
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencemeta-Terphenyl linked donor–π–acceptor dyads: intramolecular charge transfer controlled by electron acceptor group tuning†
Min-Ji Kim,
Mina Ahn,
Minjung Chae,
Sanghyun Kim,
Daehoon Kim and
Kyung-Ryang Wee *
*
Department of Chemistry, Institute of Natural Science, Daegu University, Gyeongsan 38453, Republic of Korea. E-mail: krwee@daegu.ac.kr
First published on 28th October 2021
Abstract
A series of meta-terphenyl linked donor–π–acceptor (D–π–A) dyads were prepared to understand the electronic effects of a meta-terphenyl linker according to the electron-accepting ability change. The energy band gaps of the dyads were controlled by tuning the accepting ability, which resulted in emission colors ranging from blue-green to red. In the Lippert–Mataga plots, intramolecular charge transfer (ICT) behavior was observed, which showed gradually increased ICT characteristics as the accepting ability was increased. On the other hand, in the absorption spectra, a red shift of the ICT transition was observed differently from the electron-accepting ability tendency. Thus, the experimental results show that the ICT is determined by steric hindrance rather than the acceptor ability in the ground state due to the lack of π-conjugation of the terphenyl linker by the electron node in the meta-position, whereas ICT in the excited state is controlled by electron-accepting ability.
Introduction
Diverse dipolar organic materials, consisting of electron-donor (D) and electron-acceptor (A) units with various π-linkers, have been investigated and applied to various research areas because of their unique electrochemical and photophysical properties.1–9 In particular, donor–π–acceptor (D–π–A) dyad molecules are widely used molecular systems because of the easy tailoring of the photophysical property by changing the sub-units such as donor, acceptor, and π-linker. Thus, an adequate combination of sub-units in the D–π–A dyad system is the key to achieving the desired electrochemical and photophysical properties for a wide range of applications, such as organic light-emitting diodes (OLEDs),10–17 dye-sensitized solar cells (DSSCs),18–21 organic photovoltaics (OPVs),22–24 photoelectrochemical cells (PECs),25–28 fluorescence sensors,29–31 mechanochromic materials,32–34 nonlinear optical materials,35,36 and nanostructured materials.37–39The notable features of D–π–A dyads are intramolecular charge transfer (ICT) and photoinduced electron transfer (PET) in the ground and excited states because of the large electronic dipoles. The PET and ICT properties in D–π–A dyads are entirely different phenomena. In the excited state, ICT is partial charge transfer process from the donor to the acceptor whereas PET is a unit electron transfer process.40,41 They are used in other research areas according to the difference in the ICT and PET characteristics. Generally, the characteristic ICT absorption and emission are vital in determining the optical and electronic properties.42–44 In addition, the ICT absorption and emission properties can be finely controlled by changing the donor, acceptor, π-linker units, and molecular geometry, which can result in an effective bandgap and color tuning. PET process can be called charge separation by unit electron transfer, and reverse electron transfer are called charge recombination processes.40 PET systems with high-efficiency charge separation and slow charge recombination processes play an essential role in improving the photovoltaic performance.45
Recently, ortho-, meta-, and para-terphenyl π-linker-based D–π–A dyads, comprised of triphenylamine (TPA) as a donor and 1-phenyl-1H-benzimidazole (IMI) as an acceptor, were examined to understand the effects of geometric isomerism of D–π–A dyads.46 To clarify the distinction between ICT and PET occurring in the three terphenyl isomeric D–π–A dyads, the three terphenyl backbone-based isomers were compared, and all isomers showed distinctive ICT emission rather than PET in the excited state. In particular, the non-conjugated system of the meta-terphenyl isomer D–π–A dyads also showed ICT emission and were distinguished with previously reported non-conjugated D–π–A dyads because of its unique meta-terphenyl conjugation system. meta-Terphenyl is a non-conjugated molecular system caused by the electronically nodal site at meta-position, and only two phenyl units are conjugated. Because of these electronic properties, various meta-position-linked molecules, including meta-terphenyl-containing materials, have been developed and used widely, especially in the development of wide bandgap materials.47–50 These compounds are also one of the frequently used conjugation braking methods in the development of bifunctional materials.51,52 On the other hand, the meta-position-linked molecular system is also discriminated from the conventional conjugation braking systems and not completely separated electronically between the two functional units. Although the study on the influence of a meta-linkage on the electronic coupling had been reported, information on the electronic nodal-position effect on the meta-terphenyl as a linker is still limited.53–56
In this regard, a series of meta-terphenyl linked D–π–A dyads were designed and prepared to understand the electronic effects of the meta-terphenyl linker in the D–π–A dyads. In the meta-terphenyl linked D–π–A dyads, the electron-donating group was used a TPA unit because it is one of the well-known efficient donor units.57,58 In addition, four different electron-withdrawing groups (cyanide, benzoxazole, benzothiazole, and 2,4-diphenyl-1,3,5-triazine) were selected for their varying electron affinity capability with steric effects. The electronic and steric effects on the electron-withdrawing groups were considered to clarify the meta-terphenyl electronic nodal-position effect. Basically, the 2,4-diphenyl-1,3,5-triazine moiety has strong electron-accepting ability among the four different electron-withdrawing groups and a bulky structure.59 Benzoxazole and benzothiazole moieties have relatively weaker electron-accepting ability and planar molecular structures compared to 2,4-diphenyl-1,3,5-triazine, and they were selected to compare only the electronic effect.60,61 For the cyanide moiety, the electron-acceptor ability is weakest among the electron-withdrawing groups, and it has the least steric hindrance.59 The meta-terphenyl electronic nodal-position effects with ICT control were investigated depending on the electron-withdrawing groups. The photophysical and electrochemical properties including ICT properties, were examined and density functional theory (DFT) calculations were performed. In particular, distinctive ICT emissions depending on the electron-withdrawing group were compared to understand the electronic nodal-position effects on the meta-terphenyl by changing the electron-withdrawing group.
Results and discussion
Synthesis and structure characterization
Scheme 1 shows the synthetic procedure for meta-terphenyl linked D–π–A dyads. The starting materials, (4-(diphenylamino)phenyl)boronic acid, 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile, 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)benzo[d]oxazole, (4-(benzo[d]thiazol-2-yl)phenyl)boronic acid, and 2,4-diphenyl-6-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1,3,5-triazine, were prepared according to the previously reported procedures.62 All dyads were then synthesized using two-step palladium-catalyzed Suzuki–Miyaura coupling reactions with triphenylamine (TPA) as an electron donor, and cyanide, benzoxazole, benzothiazole, and 2,4-diphenyl-1,3,5-triazine as an electron acceptor. All four compounds were purified by silica gel column chromatography and showed yields in the range of 15 to 75% due to solubility issues. The molecular structures of all products were determined by 1H- and 13C{1H}-nuclear magnetic resonance (NMR) spectroscopy (Fig. S1–S8†),63,64 elemental analysis, and mass spectrometry (Fig. S9†). Finally, the structures of Tria were authenticated by single-crystal X-ray crystallography. The Experimental section and ESI† provide details of the synthetic procedures and characterization data. | ||
| Scheme 1 Synthetic routes of Cya, Oxa, Thia, and Tria. | ||
For X-ray crystallographic analysis, a single crystal of Tria was grown by slow evaporation in a CH2Cl2/n-hexane mixture at room temperature. The Tria crystal was triclinic in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Table S1† lists the detailed crystallographic data for the molecule, and Fig. S10† shows Oak Ridge thermal ellipsoid plot (ORTEP) drawings of Tria. In the packing diagram (Fig. 1b and c), the Tria molecules were stacked in a β packing (sheet-like packing) type.65 The degree of π-conjugation of Tria in the solid-state was determined by the terphenyl torsion angles. In the X-ray structure (Fig. 1a), the angle between the central benzene and the phenyl bound to the TPA moiety was ca. 28.72° (C20–C19–C16–C15), and that between the central benzene and the phenyl bound to the triazine moiety was ca. 11.79° (C20–C21–C25–C26), suggesting that the triazine moiety is more planar than TPA in the solid state. The intramolecular distance between the N atom of TPA and the C atom of triazine bound to terphenyl was 12.2 Å (Fig. 1a), which agreed with the DFT calculated value (12.4 Å). Based on the results obtained for Tria, we assumed that the molecular structures of the dyads were reliably predicted by DFT calculation. As shown in Fig. S16,† the dihedral angle for Cya, Oxa, Thia, and Tria between the phenyl rings close to acceptor moiety were 37.2°, 36.8°, 36.6°, and 37.0°, respectively. The distance from the central benzene to the acceptor moiety increased in the order of Cya (9.7 Å) < Oxa (13.0 Å) < Thia (13.3 Å) < Tria (13.9 Å). Therefore, as a result of considering the two factors of dihedral angle and distance, it indicates bulkiness of dyads increased in the order of Cya < Oxa < Thia < Tria.
space group. Table S1† lists the detailed crystallographic data for the molecule, and Fig. S10† shows Oak Ridge thermal ellipsoid plot (ORTEP) drawings of Tria. In the packing diagram (Fig. 1b and c), the Tria molecules were stacked in a β packing (sheet-like packing) type.65 The degree of π-conjugation of Tria in the solid-state was determined by the terphenyl torsion angles. In the X-ray structure (Fig. 1a), the angle between the central benzene and the phenyl bound to the TPA moiety was ca. 28.72° (C20–C19–C16–C15), and that between the central benzene and the phenyl bound to the triazine moiety was ca. 11.79° (C20–C21–C25–C26), suggesting that the triazine moiety is more planar than TPA in the solid state. The intramolecular distance between the N atom of TPA and the C atom of triazine bound to terphenyl was 12.2 Å (Fig. 1a), which agreed with the DFT calculated value (12.4 Å). Based on the results obtained for Tria, we assumed that the molecular structures of the dyads were reliably predicted by DFT calculation. As shown in Fig. S16,† the dihedral angle for Cya, Oxa, Thia, and Tria between the phenyl rings close to acceptor moiety were 37.2°, 36.8°, 36.6°, and 37.0°, respectively. The distance from the central benzene to the acceptor moiety increased in the order of Cya (9.7 Å) < Oxa (13.0 Å) < Thia (13.3 Å) < Tria (13.9 Å). Therefore, as a result of considering the two factors of dihedral angle and distance, it indicates bulkiness of dyads increased in the order of Cya < Oxa < Thia < Tria.
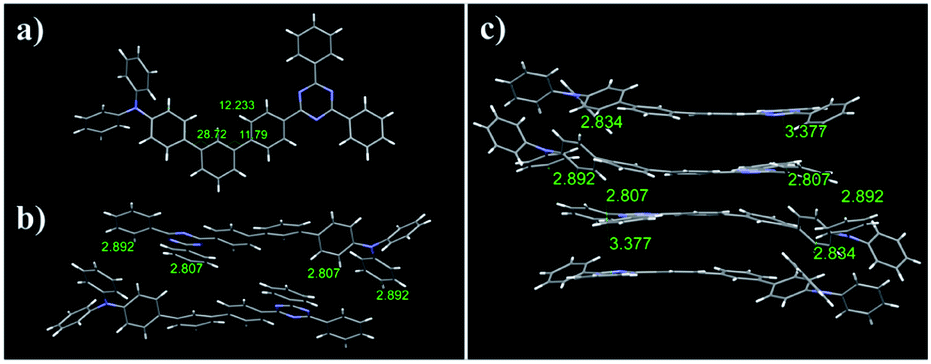 | ||
| Fig. 1 (a) Crystal structure diagram of Tria. (b) Molecular packing of the dimeric structures of Tria. (c) Crystal packing diagram of Tria. | ||
Photophysical properties
Fig. 2 shows the steady-state UV-visible absorption and emission spectra of Cya–Tria, and Table 1 lists the spectral parameters. In the UV-visible absorption spectra (Fig. S11†), all dyads showed an absorption band at 275–346 nm, which was attributed to a locally excited (LE) transition of the donor and each acceptor moieties. The shoulder hidden in the broad absorption band at ∼350 nm corresponds to the 1ICT ← S0 transition, which means an intramolecular interaction in the ground state. For Thia, the ICT transition was more red-shifted than Oxa in the absorption spectrum due to the strong acceptor ability. The red shift of the ICT transition was expected in the order of strong acceptor ability, Cya < Oxa < Thia < Tria, but the ICT transition was red-shifted in the order of Tria, Oxa < Thia < Cya. According to a previous report, compared to the well-conjugated para-terphenyl, the meta-terphenyl linker has weak electron coupling between donor and acceptor because only two phenyl units are conjugated by the electron node.40 From this point of view, this tendency suggests that ICT is determined by steric hindrance rather than the acceptor ability at the ground state, because of the lack of π-conjugation of the terphenyl linker by the electron nodes in the meta-position. The acceptor moiety of Tria has relatively stronger acceptor ability than the other compounds, but weak electron coupling between the donor and acceptor occurs because of its nonplanar molecular structure. In contrast, the acceptor ability of Cya is relatively weak, but the steric hindrance is small, resulting in strong coupling between the donor and acceptor. In the case of Oxa and Thia, the acceptor moieties have a more planar molecular structure than Tria and less steric hindrance. Hence, the ICT transition is observed with a red shift than that of Tria in the absorption spectrum. The thin-film absorption and emission spectra were also measured to examine the intermolecular interactions (Fig. S12†). The thin-film absorption and emission spectra of Cya–Tria exhibited red shifts compared to the corresponding spectra in an n-hexane solution, suggesting that all dyads formed J-aggregates.66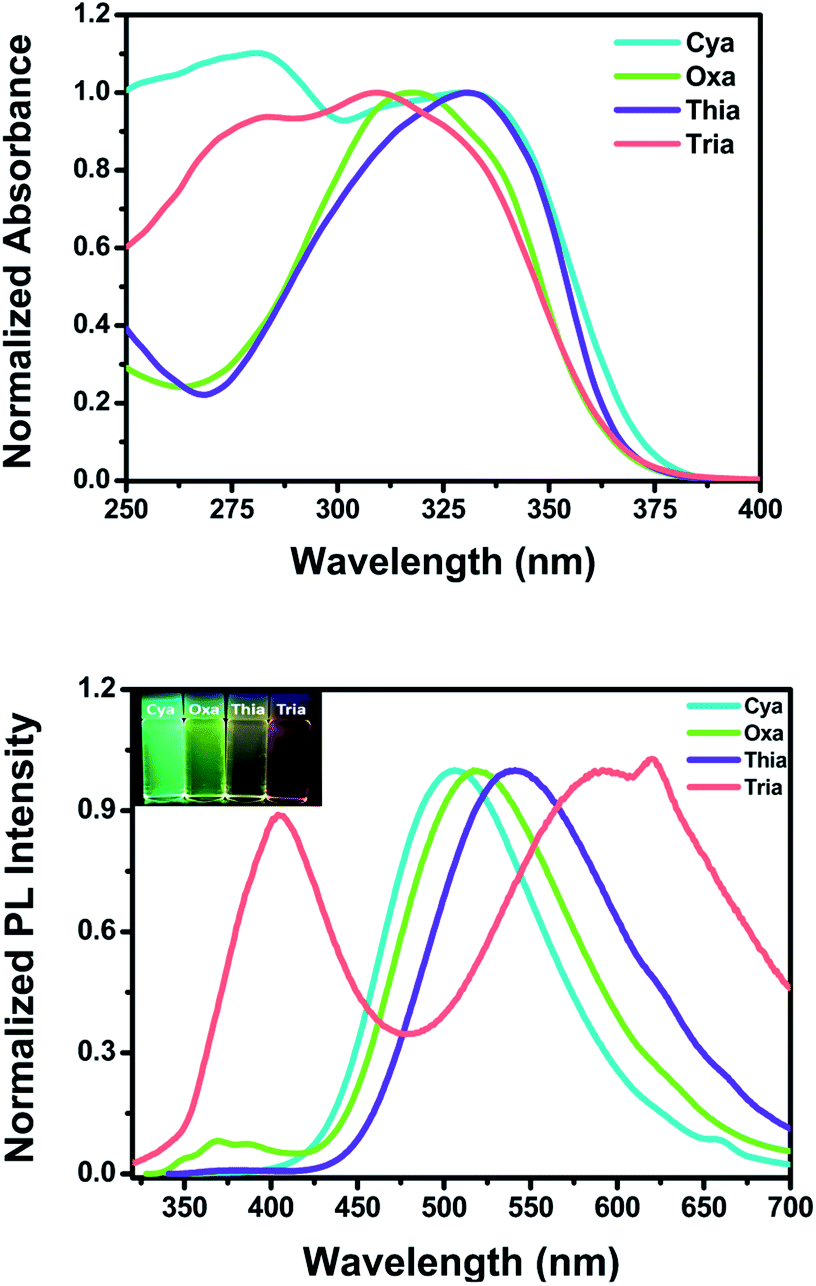 | ||
| Fig. 2 UV-vis absorption (top) and emission (bottom) spectra of Cya–Tria in CH2Cl2. Insets: photographs of Cya–Tria in CH2Cl2 solution under UV irradiation at 365 nm. | ||
| λabs (nm) | λem (nm) | Stokes shift (cm−1) | Φemc | τemd (ns) | krade × 107 (s−1) | knre × 107 (s−1) | krad/knr | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Sol (ε)a | Filmb | Sola | Filmb | |||||||
| a Measured in 1 × 10−5 M CH2Cl2 at room temperature (RT).b Measured from a drop-casted film on glass.c Fluorescence quantum yields, with 9,10-diphenylanthracene (ΦPL = 0.95, ethanol) as the standard, in CH2Cl2 at RT.d Fluorescence lifetime measured in CH2Cl2.e Values of krad and knr were calculated using krad =Φem/τem and knr =(1/τem) − krad, respectively. | ||||||||||
| Cya | 281, 329 (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 308) 308) |
351 | 506 | 441 | 10632 |
0.099 | 3.05, 22.43 | 3.24, 0.44 | 29.5, 4.02 | 0.11, 0.11 |
| Oxa | 318, 338 (76225) |
333 | 368, 517 | 442 | 10243 |
0.020 | 2.68, 22.22 | 0.75, 0.09 | 36.57, 4.41 | 0.02, 0.02 |
| Thia | 331, 346 (72182) |
357 | 375, 541 | 388, 459 | 10417 |
0.012 | 21.86 | 0.055 | 4.52 | 0.012 |
| Tria | 309, 330 (66976) |
334 | 405, 591 | 390, 486 | 13382 |
0.0096 | 5.18, 38.94 | 0.19, 0.025 | 19.12, 2.54 | 0.01, 0.01 |
Except for Cya, all dyads exhibited dual emission at approximately 375 nm and at a longer wavelength region of 506–591 nm in a CH2Cl2 solution (Fig. 2 (bottom)). The emission at short wavelengths can be assigned to the fluorescence from the locally excited state. In the case of Cya, no emission spectrum was observed at short wavelengths because electron coupling between the donor and acceptor is favored by the small steric hindrance of the cyano group. The long-wavelength emission was red-shifted in the order Cya < Oxa < Thia < Tria. The emission profiles ranged from blue-green to red, indicating that the emission color was tuned by the accepting ability. The emission at long-wavelengths was assigned to the fluorescence from the ICT state. These results suggest that the ICT is tuned by the electron-accepting ability in the excited state. In other words, the meta-position effect by the electron nodes has more influence in the ground state than in the excited state. For Tria, a greater Stokes shift of ICT transition in CH2Cl2 was observed than the other dyads, indicating relatively larger structural reorganization energy due to photo-excitation from the ground state to the excited state.
To further understand the properties of the excited state for the dyads, fluorescence quantum yield (Φem) and fluorescence lifetime (τem) measurements were performed in CH2Cl2; the results are shown in Table 1. Similar to the meta-position-based dyad reported previously, the quantum yields for the dyads were in the range of 0.099–0.0096 because of the deactivation pathways from the excited state caused by the electron nodes of terphenyl meta-positions.46 The fluorescence lifetime measurements performed on Cya, Oxa, and Tria exhibited biexponential fluorescence decays. In contrast, Thia exhibited single exponential fluorescence decay (Fig. S13†). As reported previously, for Cya and Oxa, the slow process of the biexponential fluorescence decay was attributed to charge transfer from the donor to acceptor moieties and the fast process of the biexponential fluorescence decay was attributed to charge transfer centered in and around TPA.46 In the case of Thia, the strong charge transfer from the donor to acceptor rather than the charge transfer centered in and around TPA by the strong acceptor of the planar structure would be favored. Therefore, single exponential fluorescence decay is attributed to the strong charge transfer. Because Tria has a strong acceptor with a nonplanar structure, it was assumed that the slow process of the biexponential fluorescence decay could be attributed to charge transfer from TPA to two phenyls of triazine. The rapid process of the biexponential fluorescence decay can be attributed to charge transfer from TPA to the triazine center. As the electron-accepting ability of the substituent increases, the fluorescence quantum yield decreases, and the lifetime increases. These results can be explained mainly by the radiative and nonradiative decay rate constants (krad and knr), which were calculated from the Φem and τem values (Table 1). All dyads have relatively larger knr values than krad, indicating that the non-conjugated structure of the dyads favors nonradiative decay. In addition, as the electron-accepting ability of the substituent increases, the krad value tended to decrease overall, but the knr value did not. This means that the electron-accepting ability of the substituent controls the krad value. Hence, the krad value plays a key role in controlling the emission color.
Solvatochromic properties
The absorption and emission behaviors of the meta-position-based dyads were investigated in various solvents to study the solvatochromic effect (Table S2 and Fig. S14†). The absorption spectra of all dyads showed a weak solvent polarity dependency. In contrast, the emission spectra of Cya–Tria showed a marked red shift with increasing solvent polarity from n-hexane to acetonitrile, indicating the presence of the ICT state in the excited state (Fig. 3). In addition, the solvatochromic shift in the emission spectra increased gradually from Cya to Tria, meaning that it has strong ICT properties as the electron-accepting ability of the substituent increases.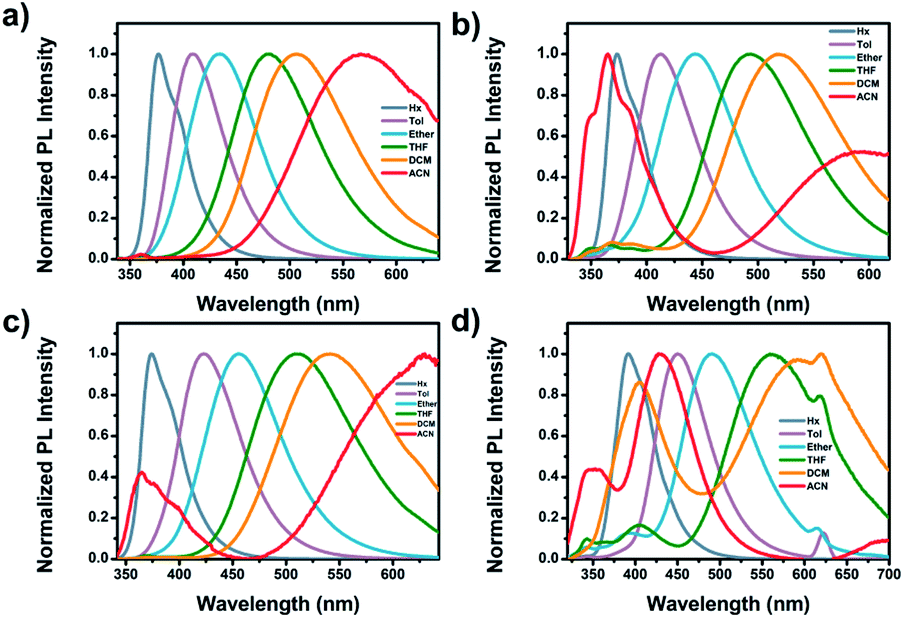 | ||
| Fig. 3 Emission spectra of (a) Cya, (b) Oxa, (c) Thia, and (d) Tria measured in various solvents. | ||
The dipole moments between the ground and excited states using a Lippert–Mataga plot were analyzed to understand the solvation effects better. The Stokes shift was plotted as a function of the solvent polarity parameter using eqn (1):67
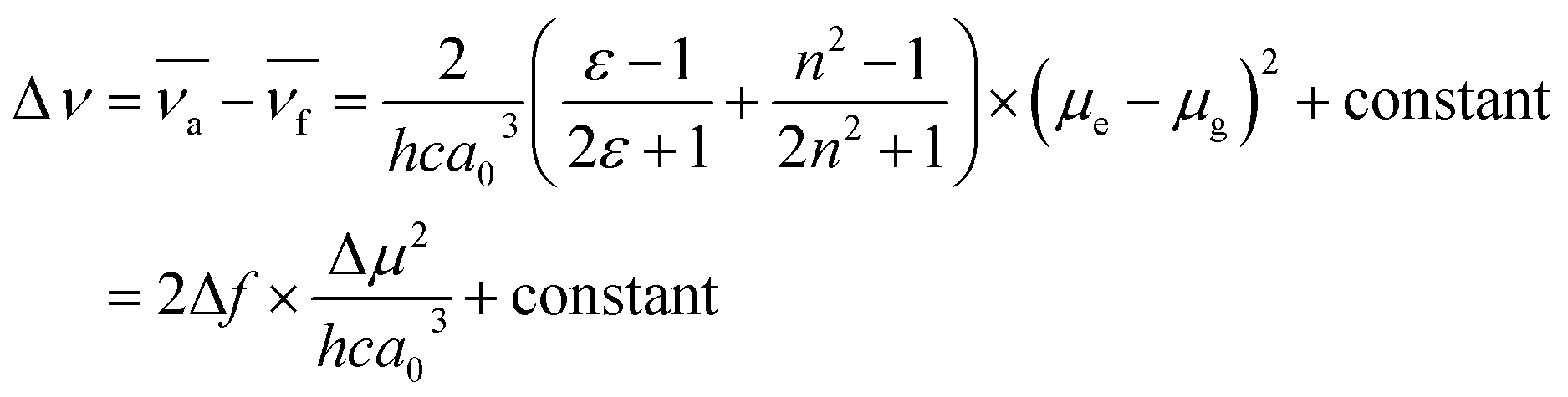 | (1) |
The ground state dipole moments for Cya, Oxa, Thia, and Tria were 5.92, 1.98, 1.52, and 0.94 D, respectively, which increases with increasing degree of asymmetry of the dyads. On the other hand, the excited state dipole moment increased in the order of Cya (40.41 D) < Oxa (61.69 D) < Thia (65.73 D) < Tria (73.39 D) as the electron-accepting ability increased. Hence, the excited state dipole moments for Cya–Tria are significantly greater than the ground state dipole moment, qualitatively showing a small charge transfer gap in the ground state and a large charge transfer gap in the excited state, as measured by steady-state UV-vis absorption and emission spectroscopy. Unlike the excited state dipole moment, the tendency of the ground state dipole moment did not coincide with the accepting ability, which suggests that ICT control by the accepting ability is controlled only in the excited state. Fig. 4 and S15† presents the plots of Δν versus Δf for four dyads, showing the linear correlation between these two factors. The trend in the slope of the Lippert–Mataga plots follows the order Cya < Oxa < Thia < Tria, and the slope of the Lippert–Mataga plot quantitatively represents the ICT process in the excited state. Hence, Tria has strong ICT character than other dyads. These results indicate that the ICT properties of meta-position-based dyads are controlled by their electron-accepting ability.
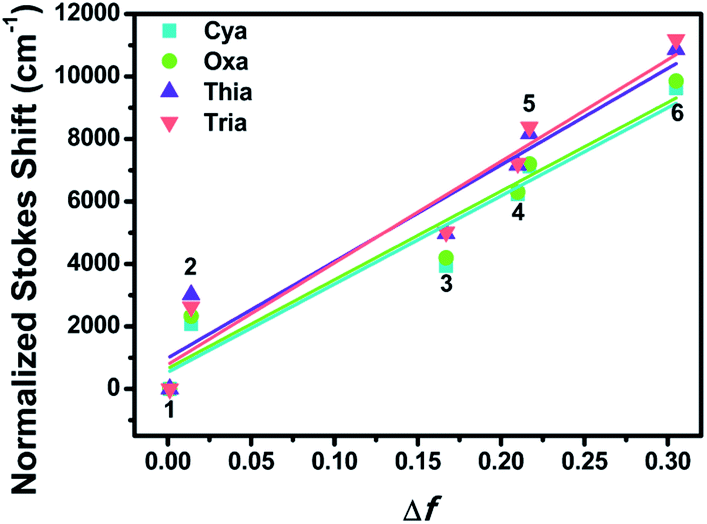 | ||
| Fig. 4 Lippert–Mataga plots of Cya–Tria in various solvents: 1, n-hexane; 2, toluene; 3, ethyl ether; 4, THF; 5, CH2Cl2; 6, CH3CN. | ||
| HOMOa (eV) | LUMOb (eV) | Eg (eV) | HOMOc (eV) | LUMOc (eV) | Ecalg (eV) | |
|---|---|---|---|---|---|---|
| a EHOMO (eV) = −e(Eoxonset + 4.8).b ELUMO (eV) = −e(Eredonset + 4.8).c Obtained by DFT calculation. | ||||||
| Cya | −5.72 | −2.91 | 2.81 | −5.08 | −1.65 | 3.43 |
| Oxa | −5.70 | −3.01 | 2.69 | −4.99 | −1.60 | 3.39 |
| Thia | −5.72 | −3.80 | 1.92 | −4.99 | −1.71 | 3.28 |
| Tria | −5.71 | −3.82 | 1.89 | −4.97 | −1.88 | 3.09 |
Electrochemical properties
Cyclic voltammetry (CV) was performed to evaluate the electrochemical properties of meta-terphenyl linked donor–π–acceptor dyads in CH2Cl2 and THF, using a three-electrode cell system, with glassy carbon, platinum wire, and SCE as the working, counter, and reference electrodes, respectively (Fig. 5). All dyads showed reversible oxidation peaks with oxidation onsets at 0.92, 0.90, 0.92, and 0.91 V for Cya, Oxa, Thia, and Tria, respectively, because the dyads had the same electron donor moiety. Hence, the dyads have a similar highest occupied molecular orbital (HOMO) energy level. In contrast, for a reversible reduction peak, the onset potentials of the first reduction were shifted anodically in the order of Cya (−1.89 V) < Oxa (−1.79 V) < Thia (−1.00 V) < Tria (−0.98 V). This tendency reflects the greater enhancement of the electron-accepting ability when going from Cya to Tria. Furthermore, for Tria, the first and second reduction peaks were close, suggesting that the lowest unoccupied molecular orbital (LUMO) and LUMO + 1 energy levels would be close.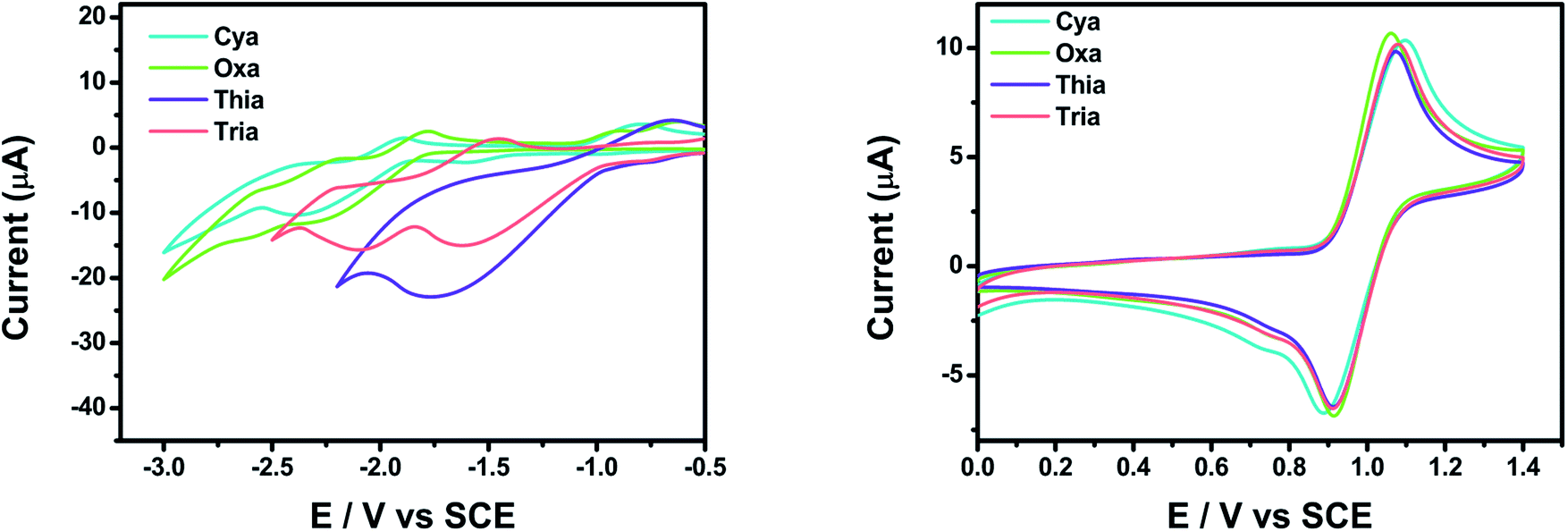 | ||
| Fig. 5 CV curves of Cya, Oxa, Thia, and Tria. The reduction potentials (left) were measured in THF and the oxidation potentials (right) in CH2Cl2. In all experiments, the electrolyte was 0.1 M TBAP and was measured at a scan rate of 0.05 V s−1. | ||
The experimental and calculated HOMO–LUMO energy levels of the meta-terphenyl linked donor–π–acceptor dyads were compared (Table 2). The experimental HOMO energy levels could be determined from the oxidation onset potentials, and the HOMO energy levels of Cya–Tria were estimated to be ca. −5.72 to −5.71 eV. The experimental LUMO energy levels were estimated to be ca. −2.91 to −3.82 eV from the reduction onset potentials. The experimental LUMO levels decreased with increasing electron-accepting ability with a maximum difference of 0.91 eV. In contrast, the experimental HOMO levels were changed slightly, indicating that the LUMO levels were susceptible to the electronic variations of the substituents. The calculated HOMO energy levels ranged from −5.08 to −4.97 eV, and the calculated LUMO energy levels ranged from −1.65 to −1.88 eV. The calculated values were higher than the experimental values for all compounds, but the tendencies were similar. These results show that an increase in the electron-accepting ability stabilizes the LUMO more than the HOMO, narrowing the bandgap and controlling the emission color.
Density functional theory (DFT)
Fig. 6 and S17–S20† presents the HOMO and LUMO orbitals of meta-terphenyl linked donor–π–acceptor dyads, which were obtained using Gaussian 16 and visualized using Chem3D and GaussView programs. The geometries were optimized by the DFT method using the B3LYP correlation function employing the 6-31G(d,p) level in the vacuum state.68–70 In all dyads studied, the HOMO was distributed mainly on the TPA moiety, and the LUMO was strongly localized on each acceptor moiety. These changes in the orbital distributions of the HOMOs and LUMOs showed that charge transfer occurs from the donor to acceptor in the excited state. The meta-terphenyl structure is conjugated at only two phenyl units because of the electronic nodes at the meta-position. In addition, the LUMOs of dyads were extended to the acceptor moiety rather than meta-terphenyl linker in the order Cya < Oxa < Thia < Tria, indicating that it is controlled by the accepting ability. Unlike the other dyads, the LUMO + 1 of Tria was centered mainly on the two phenyl groups of triazine moiety. These orbital distributions suggest that the fluorescence decay profile and electrochemical properties of Tria were determined by the nonplanar structure and strong electron-accepting ability of triazine.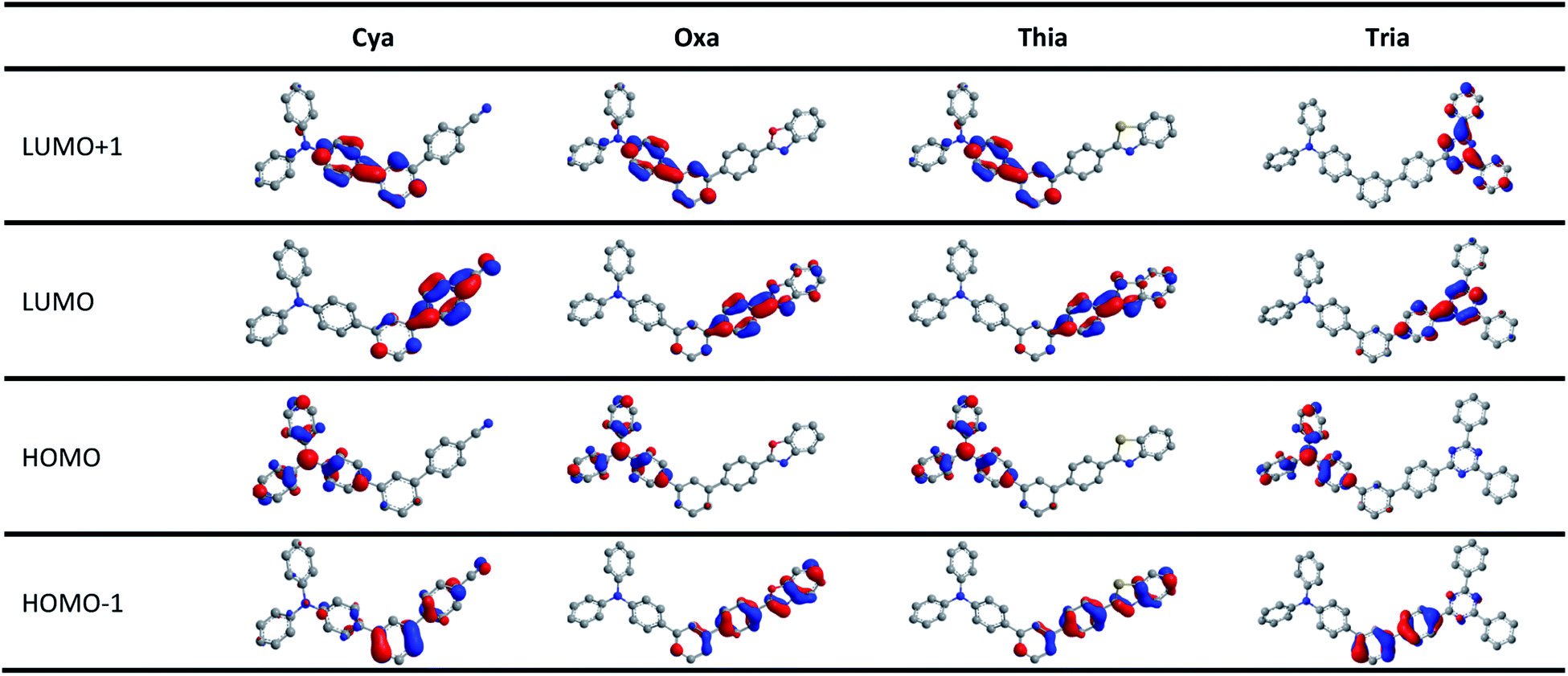 | ||
| Fig. 6 Frontier orbital distributions (HOMO − 1, HOMO, LUMO, LUMO + 1) of Cya, Oxa, Thia, and Tria calculated by DFT with the B3LYP function and the 6-31G(d,p) basis. | ||
As shown in Fig. S21–S24 and Tables S9–S12 of ESI,† time-dependent density functional theory (TD-DFT) calculations were carried out at the same functional and basis set. For all dyads, the lowest-energy singlet transition involved the LUMO ← HOMO transitions; all were ICT transitions from the donor to acceptor. These transitions of Cya, Oxa, Thia, and Tria are very weak, with oscillator strengths between 0.0156–0.0035. On the other hand, the LUMO + 1 ← HOMO transitions of Cya, Oxa, and Thia, and the LUMO + 2 ← HOMO transitions of Tria are very strong with oscillator strengths between 0.6835–1.257, showing that all are transitions centered in and around TPA. These results are because the substitution of the terphenyl linker at the meta-position causes the nodal site in the HOMO and LUMO. Furthermore, the LUMO ← HOMO transition shifts theoretically followed the order, Cya (400 nm), Oxa (402 nm), Thia (416 nm), and Tria (439 nm), which are consistent with the electron-accepting ability. Nevertheless, the experimental results were different from the theoretical results, suggesting that the properties of meta-terphenyl linked donor–π–acceptor dyads in the ground state should consider steric hindrance rather than the electron-accepting ability.
Conclusions
meta-Terphenyl linked donor–π–acceptor (D–π–A) dyads were designed and synthesized to understand the electronic effects of the meta-terphenyl linker in the D–π–A dyads. By modulating their electron-accepting ability by introducing cyanide, benzoxazole, benzothiazole, and 2,4-diphenyl-1,3,5-triazine moieties, the photophysical and electrochemical properties of the four dyads were investigated and compared. In the steady-state emission spectra, the long-wavelength emission was red-shifted in the order of strong acceptor ability, Cya < Oxa < Thia < Tria, and a bathochromic shift property was observed with increasing solvent polarity. These results showed that the ICT was tuned by the electron-accepting ability in the excited state. Similarly, the Lippert–Mataga plot and cyclic voltammetry curves were consistent with the electron-accepting ability. On the other hand, in the steady-state absorption spectra, the ICT transition was red-shifted in the order of Tria, Oxa < Thia < Cya, and was different from the electron-accepting ability tendency. Hence, ICT is determined by steric hindrance rather than the acceptor ability at the ground state due to the meta-position effect by the electron nodes. These results highlight the need to consider ICT modulation in the ground and excited states of meta-terphenyl linked donor–π–acceptor dyads with an electron node.Experimental
General information
Based on standard Schlenk techniques, all of the synthesis experimental procedures were performed under a dry argon condition. Reagents and solvents were purchased from commercial sources and used as received without further purification, unless otherwise stated. All reactions were monitored with thin layer chromatography (TLC) using commercial TLC plates (Merck Co.). Silica gel column chromatography was performed on silica gel 60 G (230–400 mesh ASTM, Merck Co.). The synthesized compounds were characterized by 1H-NMR or 13C{1H}-NMR, and elemental analysis. The 1H and proton-decoupled 13C spectra were recorded on a Bruker 500 spectrometer operating at 500 and 125 MHz, respectively, and all proton and carbon chemical shifts were measured relative to internal residual chloroform (99.5% CDCl3) from the lock solvent. The elemental analyses (C, H, N, O, S) were performed using Thermo Fisher Scientific Flash 2000 series analyzer. The GC-MS analysis was performed using a highly sensitive Gas Chromatograph/Mass Selective Detector spectrometer (Agilent, 7890B-5977B GC/MSD). The crystal structure was determined by single-crystal X-ray diffractometer at the Western Seoul Center of Korea Basic Science Institute.Synthesis of 4′′-(diphenylamino)-[1,1′:3′,1′′-terphenyl]-4-carbonitrile (Cya)
A mixture of 3′-bromo-N,N-diphenyl-[1,1′-biphenyl]-4-amine (0.38 g, 0.95 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (0.3 g, 1.33 mmol), Pd(PPh3)4 (10 mol%), K2CO3 (0.66 g, 4.75 mmol) in toluene/H2O (v/v = 40 mL/10 mL) was refluxed under argon at 110 °C for overnight. After cooling to room temperature, deionized water (50 mL) was poured and organic layer was separated using a separating funnel. The water layer was washed using methylene chloride (×3) for extracted remained organic residue. After combined all of organic solvents, the organic layer was dried over anhydrous MgSO4 and then filtered off. The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography using dichloromethane/n-hexane (v/v = 1:2) as the eluent and obtained light yellow powder. Yield: 0.22 g, 55%. 1H NMR (500 MHz, CDCl3, ppm) δ 7.76 (s, 1H), 7.74 (s, 4H), 7.63–7.61 (m, 1H), 7.53–7.52 (m, 2H), 7.50 (d, J = 9.0 Hz, 2H), 7.27 (t, J = 7.5 Hz, 4H), 7.15 (t, J = 7.0 Hz, 6H), 7.05 (t, J = 7.5 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 147.66, 147.60, 145.76, 141.74, 139.73, 134.35, 132.63, 129.53, 129.34, 127.85, 126.98, 125.68, 125.63, 124.58, 123.74, 123.15, 118.92, 111.06. GC-MS (m/z) calcd for C31H22N2: 422.53, found: 422.3 [M]+. Anal. calcd for C31H22N2: C, 88.12; H, 5.25; N, 6.63. Found: C, 88.17; H, 5.24; N, 6.59.
Synthesis of 4′′-(benzo[d]oxazol-2-yl)-N,N-diphenyl-[1,1′:3′,1′′-terphenyl]-4-amine (Oxa)
A mixture of 3′-bromo-N,N-diphenyl-[1,1′-biphenyl]-4-amine (0.45 g, 1.13 mmol), the 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)benzo[d]oxazole (0.75 g, 1.47 mmol), Pd(PPh3)4 (5 mol%), K2CO3 (5.64 g, 5.64 mmol) in toluene/H2O (v/v = 40 mL/10 mL) was refluxed under argon at 110 °C for overnight. After cooling to room temperature, deionized water (50 mL) was poured and organic layer was separated using a separating funnel. The water layer was washed using methylene chloride (×3) for extracted remained organic residue. After combined all of organic solvents, the organic layer was dried over anhydrous MgSO4 and then filtered off. The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography using all dichloromethane after that, ethyl acetate/n-hexane (v/v = 1:10) as the eluent and obtained white powder. Yield: 0.17 g, 29%. 1H NMR (500 MHz, CDCl3, ppm) δ 8.34 (d, J = 8.5 Hz, 2H), 7.85 (s, 1H), 7.82 (d, J = 9.0 Hz, 2H), 7.80–7.79 (m, 1H), 7.62–7.59 (m, 3H), 7.55–7.51 (m, 3H), 7.39–7.35 (m, 2H), 7.28 (t, J = 7.0 Hz, 4H), 7.16 (t, J = 8.5 Hz, 6H), 7.04 (t, J = 7.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 162.95, 150.84, 147.66, 147.50, 144.29, 142.24, 141.55, 140.54, 134.77, 129.38, 129.33, 128.13, 127.93, 127.68, 126.46, 126.09, 125.66, 125.65, 125.15, 124.64, 124.52, 125.86, 123.05, 120.03, 110.62. GC-MS (m/z) calcd for C37H26N2O: 514.20, found: 514.3 [M]+. Anal. calcd for C37H26N2O: C, 86.35; H, 5.09; N, 5.44; O, 3.11. Found: C, 86.53; H, 5.12; N, 5.30; O, 3.05.
Synthesis of 4′′-(benzo[d]thiazol-2-yl)-N,N-diphenyl-[1,1′:3′,1′′-terphenyl]-4-amine (Thia)
A mixture of 3′-bromo-N,N-diphenyl-[1,1′-biphenyl]-4-amine (0.67 g, 1.68 mmol), (4-(benzo[d]thiazol-2-yl)phenyl)boronic acid (0.64 g, 2.51 mmol), Pd(PPh3)4 (5 mol%), K2CO3 (1.16 g, 8.4 mmol) in THF/H2O (v/v = 40 mL/10 mL) was refluxed under argon at 110 °C for overnight. After cooling to room temperature, deionized water (50 mL) was poured and organic layer was separated using a separating funnel. The water layer was washed using methylene chloride (×3) for extracted remained organic residue. After combined all of organic solvents, the organic layer was dried over anhydrous MgSO4 and then filtered off. The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography using dichloromethane/n-hexane (v/v = 1:2) as the eluent and obtained white powder. Yield: 0.67 g, 75%. 1H NMR (500 MHz, CDCl3, ppm) δ 8.19 (d, J = 8.5 Hz, 2H), 8.09 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.84 (s, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.60–7.59 (d, J = 7.5 Hz, 2H), 7.55–7.49 (m, 4H), 7.40 (t, J = 7.0 Hz, 1H), 7.28 (t, J = 8.5 Hz, 4H), 7.16 (t, J = 9.0 Hz, 6H), 7.04 (t, J = 7.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 167.70, 154.26, 147.67, 147.49, 143.79, 141.52, 140.61, 135.11, 134.81, 132.65, 129.36, 129.32, 128.04, 127.92, 127.76, 126.39, 126.35, 125.60, 125.59, 125.23, 124.52, 123.85, 123.24, 123.05, 121.64. GC-MS (m/z) calcd for C37H26N2S: 530.18, found: 530.4 [M]+. Anal. calcd for C37H26N2S: C, 83.74; H, 4.94; N, 5.28; S, 6.04. Found: C, 83.72; H, 4.97; N, 5.26; S, 6.05.
Synthesis of 4′′-(4,6-diphenyl-1,3,5-triazin-2-yl)-N,N-diphenyl-[1,1′:3′,1′′-terphenyl]-4-amine (Tria)
A mixture of 3′-bromo-N,N-diphenyl-[1,1′-biphenyl]-4-amine (0.68 g, 1.70 mmol), 2,4-diphenyl-6-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1,3,5-triazine (1.04 g, 2.38 mmol), Pd(PPh3)4 (5 mol%), K2CO3 (1.20 g, 8.50 mmol) in THF/H2O (v/v = 40 mL/10 mL) was refluxed under argon at 110 °C for overnight. After cooling to room temperature, deionized water (50 mL) was poured and organic layer was separated using a separating funnel. The water layer was washed using methylene chloride (×3) for extracted remained organic residue. After combined all of organic solvents, the organic layer was dried over anhydrous MgSO4 and then filtered off. The solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography using dichloromethane/n-hexane (v/v = 1:5) as the eluent and obtained white powder. Yield: 0.15 g, 15%. 1H NMR (500 MHz, CDCl3, ppm) δ 8.87 (d, J = 8.5 Hz, 2H), 8.80 (d, J = 8.5 Hz, 4H), 7.89 (s, 1H), 7.86 (d, J = 8.5 Hz, 2H), 7.66–7.53 (m, 11H), 7.29 (t, J = 8.5 Hz, 4H), 7.17 (t, J = 8.5 Hz, 6H), 7.05 (t, J = 7.5 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 171.70, 171.46, 147.68, 147.49, 145.28, 141.48, 140.96, 136.32, 135.31, 134.86, 132.52, 129.52, 129.33, 129.01, 128.67, 127.94, 127.45, 126.39, 125.80, 125.75, 124.53, 123.86, 123.05. GC-MS (m/z) calcd for C45H32N4: 628.26, found: 628.5 [M]+. Anal. calcd for C45H32N4: C, 85.96; H, 5.13; N, 8.91. Found: C, 85.91; H, 5.11; N, 8.98.
Preparation of single crystal Tria
For the crystallization of dyad Tria, a vacuum-dried pure sample of Tria was taken in a vial and dissolved in CH2Cl2/n-hexane (1:1) mixed solvent; by the slow evaporation at room temperature for about 4 weeks, a white Tria single crystal was obtained. Then the crystals were picked up from the vial and a single-crystal XRD study was performed.
X-ray crystal structure analysis
The data were collected at 223(2) K using a Bruker D8 Venture equipped with IμS micro-focus sealed tube Mo Kα (λ = 0.71073 Å) and a PHOTON II 14 detector in Western Seoul Center of Korea Basic Science Institute. Preliminary unit cell constants were determined using a set of 45 narrow-frame (0.3° in ω) scans. The double pass method of scanning was used to exclude noise. Collected frames were integrated using an orientation matrix determined from narrow-frame scans. The SMART software package was used for data collection, and SAINT was used for frame integration.71 Final cell constants were determined by global refinement of xyz centroids of reflections harvested from the entire data set. Structure solution and refinement were carried out using the SHELXTL-PLUS software package.72 The crystallographic information has been deposited with the Cambridge Crystallographic Data Centre and assigned the CCDC code 2091706 for Tria.Photophysical measurements
The UV/vis absorption spectra were recorded using a Sinco Mega-2100 spectrophotometer in dual beam mode, and the fluorescence emission measurements were carried out using Shimadzu fluorometer (RF-6000) with a wavelength resolution of ∼1 nm. Fluorescence lifetimes were measured by PicoQuant FluoTime 200 that takes advantage of the time-correlated single photon counting method. A pulsed diode laser operated at 20 MHz repetition rate was used as the excitation source. The FWHM of a laser pulse was typically 45 ps, and the instrument response function was ∼190 ps when the Hamamatsu photomultiplier tube (H5783-01) was used. The emission quantum yields (ΦPL) were calculated using William's comparative method for samples of five different concentrations of (1–5) μM, using 9,10-diphenylanthracene (ΦPL = 0.95, ethanol) as a reference standard.73Cyclic voltammetry (CV)
A CH Instruments 701D potentiostat was used for electrochemical measurements, and cyclic voltammetry (CV) was performed in an electrolytic solution prepared using 1 mM of electroactive compounds and 0.1 M tetrabutylammonium perchlorate (Bu4NClO4) in deoxygenated dichloromethane and THF. A three-electrode configuration, glassy carbon, platinum wire, and SCE were used as working, counter, and reference electrodes, respectively.Density functional theory calculations
Density functional theory (DFT) calculations were performed by using Gaussian' 16 software package. Full geometry optimizations in their ground state were performed using the B3LYP functional and the 6-31G(d,p) basis set for all atoms. The excitation energies and oscillator strengths for the lowest 100 singlet–singlet transitions at the optimized geometry in the ground state were obtained in time-dependent DFT (TD-DFT) calculations using the same basis set and functional as for the ground state. All isodensity plots of the frontier orbitals were visualized by Chem3D Ultra and GaussView software. More detail DFT/TD-DFT calculation results for all dyads are described in ESI.†Author contributions
M.-J. Kim, M. Ahn, M. Chae, S. Kim, and D. Kim prepared/performed the experiment including DFT calculations and prepared figures. M.-J. Kim and K.-R. Wee planned the experiment and wrote the paper. All authors contributed to the paper with discussions and edits.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2020R1C1C1009007) and Korea Basic Science Institute (National Research Facilities and Equipment Center) grant, funded by the Ministry of Education (2020R1A6C103A028).Notes and references
- M. T. Colvin, A. B. Ricks and M. R. Wasielewski, J. Phys. Chem. A, 2012, 116, 2184–2191 CrossRef CAS PubMed.
- W. Yang and Y. Li, Dyes Pigm., 2016, 125, 100–105 CrossRef.
- M. Ito, E. Ito, M. Hirai and S. Yamaguchi, J. Org. Chem., 2018, 83, 8449–8456 CrossRef CAS PubMed.
- P. Das, A. Kumar, A. Chowdhury and P. S. Mukherjee, ACS Omega, 2018, 3, 13757–13771 CrossRef CAS PubMed.
- R. Rani, G. Kumar, K. Paul and V. Luxami, New J. Chem., 2018, 42, 12729–12736 RSC.
- J. Van Damme, O. van den Berg, J. Brancart, G. Van Assche and F. Du Prez, Tetrahedron, 2019, 75, 912–920 CrossRef CAS.
- C. Wu, Z. Wu, B. Wang, X. Li, N. Zhao, J. Hu, D. Ma and Q. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32946–32956 CrossRef CAS PubMed.
- H. Sun, X. Song, J. Xie, P. Sun, P. Gu, C. Liu, F. Chen, Q. Zhang, Z.-K. Chen and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 29924–29931 CrossRef CAS PubMed.
- J. Zhang, J. Jin, H. Xu, Q. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- E. V. Verbitskiy, Y. A. Kvashnin, P. I. Bogdanov, M. V. Medvedeva, T. S. Svalova, A. N. Kozitsina, L. G. Samsonova, K. M. Degtyarenko, D. V. Grigoryev and A. E. Kurtcevich, Dyes Pigm., 2021, 187, 109124 CrossRef CAS.
- R. Furue, K. Matsuo, Y. Ashikari, H. Ooka, N. Amanokura and T. Yasuda, Adv. Opt. Mater., 2018, 6, 1701147 CrossRef.
- X. Liang, Z. Wang, L. Wang, M. Hanif, D. Hu, S. Su, Z. Xie, Y. Gao, B. Yang and Y. Ma, Chin. J. Chem., 2017, 35, 1559–1568 CrossRef CAS.
- S. Thurakkal, K. S. Sanju, A. Soman, K. N. Unni, J. Joseph and D. Ramaiah, New J. Chem., 2018, 42, 5456–5464 RSC.
- C. Poriel and J. Rault-Berthelot, Adv. Funct. Mater., 2021, 2010547 CrossRef CAS.
- C. c. Peng, S. y. Yang, H. c. Li, G. h. Xie, L. s. Cui, S. N. Zou, C. Poriel, Z. Q. Jiang and L. s. Liao, Adv. Mater., 2020, 32, 2003885 CrossRef CAS PubMed.
- F. Lucas, O. A. Ibraikulov, C. Quinton, L. Sicard, T. Heiser, D. Tondelier, B. Geffroy, N. Leclerc, J. Rault-Berthelot and C. Poriel, Adv. Opt. Mater., 2020, 8, 1901225 CrossRef CAS.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-k. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- A. Mishra, M. K. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- W. J. Shi, T. Kinoshita and D. K. Ng, Asian J. Org. Chem., 2017, 6, 758–767 CrossRef CAS.
- Y. Ooyama, Y. Hagiwara, Y. Oda, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2336–2340 RSC.
- A. Pallikkara and K. Ramakrishnan, Int. J. Energy Res., 2021, 45, 1425–1448 CrossRef CAS.
- A. Leliège, J. Grolleau, M. Allain, P. Blanchard, D. Demeter, T. Rousseau and J. Roncali, Chem.–Eur. J., 2013, 19, 9948–9960 CrossRef PubMed.
- Y. Cui, P. Li, C. Song and H. Zhang, J. Phys. Chem. C, 2016, 120, 28939–28950 CrossRef CAS.
- Y. N. Luponosov, A. N. Solodukhin, A. L. Mannanov, V. A. Trukhanov, S. M. Peregudova, S. A. Pisarev, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun and D. Y. Paraschuk, Org. Electron., 2017, 51, 180–189 CrossRef CAS.
- E. Aslan, M. Karaman, G. Yanalak, H. Bilgili, M. Can, F. Ozel and I. H. Patir, J. Photochem. Photobiol., A, 2020, 390, 112301 CrossRef CAS.
- B. D. Sherman, M. V. Sheridan, K.-R. Wee, S. L. Marquard, D. Wang, L. Alibabaei, D. L. Ashford and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 16745–16753 CrossRef CAS PubMed.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- Q. Li, Z. Wang, W. Song, H. Ma, J. Dong, Y.-Y. Quan, X. Ye and Z.-S. Huang, Dyes Pigm., 2019, 161, 389–395 CrossRef CAS.
- S.-Y. Kim, M.-J. Kim, M. Ahn, K.-M. Lee and K.-R. Wee, Dyes Pigm., 2021, 191, 109362 CrossRef CAS.
- J.-P. Malval, M. Cranney, S. Achelle, H. Akdas-Kiliç, J.-L. Fillaut, N. Cabon, F. Robin-le Guen, O. Soppera and Y. Molard, Chem. Commun., 2019, 55, 14331–14334 RSC.
- S. Zhang, H. Zhu, J. Huang, L. Kong, Y. Tian and J. Yang, ChemistrySelect, 2019, 4, 7380–7387 CrossRef CAS.
- Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
- M.-J. Kim, M. Ahn and K.-R. Wee, Mater. Adv., 2021, 2, 5371–5380 RSC.
- P. Vineetha, A. Aswathy, E. Shiju, K. Chandrasekharan and N. Manoj, New J. Chem., 2020, 44, 6142–6150 RSC.
- C.-Z. Zhang, C. Lu, J. Zhu, G.-Y. Lu, X. Wang, Z.-W. Shi, F. Liu and Y. Cui, Chem. Mater., 2006, 18, 6091–6093 CrossRef CAS.
- K. Kannan, D. Radhika, A. Nesaraj, K. K. Sadasivuni, K. R. Reddy, D. Kasai and A. V. Raghu, Mater. Sci. Energy Technol., 2020, 3, 853–861 CAS.
- R. Prabhu, T. Jeevananda, K. R. Reddy and A. V. Raghu, Mater. Sci. Energy Technol., 2021, 4, 107–112 CAS.
- K. R. Reddy, B. Hemavathi, G. R. Balakrishna, A. Raghu, S. Naveen and M. Shankar, in Polymer Composites with Functionalized Nanoparticles, Elsevier, 2019, pp. 357–379 Search PubMed.
- Y.-J. Cho, A.-R. Lee, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 22921–22928 RSC.
- D. Thirion, M. Romain, J. Rault-Berthelot and C. Poriel, Mater. Adv., 2021, 2, 1271–1283 RSC.
- S. Delmond, J.-F. Létard, R. Lapouyade and W. Rettig, J. Photochem. Photobiol., A, 1997, 105, 135–148 CrossRef CAS.
- X. Niu, P. Gautam, Z. Kuang, P. Y. Craig, Y. Guo, H. Song, Q. Guo, J. M. Chan and A. Xia, Phys. Chem. Chem. Phys., 2019, 21, 17323–17331 RSC.
- M. Ahn, M.-J. Kim, D. W. Cho and K.-R. Wee, J. Org. Chem., 2020, 86, 403–413 CrossRef PubMed.
- M. Harrison and R. Friend, in Electronic Materials: The Oligomer Approach, Wiley-VCH, Weinheim, 1998, pp. 515–558 Search PubMed.
- M.-J. Kim, M. Ahn, J. H. Shim and K.-R. Wee, Phys. Chem. Chem. Phys., 2020, 22, 3370–3378 RSC.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li and P. Lu, Chem. Sci., 2015, 6, 3797–3804 RSC.
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2015, 54, 1176–1180 CrossRef CAS PubMed.
- C. Lu, C. Long, Y. Li, Z. Li and H. Zhu, J. Membr. Sci., 2020, 598, 117797 CrossRef CAS.
- Q. Wang, F. Lucas, C. Quinton, Y.-K. Qu, J. Rault-Berthelot, O. Jeannin, S.-Y. Yang, F.-C. Kong, S. Kumar and L.-S. Liao, Chem. Sci., 2020, 11, 4887–4894 RSC.
- S. F. Swallen, Z.-Y. Shi, W. Tan, Z. Xu, J. S. Moore and R. Kopelman, J. Lumin., 1998, 76, 193–196 CrossRef.
- C. Gao, S. K. Prasad, B. Zhang, M. Dvořák, M. J. Tayebjee, D. R. McCamey, T. W. Schmidt, T. A. Smith and W. W. Wong, J. Phys. Chem. C, 2019, 123, 20181–20187 CrossRef CAS.
- P. Guiglion and M. A. Zwijnenburg, Phys. Chem. Chem. Phys., 2015, 17, 17854–17863 RSC.
- L. Sicard, C. Quinton, J. D. Peltier, D. Tondelier, B. Geffroy, U. Biapo, R. Métivier, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem.–Eur. J., 2017, 23, 7719–7727 CrossRef CAS PubMed.
- L.-S. Cui, S.-C. Dong, Y. Liu, M.-F. Xu, Q. Li, Z.-Q. Jiang and L.-S. Liao, Org. Electron., 2013, 14, 1924–1930 CrossRef CAS.
- C. Poriel and J. Rault-Berthelot, Acc. Chem. Res., 2018, 51, 1818–1830 CrossRef CAS PubMed.
- N. Metri, X. Sallenave, C. d. Plesse, L. Beouch, P.-H. Aubert, F. Goubard, C. Chevrot and G. Sini, J. Phys. Chem. C, 2012, 116, 3765–3772 CrossRef CAS.
- J. Zhang, D. Deng, C. He, Y. He, M. Zhang, Z.-G. Zhang, Z. Zhang and Y. Li, Chem. Mater., 2011, 23, 817–822 CrossRef CAS.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, J. Mater. Chem. C, 2015, 3, 2175–2181 RSC.
- S.-H. Chen, K. Jiang, Y. Xiao, X.-Y. Cao, M. Arulkumar and Z.-Y. Wang, Dyes Pigm., 2020, 175, 108157 CrossRef CAS.
- I. M. Paczkowski, F. L. Coelho and L. F. Campo, J. Mol. Liq., 2020, 319, 114277 CrossRef.
- A. B. Morgan, J. L. Jurs and J. M. Tour, J. Appl. Polym. Sci., 2000, 76, 1257–1268 CrossRef CAS.
- A. Raghu, G. Gadaginamath, N. Mathew, S. Halligudi and T. Aminabhavi, J. Appl. Polym. Sci., 2007, 106, 299–308 CrossRef CAS.
- A. Raghu, G. Gadaginamath, M. Priya, P. Seema, H. M. Jeong and T. Aminabhavi, J. Appl. Polym. Sci., 2008, 110, 2315–2320 CrossRef CAS.
- J. E. Campbell, J. Yang and G. M. Day, J. Mater. Chem. C, 2017, 5, 7574–7584 RSC.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- B. Pramodh, P. Naresh, S. Naveen, N. Lokanath, S. Ganguly, J. Panda, S. Murugesan, A. Raghu and I. Warad, Chem. Data Collect., 2021, 31, 100587 CrossRef CAS.
- S. B. Prasad, S. Naveen, C. A. Kumar, N. Lokanath, A. Raghu, I. Daraghmeh, K. R. Reddy and I. Warad, J. Mol. Struct., 2018, 1167, 215–226 CrossRef.
- S. B. Prasad, C. Anandakumar, A. Raghu, K. R. Reddy, M. D. Urs and S. Naveen, Chem. Data Collect., 2018, 15, 1–9 Search PubMed.
- G. Sheldrick, SMART and SAINT, Bruker Analytical X-ray Division, Madison, WI, 2001 Search PubMed.
- G. Sheldrick, SHELXTL-PLUS Software Package, Bruker Analytical X-Ray Division, Madison, WI, 2002 Search PubMed.
- J. V. Morris, M. A. Mahaney and J. R. Huber, J. Phys. Chem., 1976, 80, 969–974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra; GC-MS data; X-ray crystallographic data of Tria; absorption and emission spectra of TPA donor; absorption and emission spectra in various solvents and solid state; emission lifetime profile; Lippert–Mataga plots and dipole moment values; CV; HOMO and LUMO energy; DFT/TD-DFT calculation results. CCDC 2091706 (Tria). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06602a |
| This journal is © The Royal Society of Chemistry 2021 |