
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
8,8′-(Benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(quinolin-4(1H)-one): a twisted photosensitizer with AIE properties†
Emmanouil Broumidis a,
Callum M. S. Jonesb,
Maria Koyionic,
Andreas Kourtellarisc,
Gareth O. Lloydd,
Jose Marques-Huesob,
Panayiotis A. Koutentis*c and
Filipe Vilela*a
a,
Callum M. S. Jonesb,
Maria Koyionic,
Andreas Kourtellarisc,
Gareth O. Lloydd,
Jose Marques-Huesob,
Panayiotis A. Koutentis*c and
Filipe Vilela*a
aInstitute of Chemical Sciences, School of Engineering & Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: F.Vilela@hw.ac.uk
bInstitute of Sensors, Signals and Systems, School of Engineering & Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
cDepartment of Chemistry, University of Cyprus, P.O. Box 20537, 1678 Nicosia, Cyprus
dJoseph Banks Laboratories, School of Chemistry, University of Lincoln, Brayford Pool, Lincoln, LN6 7TS, UK
First published on 1st September 2021
Abstract
A new benzothiadiazole (BTZ) luminogen is prepared via the Suzuki–Miyaura Pd-catalysed C–C cross-coupling of 8-iodoquinolin-4(1H)-one and a BTZ bispinacol boronic ester. The rapid reaction (5 min) affords the air-, thermo-, and photostable product in 97% yield as a yellow precipitate that can be isolated by filtration. The luminogen exhibits aggregated-induced emission (AIE) properties, which are attributed to its photoactive BTZ core and nonplanar geometry. It also behaves as a molecular heterogeneous photosensitizer for the production of singlet oxygen under continuous flow conditions.
Introduction
Photoluminescent materials such as organic dyes and fluorescent polymers have multiple applications in areas such as chemical sensing, organic electronics and bioimaging.1 Up until the dawn of the 21st century, most organic luminescent materials were typically large, planar and highly conjugated systems. Unsurprisingly, these materials suffer from aggregation-caused quenching (ACQ) effects, owing to solid state face-to-face π stacking interactions, that fully or partially suppress the photoluminescent (PL) properties and thus, the intended functionality of these materials.2In 2001, Tang and coworkers3 introduced a silole-based organic material which exhibited the opposite behaviour of known ACQ luminogens. Specifically, it was weakly emitting in solution but a strong emitter in the solid state. This unusual photophysical phenomenon is known as aggregation-induced emission (AIE), and there have been extensive efforts to uncover its underlying mechanism of action, as well as develop new photoactive materials with AIE properties.4 Most AIE active materials (AIEgens) have rigid, nonplanar geometries in contrast to the planar ACQ based materials. In solution, an AIEgen is subject to intermolecular rotation, vibration or stretching and, as such, any emissions occur mostly via non-radiative processes. In contrast, when in an aggregated or solid state, owing to their rigid nonplanar geometry, π–π stacking and intermolecular motions are limited. This leads to a significant enhancement of the corresponding photoluminescence quantum yield (PLQY), as radiative processes become more prevalent.5 While other mechanistic pathways exist, such as the twisted intramolecular charge transfer process (TICT)6 or crystallisation-induced emission enhancement (CIEE),7 the aforementioned restriction of intramolecular motion (RIM) process is dominant for most AIEgen materials.4 Even though in recent years there have been significant advancements in the development of new AIEgen materials, particularly in the organic light-emitting diode (OLED) industry,8 some of the challenges that remain include the need for tuneable and multifunctional AIEgens with properties such as broad excitation wavelength range, increased biocompatibility, and ability of generating reactive oxygen species (ROS).5 These characteristics are especially useful for synthesising AIEgens which can be used as probes for the elucidation of antibiotic mechanisms of action,9 or as potent agents for photodynamic therapy (PDT).10
One of the luminophores that has attracted the attention of researchers in recent years is the S,N-heterocycle 2,1,3-benzothiadiazole (BTZ). BTZ and its derivatives have an appealing set of properties which can make them useful building blocks for developing new photoactive materials.11–13 These include their biocompatibility, and their strong electron-withdrawing character that makes them useful partners in donor (D)–acceptor (A) or D–A–D photoactive systems as well as in organic semiconductor materials such as photovoltaics and OLEDs.14 As part of our work on new molecular and polymeric BTZ containing photoactive materials,15–18 we have developed a high yielding, scalable and rapid synthesis of a new quinolin-4-ol incorporating a central BTZ unit. Gratifyingly, this material has a helical, nonplanar geometry, and exhibits AIE and ROS generation properties.
Results and discussion
Synthesis and characterisation
The chemistry used to access the starting material, the antimicrobial core19 8-iodoquinolin-4(1H)-one (2), was previously developed by Koutentis and coworkers to gain access to the natural product derivative deazacanthin-4-one 3 (Scheme 1).20,21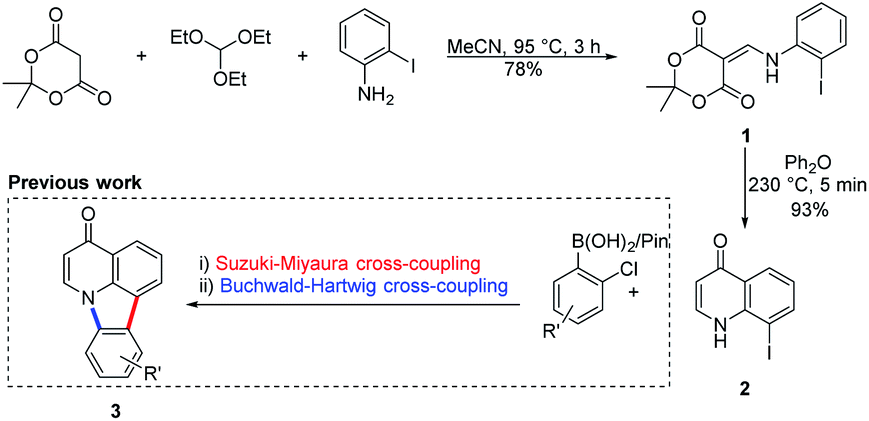 | ||
| Scheme 1 Synthetic pathway for and use of 8-iodoquinolin-4(1H)-one 2 to prepare deazacanthin-4-one 3. | ||
Heating an equimolar mixture of triethylorthoformate, Meldrum's acid and 2-iodoaniline in MeCN under reflux conditions for 3 h, gives 5-{[(2-iodophenyl)amino]methylene}-2,2-dimethyl-1,3-dioxane-4,6-dione (1), thermolysis of which affords 8-iodoquinolin-4(1H)-one (2).17 In our prior work, quinolone 2 was reacted with 2-chloroarylboronic acids or pinacol esters in a two-step, C–C and C–N bond forming process to afford the desired deazacanthin-4-ones 3.21 Herein, quinolone 2 was reacted with 4,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[c][1,2,5]thiadiazole [BTZ(Bpin)2] (4) to give the desired 8,8′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(quinolin-4-ol) (5) in up to 97% yield (Scheme 2). Product 5 precipitated from the reaction mixture and, following a simple filtration and subsequent washes with THF and EtOH, was collected as a yellow semi-crystalline powder without the need for further purification, as determined by LC-MS (ESI, S2.5 and S2.6†). Compound 5 exhibited very poor solubility in both polar and non-polar solvents (ESI, S2.7†); hot DMSO only sparingly dissolved the compound and was not suitable for obtaining satisfactory 13C NMR data. Fortunately, compound 5 dissolved in trifluoroacetic acid (TFA), forming the salt 5H22+ 2TFA− (referred to as 5H22+). X-ray quality single crystals were obtained from a hot PhCl/TFA solution that was left to slowly cool at room temperature over several days. The asymmetric unit cell was P21/n monoclinic and revealed the formation of [5H22+ 2TFA−]·2TFA. This structure represents the pyridinium dication 5H22+ co-crystallised with two molecules of TFA and two TFA anions (Fig. 1). In addition, the C–O bond lengths (dC–O = C24–O2 1.319(3) Å and C9–O1 1.326(3) Å) suggest that, in this protonated form, compound 5 exists in the enol tautomer instead as the keto tautomer, which was the case with product 3. TD-DFT calculations at the B3LYP/6-311G(d,p) level of theory confirmed this (ESI, S2.3†), as the calculated UV-vis absorption of the enol tautomer of the neutral molecule and the pyridinium form in the presence of coordinated anionic and neutral TFA molecules is in good agreement with the experimental UV-vis absorption in TFA as solvent (ESI, S2.3†). The crystal structure showed that the quinolinols deviate from the plane of the BTZ and the torsion angle between the planes defined by each quinolinol and the BTZ is 126.11(17)°. Support for this propeller-type geometry in solution came from the NOE 2D NMR spectrum (ESI, S3†) of product 5H22+, which supported a nonplanar geometry, as there was no interaction between the BTZ and quinolinol protons, showing that the two quinolinol moieties must be nearly orthogonal with respect to each other, from a top-bottom perspective. Both neutral and protonated compounds were thermally stable up to at least 400 °C as revealed by TGA analysis (ESI, S2.1†).
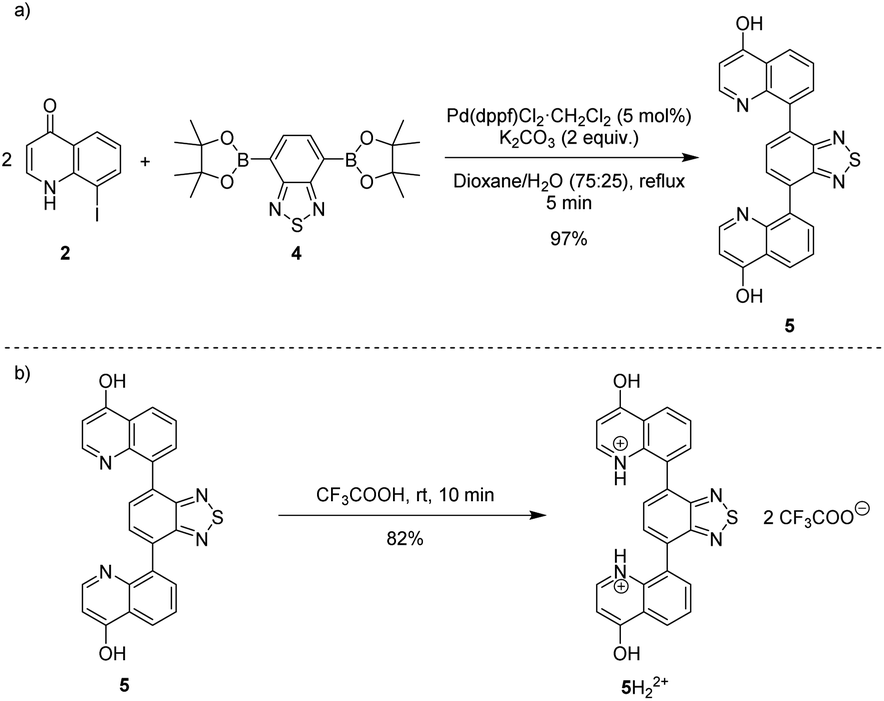 | ||
| Scheme 2 (a) Suzuki–Miyaura reaction of 8-iodoquinolin-4(1H)-one (2) with BTZ(Bpin)2 4 to afford 8,8'-(benzo[c][1,2,5]thiadiazole-4,7-diyl)bis(quinolin-4-ol) (5). (b) Synthesis of 5H22+ from 5. | ||
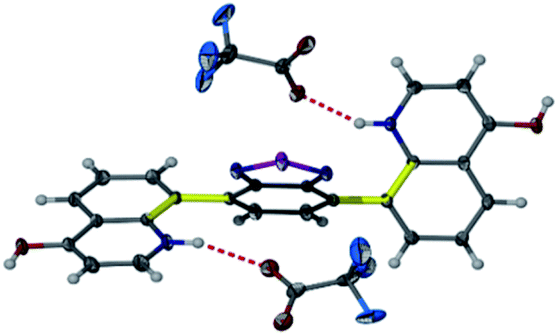 | ||
| Fig. 1 Structure of 5H22+ shown as 50% probability ellipsoids. Hydrogen bonding of the TFA molecules to 5H22+ highlights the protonation and enol tautomer of 5. Disorder of one of the triflate and both TFA molecules in the asymmetric unit are not shown for clarity. Yellow and thicker bonds indicate the torsion angle of 126.11(17)°, which represents the rotation of the two quinolinol groups to opposite sides of the BTZ. | ||
Photochemical studies
As mentioned above, the presence of the chromophore BTZ core in compound 5, prompted an investigation of its ability to act as a photosensitiser for reactive oxygen species (ROS) generation. More specifically, we decided to focus on the production of singlet oxygen (1O2), as it has a high synthetic utility22 and it is believed to be the main damaging agent in photodynamic therapy (PDT) applications.23 The formation of ascaridole via a Diels–Alder reaction between singlet oxygen (1O2) and α-terpinene was undertaken to evaluate the ability of compound 5 to act as a photosensitiser for the generation of 1O2.15 Owing to the low solubility of compound 5 (ESI, 2.7†), we carried out the reaction heterogeneously by using continuous-flow technology. This alternative enabling technology is an effective method for use with heterogeneous photosensitisers, and can offer better results compared to batch experiments, as it was recently demonstrated by our group.24 For this work, 1 mol% of compound 5 (0.0025 mmol) was mixed with 500 mg of silica (solid support). The mixture was then added into a short column reactor and was ready for use. To take full advantage of the flow instrumentation capabilities, we connected an O2 gas line to the set-up and the operating overall system pressure was set at 2 bar, allowing more O2 to be dissolved in solution.Moreover, a 60 MHz Nanalysis-60e benchtop 1H NMR instrument was connected to the set-up, which allowed us to monitor the reaction progress in real time (Fig. 2). To make the 1H NMR data acquisition possible, CDCl3 was used as the solvent, which also offers significantly increased lifetime of 1O2 in solution compared to non-deuterated and non-chlorinated solvents.25 While compound 5 absorbs mostly in the long UV region of the spectrum (ESI, S1.4†), we used an LED emitting at 390–440 nm to test whether its absorbance at 400–420 nm was enough to produce its excited state. Gratifyingly, when the reaction was tested using the aforementioned parameters, in-line 1H NMR spectroscopy indicated that after 1500 s (25 min) the reaction was complete.
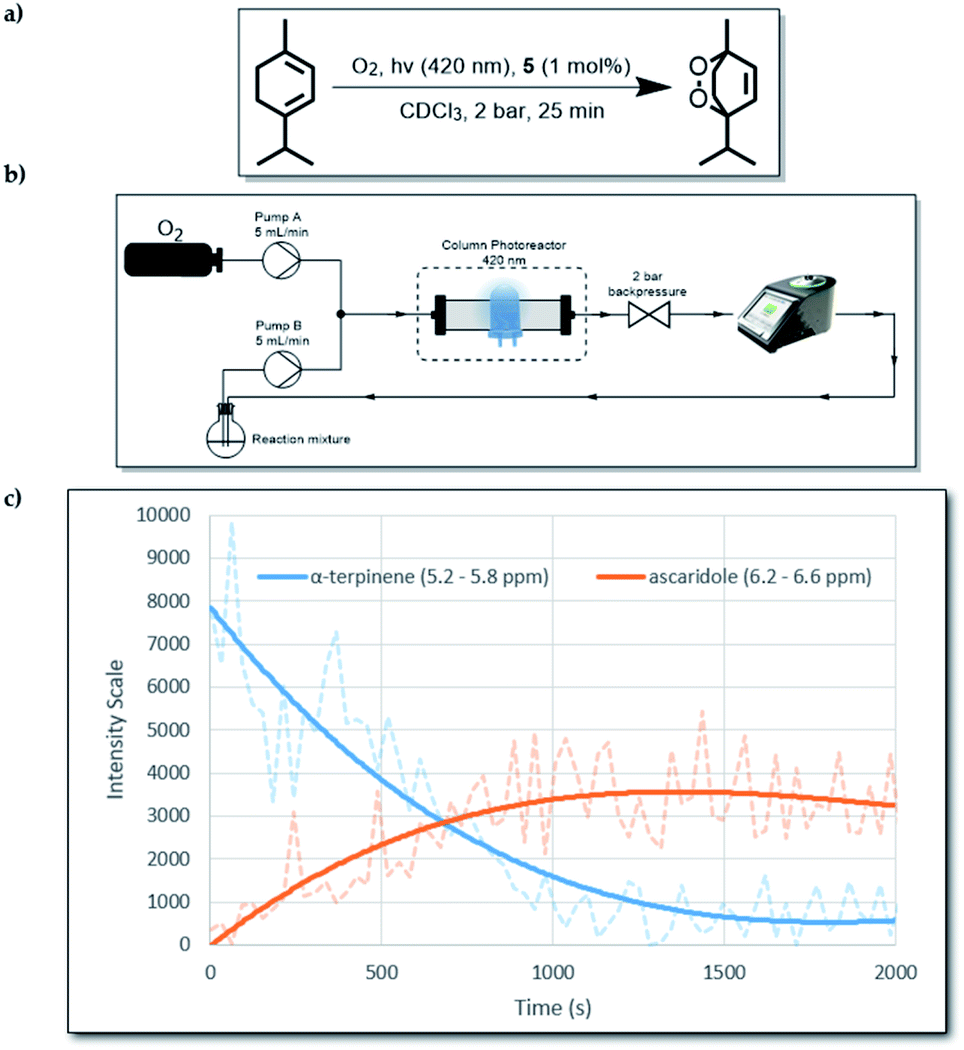 | ||
| Fig. 2 (a) The overall reaction from α-terpinene to ascaridole. (b) Schematic diagram of the flow set-up. (c) In-line 1H NMR data for the first run of the reaction. | ||
Since α-terpinene reacts selectively with 1O2,18 by calculating its conversion to ascaridole, the amount of generated 1O2 can be estimated. For the optimised reaction conditions above, this was found to be 0.54 mmol h−1 per mg of 5. This performance is comparable with previously described BTZ-based heterogeneous photocatalysts that were used under continuous flow conditions (Table 1). Even though its performance is not particularly outstanding, what makes compound 5 stand out is that it is the first example of a molecular based BTZ heterogeneous photocatalyst that has been used for 1O2 production. A comparison between the 1O2 productivities of compounds 5 and 5H22+ was also made, to test if they behave differently under identical conditions, however there was negligible difference in their performance (ESI, S2.8†).
| Entry | Photosensitiser characteristics | Reactor type | 1O2 productivitya (mmol h−1) |
|---|---|---|---|
| a Per mg of photosensitiser. | |||
| 1 (ref. 16) | Conjugated microporous BTZ polymer | Coil | 1.44 |
| 2 (ref. 18) | High internal phase emulsion BTZ polymer | Column | 0.15 |
| 3 (ref. 15) | High internal phase emulsion BTZ polymer | Column | 0.05 |
| 4 (ref. 15) | Polymer supported BTZ (microbeads) | Coil | 4.04 |
| 5-Current work | Molecular BTZ, SiO2 supported | Column | 0.54 |
To test the durability of compound 5 as a photosensitiser, we repeated the same reaction four more times without replacing the original packed column. By 1H NMR analysis, we observed that even after 5 cycles, there was quantitative consumption of α-terpinene (0.25 mmol per cycle) without any apparent photo-bleaching of 5, judging by its consistent performance. This behaviour highlights the excellent stability of 5 against 1O2. The graph (Fig. 2c) follows signal intensity for two specific spectral regions which correspond to α-terpinene and ascaridole. The actual real time data are represented by the faded dashed lines, which were used to generate a moving average trend line. The noise in the raw data is partly attributed to the flow of O2 bubbles through the bench-top 1H NMR instrument, which lowered the signal quality. Nevertheless, the signal was accurate enough to provide real-time qualitative information about the reaction progression. To confirm reaction completion, after each run the content of the collection flask was isolated by removing the solvent under reduced pressure and 1H NMR spectra using a 300 MHz instrument were obtained (ESI, S2.5†). It was also observed that when alumina was used instead of silica as a solid support, poor adsorption of compound 5 resulted in gradual leaching of solids into the reaction mixture (ESI, S2.5†).
To prove that the reaction was catalysed heterogeneously and not due to low concentrations of soluble 5, a hot suspension of compound 5 (1 mol%) in CDCl3 was stirred for 30 min, and the solids were then removed by filtration. To the resulting solution was added α-terpinene (0.25 mmol). After irradiating the solution, no ascaridole was detected, proving that luminogen 5 acts as a true molecular heterogeneous photocatalyst (ESI, S2.5†).
Finally, as we have shown that 5 can produce a stable excited triplet state for the subsequent type II energy transfer to occur, we wanted to assess whether it can also participate in type I photosensitised reactions, to produce other types of ROS, such superoxide (O2˙−). As such, we used compound 5 as a photoredox catalyst for the aerobic hydroxylation of phenylboronic acid to phenol. This reaction has been shown to be mediated by O2˙−,26 and when 1 mol% of 5 was used, there was 36% conversion to phenol, as measured by HPLC (ESI, S2.9†).
Photophysical studies
Due to the insolubility of compound 5 in organic solvents, its photoluminescence behaviour was only studied in the solid (semi-crystalline) state. A broad green emission at 512 nm was observed (ESI, S2.4†). The protonated TFA salt 5H22+ was slightly more soluble in polar organic solvents such as DMSO and thus, it was used to make dilute solutions in different ratios of DMSO/water and TFA/water (water fraction, fw ranged from 0 to 90%) and their fluorescence spectra were recorded (Fig. 3a and b). At 100% DMSO (Fig. 3d), the emission spectrum was weak, containing two peaks, a sharp one at 428 nm which can be attributed to the singlet excited state and a broad one at 446 nm. The rotatable, D–A bond between the quinolinol and BTZ moieties indicated that this broad peak may be the result of intramolecular charge transfer (ICT) or possibly twisted intramolecular charge transfer (TICT).27 TD-DFT calculations showed that in the molecular orbitals associated with the vertical excitations responsible for the absorption profile of 5H22+ the HOMO to LUMO transition has significant charge transfer character from the quinolinol moieties to the BTZ core (ESI, S2.3†), which further strengthens the case of ICT emission. A third, redshifted broad peak at 574 nm is believed to originate from an excimer, which forms a broad UV-vis absorption band at 460 nm with increasing concentrations (ESI, S2.4†). When fw was increased to 10% a fine precipitate started to appear with concomitant disappearance of the excimer peak, bathochromic and hyperchromic transformation of the ICT peak, and appearance of a new, broad peak, which continued to increase in intensity until fw reached 40% (Fig. 3a). At this point, there was a 24-fold increase in emission intensity and 10-fold increase in photoluminescence quantum yield (PLQY), which reached 8.4%. The peak continued to redshift until 490 nm when fw reached 50%. This can be attributed to ICT solvatochromism28 and shows that both AIE and ICT effects are active initially. Between fw 50–90% the emission decreased, a rarely observed behaviour seen in other AIE systems which has been ascribed to changes in the crystallisation processes and available surface area of the nanoaggregates, as more water is added.29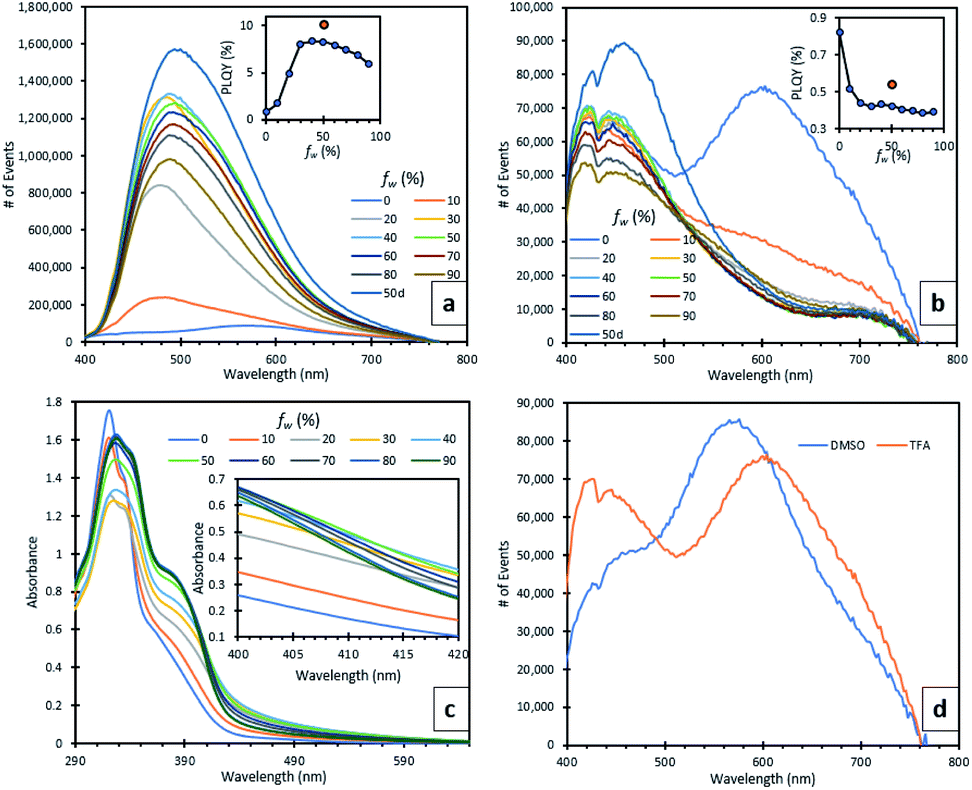 | ||
| Fig. 3 (a) DMSO/H2O series emission spectra. Inset shows relation between fw and PLQY (%). (b) TFA/H2O series emission spectrum. Inset shows relation between fw and PLQY (%). (c) DMSO/H2O series absorption spectrum. Inset shows an enlarged section between 400 nm and 420 nm. (d) Comparison of emissions between DMSO and TFA, while fw was 0. C = 2.05 × 10−4 M. Excitation wavelength was 320 nm. | ||
The UV-vis absorption spectra of the DMSO/water series show two new shoulders appearing at 371 and 410 nm (Fig. 3c). The latter follows the same behaviour as the emission spectra, showing a maximum absorbance when fw is 40% and decreasing thereafter. To verify the relationship of this absorption shoulder with the formation of nanoaggregates, we used varying excitation wavelengths spanning from 320 nm (λabs,max of 5H22+) to 430 nm to monitor the AIE emission peak at 478 nm. When λexc was 410 nm, the λem intensity was maximised. When TFA was used instead of DMSO, no aggregation was observed until fw reached 80%, confirmed by the corresponding UV-vis absorption spectra (ESI, S2.4†), showing a significant level-off tail above 400 nm, which is due to the light scattering caused by the nanoaggregate suspension.3 In spite of this, no AIE peak emerged, which can be explained by TFAs ability to form strong intermolecular H-bonds between the phenolic and pyridinium moieties of compound 5H22+, as seen by the crystal structure. These H-bonds can in turn induce non-radiative pathways, by competing with the radiative ICT processes between the BTZ and quinolinol moieties.30 To further demonstrate this point, when D2O was used instead of H2O for the solutions with fw of 50%, their fluorescence was enhanced, increasing their PLQYs for the DMSO and TFA solutions by 22 and 29%, respectively. A striking difference was seen in the TFA spectra, where the weaker ICT/AIE peak became more intense than the monomeric peak while also getting broader and bathochromically shifted to 462 nm (Fig. 3b). This effect is associated to the inferior quenching abilities of deuterium,31 and strengthens the case of H-bonding having an important role in the quenching of the AIE peak in the TFA solution. Finally, an aerated TFA/D2O (fw = 50%) solution exhibited a weak emission at 1274 nm (ESI, S2.4†), caused by 1O2 phosphorescence, which directly supported its ability to generate 1O2, even in solution.32
Conclusions
In summary, we have discovered a robust and easily accessible D–A–D chromophore with a twisted geometry. Its high insolubility allowed us to use it as a very effective molecular heterogeneous photosensitiser for 1O2 production under continuous flow conditions. In addition, we explored the photophysical properties of its TFA salt, which revealed a tuneable dual emission between a redshifted excimer and a combination of ICT/AIE which was controlled by adjusting the water fraction. When TFA was used as a solvent the AIE emission was turned off due to increased H-bonding. This study acts as a gateway for further investigation of this unique class of quinolinol-BTZ AIEgen triads, and we think the combination of their facile synthesis, tunability, heterogeneous 1O2 generating capabilities and potential antimicrobial properties make them attractive targets for future research.Author contributions
Synthesis, photochemical and photophysical investigations, E. B.; PLQY measurements and instrumentation access, C. J, J. M-H.; computational studies and SC-XRD data interpretation, M. K.; acquisition of MS data and SC-XRD data interpretation, G. O. L.; acquisition of SC-XRD data, A. K.; writing and draft presentation, E. B, M. K, G. O. L, P. A. K, and F. V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. B. would like to thank the CRITICAT Centre for Doctoral Training (EP/L016419/1) for funding. C. J and J. M-H would like to thank the Engineering and Physical Sciences Research Council (EP/T013680/1) for funding.References
- L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS PubMed.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2016, 93, 345–350 CrossRef CAS.
- J. Luo, Z. Xie, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 1, 40–42 RSC.
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1–8 Search PubMed.
- N. N. Li, J. Z. Li, P. Liu, D. Pranantyo, L. Luo, J. C. Chen, E. T. Kang, X. F. Hu, C. M. Li and L. Q. Xu, Chem. Commun., 2017, 53, 3315–3318 RSC.
- N. Alifu, X. Dong, D. Li, X. Sun, A. Zebibula, D. Zhang, G. Zhang and J. Qian, Mater. Chem. Front., 2017, 1, 1746–1753 RSC.
- J. L. Westrup, L. W. Oenning, M. M. Da Silva Paula, R. Da Costa Duarte, F. S. Rodembusch, T. E. A. Frizon, L. Da Silva and A. G. Dal-Bó, Dyes Pigm., 2016, 126, 209–217 CrossRef CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- O. A. Rakitin, Synth, 2019, 51, 4338–4347 CrossRef CAS.
- B. A. D. Neto, A. A. M. Lapis, E. N. Da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- J. M. Tobin, T. J. D. McCabe, A. W. Prentice, S. Holzer, G. O. Lloyd, M. J. Paterson, V. Arrighi, P. A. G. Cormack and F. Vilela, ACS Catal., 2017, 7, 4602–4612 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, Medchemcomm, 2019, 10, 1719–1739 RSC.
- H. A. Ioannidou, A. Martin, A. Gollner and P. A. Koutentis, J. Org. Chem., 2011, 76, 5113–5122 CrossRef CAS PubMed.
- E. Broumidis and P. A. Koutentis, Tetrahedron Lett., 2017, 58, 2661–2664 CrossRef CAS.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- L. Benov, Med. Princ. Pract., 2015, 24, 14–28 CrossRef PubMed.
- C. G. Thomson, C. M. S. Jones, G. Rosair, D. Ellis, J. Marques-Hueso, A. L. Lee and F. Vilela, J. Flow Chem., 2020, 10, 327–345 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- Y. Q. Zou, J. R. Chen, X. P. Liu, L. Q. Lu, R. L. Davis, K. A. Jørgensen and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC.
- M. Zhang, J. Li, L. Yu, X. Wang and M. Bai, RSC Adv., 2020, 10, 14520–14524 RSC.
- M. Cai, Z. Gao, X. Zhou, X. Wang, S. Chen, Y. Zhao, Y. Qian, N. Shi, B. Mi, L. Xie and W. Huang, Phys. Chem. Chem. Phys., 2012, 14, 5289–5296 RSC.
- S. Xie, S. Manuguri, O. Ramström and M. Yan, Chem. – Asian J., 2019, 14, 910–916 CrossRef CAS PubMed.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Chem. Sci., 2021, 12, 1352–1362 RSC.
- J. Baier, T. Fuß, C. Pöllmann, C. Wiesmann, K. Pindl, R. Engl, D. Baumer, M. Maier, M. Landthaler and W. Bäumler, J. Photochem. Photobiol., B, 2007, 87, 163–173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, analytical, computational, photophysical and photochemical data. CCDC 2086024. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06263h |
| This journal is © The Royal Society of Chemistry 2021 |