DOI:
10.1039/D1RA06452E
(Paper)
RSC Adv., 2021,
11, 38555-38564
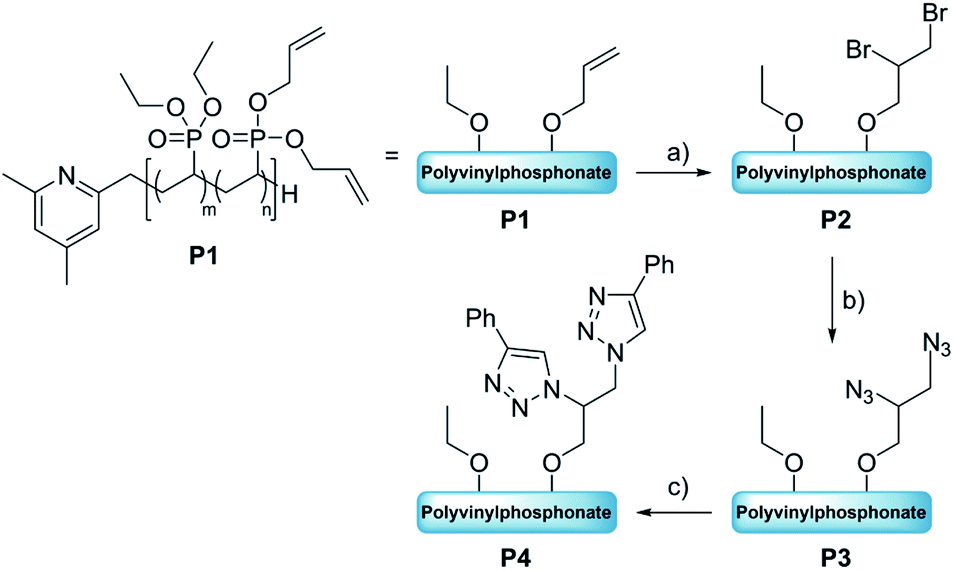
Allyl group-containing polyvinylphosphonates as a flexible platform for the selective introduction of functional groups via polymer-analogous transformations†
Received
26th August 2021
, Accepted 23rd November 2021
First published on 30th November 2021
Abstract
Polyvinylphosphonates are highly promising candidates for (bio)medical applications as they exhibit a tunable lower critical solution temperature, high biocompatibility of homo- and copolymers, and a broad foundation for post-synthetic modifications. In this work we explored polymer-analogous transformations with statistical polyvinylphosphonates comprising diethyl vinylphosphonate (DEVP) and diallyl vinylphosphonate (DAlVP). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds were used as a starting point for a cascade of organic transformations. Initially, the reactive moieties were successfully introduced via bromination, epoxidations with OXONE and mCPBA, or thiol–ene click chemistry with methyl thioglycolate (6). The obtained substrates were then employed in a variety of consecutive reactions depending on the introduced functional motif: (1) the brominated substrates were converted with sodium azide to enable the copper-mediated alkyne–azide coupling with phenylacetylene (1). (2) The epoxides were reacted with sodium azide for an alkyne–azide click coupling with 1 as well as small nucleophilic compounds (phenol (2), benzylamine (3), and 4-amino-2,1,3-benzothiadiazol (4)). Afterwards the non-converted allyl groups were reacted with thiochloesterol (5) to form complex polymer conjugates. (3) An acid-labile hydrazone-linked conjugate was formed in a two-step approach. The polymeric substrates were characterized by NMR, FTIR, and UV/Vis spectroscopy as well as elemental analysis and gel permeation chromatography to monitor the structural changes of the polymeric substrates and to prove the success of these modification approaches.
C double bonds were used as a starting point for a cascade of organic transformations. Initially, the reactive moieties were successfully introduced via bromination, epoxidations with OXONE and mCPBA, or thiol–ene click chemistry with methyl thioglycolate (6). The obtained substrates were then employed in a variety of consecutive reactions depending on the introduced functional motif: (1) the brominated substrates were converted with sodium azide to enable the copper-mediated alkyne–azide coupling with phenylacetylene (1). (2) The epoxides were reacted with sodium azide for an alkyne–azide click coupling with 1 as well as small nucleophilic compounds (phenol (2), benzylamine (3), and 4-amino-2,1,3-benzothiadiazol (4)). Afterwards the non-converted allyl groups were reacted with thiochloesterol (5) to form complex polymer conjugates. (3) An acid-labile hydrazone-linked conjugate was formed in a two-step approach. The polymeric substrates were characterized by NMR, FTIR, and UV/Vis spectroscopy as well as elemental analysis and gel permeation chromatography to monitor the structural changes of the polymeric substrates and to prove the success of these modification approaches.
Introduction
The history of polymer-analogous reactions reaches back to the 19th century, but their actual use was first established by H. Staudinger in 1939.1 By definition these reactions involve a part of the polymer, but while the degree of polymerization does not change, the molecular weight does not remain necessarily constant throughout the reaction.2 These reactions differ fundamentally from organic transformations with small molecules. As a result, purification processes are more complex, kinetic parameters can be affected, or the polymer solubility can change during the reaction.2 Prominent examples are the formation of poly(vinyl alcohol) (PVA) via saponification of poly(vinyl acetate)3 or the synthesis of linear polyethylenimine.4,5 In addition, polymer-analogous reactions are also used to form derivatives of cellulose and starch or to refine the material properties of polyolefins.6–8
Such organic transformations also allow the application of modified polymers in biomedical fields, i.e. for (bio)imaging through the conjugation of fluorescent probes,9 or radiolabeling of polymers for positron emission tomography,10 which allows advances in cancer diagnostics and monitoring of the therapeutic progress. Yet, the best known are polymer–drug conjugates, which were conceptualized by H. Ringsdorf in 1975.11,12 This rational design combines a water-soluble polymer backbone that is covalently linked to the targeting moiety and the (pro)drug motif.12 This concept was extensively used for the synthesis of a plethora of linear polymer conjugates. A broad variety of polymers was investigated ranging from poly(vinyl pyrrolidine),13,14 PVA,15 poly(ethylene glycol) (PEG),16 which is already clinically established due to its high biocompatibility,17 and N-(2-hydoxypropyl)methyl-acrylamide (HPMA).18 In the case of PEG reactive chain end-groups allow a simple and fast conjugation of small molecules. However, this approach also results in low drug loadings.19,20 Hence, branched and multi-arm PEGs were developed to overcome this limitation.21–23 In this context, dendrimeric systems are also of great interest, since the synthesis of these types of polymers can be performed with high precision and provide a high density of functional groups at the surface, thus enabling the conjugation of drugs, targeting moieties and imaging agents.12,24–26 In recent years, researchers also focused on the application of stimuli-responsive polymers, which respond to changes of external stimuli such as pH or temperatures.12 In exemplary studies doxorubicin was either linked to a HPMA-based polymer27 or to poly(ethylene oxide)28 via a pH-sensitive hydrazone linkage. In another study thermoresponsive elastin-like polypeptides were labeled with derivatives of rhodamine and were able to show an increased accumulation of polymer aggregates within the tumor tissue by exploiting hyperthermia conditions.9 The class of polyvinylphosphonates offers interesting properties for biomedical applications, e.g. a high biocompatibility29–32 and tunable cloud points between 5–92 °C.33–35 The successful synthesis of defined, high-molecular weight poly(diethyl vinylphosphonate) (PDEVP) was performed via rare earth metal-mediated group transfer polymerization (REM-GTP) for the first time in 2010 with Cp2YbMe.36 Coming from a simple homopolymerization we constantly aimed to push the boundaries of the REM-GTP by expanding the monomer scope towards new functional monomers and (co)polymer and architectures. Initial studies with these polymers also included polymer-analogous reactions such as the saponification and the transesterification of the phosphonate esters. Moreover, REM-GTP facilitated the efficient incorporation of tailor-made end-groups in these polymers at the initial step of the polymerisation.29,37–39 In this context a bipyridine-based initiator enabled the selective complexation of Re(CO)5Cl in a copolymer comprising 2-vinylpyridine (2VP) and diethyl vinylphosphonate (DEVP) for the photoreduction of carbon dioxide.40 An initiator consisting of a vinyl group allowed the precise formation of polymer–biomolecule conjugates with cholesterol and folic acid.29 Both polymer conjugates were also successfully used in localization studies with HMEC-1 cells.41 The detection of the conjugates by confocal microscopy required the presence of a fluorescent moiety and was achieved by the introduction of pyrene groups via a partial transesterification of the ethyl esters. In addition, block copolymers of 2VP, DEVP, and diallyl vinylphosphonate (DAlVP) were used in the formation of nanoparticular drug carriers.31 Introduction of this small DAlVP block in the outer shell facilitated the covalent cross-linking via thiol–ene click chemistry.
As shown in the literature allyl moieties represent flexible anchors to selectively incorporate biological moieties.42 In addition, DAlVP-containing polymers are advantageous as two ester per monomer unit can double the loading capacity compared to other copolymer systems (polyethers28,43 or polylactides44). Additionally, the water-solubility can be easily tuned by the DEVP amount. Therefore, no solubilizing motifs such as PEG or zwitterions are required.44–46 In combination with their intrinsic biocompatibility and a polymerization technique, which enables predictable and reproducible polymerization results, these copolymer types emerge as model candidates for biomedical applications. In this study, we took advantage of the allylic groups introduced through DAlVP and explored a variety of organic transformations of unsaturated carbon–carbon bonds in form of polymer-analogous reactions with statistical P(DEVP-co-DAlVP). Starting with basic transformations (bromination, epoxidation, thiol–ene click chemistry), consecutive reactions were performed to investigate the accessibility of the newly introduced, reactive groups and advanced to more complex polymeric structures. Moreover, unreacted allyl groups were converted via thiol–ene reactions to form dual-functionalized polymers and mimic the basic architecture of polymer–drug conjugates.
Experimental section
Materials and methods
General. All reactions with air- and moisture-sensitive substances were carried out under an argon atmosphere using standard Schlenk techniques or in a glovebox. Prior to use, all glassware was heat dried under a vacuum. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich, ABCR, or TCI Europe and used without further purification. Toluene, THF and DCM were dried using a MBraun SPS-800 solvent purification system and stored over 3 Å molecular sieves. The precursor complexes Y(CH2Si(CH3)3)3(THF)2 and LiCH2TMS and the catalyst Cp2Y(CH2TMS)(THF) were prepared according to procedures found in the literature.47–50 Diethyl vinylphosphonate (DEVP) was synthesized according to procedures from the literature, dried over calcium hydride, and distilled prior to use.51 Likewise, diallyl vinylphosphonate (DAlVP) was prepared according to a published procedure.31,52
Nuclear magnetic resonance spectroscopy. NMR spectra were recorded on a Bruker AV-400HD and an AVIII-500 Cryo spectrometer. 1H- and 13C-NMR spectroscopic chemical shifts δ were reported in ppm relative to the residual proton signal of the solvent. δ (1H) was calibrated to the residual proton signal, and δ (13C) to the carbon signal of the solvent. Unless otherwise stated, coupling constants J were averaged values and refer to couplings between two protons. All deuterated solvents (C6D6, MeOD-d4) were obtained from Sigma-Aldrich. For the analysis of C–H bond activation products, C6D6 was dried and stored over 3 Å molecular sieves in a glovebox. DOSY-NMR measurements were performed for the characterization of polymer conjugates.
Elemental analysis. Elemental analyses were measured on a Vario EL (Elementar) at the Laboratory for Microanalysis of the Institute of Inorganic Chemistry at the Technical University of Munich.
Infrared spectroscopy. FTIR spectra were recorded with a Vertex 70 FTIR (Bruker) using a Platinum ATR from Bruker.
UV/Vis spectroscopy. UV/Vis spectra were recorded on a Varian Cary 50 UV/Vis spectrophotometer in 40 mm × 10 mm × 2 mm quartz glass cuvettes. Methanol (HPLC grade) was used as solvent.
Dialysis. Purification via dialysis was performed with Spectra/Por 1 dialysis membranes (regenerated cellulose) with a molecular weight cut-off (MWCO) of 6–8 kDa (Spectrumlabs). Prior to use the membranes were treated with deionized water over night and then rinsed with deionized water. A 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of a dialysis fluid to sample volume was applied. Specific solvents used as dialysis fluid were given for the corresponding procedures.
:1 ratio of a dialysis fluid to sample volume was applied. Specific solvents used as dialysis fluid were given for the corresponding procedures.
Molecular weight distribution. Gel permeation chromatography was performed with samples of 5 mg mL−1 on a combination of a Shimadzu LC-10ADVP with a DGU-3A as degasser (Shimadzu) and a column thermostat CTO-10A (Shimadzu) equipped with two PL Polargel-M columns (Agilent). The eluent used was a mixture of 50% THF and 50% water, treated with tetrabutylammonium bromide (TBAB) (9 g L−1), and 340 mg L−1 3,5-di-tert-butyl-4-hydroxytoluene (BHT) as a stabilizing agent. The samples were analyzed via multiangle light scattering (MALS) using a Wyatt Dawn Heleos II in combination with a Wyatt Optilab rEX as RI detector unit.
Main synthetic procedures
Synthesis of P(DEVP-co-DAlVP) P1. A solution of sym-collidine (73.8 mg, 609 μmol, 1.00 equiv.) in absolute toluene (5.00 mL) was added to a solution of Cp2Y(CH2TMS)(THF) (23.1 mg, 60.9 μmol, 1.00 equiv.) in absolute toluene (5.00 mL), resulting in the instant yellow coloration of the reaction mixture. After stirring at room temperature overnight, full conversion was verified by proton NMR, and the mixture was diluted with additional toluene (80.0 mL). A mixture of DEVP (9.00 g, 54.8 mmol, 90 equiv.) and DAlVP (1.15 g, 6.09 mmol, 10 equiv.) in absolute toluene (5.00 mL) was then added to the solution in one portion. After four hours, the completion of the polymerization was detected by 31P-NMR spectroscopy. The reaction was quenched by the addition of MeOH (5 mL), and the polymer was precipitated in pentane. The supernatant solvent was decanted off, and the residual polymer was dissolved in water and freeze-dried to yield the pure polymer as a colorless solid.
Procedure for the bromination of P1. Bromine (50.3 mg, 315 μmol, 3.00 equiv. per allyl group) was carefully added to a solution of 100 mg of polymer P1 (npolymer = 5.00 μmol; nallyl = 105 μmol,53 1.00 equiv.) in methylene chloride (8.00 mL). After two hours of stirring at room temperature, proton NMR showed the full conversion of the allyl motifs. The brominated polymer was precipitated in ice cold hexane and washed several times with hexane to remove excess bromine.
General procedure for the epoxidation of P1 with OXONE. A solution of P1 (100 mg, npolymer = 5.00 μmol; nallyl = 105 μmol, 1.00 equiv.) in acetone (5.00 mL) was treated with sodium bicarbonate (74.1 mg, 882 μmol, 8.40 equiv. per allyl group). A solution of OXONE (220 mg, 357 μmol, 3.40 equiv. per allyl group) in deionized water (5.00 mL) was added slowly to this suspension. The suspension was stirred for 24 hours at room temperature and the conversion was determined by 1H-NMR (65%). The solids were filtrated off and the resulting polymer solution was used for further transformations.
General procedure for the epoxidation of P1 with meta-chloroperbenzoic (mCPBA). mCPBA (145 mg, 840 μmol, 8.00 equiv. per allyl group) was added to a solution of 100 mg of polymer P1 (npolymer = 5.00 μmol; nallyl = 105 μmol, 1.00 equiv.) in methylene chloride (6.00 mL). This solution was stirred for 48 hours at room temperature. The conversion (23%) of the allyl groups was checked by proton NMR and excess mCPBA was removed by filtration from the ice-cooled reaction mixture. The solution was concentrated in vacuo to half its volume and used without further purification for the consecutive modifications.
General procedure for the thermally induced thiol–ene click reaction. The polymer was dissolved in tetrahydrofuran (10 mL THF per 100 mg polymer) and treated with the respective thiol (5.00 equiv. per allyl group) as well as catalytic amounts of azobisisobutyronitrile (AIBN). The mixture was degassed via repeated evacuation and filling with argon (20 iterations) and stirred for 24 hours at 60 °C. After quantitative conversion of the allyl motifs was confirmed by 1H-NMR, volatiles were removed under reduced pressure and the residue was dissolved in water. The aqueous solution was purified by dialysis against water and freeze dried to yield the functionalized substrates.
General procedure for the photochemically induced thiol–ene click reaction. The polymer was dissolved in a mixture of tetrahydrofuran and methanol (THF/MeOH = 5/1) (10 mL solvent per 100 mg polymer) and was treated with the respective thiol (5.00 equiv. per allyl group and catalytic amounts of 2,2-dimethoxy-2-phenylacetophenone (DMPA) (0.1 equiv. per allyl group). The mixture was degassed via repeated evacuation and filling with argon (15 iterations) and irradiated (λ = 365 nm) for 18 hours. After quantitative conversion of the allyl motifs was confirmed by 1H-NMR, the volatiles were removed under reduced pressure and the residue was dissolved in water. The aqueous solution was purified by dialysis against water and freeze-dried to yield the functionalized substrates.
Results and discussion
Bromination and consecutive modifications of copolymer P1
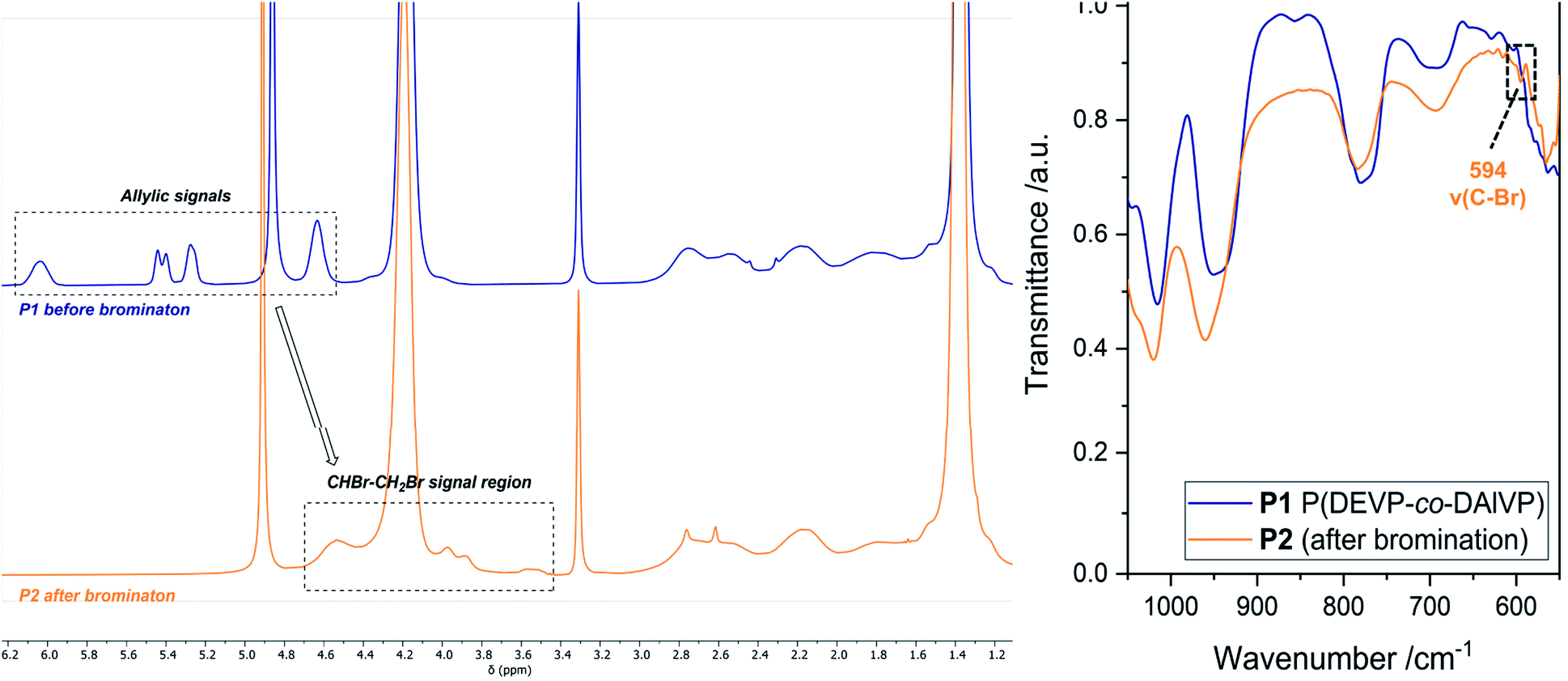
The first modification route started with the bromination of the allyl groups of P1 (Mn,NMR = 20.0 kg mol−1, nDEVP:nDAlVP = 10:1,54 Đ = 1.04) and was followed by a nucleophilic substitution with NaN3, to later facilitate an alkyne–azide click reaction between the polymer and a model compound (Scheme 1). In the present case copolymer P1 was treated with bromine and the reaction was monitored by 1H-NMR spectroscopy. After only two hours full conversion of the allyl groups was observed via 1H-NMR, since the signals of the allyl groups between 4.50 and 6.20 ppm fully disappeared (Fig. 1, left). A comparison of the integral ratios verified the integrity of P2. The section from 1.15 to 2.91 ppm (polymer backbone and POCH2CH3) was normalized to a value of I = 1036 (P1: I = 1036). Hence, an integral of 547 was determined for the section between 3.44 and 4.79 ppm (POCH2, CHBr and CH2Br). This value is equivalent to the integral of P1 between 4.50 and 6.20 ppm (I = 543). In addition to the polymer-related bands, an absorption band was found at 594 cm−1 (C–Br stretching mode) in the FTIR spectrum (Fig. 1, right). Moreover, analysis via GPC-MALS confirmed a defined copolymer with a narrow polydispersity (Mn,NMR = 23.4 kg mol−1, Đ = 1.02). P2 was then reacted with NaN3 in the presence of NH4Cl at 50 °C for 18 hours (Scheme 1) and dialyzed against water. A qualitative analysis done by IR spectroscopy provided evidence for the presence of the azide group at 2111 cm−1 (NNN stretching mode) (Fig. S8†). Approximately 53% of the bromide groups were converted during the substitution reaction. These newly introduced azide moieties were investigated in the coupling of phenylacetylene (Scheme 1). Initially, the coupling of phenylacetylene (1) to P3 was performed at 90 °C for 20 to avoid the use of a copper species as catalyst.55 Unfortunately, the analysis of P4 done by diffusion ordered spectroscopy (DOSY) revealed two distinct diffusion coefficients for the copolymer and phenylacetylene (1). Consequently, the copper-catalyzed click reaction was tested with model compound 1. Here, the use of CuSO4·H2O in the presence of sodium ascorbate presumably yielded the coupling product as a corresponding DOSY spectrum of the crude product had shown. However, the polymer remained insoluble in a variety of solvents after the work-up, preventing further characterizations. This behavior was explained with the high degree of aromaticity in the polymer side chains reducing the flexibility of the polymer chain by intra- and intermolecular interactions of the aromatic groups.
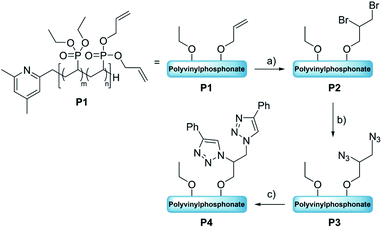 |
| | Scheme 1 Transformation of polyvinylphosphonate P1 to the click adduct P4 via bromination and conversion to the azide. Reaction conditions: (a) Br2, RT, 2 h [DCM], (b) NaN3, NH4Cl, 50 °C, 24 h [DCM/DMF], (c) phenylacetylene (1), CuSO4·H2O (cat.), Na ascorbate, RT, 18 h [THF/H2O]. | |
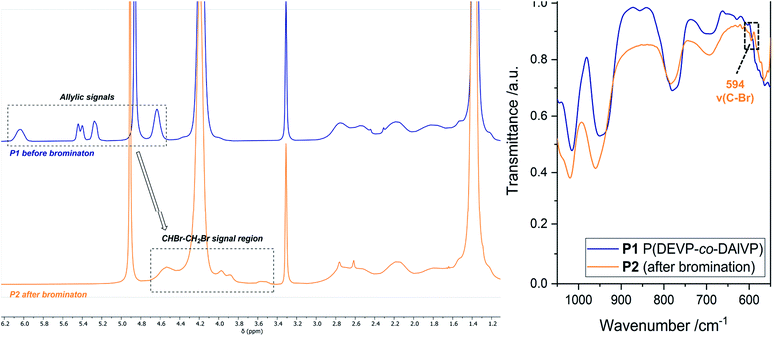 |
| | Fig. 1 Left: Comparison of the 1H-NMR spectra before (blue) and after (orange) the bromination reaction measured in MeOD-d4. Right: Section of the IR spectra of P1 (blue) and P2 (orange). | |
Epoxidation and follow-up modifications of copolymer P1
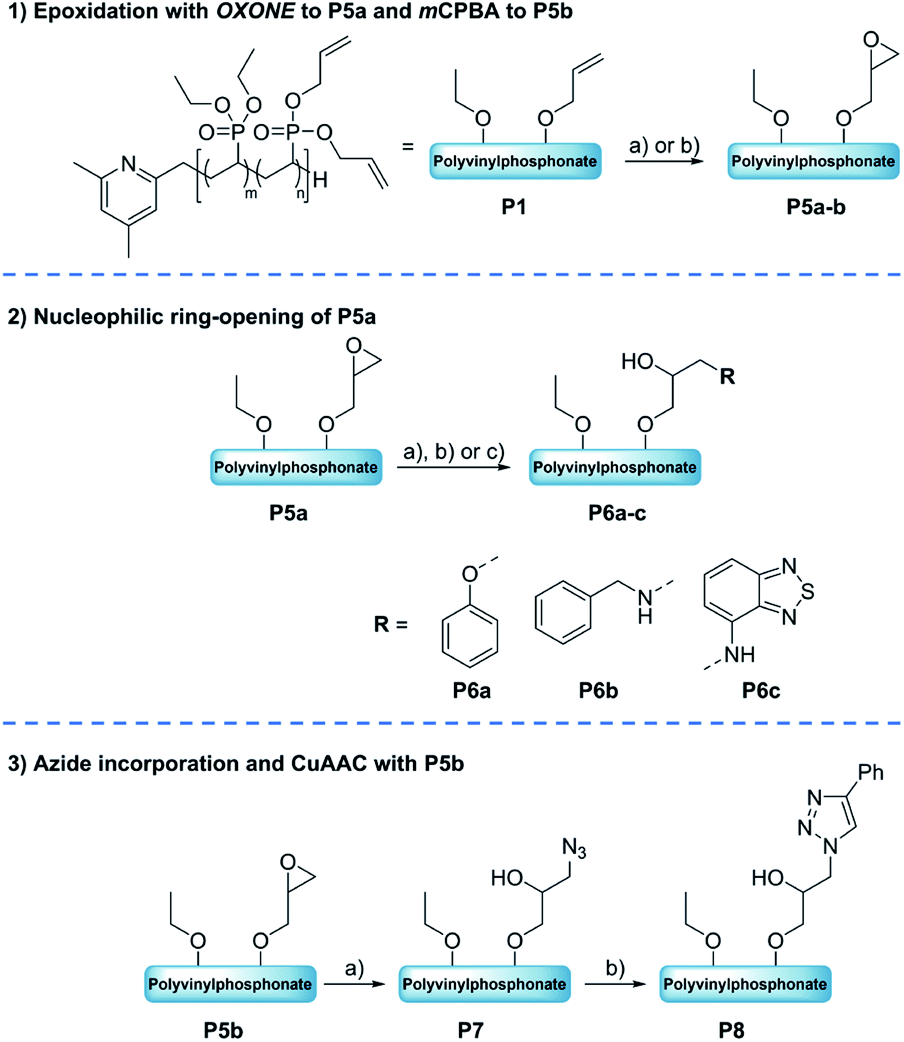
Due to the inconveniences with the alkyne–azide click reactions after the bromination of precursor P1, the synthetic strategy was focused on the epoxidation of the allyl groups (Scheme 2). Starting from the epoxide-containing polymer a variety of consecutive reactions were performed, namely the ring-opening of the epoxide motif with model nucleophilic reagents (amines, alcohols) and the investigation of the alkyne–azide click reaction after a ring-opening with sodium azide.
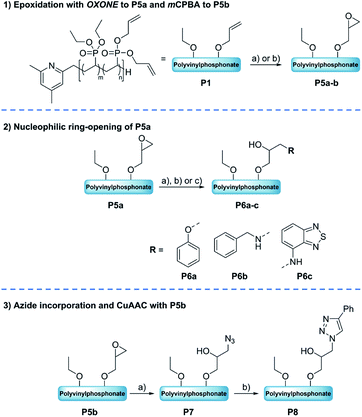 |
| | Scheme 2 Top: Epoxidation of P1 to P5a–b: (a) OXONE, NaHCO3, RT, 24 h [H2O/acetone], (b) mCPBA, RT, 48 h [DCM]. Middle: Ring-opening of the OXONE-derived substrate P5a to P6a–c with phenol (2), benzylamine (3) or 4-amino-2,1,3-benzothiadiazol (4). Reaction conditions: (a) 2, NaOH, 50 °C, 24 h [H2O/acetone], (b) 3, 50 °C, 24 h [H2O/acetone], (c) 4, 50 °C, 24 h [H2O/acetone]. Bottom: Ring-opening of P5b with NaN3 and subsequent CuAAC to click adductP8. Reaction conditions: (a) NaN3, NH4Cl, 50 °C, 24 h [DCM/DMF], (b) 1, 90 °C, 21 h [Tol]. | |
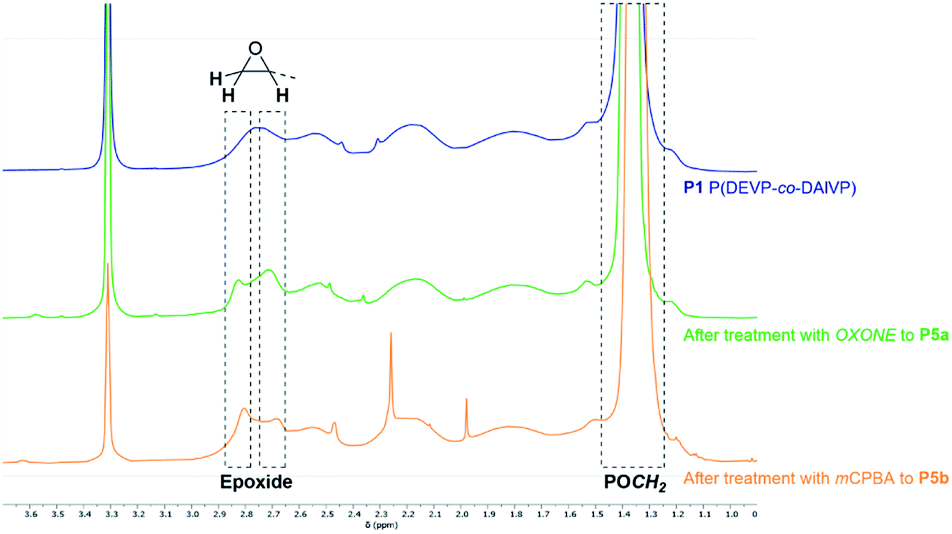
Epoxidation of the allyl motifs of P1. In a first attempt the conversion of P1 was tested with hydrogen peroxide and catalytic amounts of 2,2,2-trifluoroacetophenone.56 However, no consumption of the allyl groups was observed using these reagents. The epoxidation with OXONE as oxidizing agent in water and acetone turned out to be successful. In the corresponding 1H-NMR spectrum of P5a, new signals at 2.71 (epoxide), 2.84 (epoxide), 3.94 (POCH), and 4.48 ppm (POCH) corroborated the phosphonate-bound epoxide groups (Fig. 2, green). In comparison to precursor P1 (Fig. 2, blue) the signal intensities of the allyl groups were clearly reduced and exhibited a conversion of 65% after comparison of the integrals before and after the reaction. However, dialysis against water and lyophilization resulted in an insoluble solid. Presumably, a cross-linking process was initiated via the epoxide groups during work-up. Therefore, the reaction media containing the in situ generated epoxides were directly used in the consecutive reactions after filtration. Due to the limitation to water/acetone as solvent, the epoxidation of P1 was also investigated in the presence of meta-chloroperbenzoic acid (mCPBA) as oxidizing agent. The reaction was performed in DCM over a period of two days. Solid side products were then removed by filtration from an ice-cooled solution. A comparative analysis of the oxidized substrate with the precursor P1 by means of 1H-NMR spectroscopy showed a consumption of the allyl groups and the occurrence of the epoxide-related signals at 2.68, 2.81, 4.01 and 4.49 ppm (Fig. 2, orange). Compared to the conversion with OXONE the reaction with mCPBA was less efficient and reached a conversion of 23% with respect to the corresponding amount of allyl groups. The low conversion might be a result of the presence of random coils in solution which can hinder the oxidant from reaching the allyl groups. Moreover, mCPBA might be sterically repulsed from the polymer chains due to its bulkiness of the rigid aromatic framework.
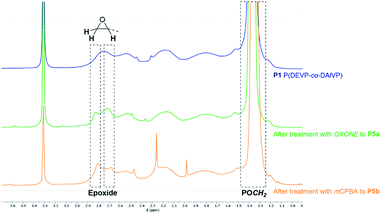 |
| | Fig. 2 Comparison of the significant shift regions of polymer P1 after epoxidation with OXONE (P5a, green) or mCPBA (P5b, orange). | |
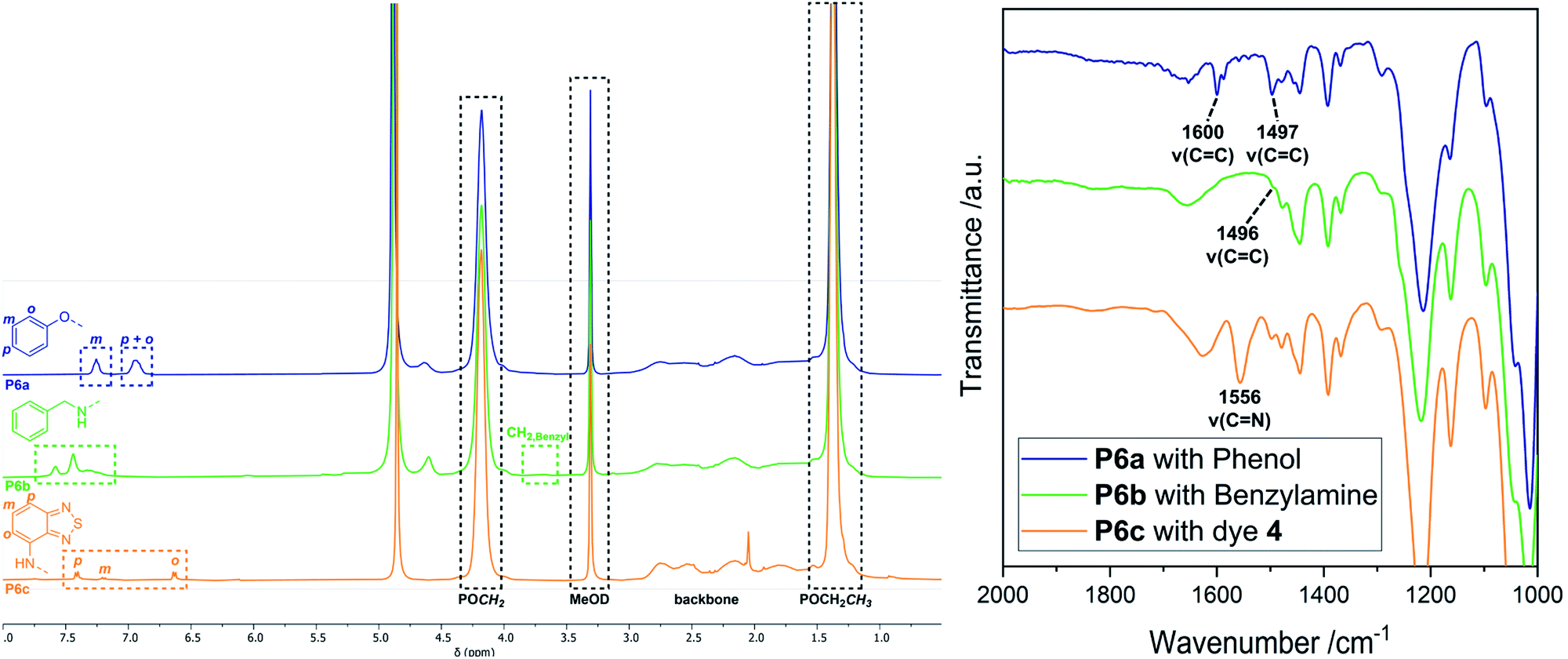
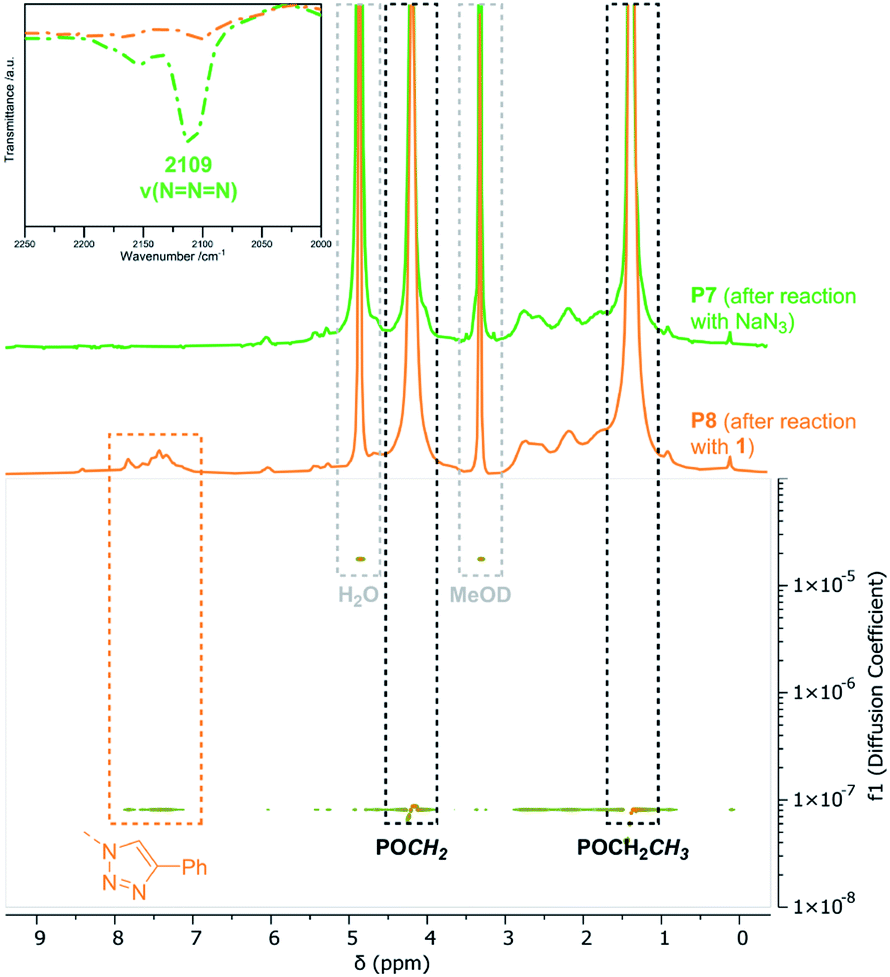
Ring-opening of the epoxide motifs. The OXONE-derived derivative of P1 was applied in the nucleophilic ring-opening with phenol (2), benzylamine (3), and 4-amino-2,1,3-benzothiadiazol (4) (Scheme 2, middle) as test substrates. Phenol was reacted with the polyvinylphosphonate-bound epoxide in the presence of sodium hydroxide using the solution with the in situ-generated epoxides (vide supra). 1H-NMR spectroscopy confirmed the vanishing of the epoxide-related signals, while the phenyl group was able to be assigned to the signals at 6.93 and 7.26 ppm (Fig. 3, blue). According to NMR 34% of the allyl groups are functionalized with phenyl ethers. DOSY-NMR further corroborated the presence of one species exclusively since the aromatic and the phosphonate signals (1.36 and 4.18 ppm) shared the same diffusion coefficient (D = 4.94 × 10−7 cm2 s−1; Fig. S13†). In addition, two absorption bands at 1497 and 1600 cm−1 (CC bending mode) corroborated the presence of the aromatic groups (Fig. 3, right; Fig. S14†). Likewise, benzylamine (3) was coupled to the epoxidized polyvinylphosphonate. The crude solution of the epoxidized copolymer P1 was treated with the amine and stirred for 24 hours at 50 °C. 1H-NMR confirmed the complete conversion of the epoxide groups and revealed the methylene at 3.65 ppm as well as the benzylic motif between 7.07 and 7.77 ppm (Fig. 3, green) which again shared the same diffusion coefficient (D = 4.94 × 10−7 cm2 s−1) as the phosphonate esters (Fig. S17†). According to a comparison of the integral values approximately 65% of the original allyl groups are bearing amine groups. However, IR spectroscopy only showed a very weak shoulder at 1495 cm−1 that can be assigned to the δ(CC) mode (Fig. 3, green). In a similar fashion, dye 4 was conjugated to the polyvinylphosphonate. Here, too, the signals of the aromatic framework (6.63, 7.20, 7.41 ppm) could be assigned in the 1H-NMR spectrum (Fig. S20†) with 26% of the allyl groups being conjugated to 4. The corresponding DOSY spectrum confirmed a covalent conjugation of the dye to the phosphonate framework (D = 1.13 × 10−6 cm2 s−1, Fig. S21†). A decisive IR band at 1556 cm−1 was able to be assigned to CN vibrations. Moreover, UV/Vis measurements featured changes in the absorption behavior of 4-amino-2,1,3-benzothiadiazol (4) since the absorption maximum shifted from 413 nm (free dye) to 422 nm (polymer conjugate). The bathochromic shift can be a result of the interaction of the aromatic motifs due to their spatial proximity, hydrogen bonding between the benzothiadiazole and the newly generated hydroxyl groups, as well as a polarity change of the surrounding environment upon conjugation.57–59 The introduction of the azide groups was performed with the crude solution containing the copolymer epoxidized by mCPBA. Similarly, sodium azide was dissolved in DMF and added to the epoxide-containing solution. The mixture was stirred at 50 °C for 24 hours (Scheme 2). As proven by 1H-NMR spectroscopy the characteristic epoxide signals (compare Fig. 2) vanished during the reaction indicating a quantitative conversion. Analysis by IR supported the success of the reaction by revealing the stretching mode of the azide at 2109 cm−1 (Fig. 4). This transformation was followed by the copper-free alkyne–azide coupling with phenylacetylene (1) at 90 °C. In contrast to the analogue reaction with copolymer P3 1H-NMR revealed a successful coupling to P7. Two new signal groups emerged between 7.28 and 7.68 ppm corresponding to the triazole motif (49% of all allyl groups). Again, DOSY corroborated a covalent incorporation of aromatic compound 1 since it shared the same diffusion coefficient as the signals of the polyvinylphosphonate (Fig. 4).
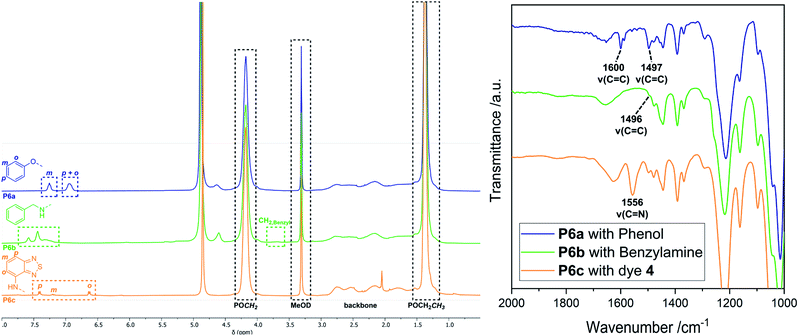 |
| | Fig. 3 Left: Comparison of the 1H-NMR spectra of the conjugation products P6a (blue), P6b (green), and P6c (orange) in MeOD-d4. Exact integral values can be found in the ESI† for each reaction. Right: Zoomed-in view of the IR spectra of the conjugation products P6a (blue), P6b (green), and P6c (orange). | |
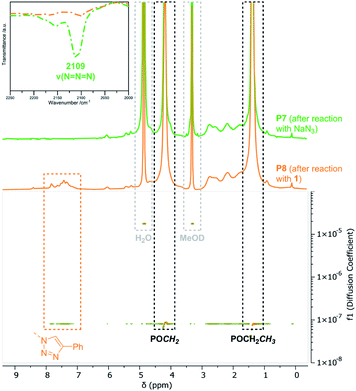 |
| | Fig. 4 Comparison of the 1H-NMR spectra recorded in MeOD-d4 of P7 (green) and P8 (orange) and corresponding region of the DOSY-NMR of P8. Top left: IR section of the azide vibration before and after azide–alkyne reaction. | |
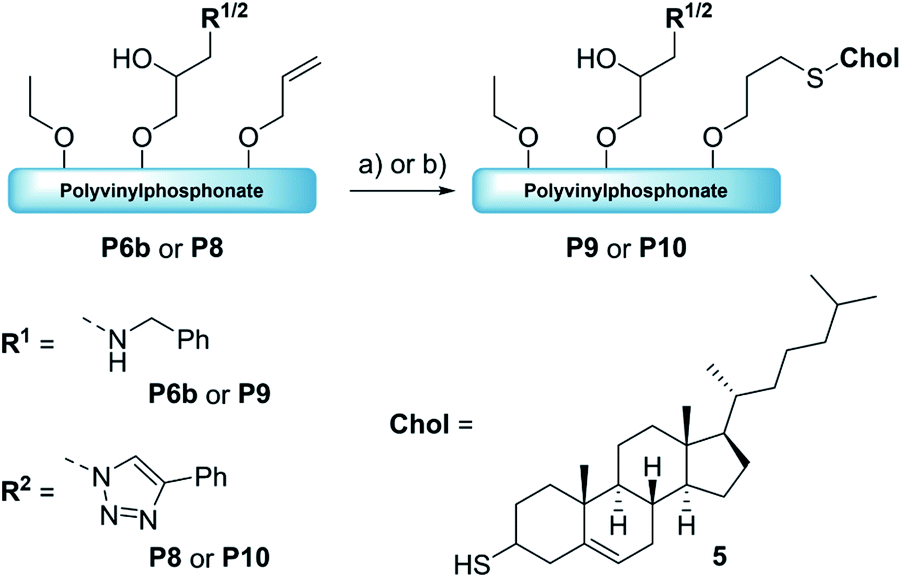
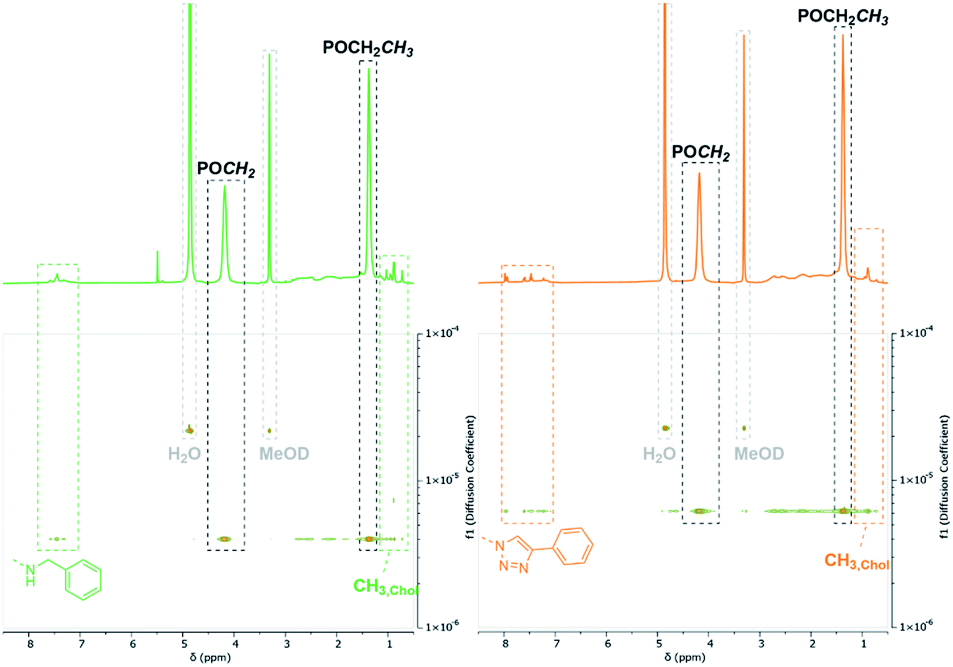
Synthesis of dual-functionalized copolymers. As presented, approximately 35% of the allyl groups remained unreacted after treatment with OXONE and 77% after using mCBPBA. These free allyl groups are easily accessible by mercaptans through the efficient thiol–ene click chemistry. This reaction enabled the introduction of additional functionalities and an imitation of the basic framework of polymer–drug conjugates. Therefore, polymers P6b (benzylamine side groups) and P8 (alkyne–azide coupling adduct) were reacted with thiocholesterol (5) (Scheme 3). All modifications were carried out under irradiation of UV light with a wavelength of 365 nm, since light-mediated thiol–ene reactions are known to be more efficient than their temperature-induced equivalents.60 The modifications of P6b were performed in a mixture of tetrahydrofuran and methanol with 2,2-dimethoxy-2-phenylacetophenone (DMPA). A comparison of the 1H-NMR spectra of P6b and the functionalized P9 provided evidence for the conversion of the remaining allyl groups (Fig. 5). A comparison of the aromatic motif and the isolated methyl group of cholesterol at 0.72 ppm gave a functionalization ratio of 77% (benzylamine) to 23% (cholesterol). The same observation was made for the conversion of P8 with thiocholesterol to P10 (Fig. 5). Moreover, DOSY measurements confirm the incorporation of 5 in the polymer chain as the cholesterol-related methyl groups between 0.80 and 1.20 ppm shared the same diffusion level as the polymer signals (Fig. 5). Similarly, a functionalization ratio of 75% (aromatic group) to 25% (cholesterol) was determined.
 |
| | Scheme 3 Formation of the dual-functionalized polymers P9andP10 with thiocholesterol (5) via thiol–ene chemistry. Reaction conditions: 5, DMPA, λ = 365 nm, RT, 18 h [THF/MeOH]. | |
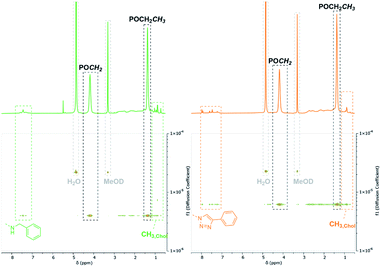 |
| | Fig. 5 Respective section of the 1H- and DOSY-NMR of P6b (green) and P8 (orange) after conversion with thiocholesterol (5) recorded in MeOD-d4. | |
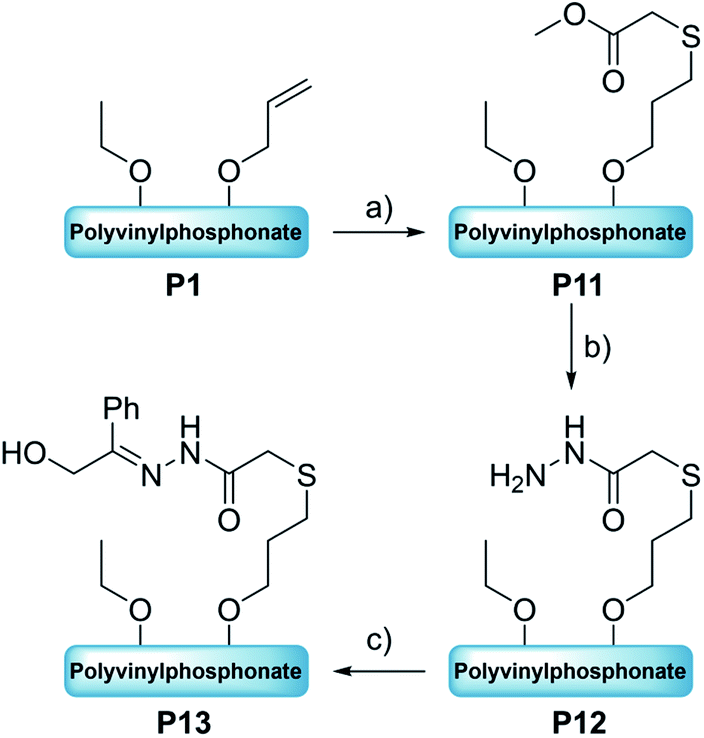
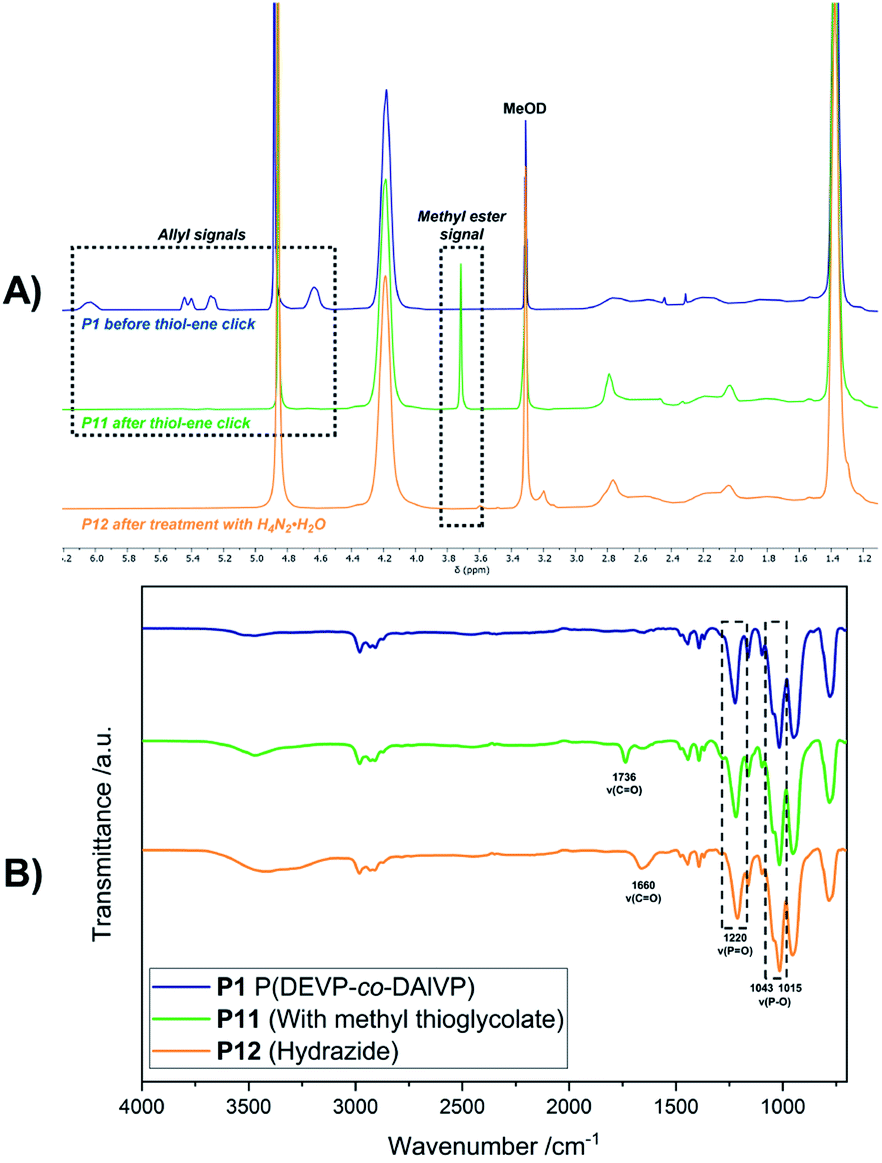
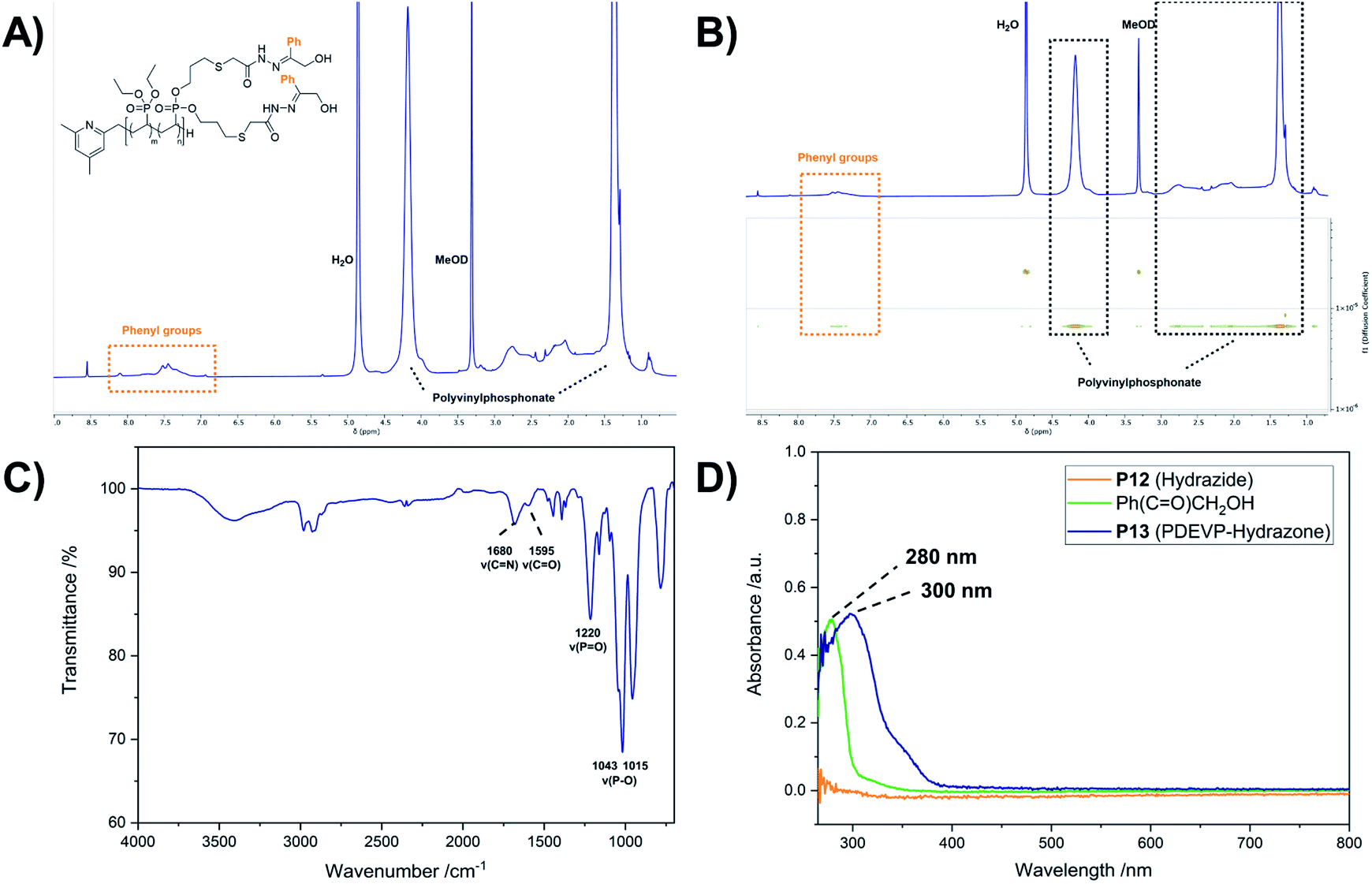
Introduction of acid-labile hydrazone linkages. In contrast to the previous functionalization strategies, which ought to insert the new, functional groups irreversibly, we also explored the introduction of a reversible hydrazone motif. Due to its pH-sensitivity, pharmaceutically active agents are frequently linked via hydrazones, which allows the pH-triggered release of the cargo from the conjugate. In this context, polyvinyl-phosphonates appear to be promising candidates because each phosphonate repeating unit carries two ester groups, which potentially double the drug-payload compared to other systems. Inspired by the work of Zhong et al.,28 2-hydroxyacetophenone (7) was conjugated to the polymer side chains in a three-step process (Scheme 4). In the first reaction methyl thioglycolate (6) was conjugated to P1 by a thiol–ene click reaction to yield P11, which was followed by the conversion to the respective hydrazide P12. In a last step, the formation of the hydrazone was studied with ketone 7 (Scheme 4). As visible in the corresponding 1H-NMR spectrum, the allyl groups of P1 between 4.50 and 6.20 ppm were consumed, while new signals appeared at 2.04 and 2.79 ppm (CH2 groups adjacent to the thioether bridge) as well as at 3.71 ppm (methyl ester) (Fig. 6A, green). Analysis by DOSY confirmed the presence of one single polymeric species (Fig. S38†). The corresponding IR spectrum corroborates a successful thiol–ene reaction. Compared to the spectrum of P1, which exhibits the characteristic absorption bands at 1016 and 1045 cm−1 (P–O stretching mode) and 1223 cm−1 (PO stretching mode),61,62 most decisive is the band at 1736 cm−1 that represents the CO stretching mode of the newly introduced ester (Fig. 6B, green). The addition of hydrazine enabled the conversion of the methyl ester to the respective hydrazide. In the corresponding proton NMR, the signal at 3.71 ppm vanished completely, while the signal structure of the polymer backbone remained untouched (Fig. 6A, orange). DOSY measurements were performed to further confirm the integrity of the polymer (Fig. S42†). The related IR spectrum supports the conclusions made by NMR analysis. The carbonyl-related stretching band shifted from 1736 cm−1 to 1660 cm−1 (Fig. 6B, orange), which can be explained by the formation of an amide motif. Moreover, the absorption at 3100 to 3600 cm−1 is more pronounced, which was accounted for by the N–H stretching mode of the hydrazide motif. However, the O–H stretching mode must also be considered, since the introduction of the polar hydrazides might cause a higher water content after freeze-drying. The hydrazone formation with 2-hydroxyacetophenone (7) revealed a new signal pattern in the aromatic region between 6.86 and 8.20 ppm. In comparison to the free ketone 7, which shows three defined multiplets at 7.54, 7.65 and 7.98 ppm (Fig. S45†), a signal broadening can be observed for the phenyl signals found in the 1H-NMR spectrum of P13 (Fig. 7A). This concluded the successful conjugation of 7 to the polymer, since the phenyl groups are now statistically distributed along the polymer chain. Again, the presence of a single polymeric species in the respective DOSY spectrum clearly corroborated the successful conjugation (Fig. 7B). A comparison of the integrals of the phenyl signals (I = 62.0) and the phosphonate signal at 4.18 ppm (I = 502) resulted in a coupling efficiency of approximately 60%. Alongside NMR analysis, FTIR measurements revealed two bands at 1595 cm−1 and 1680 cm−1, which were referred to the CO and CN stretching modes, substantiated the presence of the hydrazone motif (Fig. 7C).63,64 UV/Vis measurements of P13 in methanolic solution confirmed a peak at 300 nm, which was not observed for precursor P12 (Fig. 7D), and exhibits a bathochromic shift of approximately 20 nm compared to the peak of the free compound 7 at 280 nm.
 |
| | Scheme 4 Transformation of polyvinylphosphonate P1 to the hydrazine-bearing adduct P13 via a thiol–ene click reaction. Reaction conditions: (a) methyl thioglycolate (6), AIBN, 60 °C, 18 h [THF], (b) H4N2·H2O, reflux, 16 h [THF], (c) 2-hydroxyacetophenone (7), 4 Å molecular sieves, RT, 24 h [DMF]. | |
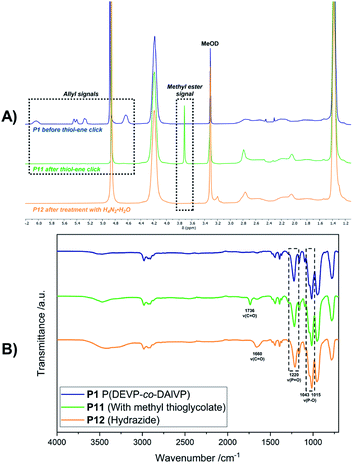 |
| | Fig. 6 1H-NMR spectra in MeOD of polymer P1, after conversion with 6, and after conversion with H4N2·H2O (A). Respective IR spectra of the polymeric species before and after the transformations (B). | |
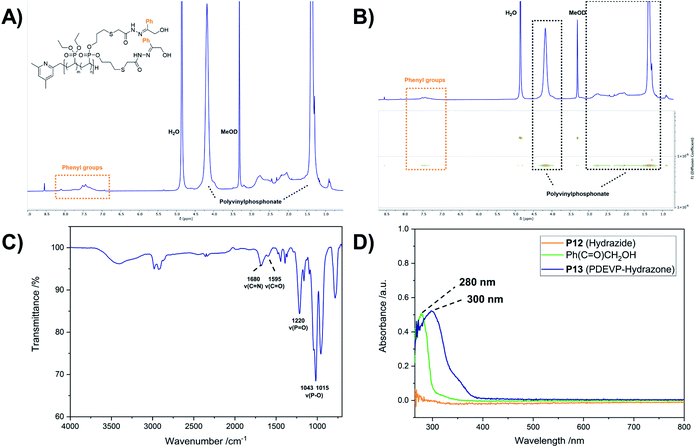 |
| | Fig. 7 1H-NMR spectrum of P13 in MeOD (A) and corresponding DOSY (B) as well as IR spectrum of P13 (C). UV/Vis spectra of the hydrazide P12 (100 μM with respect to the allyl groups), 2-hydroxyacetophenone (7) (500 μM) and the hydrazone conjugate P13 (100 μM with respect to the phenyl groups) in methanol (D). | |
Conclusions
In this study, we explored several synthetic routes to functionalize statistical P(DEVP-co-DAlVP) selectively at the position of the allyl groups. Three basic transformations (bromination, epoxidation, and thiol–ene click chemistry) were selected to insert reactive groups for the follow-up modifications. The addition of bromine was found to proceed quantitatively and was transferred into the azide with NaN3. However, solubility issues rendered the conversion of these azides via CuAAC problematic. The epoxidation of the olefines was possible using OXONE and mCPBA. In the case of OXONE, a conversion of 65% of the allyl groups was detected whereas the reaction with mCPBA was limited to 23%. The OXONE-derived substrate P5a was functionalized with phenol, benzylamine or dye 4. P5b was reacted with NaN3 followed by the conjugation of 1 in a copper-free azide–alkyne cyclization to form species P8. In all cases DOSY-NMR confirmed a successful coupling of the model substrates. Moreover, P6b and P8 were converted with thiocholesterol (5) and proved that the remaining allyl groups are still accessible to form complex polymer-conjugates potentially mimicking polymer–drug conjugates. This study was concluded by the introduction of an acid-labile hydrazone motif. Therefore, P1 was subsequently reacted with thiol 6 and hydrazine-hydrate, which was monitored via NMR and IR spectroscopy. Eventually, NMR, IR, and UV/Vis spectroscopy corroborated the successful formation of the hydrazone P13 from ketone 7.
Clearly, the polymer-bound allyl groups give access to a rich functionalization chemistry. Combined with the power of the REM-GTP, which allows the production of defined (co)polymers with predictable compositions and architectures, and our experience on their end-group modification, we established a highly flexible and versatile synthetic toolbox for the generation of smart materials. In the future this toolbox allows even more sophisticated conjugates which can comprise a variety of functional agents and are precisely attuned to their intended applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Fabian Späth, Moritz Kränzlein and Jonas Bruckmoser for revising the manuscript.
References
- H. Staudinger, Rubber Chem. Technol., 1939, 12, 117–118 CrossRef CAS.
- S. Koltzenburg, M. Maskos and O. Nuyken, in Polymer Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 407–424, DOI:10.1007/978-3-662-49279-6_15.
- L. S. Peixoto, F. M. Silva, M. A. L. Niemeyer, G. Espinosa, P. A. Melo, M. Nele and J. C. Pinto, Macromol. Symp., 2006, 243, 190–199 CrossRef CAS.
- B. Brissault, A. Kichler, C. Guis, C. Leborgne, O. Danos and H. Cheradame, Bioconjugate Chem., 2003, 14, 581–587 CrossRef CAS PubMed.
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849–853 CrossRef CAS.
- Z. Söyler and M. A. R. Meier, Green Chem., 2017, 19, 3899–3907 RSC.
- T. Heinze, O. A. El Seoud and A. Koschella, in Cellulose Derivatives: Synthesis, Structure, and Properties, Springer International Publishing, Cham, 2018, pp. 259–292, DOI:10.1007/978-3-319-73168-1_4.
- K. Jedvert and T. Heinze, J. Polym. Eng., 2017, 37, 845 CAS.
- M. R. Dreher, W. Liu, C. R. Michelich, M. W. Dewhirst and A. Chilkoti, Cancer Res., 2007, 67, 4418 CrossRef CAS PubMed.
- K. Stockhofe, J. M. Postema, H. Schieferstein and T. L. Ross, Pharmaceuticals, 2014, 7, 392–418 CrossRef CAS PubMed.
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 CrossRef CAS.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- H. Kamada, Y. Tsutsumi, Y. Yamamoto, T. Kihira, Y. Kaneda, Y. Mu, H. Kodaira, S.-i. Tsunoda, S. Nakagawa and T. Mayumi, Cancer Res., 2000, 60, 6416–6420 CAS.
- Y. Kaneda, Y. Tsutsumi, Y. Yoshioka, H. Kamada, Y. Yamamoto, H. Kodaira, S.-i. Tsunoda, T. Okamoto, Y. Mukai, H. Shibata, S. Nakagawa and T. Mayumi, Biomaterials, 2004, 25, 3259–3266 CrossRef CAS PubMed.
- T. Yasukawa, H. Kimura, Y. Tabata, H. Miyamoto, Y. Honda, Y. Ikada and Y. Ogura, Invest. Ophthalmol. Visual Sci., 1999, 40, 2690–2696 CAS.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS PubMed.
- S. Jevševar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef PubMed.
- J. Kopeček and P. Kopečková, Adv. Drug Delivery Rev., 2010, 62, 122–149 CrossRef PubMed.
- H. Zhao, C. Lee, P. Sai, Y. H. Choe, M. Boro, A. Pendri, S. Guan and R. B. Greenwald, J. Org. Chem., 2000, 65, 4601–4606 CrossRef CAS PubMed.
- E. K. Rowinsky, J. Rizzo, L. Ochoa, C. H. Takimoto, B. Forouzesh, G. Schwartz, L. A. Hammond, A. Patnaik, J. Kwiatek, A. Goetz, L. Denis, J. McGuire and A. W. Tolcher, J. Clin. Oncol., 2003, 21, 148–157 CrossRef CAS PubMed.
- Y. Nojima, Y. Suzuki, K. Yoshida, F. Abe, T. Shiga, T. Takeuchi, A. Sugiyama, H. Shimizu and A. Sato, Pharm. Res., 2009, 26, 2125–2132 CrossRef CAS PubMed.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, J. Am. Chem. Soc., 2009, 131, 4783–4787 CrossRef CAS PubMed.
- H. Zhao, B. Rubio, P. Sapra, D. Wu, P. Reddy, P. Sai, A. Martinez, Y. Gao, Y. Lozanguiez, C. Longley, L. M. Greenberger and I. D. Horak, Bioconjugate Chem., 2008, 19, 849–859 CrossRef CAS PubMed.
- N. Vijayalakshmi, A. Ray, A. Malugin and H. Ghandehari, Bioconjugate Chem., 2010, 21, 1804–1810 CrossRef CAS PubMed.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171–185 CrossRef CAS PubMed.
- I. J. Majoros, C. R. Williams, A. Becker and J. R. Baker Jr, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 502–510 CAS.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelínková and B. Říhová, J. Controlled Release, 2003, 87, 33–47 CrossRef CAS PubMed.
- L. Zhou, R. Cheng, H. Tao, S. Ma, W. Guo, F. Meng, H. Liu, Z. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 1460–1467 CrossRef CAS PubMed.
- C. Schwarzenböck, A. Schaffer, P. Pahl, P. J. Nelson, R. Huss and B. Rieger, Polym. Chem., 2018, 9, 284–290 RSC.
- C. Schwarzenböck, S. I. Vagin, W. R. Heinz, P. J. Nelson and B. Rieger, Macromol. Rapid Commun., 2018, 39, 1800259 CrossRef PubMed.
- C. Schwarzenböck, P. J. Nelson, R. Huss and B. Rieger, Nanoscale, 2018, 10, 16062–16068 RSC.
- P. T. Altenbuchner, P. D. L. Werz, P. Schöppner, F. Adams, A. Kronast, C. Schwarzenböck, A. Pöthig, C. Jandl, M. Haslbeck and B. Rieger, Chem.–Eur. J., 2016, 22, 14576–14584 CrossRef CAS PubMed.
- F. Adams, P. T. Altenbuchner, P. D. L. Werz and B. Rieger, RSC Adv., 2016, 6, 78750–78754 RSC.
- N. Zhang, S. Salzinger and B. Rieger, Macromolecules, 2012, 45, 9751–9758 CrossRef CAS.
- S. Salzinger, U. B. Seemann, A. Plikhta and B. Rieger, Macromolecules, 2011, 44, 5920–5927 CrossRef CAS.
- U. B. Seemann, J. E. Dengler and B. Rieger, Angew. Chem., 2010, 122, 3567–3569 CrossRef.
- B. S. Soller, S. Salzinger, C. Jandl, A. Pöthig and B. Rieger, Organometallics, 2015, 34, 2703–2706 CrossRef CAS.
- P. Pahl, C. Schwarzenböck, F. A. D. Herz, B. S. Soller, C. Jandl and B. Rieger, Macromolecules, 2017, 50, 6569–6576 CrossRef CAS.
- A. Schaffer, M. Kränzlein and B. Rieger, Macromolecules, 2020, 53, 4345–4354 CrossRef CAS.
- F. Adams, M. Pschenitza and B. Rieger, ChemCatChem, 2018, 10, 4309–4316 CrossRef CAS.
- C. Schwarzenböck, A. Schaffer, E. Nößner, P. J. Nelson, R. Huss and B. Rieger, Chem.–Eur. J., 2018, 24, 2584–2587 CrossRef PubMed.
- H. Yang, A. Sun, C. Chai, W. Huang, X. Xue, J. Chen and B. Jiang, Polymer, 2017, 121, 256–261 CrossRef CAS.
- X. Guan, X. Hu, S. Liu, Y. Huang, X. Jing and Z. Xie, RSC Adv., 2014, 4, 55187–55194 RSC.
- H. Sun, L. Yan, M. Y. Z. Chang, K. A. Carter, R. Zhang, L. Slyker, J. F. Lovell, Y. Wu and C. Cheng, Nanoscale Adv., 2019, 1, 2761–2771 RSC.
- H. Sun, M. Y. Z. Chang, W.-I. Cheng, Q. Wang, A. Commisso, M. Capeling, Y. Wu and C. Cheng, Acta Biomater., 2017, 64, 290–300 CrossRef CAS PubMed.
- H. Sun, L. Yan, R. Zhang, J. F. Lovell, Y. Wu and C. Cheng, Biomater. Sci., 2021, 9, 5000–5010 RSC.
- K. C. Hultzsch, P. Voth, K. Beckerle, T. P. Spaniol and J. Okuda, Organometallics, 2000, 19, 228–243 CrossRef CAS.
- G. D. Vaughn, K. A. Krein and J. A. Gladysz, Organometallics, 1986, 5, 936–942 CrossRef CAS.
- C.-X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131–136 CrossRef CAS.
- S. Salzinger, B. S. Soller, A. Plikhta, U. B. Seemann, E. Herdtweck and B. Rieger, J. Am. Chem. Soc., 2013, 135, 13030–13040 CrossRef CAS PubMed.
- S. Salzinger, PhD thesis, Technical University of Munich, 2013.
- L. Rigger, R. L. Schmidt, K. M. Holman, M. Simonović and R. Micura, Chem.–Eur. J., 2013, 19, 15872–15878 CrossRef CAS PubMed.
- The uneven number of allyl groups is a result of their calculation. On a molecular level each individual chain contributes an even number of allyl groups. However, the number mean of the copolymer composition averaged out to be an uneven number on a macroscopic level.
- Compared to the feed ration the polymer composition tended slightly in favour of DEVP. The propagation rate is dependent from the bulkiness of the phosphonate ester. Here, DEVP is slightly less bulky than DAlVP leading to a faster consumption which is represented by a slightly higher content in the resulting copolymer. Same observations can be found in the literature for the conversion of methacrylates derivatives with zirconocenes.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef.
- D. Limnios and C. G. Kokotos, J. Org. Chem., 2014, 79, 4270–4276 CrossRef CAS PubMed.
- D. Seo, J. Park, T. J. Shin, P. J. Yoo, J. Park and K. Kwak, Macromol. Res., 2015, 23, 574–577 CrossRef CAS.
- K. K. Bansal, E. Özliseli, A. Rosling and J. M. Rosenholm, Adv. Funct. Mater., 2021, 31, 2101998 CrossRef CAS.
- K. Takagi, H. Takao and T. Nakagawa, Polym. J., 2013, 45, 396–400 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- N. Zhang, S. Salzinger, F. Deubel, R. Jordan and B. Rieger, J. Am. Chem. Soc., 2012, 134, 7333–7336 CrossRef CAS PubMed.
- Q. Wang, S. Chen, Y. Liang, D. Dong and N. Zhang, Macromolecules, 2017, 50, 8456–8463 CrossRef CAS.
- D. Sadhukhan, A. Ray, G. Pilet, G. M. Rosair, E. Garribba, A. Nonat, L. J. Charbonnière and S. Mitra, Bull. Chem. Soc. Jpn., 2011, 84, 764–777 CrossRef CAS.
- Z.-Y. Yin, J.-H. Hu, Q.-Q. Fu, K. Gui and Y. Yao, Soft Matter, 2019, 15, 4187–4191 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures of the follow-up functionalisations. Detailed characterisation data (1H-, 13C-, 31P-, and DOSY-NMR spectra, FTIR spectra, UV/Vis spectra, elemental analysis, as well as GPC traces). See DOI: 10.1039/d1ra06452e |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*