
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBacterial virulence factors: a target for heterocyclic compounds to combat bacterial resistance
Rehab H. Abd El-Aleama,
Riham F. George *b,
Hanan H. Georgeybc and
Hamdy M. Abdel-Rahmande
*b,
Hanan H. Georgeybc and
Hamdy M. Abdel-Rahmande
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Modern University for Technology and Information MTI, Cairo 11571, Egypt
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo 11562, Egypt. E-mail: rihamfgeorge@gmail.com
cPharmaceutical Chemistry Department, Faculty of Pharmacy and Drug Technology, Egyptian Chinese University, Cairo 11786, Egypt
dMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
ePharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University, Beni Suef, Egypt
First published on 19th November 2021
Abstract
Antibiotic resistance is one of the most important challenges of the 21st century. However, the growing understanding of bacterial pathogenesis and cell-to-cell communication has revealed many potential strategies for the discovery of drugs that can be used for the treatment of bacterial infections. Interfering with bacterial virulence and/or quorum sensing could be a particularly interesting approach, because it is believed to exert less selective pressure on the bacterial resistance than with traditional strategies, geared toward killing bacteria or preventing their growth. Here, we discuss the mechanism of bacterial virulence, presenting promising strategies and recently synthesized heterocyclic compounds to combat future bacterial infections.
1 Introduction
The incidence of microbial infections has increased in alarming levels over the world because of antimicrobial resistance.1 Due to the potential impact of microbes on morbidity, mortality, and health care costs, they have become a serious fear.2 Infections caused by microorganisms are one of the leading causes of death worldwide. The limited number of antibiotics used to treat infections and the continuous development of resistance to recently used antimicrobial agents represent a serious challenge.3,4Antimicrobial resistance (AMR) occurs when bacteria, viruses, fungi, and parasites mutate over time and no longer respond to drugs, which makes the infections more difficult to be treated and increases the spread of disease, serious illness, and the death risk.5
WHO's 2020 update of the global review of antibacterial products in preclinical development captures 292 antibacterial products that are being developed in 162 institutions around the world. These products target WHO priority pathogens, Mycobacterium tuberculosis and Clostridium difficile. The current clinical antibacterial pipeline contains 43 antibiotics and combinations with a new therapeutic entity and 27 non-traditional antibacterial agents (agents that are not small molecule drugs and/or do not act by directly targeting bacterial components necessary for bacterial growth) (Fig. 1).6 However, few antibacterial agents either traditional or non-traditional reach phase 3. Therefore, there is an urgent need to develop new antibacterial agents to overcome the emergence and spread of bacterial resistance.
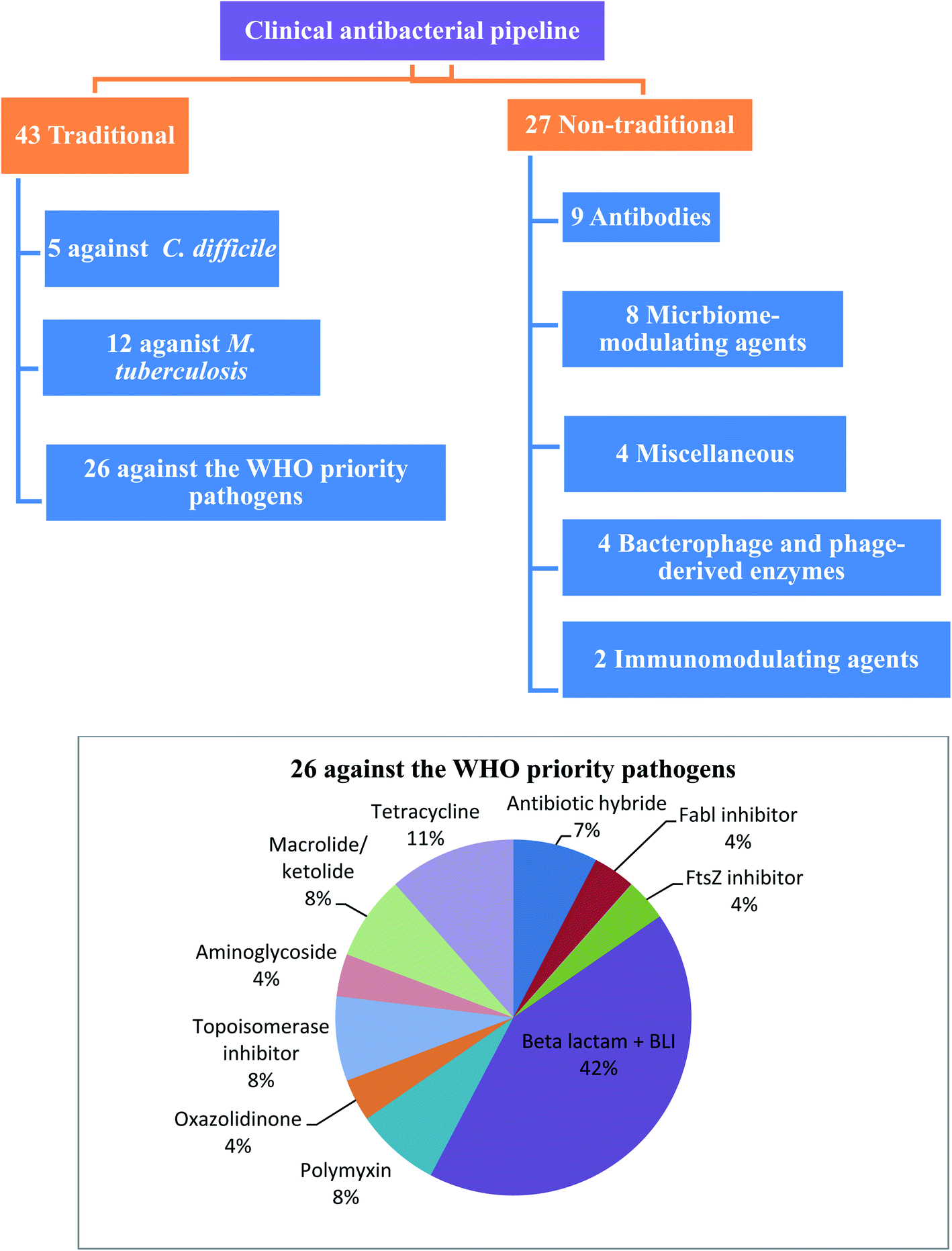 | ||
| Fig. 1 Traditional and non-traditional antibacterial in clinical phases. | ||
2 Mechanisms of antibiotic resistance
Bacteria develop resistance to chemotherapeutics through five mechanisms:7 enzyme inactivation and modification, mutation of antibiotic target sites, overproduction of modified target sites, replacement of target sites, and efflux or reduced permeability. Bacteria can develop one or more mechanisms of resistance as shown in (Fig. 2).8 | ||
| Fig. 2 Different mechanisms of antibiotic resistance in Gram-positive and Gram-negative bacteria. | ||
2.1. Inactivating and modifying enzymes
Destruction or modification of the structure of antibiotics is one of the most common resistance mechanisms involving enzymes. Depending on the type of reaction they catalyze, the enzymes involved in this resistance mechanism are subdivided into hydrolases, transferases and oxidoreductases (Fig. 3).9–12 | ||
| Fig. 3 The main classes of enzymes modifying antimicrobial drugs. | ||
2.2. Mutation of the antibiotic target site
Since antimicrobial drugs have very specific targets, structural changes to those targets can prevent drug binding, rendering the drug ineffective. Through spontaneous mutations in the gene encoding antibacterial drug targets, bacteria have an evolutionary advantage that permit them to develop resistance to drugs. This mechanism of resistance development is quite common. For example, genetic changes impacting the active site of penicillin binding proteins (PBPs) can inhibit the binding of β-lactam drugs and provide resistance to multiple drugs within this class.13 This mechanism is very common among strains of Streptococcus pneumonia, which alter their own PBPs through genetic mechanisms.14Similarly, the resistance of many bacterial pathogens to fluoroquinolone antibiotics (such as ciprofloxacin) is mediated by mutations in the ciprofloxacin-targeted DNA gyrase and DNA topoisomerase IV genes.15
2.3. The target site is changed
Although bacteria such as Streptococcus pneumoniae have mutated antibiotic targets, another similar mechanism for resistance is to obtain additional copies of a gene that encodes a protein that retains activity, but antibiotics cannot bind to it. This is how the pathogen Staphylococcus aureus becomes resistant to most β-lactam antibiotics (such as penicillin). Methicillin-resistant Staphylococcus aureus (MRSA) are resistant to β-lactam antibiotics, which is changed by acquiring additional penicillin binding protein 2 (target of β-lactam) making it resistant to antibiotics. In the presence of β-lactam antibiotics, this additional version called penicillin-binding protein 2a (PBP2a) that cannot function as a target for penicillin binding helping the bacteria to survive.14,16Furthermore, a resistance mechanism called target mimicry was discovered, which involves the production of proteins that bind and sequester drugs, thus preventing the drug from binding to its target. For example, Mycobacterium tuberculosis produces a protein that has a regular pentapeptide repeating sequence that appears to mimic the structure of DNA. The protein binds to fluoroquinolones, isolates them, and prevents it from binding to DNA, making Mycobacterium tuberculosis resistant to fluoroquinolones. Proteins that mimic the A site of bacterial ribosomes have also been found to play a role in aminoglycoside resistance.17
2.4. Overproduction of the target
Bacteria can also overproduce antibiotic targets, which means that the protein targets of antibiotics are excessive compared to the antibiotics themselves. This means that, in the presence of antibiotics, there is enough target protein to continue to function in the cell; this is the mechanism by which E. coli and Haemophilus influenzae are resistant to trimethoprim. Overexpression is sometimes found to combine with mutations that reduce the ability of the antibiotic to bind to its target.182.5. Efflux and reduced permeability
Bacteria are inherently resistant to certain antibiotics through either reduction of bacterial membrane permeability or activation of efflux pumps to these antibiotics. Efflux pumps (EPs) are one of the most famous examples of bacterial mechanisms that confer cross-resistance to different antibiotics.19 This resistance mechanism involves antibiotics that exert their antibacterial activity by inhibiting the bacterial protein and DNA synthesis, especially tetracyclines, macrolides and fluoroquinolones.20 The efflux system can actively squeeze out conventional antibiotics, leading to an increase in their minimum inhibitory concentration (MIC) or, in some cases, a loss of their antibacterial activity. These systems can not only eradicate antibiotics, but also non-antibiotic substrates, such as detergents and heavy metals.21–23 Additionally, bacteria can get additional efflux pumps, which are specifically designed to pump an antibiotic, such as the TetA efflux pump, which specifically removes tetracycline out the cell. Similarly, by obtaining mutations in porins (protein channels through the cell membrane), the permeability of the cell can be changed. These mutations can include porin deletions, changes in the size or conductivity of porin channels, or lower levels of porin expression. Finally, two mechanisms, namely the efflux pump and reduced permeability, reduce the concentration of intracellular antibiotics inside the bacterial cells by exporting antibiotics or by not allowing their import, respectively.24,25Therefore, the discovery of innovative and effective antibacterial agents may be the only way to solve the resistance problem and develop successful methods to treat infectious diseases.26 In this review, we will compare potential synthetic small molecules that can prevent bacterial virulence formation or eradicate pre-existing virulence factors from clinically relevant Gram-positive and Gram-negative pathogens. In addition, we also provide a comprehensive list of potential targets to counteract the formation and development of virulence factors.
3 Antibiotic adjuvant
Therefore, to overcome the emerging of antibiotic resistance, combination therapies of antibiotic with potentiating adjuvants may be used. These adjuvants include drugs that block the mechanisms of resistance for the antibiotics as: (a) efflux pump inhibitors, (b) β-lactamase inhibitors, (c) outer membrane permeability enhancer and (d) anti-virulence compounds.27Anti-virulence agents suppress virulence phenotypes without affecting bacterial growth and therefor enhance the antibacterial effect of drugs.28 This method consists of identifying the proteins, genes and other biological macromolecules that cause the virulence of the bacteria, thus, their inhibition will reduce the fitness of the bacteria, making them more vulnerable to the immune system and the use of antibiotics. In fact, such targets have been proposed, although they are not important for survival per se, they are unlikely to produce mutations. Furthermore, to expand drug targets, small molecules that target these “nonessential” genes can be combined with existing antibiotics29 (Fig. 4).
 | ||
| Fig. 4 Antibiotic adjuvants classes. | ||
4 Targeting virulence factor
In the process of identifying new antibiotics that are active against drug-resistant strains, the motivation to consider alternative cellular pathways as the source of targets for the development of new antimicrobial adjuvants represents an interesting alternative to traditional methods.Antibiotic resistance usually leads to latent and persistent infections that are difficult to treat. During the continuous existence of the host, the pathogen faces an extremely harsh environment, and requires extensive reprogramming of bacterial metabolic functions to survive under such unfavorable conditions. Therefore, targeting key metabolic functions related to pathogen survival under such conditions may lead to better antibiotic treatment and increase sensitivity to traditional antibiotics.30
In this case, the main factor leading to the deterioration of the patient's health in the process of bacterial infection is bacterial virulence. In the last decade, a new method has emerged to combat the virulence or pathogenicity of bacteria.31–34 Unlike traditional antimicrobial drugs that work by killing bacteria or preventing bacterial growth, anti-virulence drugs can reach specific targets called virulence factors, which are only expressed in bacteria when infected. They are not required for the basic bacterial cell cycle, but are essential for pathogenesis, and their pharmacological inactivation prevents bacteria from causing pathological infections in the host. In this case, the host's immune system can quickly and easily resist less toxic pathogens more effectively.35 Furthermore, virulence inhibitors do not target essential factors for the pathogen's life cycle, it is assumed that the selection pressure for resistant mutants will be less relevant.36 Instead, anti-virulence agents will target examples of pathway enriched with nonessential targets include the sulfur assimilation pathways, quorum sensing, and biofilms.37
4.1. Targeting cysteine biosynthesis
The rationale behind the exploitation of amino acid biosynthesis as a target for antimicrobial adjuvant development is the observation that some pathogens spend part of their life cycle in extremely harsh conditions, such as macrophages or the gastric mucosa, where survival and proliferation require powerful adaptation mechanisms involving metabolic pathways.38,39 In this case, interference with pathogen adaptation strategies can lead to increased sensitivity to antibiotics. Among potential new drugs targets is an enzyme involved in cysteine biosynthesis.It has been observed that the importance of cysteine biosynthesis enzymes differs during the life cycle of pathogens: their activity can be dispensable during growth in vitro or acute infections but becomes indispensable during the persistence phase.40,41 Compared with traditional antibiotics, molecules developed for cysteine biosynthesis and other biosynthetic pathways may have the potential advantage of being more effective against persistence in the host, helping to prevent the development of drug resistance during the clinical incubation period.42,43
Many studies on the response of microorganisms to environmental stress (such as lack of nutrients, hypoxia, and oxidative stress) have shown that many genes of the cysteine regulator have a positive regulatory effect.44
Therefore, the role of cysteine biosynthesis in the development of antibiotic resistance has recently been pointed out.45,46 The study of mutants lacking the cysteine biosynthesis pathway in Salmonella typhimurium concluded that, due to inhibition of cysteine biosynthesis, unpaired oxidative stress can lead to decrease in the antibiotic resistance, in both vegetative and swarm cell populations. Antibiotic-induced oxidative stress has been widely recognized as a general mechanism of action for many antibacterial drugs,47 which may explain the reduction in the rate of resistance observed in bacteria with impaired cysteine biosynthesis. These findings indicate that inhibitors of cysteine biosynthesis may therefore improve the efficacy of antibiotic treatment and reduce the spread of resistance.40
Most bacteria and plants carry out cysteine biosynthesis through the reductive sulfate assimilation pathway (RSAP), which is a multi-step sulfate reduction process that culminates in the incorporation of bisulfide into activated form of serine to obtain cysteine48 (Fig. 5).
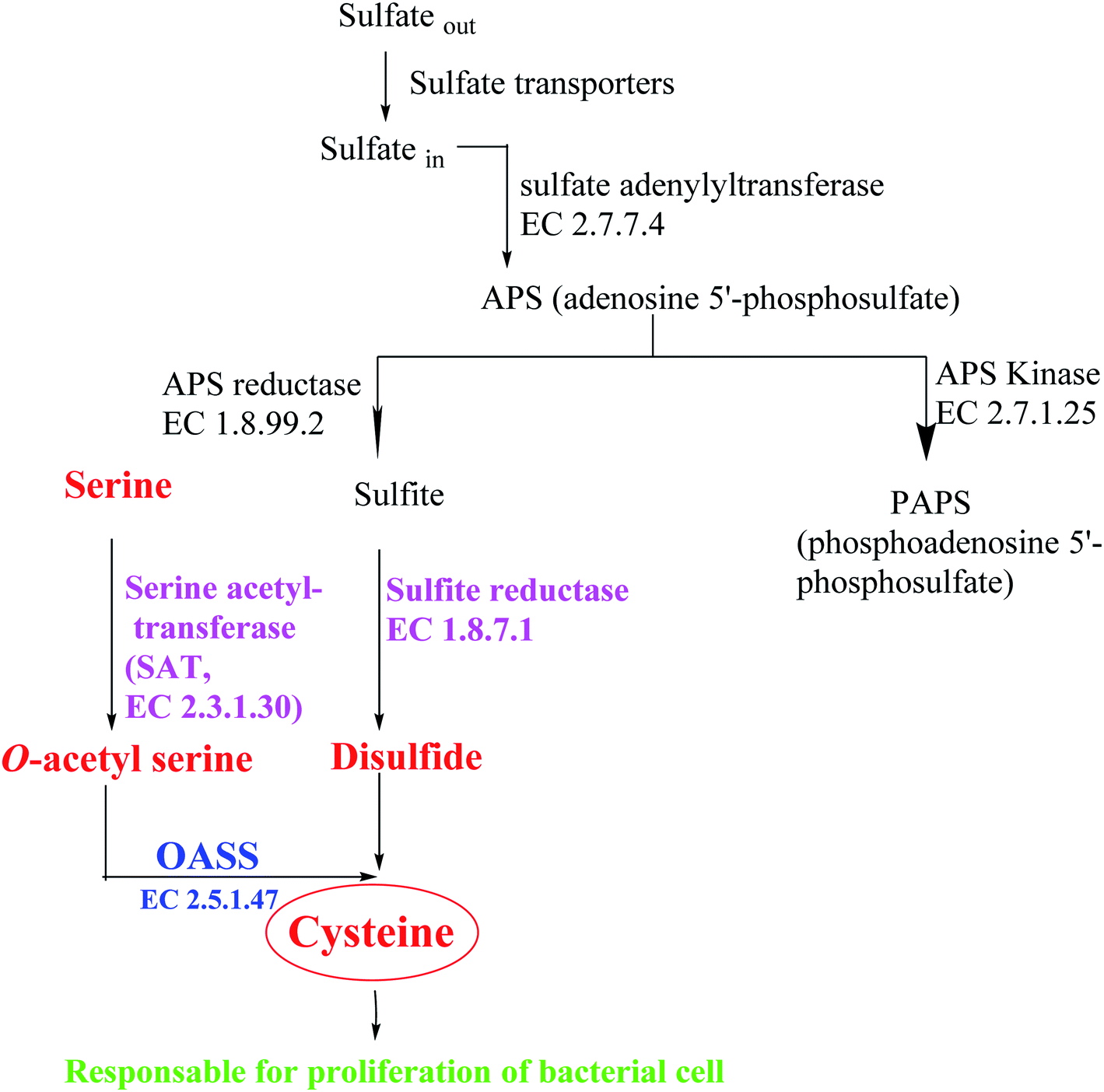 | ||
| Fig. 5 Overview of sulfur assimilation and related biochemical pathways. | ||
RSAP starts with the transport of sulfate in the cell and then reducing it to disulfide. This process consumes a lot of energy and is suitable for cellular needs. Bacteria find sulfur in the environment in the form of sulfate and actively transport it across the plasma membrane. After sulfate is reduced to disulfide, the latter is incorporated into cysteine by a member of a large enzyme family called cysteine synthase complex (CSC). The CSC is composed of serine acetyl transferase (SAT, EC 2.3.1.30) and O-acetylserine sulfhydralase (OASS; EC 2.5.1.47), which catalyzes the final step of cysteine biosynthesis.49
SAT has a catalytic activity in this complex where the acetyl group of acetyl-CoA is transferred to the hydroxyl group of serine to form O-acetylserine (OAS) and CoA. OAS is unstable and will be spontaneously converted to N-acetyl serine (NAS), which is a natural inducer of cysteine regulation signaling. In fact, high concentrations of bisulfide stabilize cysteine synthase complex (CSC), whereas OAS promotes its dissociation; on the other hand, increasing levels of cysteine can inhibit SAT catalytic activity by feedback control, triggered by binding to its active site.50
Depending on the organism and the growth conditions, the last step of cysteine biosynthesis is catalyzed by different thiol lyases which share high homology, but they also show some functions and structures difference. The first identified OASS subtype (OASS-A) was first isolated and identified from Salmonella typhimurium.51 Subsequently, it was observed how many pathogens have two isoforms of the enzyme: O-acetylserine sulfhydralase (OASS-A encoded by cysK) and O-phosphoserine sulfhydrylase (OASS-B encoded by cysM, EC 2.5.1.65). Which differ in functional and structural properties. In Salmonella typhimurium, the OASS-A and B isomers use O-acetylserine (OAS) as the substrate and S2− as the main source of sulfur. The expression of OASS isozymes depends on environmental conditions (OASS-A under aerobic conditions, OASS-B under anaerobic conditions).49 Both are Pyridoxal phosphate PLP-dependent enzymes, belonging to the type II subfamily of the PLP superfamily. They also have the same Bi–Bi ping pong-like reaction mechanism, in which the first half of the reaction is through β-elimination of the β-substituted L-serine external aldimine to generate the intermediate Schiff base of α-amino acrylate. In the second half-reaction, the sulfur source attacks the α-amino acrylate to form L-Cys.52 Since its discovery, OASS has been thought to have multiple functions, such as swarming motility in Proteus mirabilis,53 the transcriptional regulation of certain bacteria and nematodes,54 and the activation of toxins in Escherichia coli,55 and antibiotic resistance in Salmonella typhimurium.45,46
Based on these facts and considering the need to inhibit both enzymes to prevent the biosynthesis of L-cysteine,50 the identification of small molecules that can inhibit these two subtypes of OASS is of great importance for research.
In 2016, Pieroni et al.,56,57 starting from the evidence that SAT competitively inhibits OASS-A, based on the structural characteristics of the OASS-SAT interaction, a rational design of the first sulfydrylase inhibitor was developed. Taking into account their previous research data,58 combined with computational methods59,60 and spectroscopic methods, such as saturation transfer difference (STD) and nuclear magnetic resonance (NMR), rational design and synthesis a series of 2-phenylcyclopropanecarboxylic acid derivatives targeting two isoforms of St-OASS.61 In fact, they demonstrated that compounds that bind to the enzyme's active site effectively inhibit the OASS-A and B isoforms by competing with SAT. These findings provide a proof of principle and support the idea that it is possible to develop molecules that can inhibit both OASS-A and B enzymes and pave the way for the development of pharmacological tools to overcome bacterial virulence and resistance (Fig. 6).
 | ||
| Fig. 6 A schematic diagram of the rational design of the first sulfhydrylase inhibitor based on the structural characteristics of the OASS-SAT interaction. | ||
In 2018, a small series of 1,2-substituted-1H-benzo[d]imidazole derivatives was synthesized, which have a submicromolar range for all forms of leishmaniasis (Ki = 0.15–0.69 μM). The affinity for Leishmania, Mexico CPB2. 8ΔCTE, one of the most promising targets for anti-detox drug design. These compounds confirmed their in vitro activity against the intracellular amoeba of Leishmania infantum. Although they have a certain degree of cytotoxicity (CC50 = 8.0 μM in PMM, CC50 = 32.0 μM in MCR-5), compound 1d (IC50 = 6.8 μM) gave the best results. The molecular coupling and prediction studies of ADME-Tox properties were carried out on the computer to verify the expected target interaction hypothesis and evaluate the drug similarity of these derivatives62 (Fig. 7).
 | ||
| Fig. 7 Structure of 1,2-substituted-1H-benzo[d]imidazole derivatives 1 and compounds 2a,b. | ||
Besides, type I signal peptidase plays an important role in bacterial viability, and it is a promising but rarely developed target for antibacterial drugs. In view of the increasing rate of antimicrobial resistance, a new macrocyclic lipopeptide that combines P2 and P10 with a borate warhead was developed and found to inhibit E. coli type I signal peptidase (EcLepB) and show good antibacterial activity. The structural modification of the macrocycle, peptide sequence and lipophilic tail allowed to obtain fourteen new macrocyclic boronic esters. It can be shown that macrocyclization is well tolerated in terms of EcLepB inhibition and antibacterial activity. Among the synthesized macrocyclic compounds, effective enzyme inhibitors were also identified in the low nanomolar range (for example, compound 2a, EcLepB IC50 = 29 nM), and they also showed good antibacterial activity (for example, compound 2b, Escherichia coli WT MIC = 16 mg mL−1) (Fig. 7). The unique macrocyclic boronic ester described in this article is based on the previously announced EcLepB linear lipopeptide inhibitor, which attempts to solve the problem of cytotoxicity and hemolysis. This article revealed that the structural changes of the macrocycle can affect the cytotoxicity and hemolytic activity, which indicates that the P2 to P1′ linker provides a way to optimize off-target effects. However, for the current compounds, it cannot be distinguished between antibacterial activity and cytotoxicity.63
4.2. Targeting quorum sensing
In many bacterial pathogens, population growth is controlled by quorum sensing (QS), which is an intercellular communication mechanism that controls phenotypic manifestations (such as virulence).64 Bacteria use this system to communicate with each other in a given population.65 It consists of a signal molecule (called autoinducer) continuously secreted by each type of bacteria, and when the defined concentration of this molecular messenger reaches a threshold, it will activate the QS control process. Several aspects of virulence are affected by QS, so that, the identification of small molecules that can interfere with this cell–cell mechanism is currently a field of great interest.The most frequently studied QS autoinducers are N-acyl homoserine lactone (AHL) and peptides. AHL is produced by Gram-negative bacteria while peptides are formed by Gram-positive bacteria.66 AHL, also known as autoinducer-1 (AI-1), consists of a homoserine lactone ring with additional fatty acid side chains and passively diffuse into and out of Gram-negative bacterial cell.67 Although the AHL signal has traditionally been considered as intraspecific communication, studies have shown that AHL can also be used to detect potential environmental competitors.68
Peptides (auto-inducible peptides or quorum sensing peptides) are synthesized on the ribosome of Gram-positive bacteria and exhibit post-translational modifications in terms of their stability and functionality.69,70 When the extracellular concentration of signal peptide accumulates to a trough value, it will be detected by the protein (histidine-sensing kinase).66,67
Moreover, autoinducer 2 (AI-2) is another type of a signaling molecule that can be found in both Gram-positive and Gram-negative bacteria. AI-2 is widely used for interspecies communication, which is why it is considered as a universal communication signal between different bacterial species.71 Pseudomonas Quinolone Signal (PQS) autoinducer, also known as quinolone, was identified in Pseudomonas aeruginosa, produced by the protein encoded by pqsABCDH gene, and together with other AHL autoinducers, it controls biofilm formation and the production of virulence factors, Such as lecithin.72,73 Recently, auto-inducible IQS produced by the protein encoded by ambBCDE has been described in Pseudomonas aeruginosa. This molecule controls the expression of genes related to the production of pyocyanin, rhamnolipid, and elastase.74 Thus, QS has been identified in a wide range of Gram-negative and Gram-positive bacteria and is important in many disease-causing species as it has been shown to play a role in biofilm formation.75
Fig. 8 shows some autoinducers as AI-1, autoinducing peptide (AIP-1), PQS, γ-butyrolactone and CAI-1 along with their chemical structure. Whereas Fig. 9 illustrates some of the chiral signaling molecules belonging to the acyl-homoserine lactone (AHSLs) group (3–5) used by Gram-negative bacteria, furanosyl borate diester (6) used by Gram-positive and Gram-negative bacteria, and (S)-3-hydroxytridecan-4-one) (7) found in Vibrio species.71
 | ||
| Fig. 8 Structures of selected quorum sensing auto-inducers. | ||
 | ||
| Fig. 9 Some structures of chiral quorum sensing signaling molecules in bacteria. | ||
Acinetobacter baumannii,76 Pseudomonas aeruginosa77,78 and Escherichia coli79 are examples of Gram-negative bacteria that have QS systems. QS systems from other Gram-negative bacteria generate signal molecules through LuxI homologous protein synthase. These proteins produce specific AHL for each bacterial species.69 The change of AHL depends on the length of the carbon chain.71 Escherichia coli and Salmonella do not have LuxI protein and therefore do not synthesize AHL.80 However, both synthesize the SdiA protein that recognizes and binds to AHL produced by other bacteria.75 Pseudomonas aeruginosa has four known QS systems: las, rhl, PQS and integrated QS (IQS). The first two are mediated by homoserine N-acyl lactone (AHL) autoinducers, the third is mediated by quinolone (PQS signal), and the last is mediated by IQS signal74,81 (Fig. 10).82
 | ||
| Fig. 10 A hypothetical model explaining transcriptional regulation of QSI in P. aeruginosa.82 | ||
The innate receptors of these auto inducers belong to the LuxR type which act as transcriptional regulators upon activation by their native agonists. In P. aeruginosa, the so-called las and rhl systems have been identified to be the key AHL-based QS systems. The native ligands of the involved cytoplasmic receptors LasR and RhlR are N-(3-oxo-dodecanoyl)-L-homoserine lactone (3-oxo-C12-HSL or OdDHL) and N-butanoyl-L-homoserine lactone (C4-HSL or BHL), respectively (Fig. 10). Due to these similarities, bacteria can spread between species through this mechanism, which is useful for bacterial coinfection. Generally, in a species, there are multiple communication systems that are interconnected and affect each other.83 In the literature, there are reports describing various quorum sensing systems.84,85
This mechanism of QS can be found in different bacteria such as Staphylococcus aureus,86 Bacillus subtilis,87 Clostridium botulinum,88 and S. pneumoniae,89 and can regulate different phenotypes.
Mechanisms of QS inhibition in controlling bacterial biofilm formation are shown in Fig. 11: (1) inhibit AIs synthesis; (2) degrade or inactivate AIs by AHL-lactonases, oxidoreductases, antibodies, etc.; (3) interfere with the signal receptors using AI antagonists; (4) interfere with the response regulators thus disturbing signaling cascade; (5) reduce the extracellular AIs accumulation by inhibiting AIs efflux hence inhibited cell-to-cell signaling.82,90
 | ||
| Fig. 11 The mechanisms of QS inhibiting agents in controlling bacterial biofilm formation. | ||
Many researchers have focused on the development of LasR antagonists to disrupt the AHL signal in Pseudomonas aeruginosa. The ideal cellular effect of QSI targeting LasR and/or RhlR would be the reduction in the production of virulence factors (such as elastase, hydrogen cyanide, pyocyanin, pyoverdin, rhamnolipid or alkaline protease) as well as the attenuation of biofilm formation.82–90 The following section provides a detailed but not exhaustive overview of the structures covered by the synthetic QSI that interferes with AHL signals. Generally, compounds targeting LasR and Rh1R can be divided into two categories: structural mimics of AHL and structurally unrelated substances.84,85 Many synthetic agonists and antagonists of the LuxR-like receptor have structural characteristics that are very similar to natural ligands, or are directly synthesized derivatives thereof (Fig. 12 and 13). Obviously, the AHL scaffold can be divided into two parts. The head group is composed of a 5-membered homoserine lactone part, while the tail region contains linear N-acyl residue of different lengths. The amide-based linker between the two fragments facilitates a modular approach to direct synthesis and derivatization. Many reports in the literature dealing with synthetic AHL analogs focus on the structural modification of one of these two parts while keeping the other part of the molecule unchanged. Also, unnatural head and tail module combinations are described.91–103
 | ||
| Fig. 12 Some reported substituents as head group replacements in AHL to obtain QSI. | ||
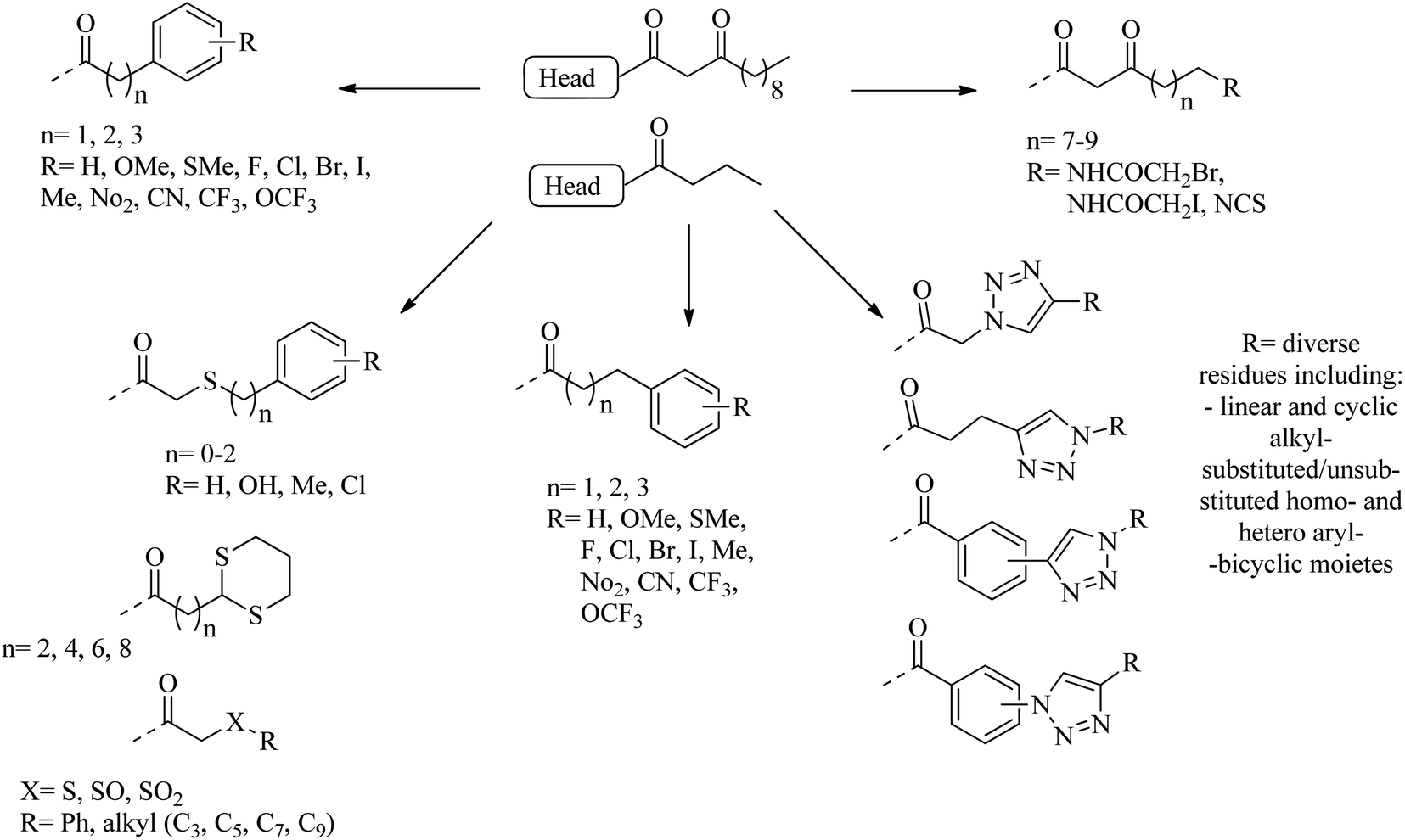 | ||
| Fig. 13 A diagram of some substituents as tail section replacements to obtain QSI. | ||
The combination of unnatural favorable head and tail groups with each other can produce quite unexpected results. Spring and colleagues have shown that many of these “chimeric” compounds are low or essentially inactive.98 Therefore, the conservation of natural 3-oxododecanoyl chains in antagonists with unnatural head groups may be mandatory for strong QS inhibition. Fig. 14 shows some promising AHL-simulated LasR and RhlR antagonists and their effects on Pseudomonas aeruginosa.92–94,98–100,103–107
 | ||
| Fig. 14 Some LasR and/or RhlR antagonists with reported effects on P. aeruginosa cells. | ||
Various methods of developing quorum sensing inhibitors started by mimicking the chemical structure of quorum sensing signaling molecules. Pseudomonas aeruginosa can be considered as a potential target. The biosynthesis of PQS signal requires the action of a set of enzymes PqsABCDEH and its autocatalytic receptor PqsR (MvfR). Later, many compounds were active in infection models, such as compound 20 which inhibits QS system and biofilm formation108,109 (Fig. 15).
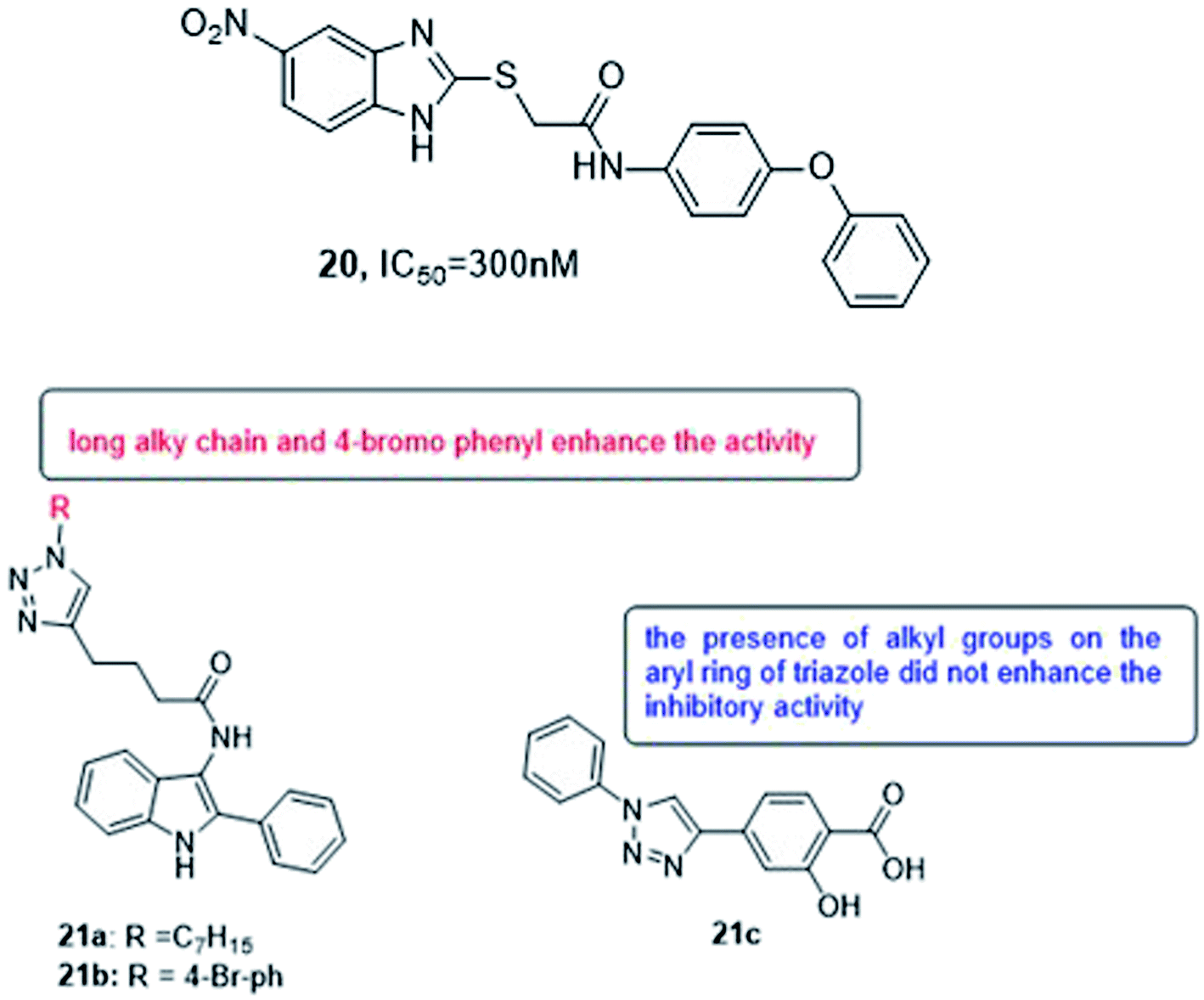 | ||
| Fig. 15 Structure of compounds 20 and 21a–c. | ||
In 2018, Srinivasarao et al.110 designed and synthesized 2-phenylindole-amide-triazole and salicyclic acid-triazole analogues (21a–c). These compounds were screened for in vitro quorum sensing inhibitory (QSI) activity against Pseudomonas aeruginosa. The QSI activity was determined in the LasR expressing P. aeruginosa MH602 reporter strain by measuring green fluorescent protein (GFP) production. 4-(1-heptyl-1H-1,2,3-triazol-4-yl)-N-(2-phenyl-1H-indol-3-yl)butanamide (21a) and 4-(1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)-N-(2-phenyl-1H-indol-3-yl)butanamide (21b) exhibited promising QSI activity with 58.89 and 54.34% at 250 μM, respectively. 1,2,3-Triazole based salicylic acid derivatives exhibited moderate to good activity that 2-hydroxy-4-(1-phenyl-1H-1,2,3- triazol-4-yl)benzoic acid (21c) was the most promising QS inhibitor with 40.28% inhibition at 250 μM (Fig. 15).
In 2018, Onem et al.111 studied the quorum sensing effect of synthetic benzimidazole derivatives (N-acyl homoserine lactone analogs). In Pseudomonas aeruginosa PA01, the anti-virulence ability of all synthetic compounds was investigated using different tests as elastase, pyocyanin, and group movement. Thus, 1,3-dihydro-2H-benzimidazol-2-one (22a), 5-methyl-1,3-dihydro-2H-benzimidazol-2-one (22b) and 1,3-diacetyl-1,3-dihydro-2H-benzimidazol-2-one (22c) were found to have anti-QS activity and a significant inhibitory effect on elastase, pyocyanin production and mass movement. In summary, the results indicated that synthetic benzimidazole derivatives can inhibit the spread of bacteria in Pseudomonas aeruginosa, which is essential for infection, and the role of these molecules in other bacteria should be further studied (Fig. 16).
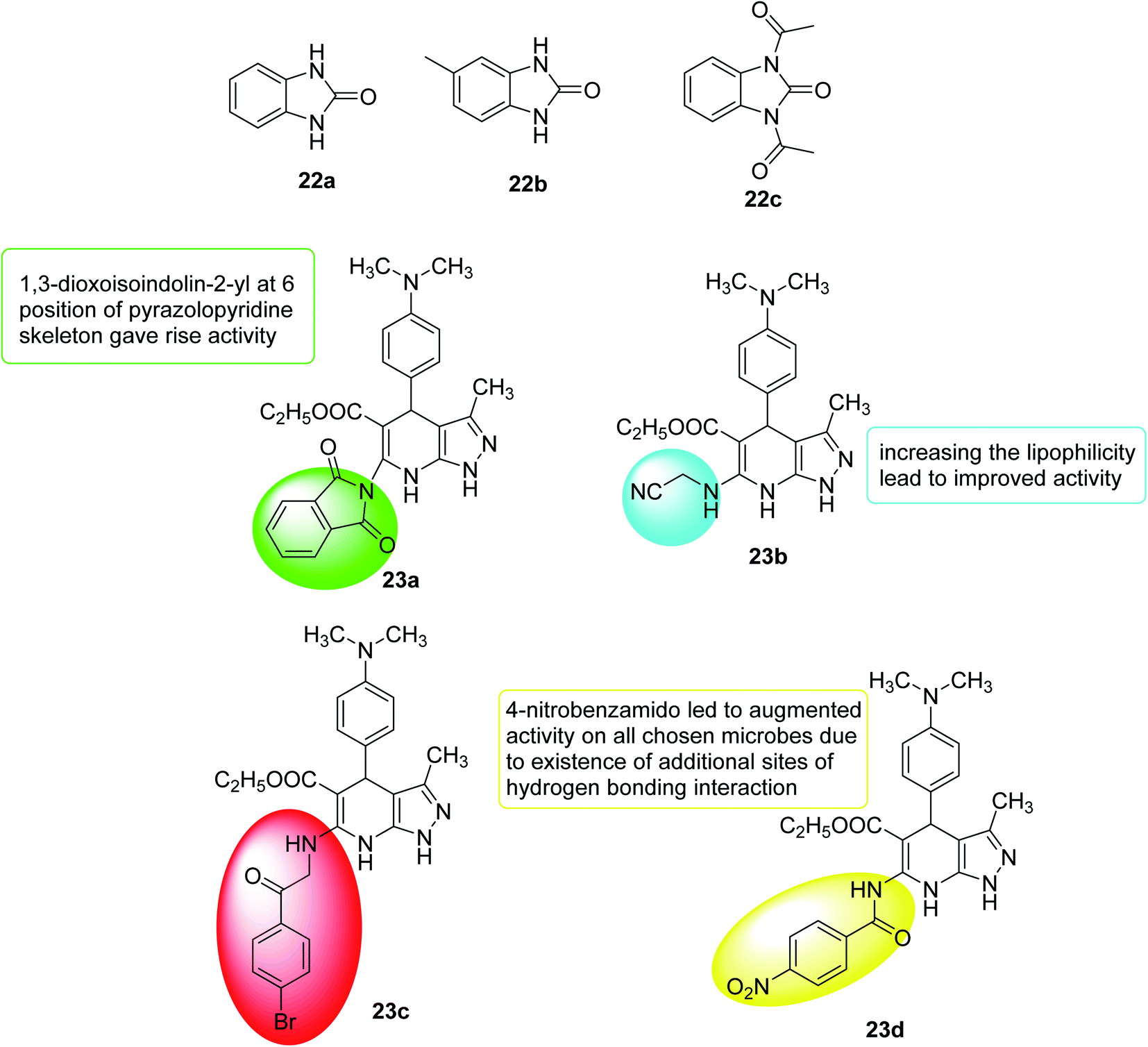 | ||
| Fig. 16 Structure of compounds 22a–c and 23a–d. | ||
In 2019, a pyrazolo[3,4-b]pyridine analogs were synthesized, and antibacterial tests were performed on Gram-negative bacteria, Gram-positive bacteria and selected fungi. Compound 23a showed broad spectrum and effective antimicrobial activity. In addition, 23b and 23c showed significant broad-spectrum antibacterial activity. The anti-quorum detection activity of the new members was tested on C. violaceum, where 23c showed high efficacy (pigment inhibition diameter 17 nm), while 23a and 23d revealed moderate efficacy (pigment inhibition diameter 10–15 nm)112 (Fig. 16).
In 2019, four series of L-homoserine lactone analogs were designed, synthesized and their inhibitory activity against Pseudomonas aeruginosa QS was evaluated. The results showed that compounds 24a, 24b of series I and 24c, 24d of series II represent L-homoserine lactone analogs with benzothiazolyl and 4-chlorophenyl substituents, respectively, and have a high inhibitory activity on CV026 and PAO1. Furthermore, most of the compounds of series III with side chains containing phenylurea and dithiocarbamate groups exhibited significant inhibition of QS at CV026 and PAO1. The results of the mechanism of action study confirmed that the analogs 24a, 24b, 24c, 24d, 24f and 24g inhibited the QS system of Pseudomonas aeruginosa by selectively reducing the expression of virulence factors, especially the biofilm formed by PAO1 by 28.6, 38.0, 34.1, 35.3, 36.2 and 40.3%, respectively. The activity of compound 24f is better than all other analogs, and the molecular coupling results showed that compound 24f competes with OdDHL for binding to LasR. This compound inhibited the expression of las system genes, PQS system genes and rhl system related genes, thereby inhibiting the production of virulence factors and the formation of PAO1 biofilms113 (Fig. 17).
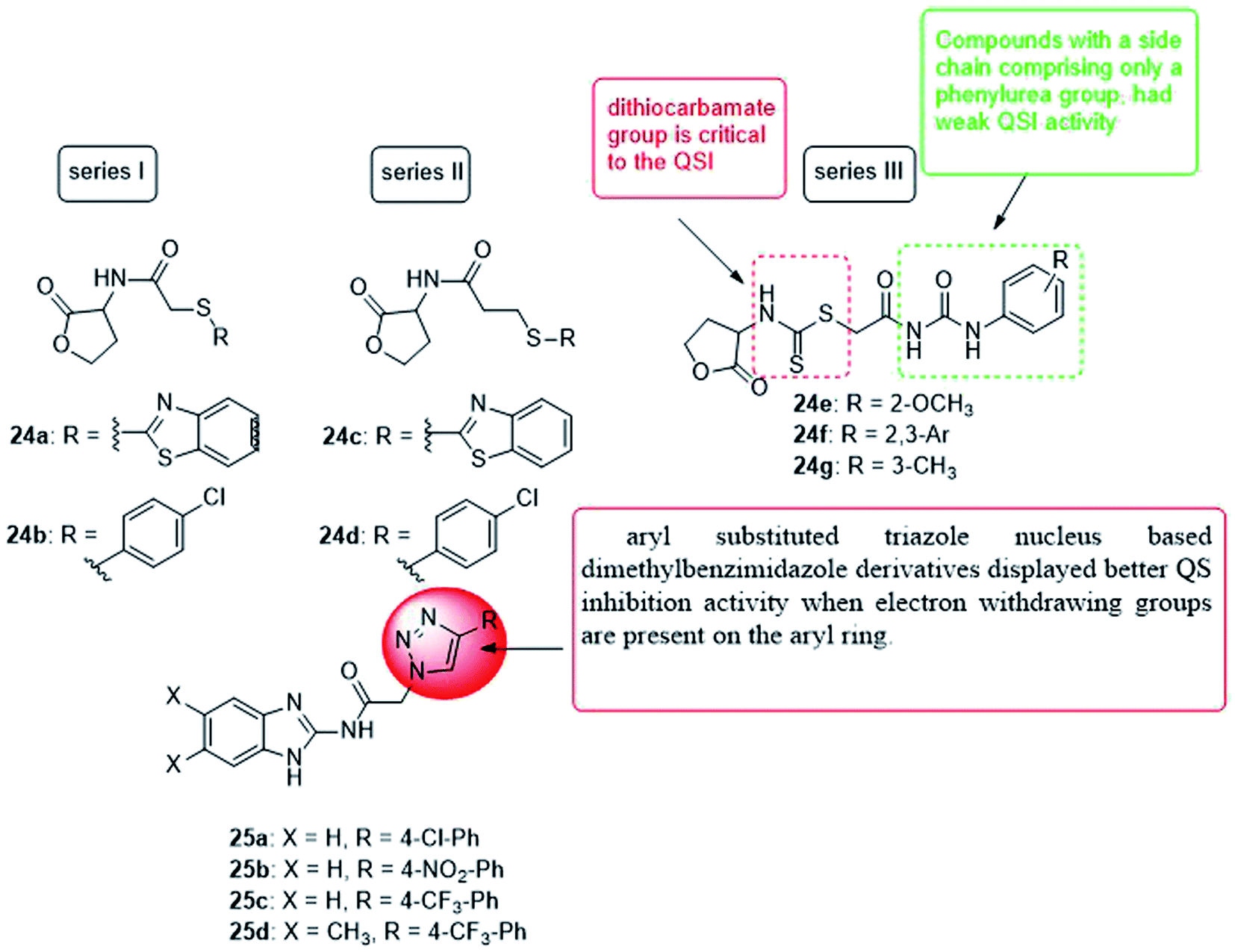 | ||
| Fig. 17 Structure of compounds 24a–g and 25a–d. | ||
In 2019, a series of 2-aminobenzimidazoles based on 1,2,3-triazole was synthesized, and the in vitro quorum sensing inhibitory activity against Pseudomonas aeruginosa was evaluated. By measuring the production of green fluorescent protein, the quorum sensing inhibitory activity was determined in the LasR-expressing Pseudomonas aeruginosa reporter strain MH602. The synthesized compound, N-(1H-benzo[d]imidazol-2-yl)-2-(4-(4-chlorophenyl)-1H-1,2,3-triazol-1-yl)acetamide (25a) showed a good quorum sensing inhibitory activity of 64.99% at 250 mM. Whereas, N-(1H-benzo[d]imidazol-2-yl)-2-(4-(4-nitrophenyl)-1H-1,2,3-triazol-1-yl)acetamide (25b) was the most promising quorum inhibitor at 250, 125, and 62.5 mM, and the inhibitory rates were 68.23, 67.10, and 63.67%, respectively. Moreover, the trifluoromethyl phenyl analogs 25c and 25d also showed a quorum sensing inhibition of 64.25% and 65.80% at 250 mM, respectively. Compound 25b was the most active quorum sensing inhibitor and revealed low cytotoxicity to normal human embryonic kidney cell lines at the concentrations tested (25, 50 and 100 mM)114 (Fig. 17).
In 2020, Hossain et al.115 designed, synthesized and evaluated some compounds capable of reducing the virulence of Pseudomonas aeruginosa. Compounds 26a, 26b and 26c exhibited the best activity, where compounds 26a and 26b represented the new QSI scaffolds. As anti-virulence agents, these compounds have been shown to reduce the production of all tested PA virulence factors: biofilm, pyocyanin, and rhamnolipids in the range of 40–60%. Furthermore, these compounds have been found to reduce the pro-virulence of ciprofloxacin at sub-inhibitory concentrations (Fig. 18).
 | ||
| Fig. 18 Structure of compounds 26a–c, 27 and 28. | ||
In 2020, Sabir et al.116 designed and synthesized fourteen anthranilyl-AMP mimics, which contained triazole linkers as potential inhibitors of Pseudomonas quinolone biosynthesis. However, with the exception of deoxycytidine analogs (27), most of these analogs showed no PQS inhibitory activity against the Pseudomonas aeruginosa reporter chain, which inhibited 30% of the PQS activity at 125 μM. Interestingly, the biofilm morphology observed by these compounds under the microscope changes significantly, indicating that they can inhibit PQS or other signaling systems that cause bacterial aggregation. Therefore, future efforts should be focused on modifying these analogs to increase penetration of bacterial cells.
In 2020, twenty-two quorum sensing inhibitors were synthesized, mimicking the structure of autoinducers of Acinetobacter, and 28 of them inhibited biofilms by up to 34%. By inhibiting extracellular polysaccharides and synergistic effects with gentamicin sulfate, the biofilm inhibition effect is further proved117 (Fig. 18).
In 2020, a series of 3-amino-2-oxazolidinone derivatives was designed and synthesized. C. violaceum CV026 was used as the reporter strain to evaluate the inhibitory activity of new oxazolidinone compounds on QS. Thirteen compounds showed good activity (IC50 range 3.69–63.58 mM), where compound 29 inhibition of biofilm formation was the most significant (IC50 = 3.686 ± 0.5790 mM). In vitro, compound 29 significantly inhibited PAO1 biofilm formation (range 42.98–7.67%), virulence factor production (pyocyanin, elastase, rhamnolipid, and protease), and bacterial movement. Furthermore, the combination of compound 29 and antibiotics (meropenem trihydrate) can significantly improve the antibiotic sensitivity of PAO1 cells of the Pseudomonas aeruginosa biofilm. In vivo, compound 29 significantly prolonged the lifespan of wild-type C. elegans N2 infected by Pseudomonas aeruginosa PAO1. In summary, compound 29 can be considered as a drug candidate for drug-resistant Pseudomonas aeruginosa PAO1, providing a tool for finding new antibacterial drug118 (Fig. 19).
 | ||
| Fig. 19 Structure of compounds 29 and 30a–f. | ||
In 2021, the 1,2,4-triazole derivatives were designed, synthesized and selected for in vitro evaluation of antimicrobial activity and anti-QS activity. Out of fourteen derivatives, compound 30f selectively possessed antibacterial activity against C. violaceum. Further derivatives that possessed an electron withdrawing group and halogen atoms in N-phenylacetamide moiety were moderately active against Chromobacterium violaceum and Xanthomonas campestris pv. Campestris (Xcc). After observing the reduction of violacein production through plate assay, compounds 30a, 30b, 30c, 30d and 30e were subjected to quantification of quorum sensing inhibition. Compounds with the electron-withdrawing group in N-phenylacetamide moiety showed admirable activity with >80% inhibition of violacein. Mainly compound 30b which had no effect on the growth of bacteria was identified as excellent QSI which could be a lead compound for further development119 (Fig. 19).
4.3. Targeting biofilms
In nearly 65% of infections, bacteria grow in the form of clusters which known as biofilms, that stick and grow on the surface of plants (roots) or animals (epithelium). During the growth period, under infection conditions, the resistance of bacteria to antibiotics increases 10 to 1000 times.37,118Current biofilm targeting methods can be divided into two categories: the first is a physical-mechanical method designed to interrupt and eliminate the biofilm, and the second method involves the use of antibiotics or antibiotics in the matrix to prevent the formation of biofilm.120
DR-1018 (or just 1018) is a small cationic synthetic peptide (VRLIVAVRIWRR-NH2) that has been developed based on bactenecin, a peptide antibiotic isolated from bovine neutrophil granules. This peptide has numerous biological activities that targets both eukaryotic and bacterial cells. By targeting both Gram-negative (e.g. Pseudomonas aeruginosa and Escherichia coli) and Gram-positive (e.g. Staphylococcus aureus) bacteria 1018 acts as a potent antibacterial: it kills bacteria, disperses biofilms and inhibits bacterial swarming. As a result, peptide 1018 can be used as a new type of antibiotic adjuvant, which not only has broad-spectrum activity,121 but also can be combined with commonly used antibiotics such as tobramycin, ceftazidime, imipenem and ciprofloxacin.122
However, the field of therapeutics targeting biofilms is rapidly evolving and currently includes different strategies aimed at interfering with this bacterial cell–cell communication mechanism.123–127
In 2017, thirty-three Schiff bases of 2-amino-5-chlorobenzophenone were variably substituted, and their effects on Klebsiella pneumoniae, Pseudomonas mirabilis, Staphylococcus aureus and mutants were evaluated. Twelve compounds were found to have biofilm inhibitory activity when the concentration was less than 100 μg mL−1. Three compounds 31a, 31b and 31c also showed destructive properties on the biofilm. Fluorescence microscopy further confirmed the inhibitory effect of selected compounds on the isolate biofilm. The activity of these compounds depends mainly on the different substituents on the aryl part of the molecule. These compounds may have the potential to develop new anti-biofilm agents against different bacterial strains128 (Fig. 20).
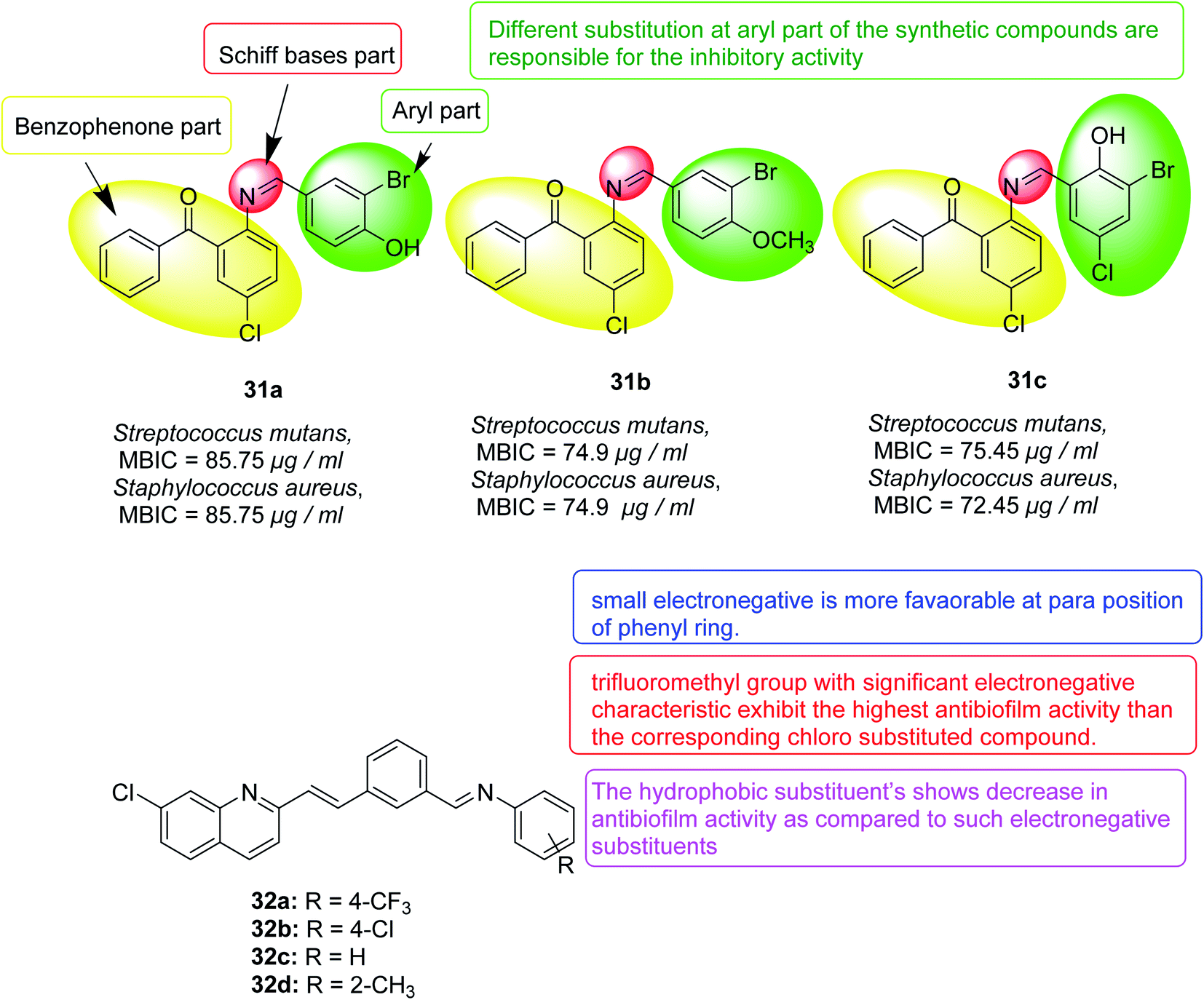 | ||
| Fig. 20 Structure of compounds 31a–c and 32a–d. | ||
In 2018, a chloroquinoline derivative with a vinylbenzylidene aniline substituent at position 2 was synthesized and provided biofilm inhibition, antifungal and antibacterial activities. The Candida albicans biofilm inhibition results showed that, in comparison to the standard antifungal drug fluconazole (IC50 = 40.0 μM), compounds 32a (IC50 = 51.2 μM) and 32b (IC50 = 66, 2 μM) had good inhibition of the anti-bio membrane. Compounds 32b (MIC = 94.2 μg mL−1) and 32c (MIC = 98.8 μg mL−1) also revealed good antifungal activity comparable to that of the standard drug fluconazole (MIC = 50.0 μg mL−1). Antibacterial activity detection against four types of bacteria, Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus showed that they have potential antibacterial activity where all compounds except 32d were better than the standard drug ciprofloxacin for Bacillus subtilis129 (Fig. 20).
In 2018, a series of pyrazole-thiazole hybrids was designed, synthesized and evaluated for their in vitro antibacterial activity against Gram-positive and Gram-negative pathogenic bacteria and fungal strains. Compounds derived from p-methylphenyl (33a), p-bromophenyl (33b), 8-bromocoumarin-based (33c) and 6,8-dibromocoumarin-based (33d) showed promising inhibitory activity/minimum bactericidal concentration (MBC) spectrum from 1.9/7.8 μg mL−1 to 3.9/7.8 μg mL−1. Compounds substituted with p-methylphenyl (33a), benzo[f]coumarin group (33e), 8-bromocoumarin group (33c) revealed MIC/minimum fungicide concentration of 3.9/7.8 μg mL−1 against various Candida strains. In addition, the toxicity and anti-biofilm properties of these compounds were also tested. The results of biofilm inhibition showed that compound 33e showed good activity with an IC50 value of 11.8 μM against S. aureus MTCC 96. Compound 33c exhibited significant activity against S. aureus MLS16 MTCC 2940, K. planticola MTCC 530 and C. albicans MTCC 3017, with IC50 values of 12, 14, and 16 μM, respectively130 (Fig. 21).
 | ||
| Fig. 21 Structure of compounds 33a–e, 34a–d, 35a–e, 36 and 37. | ||
In the same year, a series of phenyl thiazoles was synthesized with lipophilic t-butyl substituent and their antibacterial activity was evaluated against a group of multidrug resistant bacterial pathogens. Five compounds showed promising antibacterial activity against methicillin-resistant Staphylococcus strains and various vancomycin-resistant Staphylococcus and Enterococcus species. Furthermore, the four derivatives 34a, 34b, 34c and 34d exhibited rapid bactericidal activity with MIC ranging 4–16 μg mL−1 and a remarkable ability to destroy the mature biofilm (64%, 48.2%, 28.3%, and 49%, respectively) produced by MRSA USA300 (Fig. 21).131
In 2018, a series of amide chalcones coupled with different secondary amines was synthesized and tested for antibacterial activity in vitro. Compounds 35a–e are the most active of the synthetic series, with MIC values of 2.0–10.0 μg mL−1 for different bacterial strains. Compound 35a is equivalent to the standard drug ampicillin, which shows MBC value of 2.0 μg mL−1, against the bacterial strain Staphylococcus aureus. Compounds 35a, 35b and 35c showed promising anti-biofilm activity with IC50 values ranging from 2.4 to 8.6 μg (Fig. 21).132
In 2018, a benzimidazole derivative (36) was identified to rescue nematodes from Staphylococcus aureus infection. It can prevent the formation of biofilms in a dose-dependent manner without interfering with the viability of bacteria, thus, supplementing compound from 0.78 μM to 100 μM can reduce biofilm formation by 8.9% to 68%. According to these data, compound 36 at 6.25 μM inhibited biofilm formation by 50%. To test the effect of its expression on the Staphylococcus aureus virulence genes, transcriptome analysis of the entire genome of the pathogen was performed. The data showed that genes related to biofilm formation, especially genes related to bacterial attachment, are eliminated in treated bacteria. In the presence of compound 36, the virulence factor of Staphylococcus aureus was also down-regulated. In summary, these findings indicate that compound 36 can be considered as a promising compound for further development as an anti-virulence agent to control Staphylococcus aureus infections133 (Fig. 21).
In 2019, twenty-two new compounds were prepared and evaluated for their activity against methicillin-resistant Staphylococcus aureus. The compound 37 with 3-hydroxyazetidine showed moderate antibacterial activity against all methicillin-sensitive Staphylococcus aureus (MSSA) and MRSA tested strains in a concentration range of 4 to 16 μg mL−1. Their MBC value was higher than 64 μg mL−1, which was more than three times higher than their MIC value for the tested strain and proved that the compound was a bacteriostatic agent. Other advantages of this compound was its ability to eradicate staphylococcal biofilm clusters in a dose-dependent manner, high metabolic stability after oral administration of 25 mg kg−1, biological half-life of more than 5 hours, and plasma concentration (Cmax) that exceeds the MIC value134 (Fig. 22).
 | ||
| Fig. 22 Structure of compounds 38a–d and 39a–i. | ||
In 2019, Tandon et al. developed an ecological method using water as a solvent to synthesize a series of aminobenzoquinones. Subsequently, the in vitro antibacterial potential of all PQ analogs was evaluated in a set of seven bacterial strains (three Gram-positive bacteria and four Gram-negative bacteria) and three fungi. The antifungal properties of all PQ analogs indicated that all four analogs had effective antifungal activity (38a–c were effective against Candida tropicalis and 38d was effective against Candida albicans). The results illustrated that in comparison to the popular clinical antibacterial drug cefuroxime, 38b showed similar antibacterial activity against Staphylococcus epidermidis. While compound 38d revealed the highest antibacterial activity against Staphylococcus aureus. Due to their outstanding activity, 38b and 38d were selected for additional studies in the evaluation of biofilm and cytotoxicity. According to the performed tests, there was a significant positive correlation between inhibition of biofilm adhesion and time. Furthermore, compounds 38b and 38d exerted cytotoxic effects on Balb/3T3, HaCaT, HUVEC and NRK-52E cells at high concentrations (>24 and >18 μg mL−1, respectively). Therefore, these two analogs (38b and 38d) were identified as the hits with the strong antibacterial efficiency against the S. epidermidis with low MIC value.135 (Fig. 22).
In 2019, a series of new 2-(4-(acridin-9-amino) phenyl) isoindoline-1,3-dione derivatives was synthesized and tested for anti-biofilm and anti-quorum detection. Among all compounds, compound 39 inhibited approximately 70% of Pseudomonas aeruginosa biofilms while the remaining compounds showed mild inhibition of biofilm. Considering all these compounds, compound 39f with nitro substitution may be important to eradicate biofilms136 (Fig. 22).
In 2020, a series of pyran derivatives was synthesized and characterized, and their anti-biofilm activity was measured in vitro against Staphylococcus aureus and Pseudomonas aeruginosa by determining the minimum inhibitory concentration of biofilm. A mouse model of tissue cage infection induced by Staphylococcus aureus and rat model of Pseudomonas aeruginosa-induced urinary catheter infection were used. Several pyran derivatives showed anti-biofilm activity in vitro against Staphylococcus aureus and Pseudomonas aeruginosa, and the activity of these compounds was not through the accessory gene of Staphylococcus aureus system-mediated quorum sensing regulator (agr). One of the pyran derivatives, 40, can effectively inhibit the biofilm formation of Staphylococcus aureus and Pseudomonas aeruginosa in vivo. The results showed that the minimum biofilm inhibitory concentrations (MBIC) of compound 40 in Staphylococcus aureus (US 300) and Pseudomonas aeruginosa (ATCC 27853) were greater than 200 μg mL−1 and 256 μg mL−1, respectively137 (Fig. 23).
 | ||
| Fig. 23 Structure of compounds 40, 41a–d, 42, 43a–c and 44. | ||
In 2020, a series of new triazole-thiazole hybrids was designed and synthesized using a multicomponent reaction method, and antibacterial activity was evaluated in vitro. Most of the tested compounds showed promising inhibitory activity against bacterial strains, with values ranging from 2.8 to 15.7 μM. Moreover, compounds 41a–c showed potential Candida activity against various Candida strains, with spectrum values ranging from 5.9 to 14.2 μM. Furthermore, the anti-biofilm and toxicity characteristics of the effective compounds were also tested. Compounds 41a, 41b, and 41d were found to inhibit biofilm formation with IC50 values of 6.6, 15.9, and 16.6 μM, respectively, against Bacillus subtilis MTCC. 121. Furthermore, 41b and 41d also showed promising biofilm formation inhibitory activity against Staphylococcus aureus MTCC 96, with IC50 values of 12.0 and 13.5 μM, respectively (Fig. 23). In summary, the activity results highlighted compounds 41b and 41d as possible drivers for the further development of antibacterial, anti-Candida and anti-biofilm agents.138
In 2020, a new series of seventeen 1,2,4-oxadiazole derivatives were synthesized and efficiently screened as potential new antitoxic agents. The ability to inhibit biofilm formation against Gram-positive and Gram-negative pathogens was evaluated. It should be noted that all these compounds inhibited the formation of Staphylococcus aureus and Pseudomonas aeruginosa biofilms in a dose-dependent manner. For the most active compound 42 (Fig. 23), the 50% biofilm inhibitory concentration (BIC50) was equal to 10 μM. The inhibition of the catalytic activity of transpeptidase sortasse A (SrtA) was verified as one of the possible mechanisms of action of these new 1,2,4-oxadiazole derivatives in the tested Gram-positive pathogens, using the recombinant Staphylococcus aureus SrtA. The BIC50 values of the three most active compounds against Staphylococcus aureus ATCC 25923 are between 0.7 and 9.7 μM, showing good enzymatic activity, with IC50 values between 2.2 and 10.4 μM.139
In 2021, El-Malah et al.140 synthesized an alkyne-functionalized pyridine derivatives and evaluated in vitro their antibacterial and anti-biofilm activity against methicillin-resistant Staphylococcus aureus (MRSA) in sessile and planktonic cells. Compounds 43a–c illustrated good growth inhibitory activity against planktonic and sessile MRSA cells, with IC50 of 34.94, 37.91 and 43.88 mM, respectively. Vancomycin (Van) was used as a standard reference drug (IC50 = 186.0 mM) to evaluate the anti-biofilm activity of the new compound (Fig. 23).
In 2021, Paul et al.141 tested the antibacterial and anti-biofilm activity of 1,4-naphthoquinone (44) against Staphylococcus aureus (Fig. 23). The results showed that the minimum bactericidal concentration (MBC) of this compound against Staphylococcus aureus is 100 μg mL−1. In this sense, a series of experiments related to biofilm determination (crystal violet, biofilm protein measurement and microanalysis) was carried out, in which the concentration of 1.4-naphthoquinone sub-MBC (1/20 and 1/10 MBC were used). All biofilm test results revealed that the compounds (1,4-naphthoquinone) at these test concentrations (1/20 and 1/10 MBC) significantly reduced the formation of Staphylococcus aureus biofilm. Furthermore, the proven concentrations (1/20 and 1/10 MBC) of the compound (1,4-naphthoquinone) can reduce microbial movement of Staphylococcus aureus which can affect biofilm development. Other studies have shown that treatment of organisms with 1,4-naphthoquinone will increase cellular accumulation of reactive oxygen species (ROS), thus inhibiting the formation of Staphylococcus aureus biofilms. Therefore, it can be concluded that 1,4-naphthoquinone can be considered a promising compound to inhibit the biofilm caused by Staphylococcus aureus.
5 Conclusion
Antibiotic treatment will produce a kind of selective pressure to kill sensitive microorganisms by inhibiting growth-related cellular activities (such as DNA, RNA, and protein synthesis). However, the resistant pathogenic bacteria can survive in the presence of antibiotics using different virulence factors that help the infection to spread within the host. A clear understanding of the underlying mechanisms of virulence can help to design new agents that inhibit the virulence factors and overcome the bacterial resistance. Among virulence factors are quorum sensing (QS) and biofilm formation which are recently studied where different heterocyclic compounds were synthesized and screened for their inhibition of these virulence factors.The discovery of the QS and QSI system opened the door for the development of these new therapies that can be used as antibiotic adjuvants. However, there is a lot to discover and much more investigation should be performed as the published anti-virulence agents can be considered only as hits for further optimization to obtain novel anti-virulence therapies.
Author contributions
Rehab H. Abd El-Aleam: data curation and writing the original draft of this review article. Riham F. George: supervision and writing-review & editing. Hanan H. Georgey: supervision and visualization. Hamdy M. Abdel-Rahman: conceptualization and supervision.Conflicts of interest
There are no conflicts to declare.References
- L. Santos and F. Ramos, Int. J. Antimicrob. Agents, 2018, 52(2), 135–143, DOI:10.1038/nm.4465.
- M. Boolchandani, A. W. D`Souza and G. Dantas, Nat. Rev. Genet., 2019, 20(6), 356–370, DOI:10.1038/s41576-019-0108-4.
- A. Gupta, R. Singh, P. K. Sonar and S. K. Saraf, Biochem. Res. Int., 2016, 2016, 8086762, DOI:10.1155/2016/8086762.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Pathog. Glob. Health, 2015, 109(7), 309–318, DOI:10.1179/2047773215y.0000000030.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, S14459, DOI:10.4137/pmc.S14459.
- World Health Organization, 2020 antibacterial agents in clinical and preclinical development: an overview and analysis, 2020 antibacterial agents in clinical and preclinical development: an overview and analysis, 2021, https://cdn.who.int/media/docs/default-source/antimicrobial-resistance/amr-gcp-irc/2020-antibacterial-agents-in-clinical-and-preclinical-development.pdf?sfvrsn=2c480edd_5 Search PubMed.
- I. M. da Silva, J. da S. Filho, P. B. G. da Silva Santiago, M. S. do Egito, C. A. de Souza, F. L. Gouveia, R. M. Ximenes, K. X. da Fonseca Ribeiro de Sena, A. Rodolfo de Faria, D. J. Brondani and J. F. C. de Albuquerque, Biomed Res. Int., 2014, 316082, DOI:10.1155/2014/316082.
- H. Lade and J. Kim, Antibiotics, 2021, 10(4), 398, DOI:10.3390/antibiotics10040398.
- M. Bassetti, E. Righi and C. Viscoli, Expert Opin. Invest. Drugs, 2008, 17(3), 285–296, DOI:10.1517/13543784.17.3.285.
- S. R. Ali, S. Pandit and M. De, ACS Appl. Bio Mater., 2018, 1(4), 967–974, DOI:10.1021/acsabm.8b00105.
- G. Maravic, Curr. Drug Targets Infect. Disord., 2004, 4(3), 193–202, DOI:10.2174/1568005043340777.
- K. T. Welch, K. G. Virga, N. A. Whittemore, C. Özen, E. Wright, C. L. Brown, R. E. Lee and E. H. Serpersu, Bioorg. Med. Chem., 2005, 13(22), 6252–6263, DOI:10.1016/j.bmc.2005.06.059.
- P. A. Lambert, Adv. Drug Deliv. Rev., 2005, 57(10), 1471–1485, DOI:10.1016/j.addr.2005.04.003.
- G. Laible, B. G. Spratt and R. Hakenbeck, Mol. Microbiol., 1991, 5(8), 1993–2002, DOI:10.1111/j.1365-2958.1991.tb00821.x.
- D. C. Hooper, Emerg. Infect. Dis., 2001, 7(2), 337, DOI:10.3201/eid0702.010239.
- I. A. Hashim, Q. H. Wdaah and A. A. Atya, UT sci, 2019, 7(1), 7–14 CrossRef /https://jsci.utq.edu.iq/index.php/main/article/view/241.
- R. Singh, S. P. Dwivedi, U. S. Gaharwar, R. Meena, P. Rajamani and T. Prasad, J. Appl. Microbiol., 2020, 128(6), 1547–1567, DOI:10.1111/jam.14478 , Epub 2019 Oct 29. PMID: 31595643..
- V. de Lastours, T. Goulenok, F. Guérin, H. Jacquier, C. Eyma, F. Chau, V. Cattoir and B. Fantin, Eur. J. Clin. Microbiol. Infect. Dis., 2018, 37(3), 417–421, DOI:10.1007/s10096-018-3186-x.
- H. Nikaido and H. I. Zgurskaya, Curr. Opin. Infect. Dis., 1999, 12, 529–536, DOI:10.1097/00001432-199912000-00001.
- M. A. Webber and L. J. V. Piddock, J. Antimicrob. Chemother., 2003, 51, 9–11, DOI:10.1093/jac/dkg050.
- J. L. Ramos, E. Duque, M. T. Gallegos, P. Godoy, M. I. Ramos-Gonzalez, A. Rojas, W. Terán and A. Segura, Annu. Rev. Microbiol., 2002, 56, 743–768, DOI:10.1146/annurev.micro.56.012302.161038.
- H. I. Zgurskaya and H. Nikaido, Mol. Microbiol., 2000, 37, 219–225, DOI:10.1046/j.1365-2958.2000.01926.x.
- D. H. Nies, FEMS Microbiol. Rev., 2003, 27, 313–339, DOI:10.1016/S0168-6445(03)00048-2.
- P. Fernandes, B. S. Ferreira and J. M. S. Cabral, Int. J. Antimicrob. Agents, 2003, 22(3), 211–216, DOI:10.1016/S0924-8579(03)00209-7.
- L. McMurry, R. E. Petrucci and S. B. Levy, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 3974–3977, DOI:10.1073/pnas.77.7.3974.
- O. Bozdağ-Dündar, Ö. Özgen, A. Menteşe, N. Altanlar, O. Atlı, E. Kendi and R. Ertan, Bioorg. Med. Chem., 2007, 15(18), 6012–6017, DOI:10.1016/j.bmc.2007.06.049.
- J. D. Hartzell, R. Neff, J. Ake, R. Howard, S. Olson, K. Paolino, M. Vishnepolsky, A. Weintrob and G. Wortmann, Clin. Infect. Dis., 2009, 48, 1724–1728, DOI:10.1086/599225.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548, DOI:10.1038/nchembio.2007.24.
- M. Garland, S. Loscher and M. Bogyo, Chem. Rev., 2017, 117(5), 4422–4461, DOI:10.1021/acs.chemrev.6b00676.
- B. Campanini, M. Pieroni, S. Raboni, S. Bettati, R. Benoni, C. Pecchini, G. Costantino and A. Mozzarelli, Curr. Med. Chem., 2015, 22(2), 187–213 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548, DOI:10.1038/nchembio.2007.24.
- S. W. Dickey, G. Y. C. Cheung and M. Otto, Nat. Rev. Drug Discovery, 2017, 16, 457–471, DOI:10.1038/nrd.2017.23.
- A. Larsson, S. M. C. Johansson, J. S. Pinkner, S. J. Hultgren, F. Almqvist, J. Kihlberg and A. Linusson, Nat. Rev. Microbiol., 2008, 6, 17–27, DOI:10.1021/jm040818l.
- G. Annunziato, L. Giovati, A. Angeli, M. Pavone, S. D. Prete, M. Pieroni, C. Capasso, A. Bruno, S. Conti, W. Magliani, C. T. Supuran and G. Costantino, J. Enzyme Inhib. Med. Chem., 2018, 33, 1537–1544, DOI:10.1080/14756366.2018.1516652.
- D. A. Raskoand and V. Sperandio, Nat. Rev. Drug Discovery, 2010, 9, 117–128, DOI:10.1038/nrd3013.
- R. C. Allen, R. Popat, S. P. Diggle and S. P. Brown, Nat. Rev. Microbiol., 2014, 12, 300–308, DOI:10.1038/nrmicro3232.
- J. Fernebro, Drug Resist. Updates, 2011, 14, 125–139, DOI:10.1016/j.drup.2011.02.001.
- R. J. Youle and M. Karbowski, Nat. Rev. Mol. Cell Biol., 2005, 6, 657–663, DOI:10.1038/nrm1697.
- R. M. Roop II, J. M. Gaines, E. S. Anderson, C. C. Caswell and D. W. Martin, Med. Microbiol. Immunol., 2009, 198, 221–238, DOI:10.1007/s00430-009-0123-8.
- D. P. Bhave, B. Wilson III and K. S. Carroll, Infect. Disord. Drug Targets, 2007, 7, 140–158, DOI:10.2174/187152607781001772.
- D. Becker, M. Selbach, C. Rollenhagen, M. Ballmaier, T. F. Meyer, M. Mann and D. Bumann, Nature, 2006, 440, 303–307, DOI:10.1038/nature04616.
- A. Coates, Y. Hu, R. Bax and C. Page, Nat. Rev. Drug Discovery, 2002, 1, 895–910, DOI:10.1038/nrd940.
- A. R. M. Coates and Y. Hu, Br. J. Pharmacol., 2007, 152, 1147–1154, DOI:10.1038/sj.bjp.0707432.
- H. E. Seifried, D. E. Anderson, E. I. Fisher and J. A. Milner, J. Nutr. Biochem., 2007, 18(9), 567–579, DOI:10.1016/j.jnutbio.2006.10.007.
- A. L. Turnbull and M. G. Surette, Microbiol, 2008, 154, 3410–3419, DOI:10.1099/mic.0.2008/020347-0.
- A. L. Turnbull and M. G. Surette, Res. Microbiol., 2010, 161, 643–650, DOI:10.1016/j.resmic.2010.06.004.
- M. A. Kohanski, D. J. Dwyer, B. Hayete, C. A. Lawrence and J. J. Collins, Cell, 2007, 130(5), 797–810, DOI:10.1016/j.cell.2007.06.049.
- H. B. Krishnan, B. Song, N. W. Oehrle, J. C. Cameron and J. M. Jez, Sci. Rep., 2018, 8, 2367, DOI:10.1038/s41598-018-20919-8.
- A. Mozzarelli, S. Bettati, B. Campanini, E. Salsi, S. Raboni, R. Singh, F. Spyrakis, V. P. Kumar and P. F. Cook, Biochim. Biophys. Acta, 2011, 1814(11), 1497–1510, DOI:10.1016/j.bbapap.2011.04.011.
- M. Marchetti, F. Saverio De Angelis, G. Annunziato, G. Costantino, M. Pieroni, L. Ronda, A. Mozzarelli, B. Campanini, S. Cannistraro, A. R. Bizzarri and S. Bettati, Catalysts, 2021, 11, 700, DOI:10.3390/catal11060700.
- C. M. Gossner, D. van Cauteren, S. Le Hello, F. X. Weill, E. Terrien, S. Tessier, C. Janin, A. Brisabois, V. Dusch, V. Vaillant and N. Jourdan-da Silva, Eurosurveillance, 2011, 17(5), 20071, DOI:10.2807/ese.17.05.20071-en.
- C. H. Tai, S. R. Nalabolu, T. M. Jacobson, D. E. Minter and P. F. Cook, Biochemistry, 1993, 32(25), 6433–6442, DOI:10.1021/bi00076a017.
- M. M. Pearson, A. Yep, S. N. Smith and H. L. T. Mobley, Infect. Immun., 2011, 79(7), 2619–2631, DOI:10.1128/IAI.05152-11.
- C. Tanous, O. Soutourina, B. Raynal, M. F. Hullo, P. Mervelet, A. M. Gilles, P. Noirot, A. Danchin, P. England and I. M. Verstraete, J. Biol. Chem., 2008, 283(51), 35551–35560, DOI:10.1074/jbc.M805951200.
- E. J. Diner, C. M. Beck, J. S. Webb, D. A. Low and C. S. Hayes, Genes Dev., 2012, 26(5), 515–525, DOI:10.1101/gad.182345.111.
- M. Pieroni, G. Annunziato, C. Beato, R. Wouters, R. Benoni, B. Campanini, T. A. Pertinhez, S. Bettati, A. Mozzarelli and G. Costantino, J. Med. Chem., 2016, 59(6), 2567–2578, DOI:10.1021/acs.jmedchem.5b01775.
- G. Annunziato, M. Pieroni, R. Benoni, B. Campanini, T. A. Pertinhez, C. Pecchini, A. Bruno, J. Magalhães, S. Bettati, N. Franko, A. Mozzarelli and G. Costantino, J. Enzyme Inhib. Med. Chem., 2016, 31(sup4), 78–87, DOI:10.1080/14756366.2016.1218486.
- L. Amori, S. Katkevica, A. Bruno, B. Campanini, P. Felici, A. Mozzarelli and G. Costantino, MedChemComm, 2012, 3(9), 1111–1116, 10.1039/c2md20100c.
- J. Magalhães, G. Annunziato, N. Franko, M. Pieroni, B. Campanini, A. Bruno and G. Costantino, J. Chem. Inf. Model., 2018, 58(3), 710–723, DOI:10.1021/acs.jcim.7b00733.
- J. Magalhães, N. Franko, G. Annunziato, M. Welch, S. K. Dolan, A. Bruno, A. Mozzarelli, S. Armao, A. Jirgensons, M. Pieroni, G. Costantino and B. Campanini, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 1444–1452, DOI:10.1080/14756366.2018.1512596.
- J. Magalhães, N. Franko, G. Annunziato, M. Pieroni, R. Benoni, A. Nikitjuka, A. Mozzarelli, S. Bettati, A. Karawajczyk, A. Jirgensons, B. Campanini and G. Costantino, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 31–43, DOI:10.1080/14756366.2018.1518959.
- L. De Luca, S. Ferro, M. R. Buemi, A. M. Monforte, R. Gitto, T. Schirmeister, L. Maes, A. Rescifina and N. Micale, Chem. Biol. Drug Des., 2018, 92, 1585–1596, DOI:10.1111/cbdd.13326.
- N. Szałaj, L. Lu, A. Benediktsdottir, E. Zamaratski, S. Cao, G. Olanders, C. Hedgecock, A. Karlen, M. Erdelyid, D. Hughes, S. L. Mowbray and P. Brandt, Eur. J. Med. Chem., 2018, 157, 1346–1360, DOI:10.1016/j.ejmech.2018.08.086.
- S. T. Rutherford and B. L. Bassler, Cold Spring Harb. Perspect. Med., 2012, 2, 1–25, DOI:10.1101/cshperspect.a012427.
- B. L. Bassler and R. Losick, Cell, 2006, 125(2), 237–246, DOI:10.1016/j.cell.2006.04.001.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55(1), 165–199, DOI:10.1146/annurev.micro.55.1.165.
- K. Reuter, A. Steinbach and V. Helms, Med. Chem., 2016, 8, 1–15, DOI:10.4137/PMc.s13209.
- V. C. Kalia, Biotechnol. Adv., 2013, 31(2), 224–245, DOI:10.1016/j.biotechadv.2012.10.004.
- S. R. Kim and K. M. Yeon, Compr. Anal. Chem., 2018, 81, 57–94, DOI:10.1016/bs.coac.2018.03.010.
- R. G. Abisado, S. Benomar, J. R. Klaus, A. A. Dandekar and J. R. Chandler, MBio, 2018, 9(3), 1–13, DOI:10.1128/mBio.02331-17e02331-17.
- N. B. Turan, D. S. Chormey, Ç. Büyükpınar, G. O. Engin and S. Bakirdere, TrAC Trends Anal. Chem., 2017, 91, 1–11, DOI:10.1016/j.trac.2017.03.007.
- J. T. Hodgkinson, J. Gross, Y. R. Baker, D. R. Spring and M. Welch, Chem. Sci., 2016, 7(4), 2553–2562, 10.1039/c5sc04197j.
- K. Papenfortand and B. L. Bassler, Nat. Rev. Microbiol., 2016, 14(9), 576–588, DOI:10.1038/nrmicro.2016.89.
- J. Lee, J. Wu, Y. Deng, J. Wang, C. Wang, J. Wang, C. Chang, Y. Dong, P. Williams and L. H. Zhang, Nat. Chem. Biol., 2013, 9(5), 339–343, DOI:10.1038/nchembio.1225.
- L. C. M. Antunes, R. B. R. Ferreira, M. M. C. Buckner and B. B. Finlay, Microbiol, 2010, 156(8), 2271–2282, DOI:10.1099/mic.0.038794-0.
- Y. Dou, F. Song, F. Guo, Z. Zhou, C. Zhu, J. Xiang and J. Huan, Mol. Med. Rep., 2017, 15(6), 4061–4068, DOI:10.3892/mmr.2017.6528.
- Y. H. Li and X. Tian, Sensors, 2012, 12(3), 2519–2538, DOI:10.3390/s120302519.
- R. G. Contreras, Front. Microbiol., 2016, 7, 1–7, DOI:10.3389/fmicb.2016.01454.
- E. Rossi, A. Cimdins, P. Lüthje, A. Brauner, Å. Sjöling, P. Landini and U. Römling, Crit. Rev. Microbiol., 2017, 44(1), 1–30, DOI:10.1080/1040841x.2017.1303660.
- M. Walters and V. Sperandio, Int. J. Med. Microbiol., 2006, 296, 125–131, DOI:10.1016/j.ijmm.2006.01.041.
- J. Lee and L. Zhang, Protein Cell, 2015, 6(1), 26–41, DOI:10.1007/s13238-014-0100-x.
- V. K. Singh, A. Mishra and B. Jha, Front. Cell. Infect. Microbiol., 2017, 7, 337, DOI:10.3389/fcimb.2017.00337.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016, 59(13), 5929–5969, DOI:10.1021/acs.jmedchem.5b01698.
- W. R. J. D. Galloway, J. T. Hodgkinson, S. D. Bowden, M. Welch and D. R. Spring, Chem. Rev., 2011, 111, 28–67, DOI:10.1021/cr100109t.
- G. D. Geske, J. C. O'Neill and H. E. Blackwell, Chem. Soc. Rev., 2008, 37, 1432–1447, 10.1039/b703021p.
- I. C. Juárez, T. Maeda, E. A. M. Tinoco, M. Tomás, B. P. Eretza, S. J. G. Contreras, T. K. Wood and R. García-Contreras, World J. Clin. Cases, 2015, 3(7), 575–598, DOI:10.12998/wjcc.v3.i7.575.
- I. Dogsa, A. Oslizlo, P. Stefanic and I. M. Mulec, Food Technol. Biotechnol., 2014, 52(2), 149–157 Search PubMed.
- J. Li, J. Chen, J. E. Vidal and B. A. McClane, Infect. Immun., 2011, 79(6), 2451–2459, DOI:10.1128/iai.00169-11.
- M. K. Yadav, J. E. Vidal, Y. Y. Go, S. H. Kim, S. W. Chae and J. J. Song, Front. Cell. Infect. Microbiol., 2018, 8, 138, DOI:10.3389/fcimb.2018.00138.
- L. Zhou, Y. Zhang, Y. Ge, X. Zhu and J. Pan, Front. microbiol., 2020, 11, 2558, DOI:10.3389/fmicb.2020.589640.
- C. E. McInnis and H. E. Blackwell, Bioorg. Med. Chem., 2011, 19(16), 4820–4828, DOI:10.1016/j.bmc.2011.06.071.
- G. J. Jog, J. Igarashi and H. Suga, Chem. Biol., 2006, 13(2), 123–128, DOI:10.1016/j.chembiol.2005.12.013.
- T. Ishida, T. Ikeda, N. Takiguchi, A. Kuroda, H. Ohtake and J. Kato, Appl. Environ. Microbiol., 2007, 73(10), 3183–3188, DOI:10.1128/Aem.02233-06.
- K. M. Smith, Y. Bu and H. Suga, Chem. Biol., 2003, 10(6), 563–571, DOI:10.1016/S1074-5521(03)00107-8.
- C. Kim, J. Kim, H. Y. Park, J. H. Lee, H. J. Park, C. K. Kim and J. Yoon, Appl. Microbiol. Biotechnol., 2009, 83(6), 1095–1103, DOI:10.1007/s00253-009-1954-3.
- T. Persson, T. H. Hansen, T. B. Rasmussen, M. E. Skindersø, M. Givskov and J. Nielsen, J. Org. Biomol. Chem., 2005, 3(2), 253–262, 10.1039/B415761c.
- B. Morkunas, W. R. J. D. Galloway, M. Wright, B. M. Ibbeson, J. T. Hodgkinson, K. M. G. O'Connell, N. Bartolucci, M. D. Valle, M. Welch and D. R. Spring, Org. Biomol. Chem., 2012, 10(42), 8452–8464, 10.1039/c2ob26501j.
- J. T. Hodgkinson, W. R. J. D. Galloway, M. Wright, I. K. Mati, R. L. Nicholson, M. Welch and D. R. Spring, Org. Biomol. Chem., 2012, 10(30), 6032–6044, 10.1039/C2ob25198a.
- C. T. O'Loughlin, L. C. Miller, A. Siryaporn, K. Drescher, M. F. Semmelhack and B. L. Bassler, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(44), 17981–17986, DOI:10.1073/pnas.1316981110.
- G. D. Geske, Ra. J. Wezeman, A. P. Siegel and H. E. Blackwel, J. Am. Chem. Soc., 2005, 127(37), 12762–12763, DOI:10.1021/Ja0530321.
- G. D. Geske, J. C. O'Neill, D. M. Miller, M. E. Mattmann and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129(44), 13613–13625, DOI:10.1021/Ja074135h.
- G. D. Geske, M. E. Mattmann and H. E. Blackwell, Bioorg. Med. Chem. Lett., 2008, 18(22), 5978–5981, DOI:10.1016/j.bmcl.2008.07.089.
- D. M. Stacy, S. T. L. Quement, C. L. Hansen, J. W. Clausen, T. T. Nielsen, J. W. Brummond, M. Givskov, T. E. Nielsen and H. E. Blackwell, Org. Biomol. Chem., 2013, 11(6), 938–954, 10.1039/c2ob27155a.
- U. Müh, M. Schuster, R. Heim, A. Singh, E. R. Olson and E. Peter Greenberg, Antimicrob. Agents Chemother., 2006, 50(11), 3674–3679, DOI:10.1128/aac.00665-06.
- K. S. Musthafa, K. Balamurugan, S. K. Pandian and A. V. Ravi, J. Basic Microbiol., 2012, 52(6), 679–686, DOI:10.1002/jobm.201100292.
- M. Hentzer, H. Wu, J. B. Andersen, K. Riedel, T. B. Rasmussen, N. Bagge, N. Kumar, M. A. Schembri, Z. Song, P. Kristoffersen, M. Manefield, J. W. Costerton, S. Molin, L. Eberl, P. Steinberg, S. Kjelleberg, N. Hùiby and M. Givskov, Embo J, 2003, 22(15), 3803–3815, DOI:10.1093/emboj/cdg366.
- H. Wu, Z. Song, M. Hentzer, J. B. Andersen, S. Molin, M. Givskov and N. Høiby, J. Antimicrob. Chemother., 2004, 53(6), 1054–1061, DOI:10.1093/jac/dkh223.
- M. Starkey, F. Lepine, D. Maura, A. Bandyopadhaya, B. Lesic, J. He, T. Kitao, V. Righi, S. Milo, A. Tzika and L. Rahme, PLoS Pathog., 2014, 10(8), e1004321, DOI:10.1371/journal.ppat.1004321.
- A. Ilangovan, M. Fletcher, G. Rampioni, C. Pustelny, K. Rumbaugh, S. Heeb, M. Cámara, A. Truman, S. R. Chhabra, J. Emsley and P. Williams, PLoS Pathog., 2013, 9(7), e1003508, DOI:10.1371/journal.ppat.1003508.
- S. Srinivasarao, S. Nizalapur, T. T. Yu, D. S. Wenholz, P. Trivedi, B. Ghosh, K. Rangan, N. Kumar and K. V. G. C. Sekhar, Chemistry Select, 2018, 3(32), 9170–9180, DOI:10.1002/slct.201801622.
- E. Onem, Y. Dundar, S. Ulusoy, N. Noyanalpan and G. B. Tinaz, Fresenius Environ. Bull., 2018, 27(12 B), 9906–9912 CAS.
- N. S. El-Goharya, M. T. Gabrab and M. I. Shaabanc, Bioorg. Chem., 2019, 89, 102976, DOI:10.1016/j.bioorg.2019.102976.
- H. Liu, Q. Gong, C. Luo, Y. Liang, X. Kong, C. Wu, P. Feng, Q. Wang, H. Zhang and M. A. Wireko, Chem. Pharm. Bull., 2019, 67(10), 1088–1098, DOI:10.1248/cpb.c19-00359.
- S. Srinivasarao, A. Nandikolla, S. Nizalapur, T. T. Yu, S. Pulya, B. Ghosh, S. Murugesan, N. Kumar and K. V. G. C. Sekhar, RSC Adv., 2019, 9(50), 29273–29292, 10.1039/c9ra05059k.
- M. A. Hossain, N. Sattenapally, H. I. Parikh, W. Li, K. P. Rumbaugh and N. A. German, Eur. J. Med. Chem., 2020, 185, 111800, DOI:10.1016/j.ejmech.2019.111800.
- S. Sabir, S. Subramoni, T. Das, D. S. C. Black, S. A. Rice and N. Kumar, Molecules, 2020, 25(13), 3103, DOI:10.3390/molecules25133103.
- G. Yang, C. Cheng, G. B. Xu, L. Tang, K. L. Chua and Y. Y. Yang, Bioorg. Med. Chem., 2020, 28(16), 115606, DOI:10.1016/j.bmc.2020.115606.
- K. Jiang, X. Yan, J. Yu, Z. Xiao, H. Wu, M. Zhao, Y. Yue, X. Zhou, J. Xiao and F. Lin, Eur. J. Med. Chem., 2020, 194, 112252, DOI:10.1016/j.ejmech.2020.112252.
- R. Sathyanarayana, S. K. Bajire, B. Poojary, R. P. Shastry, V. Kumar and R. B. Chandrashekarappa, J. Iran. Chem. Soc., 2021, 18(5), 1051–1066, DOI:10.1007/s13738-020-02093-9.
- S. Mistry, S. Roy, N. J. Maitra, B. Kundu, A. Chanda, S. Datta and M. Joy, J. Control. Release., 2016, 239, 169–181, DOI:10.1016/j.jconrel.2016.08.014.
- C. D. l. F. Núñez, F. Reffuveille, E. F. Haney, S. K. Straus and R. E. W. Hancock, PLoS Pathog., 2014, 10, e1004152, DOI:10.1371/journal.ppat.1004152.
- F. Reffuveille, C. D. L. F. Núñez, S. Mansour and R. E. W. Hancock, Antimicrob. Agents Chemother., 2014, 58, 5363–5371, DOI:10.1128/aac.03163-14.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. H. Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755, DOI:10.1038/nrmicro.2017.99.
- C. d. l. F. Núñez, F. Reffuveille, L. Fernández and R. E. W. Hancock, Curr. Opin. Microbiol., 2013, 16, 580–589, DOI:10.1016/j.mib.2013.06.013.
- H. C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575, DOI:10.1038/nrmicro.2016.94.
- H. V. Acker, P. V. Dijck and T. Coenye, Trends Microbiol., 2014, 22, 326–333, DOI:10.1016/j.tim.2014.02.001.
- D. Lebeaux, J. M. Ghigo and C. Beloin, Microbiol. Mol. Biol. Rev., 2014, 78, 510–543, DOI:10.1128/mmbr.00013-14.
- A. H. Arshia, A. K. Khan, K. M. Khan, A. Ahmed, M. Taha and S. Perveen, Microb. Pathog, 2017, 110, 497–506, DOI:10.1016/j.micpath.2017.07.040.
- F. A. Kalam Khan, R. N. Kaduskar, R. Patil, R. H. Patil, S. Akber Ansari, H. M. Alkahtani, A. A. Almehizia, D. B. Shinde and J. N. Sangshetti, Bioorg. Med. Chem. Lett., 2019, 29(4), 623–630, DOI:10.1016/j.bmcl.2018.12.046.
- R. Gondru, K. Sirisha, S. Raj, S. K. Gunda, C. G. Kumar, M. Pasupuleti and R. Bavantula, Chemistry Select, 2018, 3(28), 8270–8276, DOI:10.1002/slct.20180139.
- A. Kotb, N. S. Abutaleb, M. A. Seleem, M. Hagras, H. Mohammad, A. Bayoumi, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 151, 110–120, DOI:10.1016/j.ejmech.2018.03.044.
- S. M. El-Messery, E. S. E. Habib, S. T. A. Al-Rashood and G. S. Hassan, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 818–832, DOI:10.1080/14756366.2018.1461855.
- C. Kong, C. F. Chee, K. Richter, N. Thomas, N. A. Rahman and S. Nathan, Sci. Rep., 2018, 8(1), 1–16, DOI:10.1038/s41598-018-21141-2.
- A. Kotb, N. S. Abutaleb, M. Hagras, A. Bayoumi, M. M. Moustafa, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, RSC Adv., 2019, 9(12), 6770–6778, 10.1039/c8ra10525a.
- A. F. Tuyun, M. Yıldız, N. Bayrak, H. Yıldırım, E. M. Kara, A. T. Jannuzzi and B. O. Celik, Drug Dev. Res., 2019, 80(8), 1098–1109, DOI:10.1002/ddr.21591.
- S. G. Mane, K. S. Katagi, P. Bhasme, S. Pattar, Q. Wei and S. D. Joshi, Chem. Data Collect., 2019, 20, 100198, DOI:10.1016/j.cdc.2019.100198.
- S. Su, P. Yin, J. Li, G. Chen, Y. Wang, D. Qu, Z. Li, X. Xue, X. Luo and M. Li, J. Infect. Public Health, 2020, 13(5), 791–799, DOI:10.1016/j.jiph.2019.10.010.
- R. Gondru, S. Kanugala, S. Raj, C. G. Kumar, M. Pasupuleti, J. Banothu and R. Bavantula, Bioorg. Med. Chem. Lett., 2021, 33, 127746, DOI:10.1016/j.bmcl.2020.127746.
- B. Parrinoa, D. Carbonea, S. Cascioferro, C. Pecoraro, E. Giovannetti, D. Deng, V. D. Sarno, S. Musella, G. Auriemma, M. Grazi, M. G. Cusimano, D. Schillaci, G. Cirrincione and P. Diana, Eur. J. Med. Chem., 2021, 209, 112892, DOI:10.1016/j.ejmech.2020.112892.
- T. El Malah, H. A. Soliman, B. A. Hemdan, R. E. Abdel Mageid and H. F. Nour, New J. Chem., 2021, 45, 10822–10830, 10.1039/d1nj00773d.
- P. Paul, P. Chakraborty, A. Chatterjee, R. K. Sarker, D. G. Dastidar, T. Kundu, N. Sarkar, A. Das and P. Tribedi, Arch. Microbiol., 2021, 203(3), 1183–1193, DOI:10.1007/s00203-020-02117-1.
| This journal is © The Royal Society of Chemistry 2021 |