrossRef CAS.
- L. Kampermann, J. Klein, J. Korte, O. Kowollik, O. Pfingsten, T. Smola, S. Saddeler, T. H. Piotrowiak, S. Salamon, J. Landers, H. Wende, A. Ludwig, S. Schulz and G. Bacher, J. Phys. Chem. C, 2021, 125, 14356–14365 CrossRef CAS.
- Z. Chen, C. X. Kronawitter, I. Waluyo and B. E. Koel, J. Phys. Chem. B, 2018, 122, 810–817 CrossRef PubMed.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. García-Mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116, 21077–21082 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Chen and A. Selloni, J. Phys. Chem. Lett., 2012, 3, 2808–2814 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06228j |
| This journal is © The Royal Society of Chemistry 2021 |