
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAl-doped α-MnO2 coated by lignin for high-performance rechargeable aqueous zinc-ion batteries†
Jingliang Xuabc,
Xinhang Hua,
Md Asraful Alam a,
Gul Muhammada,
Yongkun Lva,
Minghai Wanga,
Chenjie Zhud and
Wenlong Xiong*a
a,
Gul Muhammada,
Yongkun Lva,
Minghai Wanga,
Chenjie Zhud and
Wenlong Xiong*a
aSchool of Chemical Engineering, Zhengzhou University, Zhengzhou 450001, China. E-mail: xiongwenlong@zzu.edu.cn
bZhengzhou Tuoyang Industrial Co., Ltd, Zhengzhou, China
cZhengzhou University Industrial Technology Research Institute Co., Ltd, Zhengzhou, China
dCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 211816 Nanjing, China
First published on 1st November 2021
Abstract
Zn/MnO2 batteries, one of the most widely studied rechargeable aqueous zinc-ion batteries, suffer from poor cyclability because the structure of MnO2 is labile with cycling. Herein, the structural stability of α-MnO2 is enhanced by simultaneous Al3+ doping and lignin coating during the formation of α-MnO2 crystals in a hydrothermal process. Al3+ enters the [MnO6] octahedron accompanied by producing oxygen vacancies, and lignin further stabilizes the doped Al3+ via strong interaction in the prepared material, Al-doped α-MnO2 coated by lignin (L + Al@α-MnO2). Meanwhile, the conductivity of L + Al@α-MnO2 improves due to Al3+ doping, and the surface area of L + Al@α-MnO2 increases because of the production of nanorod structures after Al3+ doping and lignin coating. Compared with the reference α-MnO2 cathode, the L + Al@α-MnO2 cathode achieves superior performance with durably high reversible capacity (∼180 mA h g−1 at 1.5 A g−1) and good cycle stability. In addition, ex situ X-ray diffraction characterization of the cathode at different voltages in the first cycle is employed to study the related mechanism on improving battery performance. This study may provide ideas of designing advanced cathode materials for other aqueous metal-ion batteries.
1. Introduction
With the rapid development of the utilization of renewable energy (e.g., solar, wind, tide, and biomass), advanced batteries for energy storage have attracted extensive consideration.1–3 Aqueous metal-ion batteries with the advantages of good safety and high ion conductivity are considered to be the next generation of large-scale energy storage devices.4,5 Among numerous contenders of rechargeable aqueous metal-ion batteries,6,7 aqueous Zn-ion batteries (AZIBs) appear to be tremendously promising because of their superior attributes,8 including high theoretical capacitance of the Zn anode (820 mA h g−1), low electrochemical potential of Zn2+/Zn (−0.763 V, SHE), low cost, and environmental friendliness.9–11As for AZIBs, the commonly used cathode materials include Mn-based oxides,12,13 V-based compounds,14,15 Prussian blue analogs,16,17 spinel ZnCo2O4 materials,18 transition metal dichalcogenides,19 and quinone and ketone compounds.20 MnO2 with various polymorphs is the most explored material in the family of cathode materials due to its advantages, such as high theoretical capacity (308 mA h g−1 based on single-electron transfer or 616 mA h g−1 based on two-electron transfer), low toxicity, scalable industrial manufacturing, and abundance.21–23 α-MnO2 with a typical 2 × 2 tunnel structure made of [MnO6] octahedral units are more beneficial for playing theoretical capacity than MnO2 with other crystal forms.24–26
However, α-MnO2 electrodes still suffer from severe capacity fading because of structure collapse, which is caused by the formation of Zn4(OH)6(SO4)·5H2O (ZHS)27 and the dissolution of Mn2+ in the electrolyte during charge–discharge process.28–30 Thus far, some mitigation strategies focusing on the optimization of crystal structure of MnO2 have been proposed.31–35 For example, Cao's group prepared α-MnO2/graphite nanosheet hybrids via ball milling method. The chemical bonding between MnO2 and graphite nanosheets strengthened the internal stability and interfacial adhesion and enhanced wettability and conductivity, thereby promoting charge transfer rate.36 Fang et al. studied a hypoxic α-MnO2 (K0.8Mn8O16) with potassium ions inserted into the tunnel as a highly active cathode for Zn-ion batteries. They showed that oxygen defects promoted conductivity and opened the polyhedral wall of [MnO6] to enhance ion diffusion, which is beneficial to accelerate the reaction kinetics and increase the capacity of K0.8Mn8O16.37 In particular, Xu et al. reported a new nanocomposite material, which was implemented via pre-intercalation of Fe3+ during the formation of α-MnO2 crystals and the coating of a polypyrrole layer on the surface of α-MnO2. Fe3+ enlarged the lattice spacing and decreased the hindrance for Zn2+ insertion/extraction. Meanwhile, polypyrrole prevented the dissolution of α-MnO2 during charge–discharge process.38 These meaningful studies led to the study of the link between polymer coating and metal doping for the modification of MnO2.
Herein, Al-doped α-MnO2 coated by lignin (L + Al@α-MnO2) was prepared via hydrothermal method to stabilize the structure of α-MnO2. Lignin, a renewable and abundant natural material, was selected as the target polymer because of its strong interaction with various metal ions for the electrostatic and cation–π interaction.39,40 Al3+ doping combined with lignin coating effectively inhibited the production of ZHS on the electrode surface and prevented rapid collapse of the MnO2 structure. L + Al@α-MnO2 displayed better electrical conductivity and structural stability than α-MnO2. In particular, the L + Al@α-MnO2 cathode achieved a durably higher reversible capacity of 188 mA h g−1 at 1.5 A g−1 and good cycle stability with lower fluctuations. Changes in doping and coating on the surface compositions of materials were analyzed by X-ray photoelectron spectroscopy (XPS). The synergistic effect of lignin on metal ions allowed Al3+ to enter the MnO2 lattice more fully. In addition, the reason behind the stability of the L + Al@α-MnO2 electrode was clarified by studying the evolution of electrodes during charge–discharge process.
2. Experimental section
2.1. Materials
NH4F (≥99.99% metal basis) was purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). KMnO4 (GR) was purchased from Nanjing Chemical Reagent Co., Ltd (Nanjing, China). Sodium lignosulfonate (Vanisperse A, battery-grade) was provided by Shandong Jinkeli Power Sources Technology Co., Ltd (Zibo, China). Al2(SO4)3 (AR) was purchased from Shanghai Yien Chemical Technology Co., Ltd (Shanghai, China). ZnSO4·7H2O (AR) and MnSO4·H2O (AR) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China).2.2. Synthesis of α-MnO2, Al@α-MnO2, L@α-MnO2 and L + Al@α-MnO2
α-MnO2 was synthesized via a hydrothermal method.41 Typically, 3.6 g of NH4F was dissolved in 160 mL of deionized water, with continuous stirring. Then, 0.4 g KMnO4 was added to the obtained aqueous solution of NH4F. After mixing was conducted by magnetic stirring for 15 min at room temperature, the prepared reaction mixture was transferred into a Teflon-lined autoclave with an inner volume of 200 mL. Finally, the autoclave was sealed, slowly heated to 200 °C, maintained at 200 °C for 24 h, and cooled to room temperature. The prepared brown flocculent precipitates were filtered, sufficiently rinsed with deionized water, and then dried at 80 °C for 12 h.Al@α-MnO2 was synthesized using the same method as that of α-MnO2. All the reaction conditions were the same except that the reaction mixture was changed to an aqueous solution containing 3.6 g NH4F, 0.2164 g Al2(SO4)3 and 0.4 g KMnO4.
L@α-MnO2 was synthesized using the same method as that of α-MnO2. All the reaction conditions were the same except that the reaction mixture was changed to an aqueous solution containing 3.6 g NH4F, 0.022 g sodium lignosulfonate, and 0.4 g KMnO4.
L + Al@α-MnO2 was also synthesized using the same method as that of α-MnO2. All the reaction conditions were the same except that the reaction mixture was changed to an aqueous solution containing 3.6 g NH4F, 0.2164 g Al2(SO4)3, 0.022 g sodium lignosulfonate, and 0.4 g KMnO4. The preparation process for L + Al@α-MnO2 is shown in Fig. 1.
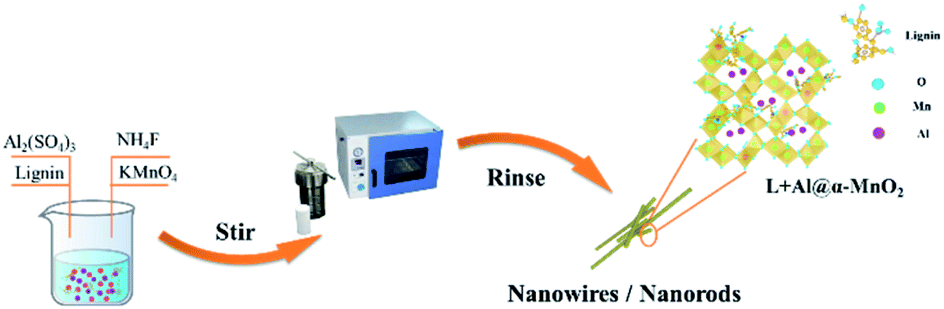 | ||
| Fig. 1 Schematic diagram of the preparation of L + Al@α-MnO2. | ||
2.3. Material characterizations
The α-MnO2 based materials were characterized by X-ray diffraction (XRD, Empyrean, Netherlands) equipped with a Cu Kα radiation source (λ = 1.5406 Å). The scanning was conducted within the range of 2θ = 5–90° with intervals of 0.02°.A vacuum coater (Leica EM ACE600, Germany) was used to enhance the conductivity of α-MnO2-based materials via gold coating. Then, morphologies were characterized by field-emission scanning electron microscopy (SEM, Zeiss/Auriga-bu, Germany).
High-resolution transmission electron microscopy (HRTEM, FEI TalosF200S, Czech Republic) and energy dispersive X-ray spectroscopy (EDX) mapping were used to investigate the microstructures and element distribution of the materials.
The composition and surface element state of the material samples were analyzed by XPS (AXIS Supra, UK).
Inductively coupled plasma emission spectrum (ICP-OES, Agilent 5110, USA) was used to measure the element content in the sample.
A Fourier transform infrared spectrometer (Bruker, Tensor II, USA) and STA8000 Synchronous Thermal Analyzer (PerkinElmer, USA) were used to evaluate the effect of doping and coating on the α-MnO2 based materials.
Raman spectroscopy was conducted on a Raman spectrometer (LabRAM HR Evo, France).
2.4. Electrochemical measurements
A homogeneous slurry consisted of active material (α-MnO2, Al@α-MnO2, or L + Al@α-MnO2), acetylene black, and polyvinylidene fluoride was prepared by adequately mixing them at a mass ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 in N-methyl-2-pyrrolidone. The prepared slurry was coated on a conductive polyethylene film, subsequently dried in a vacuum oven at 60 °C, and finally cut to a 12 mm-diameter disc to be used as the cathode. The mass loading of the active material in the cathode was approximately 1–2 mg cm−2. A polished Zn metal foil with a diameter of 12 mm and a thickness of 0.3 mm was used as the anode. The battery was assembled by packing the cathode, anode, separator (absorbed glass mat), and aqueous electrolyte (2 M ZnSO4 + 0.2 M MnSO4) in a CR 2025-type coin cell base.
:20:10 in N-methyl-2-pyrrolidone. The prepared slurry was coated on a conductive polyethylene film, subsequently dried in a vacuum oven at 60 °C, and finally cut to a 12 mm-diameter disc to be used as the cathode. The mass loading of the active material in the cathode was approximately 1–2 mg cm−2. A polished Zn metal foil with a diameter of 12 mm and a thickness of 0.3 mm was used as the anode. The battery was assembled by packing the cathode, anode, separator (absorbed glass mat), and aqueous electrolyte (2 M ZnSO4 + 0.2 M MnSO4) in a CR 2025-type coin cell base.
Electrochemical impedance spectroscopy (EIS, from 100 kHz to 0.01 Hz) and cyclic voltammetry (CV) were performed on the CHI604E electrochemical workstation (Shanghai Chenhua Instrument Co., Ltd., China). The CV scanning voltage was 1–1.9 V. The cycle performance of the battery was tested using the NEWARE battery tester (Neware Co. Ltd., China) at room temperature, and the current density was 1.5 A g−1. The rate performance of the battery was tested under the same instrument and environmental conditions as the cycle performance test, and the constant current charge and discharge current density was set to 0.1, 0.2, 0.5, 1, 2, 3, and 5 A g−1.
The evolution of cathode during the first charge–discharge process was characterized by ex situ XRD. Before characterization, the tested cathodes were fully rinsed with deionized water to remove the residual electrolyte.
3. Results and discussion
3.1. Structural analysis of materials
The microstructure and morphology of the as-prepared α-MnO2-based materials were characterized by XRD, SEM, TEM, and energy-dispersive spectroscopy (EDS) mapping. Fig. 2a shows the XRD patterns of the as-prepared α-MnO2-based materials. The diffraction peaks clearly revealed that α-MnO2 (JCPDS 44-0141) was successfully synthesized, and Al3+ doping and lignin coating did not change the major crystalline phase of α-MnO2. SEM was used to observe the morphology of α-MnO2, Al@α-MnO2, and L + Al@α-MnO2. The SEM image in Fig. 2b presents a major one-dimensional (1D) nanowire structure of α-MnO2. Single Al3+ doping slightly changed the morphology of α-MnO2 according to the SEM images displayed in Fig. 2b and c, in which a small amount of 1D nanorod structures appeared. However, more 1D nanorod structures also appeared in the SEM image (Fig. 2d) of L + Al@α-MnO2. This finding indicated that more α-MnO2 transformed from 1D nanowire structure to 1D nanorod structure during the formation process participated by Al3+ and lignin. The possible reason is because in the synthesis process of α-MnO2, lignin accelerates the crystal nucleation rate (Fig. S1a†), and the force between Al3+ and lignin further accelerates the crystallization process of the material, thereby limiting the growth of MnO2 particles, eventually resulting in a shortened length of the nanowire. Even when the Al3+ doping amount was doubled, the Al@α-MnO2 doped alone still only had few nanorods (Fig. S1b†), and the electrochemical performance worsened. The shorter rod structure effectively increased the specific surface area of the material, thus providing more electrochemically active sites for redox reactions. Examination of the diffraction rings obtained from SAED analysis (Fig. S2b†) showed the polycrystalline nature of the sample. According to the EDS mappings (Fig. 2f), except for the obvious Mn and O elements in the region, the distribution of Al elements reflected that Al3+ was uniformly doped in α-MnO2 and lignin coating caused C to overlap with MnO2 in the region. The content of C atom in L + Al@α-MnO2 was 4.42% based on the EDS mappings. The results of ICP-OES showed that the contents of Al and Mn element in L + Al@α-MnO2 were 1.41 wt% and 63.27 wt%. Thus, the atomic ratio of Mn to Al (22:1) was calculated. Although the ratio of Mn:Al used to prepare the material was 1:0.5, the amount of Al3+ that could enter MnO2 was limited due to the limitation of the number of active sites of MnO2.33 In addition, the α-MnO2 based materials were investigated using Raman spectroscopy (Fig. S3†) and FT-IR spectroscopy (Fig. S4†) to confirm the change of crystal lattice. The Mn–O vibration of the crystal caused peaks shift under the effect of doping, further indicating that Al3+ had entered the MnO2 crystal.38,42
 | ||
| Fig. 2 Structural analyses of as-prepared α-MnO2-based materials. (a) XRD patterns of α-MnO2, Al@α-MnO2, and L + Al@α-MnO2. (b–d) SEM images of α-MnO2, Al@α-MnO2, and L + Al@α-MnO2. (e and f) EDS elemental mapping images of Mn, O, Al, and C elements of L + Al@α-MnO2. | ||
Fig. 3 depicts the wide-scan XPS spectrum of the MnO2-based materials, in which the positions of all peaks were obtained after calibrating the position peak of C 1s (284.8 eV). XPS detection of the material surface could analyze the structural composition and element states. The Mn 2p of MnO2 spectrum is shown in Fig. 3a. The peak positions were 642.1 and 653.8 eV, which were assigned to Mn 2p3/2 and Mn 2p1/2, respectively.43 In addition, the occurrence of multiple splitting led to several contributions to the spectra of MnO2 (Mn 2p), caused by the presence of multiple oxidation states of Mn. For the Mn 2p peak of L + Al@α-MnO2 in Fig. 3b, the peak position of Mn 2p3/2 was 641.75 eV, and the peak position of Mn 2p1/2 was 653.5 eV. The peak position of Mn 2p obviously shifted to low binding energy, indicating that the average oxidation state of Mn is lower than that of α-MnO2. This situation was due to the fact that a part of Al3+ replaced Mn4+ in [MnO6] units, resulting in the generation of oxygen vacancies. The introduction of oxygen defects in MnO2 reduces the Gibbs free energy of ion intercalation,44 which enhances the structural stability in repeated cycles, thereby improving electrochemical reversibility.
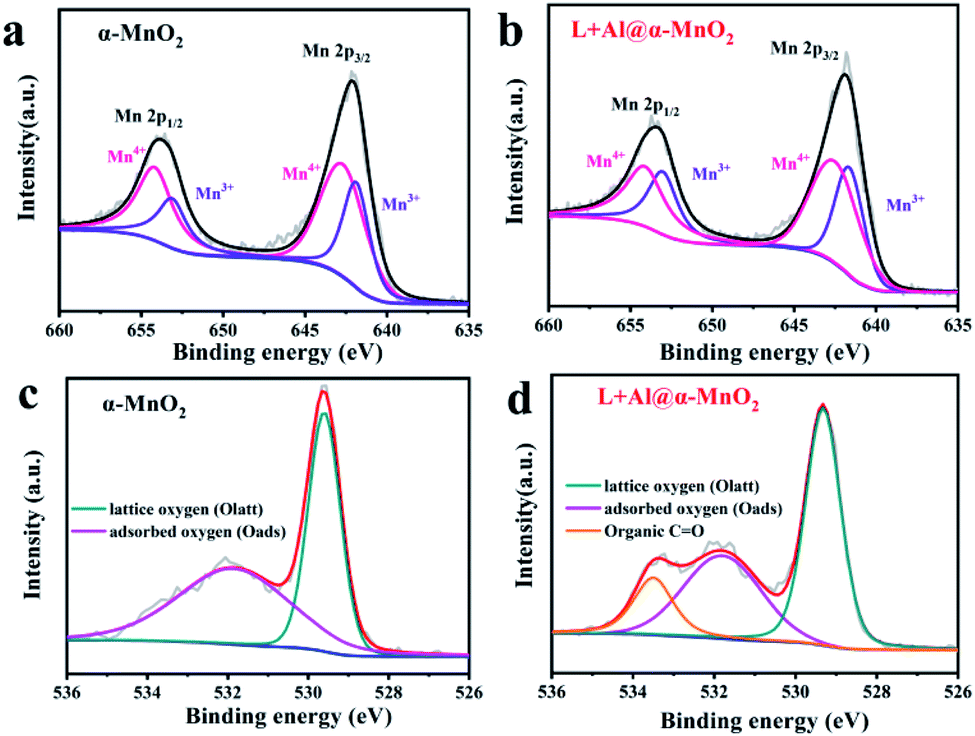 | ||
| Fig. 3 XPS spectra of as-prepared α-MnO2-based materials. (a and b) Mn 2p spectrum of α-MnO2 and L + Al@α-MnO2. (c and d) O 1s spectrum of α-MnO2 and L + Al@α-MnO2. | ||
As shown in Fig. 3c, the O 1s of the MnO2 spectrum exhibited two fractional peaks. The peak position of lattice oxygen (Mn–O–Mn) was 529.6 eV, and the peak position of surface-adsorbed oxygen (H–O–H) was 531.7 eV. However, with the coating of lignin, L + Al@MnO2 showed a new type of fractional peak, which was organic carbon–oxygen double bond oxygen (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) at 533.6 eV.45 Compared with L + Al@MnO2, the new peak does not exist in the O 1s spectrum of Al@MnO2 (Fig. S5e†). Thus, the XPS spectrum proved that lignin was successfully coated on the surface of MnO2. Thermogravimetric analysis (Fig. S6†) of α-MnO2 based materials further indicated that the average oxidation state of Mn decreased and the existence of lignin in L + Al@α-MnO2. In addition, the weight loss of L + Al@α-MnO2 was more than α-MnO2 within 100–300 °C, which indicated that the content of structural water in L + Al@α-MnO2 increased significantly. This was probably due to the hydrophilic groups of sodium lignosulfonate adsorbed more water molecules into L + Al@α-MnO2. The rich of structural water in the material effectively improved the interfacial kinetics of ions, which leaded to that the battery using L + Al@α-MnO2 presented superior cyclability than that of the battery using α-MnO2 after activation.46
C) at 533.6 eV.45 Compared with L + Al@MnO2, the new peak does not exist in the O 1s spectrum of Al@MnO2 (Fig. S5e†). Thus, the XPS spectrum proved that lignin was successfully coated on the surface of MnO2. Thermogravimetric analysis (Fig. S6†) of α-MnO2 based materials further indicated that the average oxidation state of Mn decreased and the existence of lignin in L + Al@α-MnO2. In addition, the weight loss of L + Al@α-MnO2 was more than α-MnO2 within 100–300 °C, which indicated that the content of structural water in L + Al@α-MnO2 increased significantly. This was probably due to the hydrophilic groups of sodium lignosulfonate adsorbed more water molecules into L + Al@α-MnO2. The rich of structural water in the material effectively improved the interfacial kinetics of ions, which leaded to that the battery using L + Al@α-MnO2 presented superior cyclability than that of the battery using α-MnO2 after activation.46
3.2. Electrochemical performance and behavior
Fig. 4 shows the electrochemical performance of the α-MnO2-based batteries. The change in the structure of α-MnO2 improved the electrochemical performance. Fig. 4a and b show the CV data of two electrodes. The α-MnO2 electrode exhibited an obvious cathodic peak near 1.17 V during the first cathodic sweep. However, the reduction peak disappeared in the second sweep. New reduction peaks appeared near 1.26 and 1.38 V, while the anode sweep had corresponding anode peaks near 1.57 and 1.61 V. The appearance of two peaks in the working window from 1 V to 1.9 V implied a two-step insertion process. In the following sweeps, the strengths of the redox peak at 1.57 V decreased, while the oxidation peak at 1.61 V gradually increased. Unlike in α-MnO2, with the following sweep, the reaction at higher voltage (1.38/1.61 V) occupied the dominant position in the charge–discharge process of L + Al@α-MnO2. By studying the evolution of the electrode, this phenomenon was determined to correspond to the insertion/extraction of H+. According to the CV curves, the battery using L + Al@α-MnO2 had much lower current responses than that of the battery using α-MnO2. This was because L + Al@α-MnO2 underwent an activation process. The peak at ∼1.57 V was related to the insertion of Zn2+,47 which was hindered by Al3+ in the tunnel of α-MnO2. Hence, the peak at ∼1.57 V presented attenuation trend incipiently. H+ was not as much affected as that of Zn2+ due to its small ion radius. The same trend appeared in the battery using Al@α-MnO2 (Fig. S7a†), showing the dominated insertion/extraction of H+ at the beginning. The CV curve of battery using L@α-MnO2 (Fig. S7b†) was similar to that of battery using α-MnO2. These phenomena illustrated that the activation process of L + Al@α-MnO2 was due to the Al3+ doping.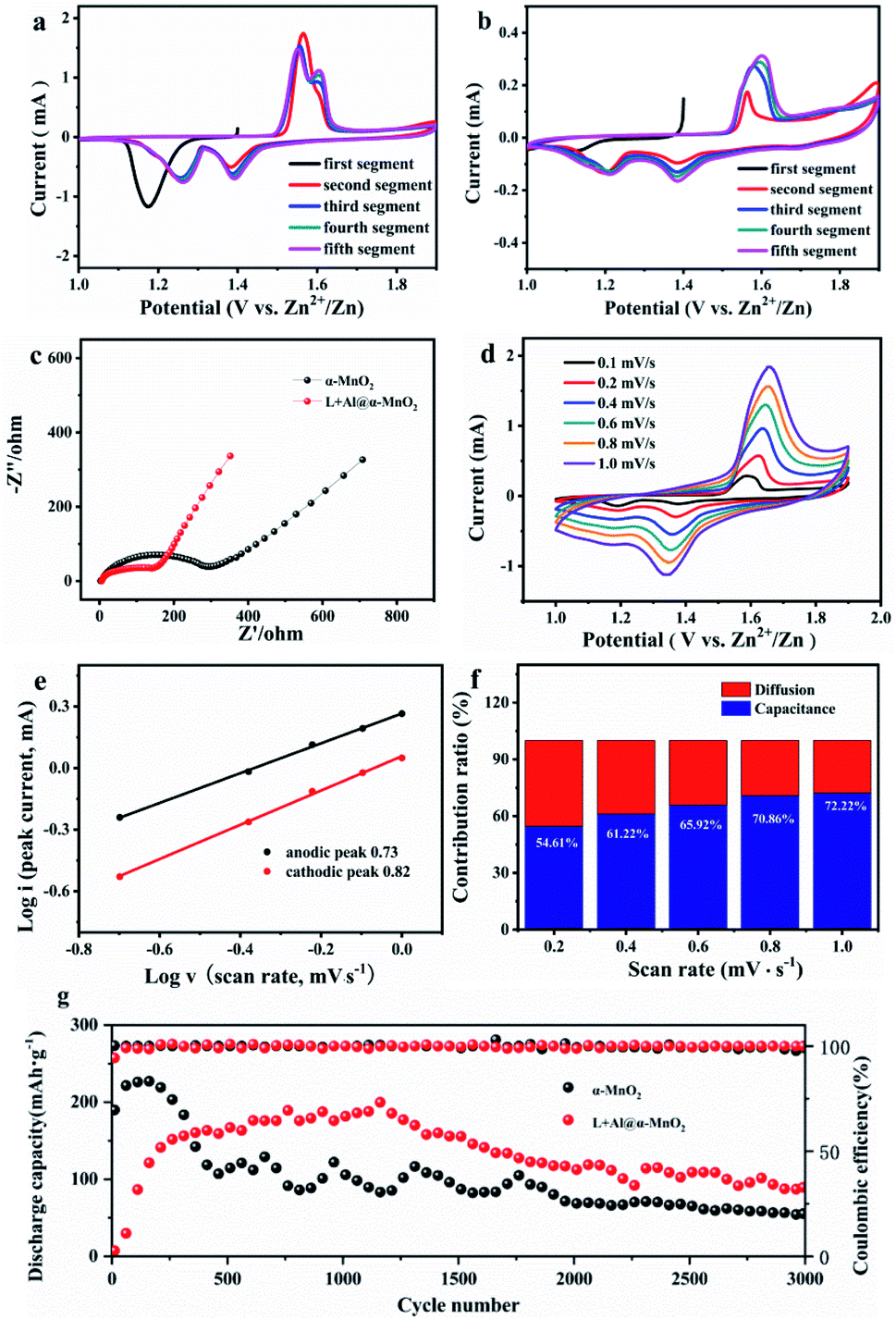 | ||
| Fig. 4 Electrochemical performance of MnO2 and L + Al@α-MnO2 electrodes. (a and b) Cyclic voltammetry curves of α-MnO2 and L + Al@α-MnO2 at 0.1 mV s−1. (c) Electrochemical impedance spectroscopy plots of α-MnO2 and L + Al@α-MnO2. (d) CV curves of L + Al@α-MnO2 at different sweep rates. (e) Log (i, peak current) versus log (v, scan rate) plots of two peaks in the CV curves of L + Al@α-MnO2. (f) Percentage of capacitance of L + Al@α-MnO2 electrode. (g) Cycling performance of α-MnO2 and L + Al@α-MnO2 and corresponding coulombic efficiency at a current density of 1.5 A g−1. | ||
The resistance and conductivity of α-MnO2 and L + Al@α-MnO2 were obtained by EIS to further explore the resistance and diffusion behavior of the electrode. Fig. 4c illustrates the Nyquist plots of two electrodes. The semicircle of the charge transfer resistance (Rct) was located in the high frequency region, and the oblique line in the low frequency region was associated with the Warburg-type impedance of diffusion process. The semicircle of L + Al@α-MnO2 was smaller than that of α-MnO2, indicating the improvement of electron transfer kinetics after doping, resulting in lower Rct of the material. In the low frequency region, the line slope of L + Al@α-MnO2 was higher than that of α-MnO2, indicating that the diffusion rate of ions in L + Al@α-MnO2 was faster. The short nanorod tunnel structure of L + Al@α-MnO2 shortened the electron transport path effectively and promoted the diffusion of active materials. In addition, the oxygen defects produced by Al3+ doping improved the conductivity and reversibility of the material, increasing the rate of ions transmission effectively.44,48 Thus, the reaction kinetics was enhanced.
The CV profiles of L + Al@α-MnO2 electrode at various scan rates from 0.1 mV s−1 to 1.0 mV s−1 are shown in Fig. 4d. As the scanning speed increased, the cathode and anode peaks shifted to higher and lower potentials, respectively, due to the increased polarization at higher scan rates. The pair of reduction and oxidation peaks gradually disappeared in the L + Al@α-MnO2 electrode compared with that in the α-MnO2 electrode (Fig. S7c†). The linear relationship was calculated using the following equation:38
| i = avb | (1) |
|
lg(i) = blg(v) + lg(a)
| (2) |
| i(v) = k1v + k2v1/2 | (3) |
Fig. 4g shows that the α-MnO2-based battery delivered 225 mA h g−1 at the current density of 1.5 A g−1 in the first 100 cycles. However, this capacity was only maintained for about 300 cycles, and soon began to decline significantly. After approximately 500 cycles the capacity only remained around 107 mA h g−1 and gradually decreased to 54 mA h g−1 after 3000 cycles. In addition, the capacity fading of α-MnO2-based-battery was accompanied by obvious fluctuations, indicating that the structure of α-MnO2 was unstable. When tested at the same current density, Al3+ doped electrodes showed better stability. After undergoing an activation process about 400 cycles, the Al@α-MnO2-based battery (Fig. S8a†) showed a stable discharge capacity of 140 mA h g−1. After 1400 cycles, the capacity began to decline slowly. The L@α-MnO2-based battery (Fig. S8a†) began to decay after 50 cycles when the capacity reached a peak of 230 mA h g−1, and showed a capacity platform of 155 mA h g−1 at 200 cycles. The capacity faded after 1300 cycles, and only remained at 47 mA h g−1 after 3000 cycles. On the basis of Al3+ doping and lignin coating, lignin did not change the cycle trend of the L + Al@α-MnO2-based battery, which presented the similar trend with the Al@α-MnO2-based battery. Specifically, although the capacity before 300 cycles was not as high as that of α-MnO2-based battery, the L + Al@α-MnO2 based battery showed highest capacity of 188 mA h g−1 after 400 cycles of activation process. After 1400 cycles, battery using the L + Al@α-MnO2 also showed capacity fading, but the process was much more stable than that of battery using the α-MnO2. After 3000 cycles, battery using the L + Al@α-MnO2 still maintained a higher capacity (∼90 mA h g−1) than the other batteries, and the capacity was 66.7% more than that of battery using the α-MnO2. This finding is largely attributable to the synergistic effect of lignin and metal ions, which made Al3+ better doped and enhances the reversibility of ion insertion/extraction, thus avoiding collapse of the MnO2 structure during charge–discharge process.
Compared with α-MnO2, the materials after doping needed to undergo an activation process. The reason for this trend may be that the ionic radius of Al3+ is similar to that of Mn4+, which replaces part of Mn4+ in the [MnO6] octahedron. The result of doping strengthened the structure of the MnO2 host; however, a part of Al3+ entered the tunnel.49 Thus, the insertion of ions into the main body of MnO2 was hindered, particularly the Zn2+ with a large ion radius. This was consistent with the CV curves of battery using the L + Al@α-MnO2 cathode (Fig. 4b), which presented low current responses of the insertion/extraction of Zn2+ and H+. Moreover, the current response of Zn2+ was much lower than that of H+. As the reversible insertion/extraction of ions (H+ and a small amount Zn2+) in the cathode material, the Al3+ in the tunnel was gradually released. Therefore, the amounts of Zn2+ and H+ that entered the cathode material rose, resulting in an increase in capacity over cycle number.
The charge–discharge curve is shown in Fig. 5a and d to explore the transformation of α-MnO2 and L + Al@α-MnO2 in the charge–discharge process. The voltage corresponding to the obvious change in the curve was recorded, and the ex situ XRD patterns of cathode were tested at selected voltage states. Fig. 5b shows the XRD patterns of α-MnO2 during the discharge process. A notable detail that the strong diffraction peak at 21–24°, which belongs to the current collector made of PE material, was shielded. In the following discharge process, the characteristic peaks of α-MnO2 were unchanged. However, a new phase was generated when the electrode was discharged from the initial voltage to 1.25 V, because the insertion of H+ increased the pH in the electrolyte, and more OH− combined with other components to form this sheet-like ZHS,50 which adheres to the surface of the electrode. Fig. 5c shows the evolution of α-MnO2 during the charge process. Except the still unchanged characteristic peaks of α-MnO2, the diffraction peak of ZHS weakened rapidly at the first charging platform, and a small amount of the product remained on the cathode of α-MnO2 when charged to 1.9 V. However, as shown in Fig. 5e, the generation of ZHS was significantly suppressed during the discharge process of L + Al@α-MnO2. In the charge process, ZHS basically disappeared when charged to 1.5 V, and its diffraction peak completely disappeared after charging to 1.6 V.
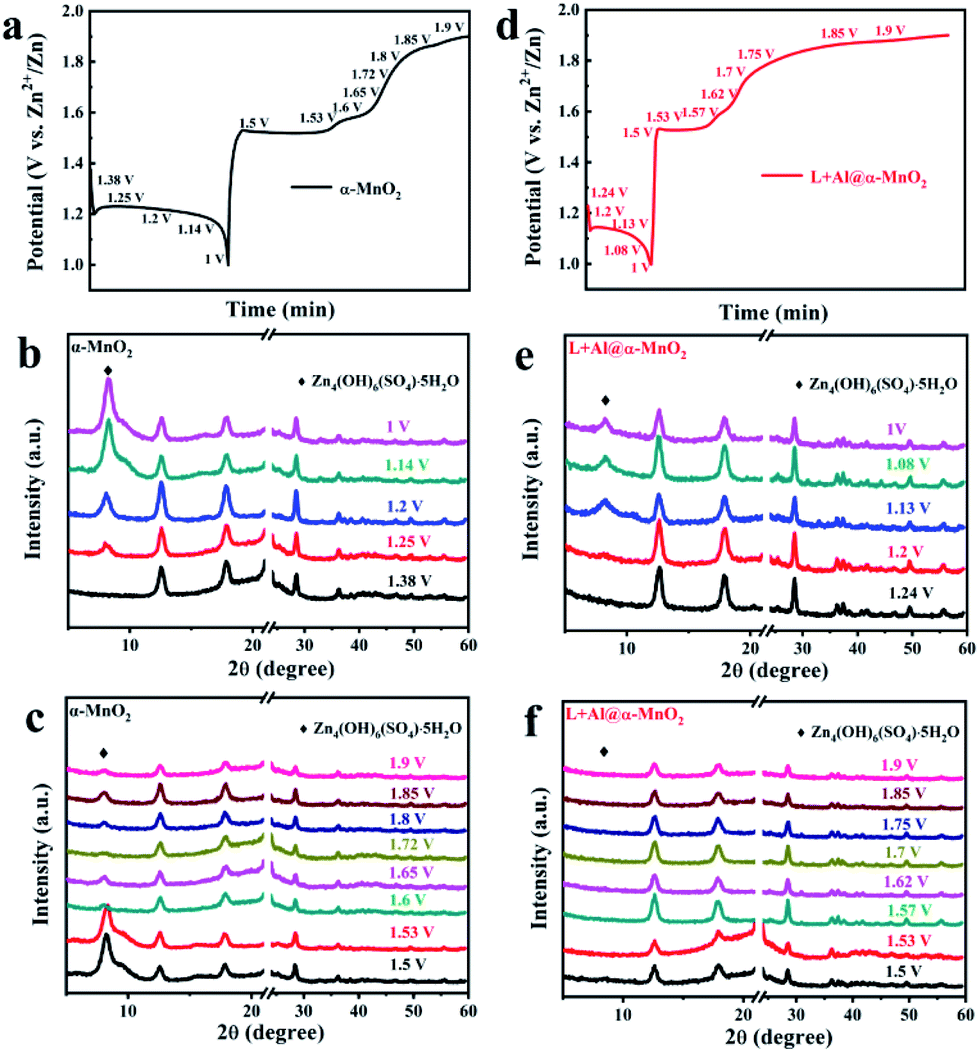 | ||
| Fig. 5 Structure evolution of cathodes in ex situ X-ray diffraction patterns during charge–discharge process at 100 mA g−1. (a–c) Charge–discharge curve of α-MnO2 and XRD patterns at selected voltage. (d–f) Charge–discharge curve of L + Al@α-MnO2 and XRD patterns at selected voltage. | ||
In a word, a new phase was formed during the discharge process, and it disappeared during the charge process. The CV data and XRD pattern showed that the behavior of H+ and Zn2+ during the initial electrochemical process could be determined. After the electrode was discharged from the initial voltage, ZHS did not appear immediately. Subsequently, during the charge process, the ZHS on the electrode surface did not decrease significantly at first but began to disappear rapidly when the voltage reached 1.6 V, indicating the extraction of H+. Thus, the higher peak at 1.38/1.61 V corresponded to H+ insertion/extraction and the lower peak at 1.26/1.57 V was assigned to Zn2+ insertion/extraction. However, after charging to 1.9 V, visible ZHS still remained on the electrode surface. The presence of ZHS was accompanied by the disproportionation of a part of Mn3+ into Mn2+ causing the dissolution of Mn,51 leading to the collapse of the MnO2 host structure. According to previous reports, these residual sulfates gradually recombine into ZnMn2O4,23 which reduces the electrochemical activity of the electrode. ZHS obviously showed better reversibility of L + Al@α-MnO2 than α-MnO2 due to the effect of lignin on Al3+ during the doping process, which accelerated the nucleation of MnO2 and promoted the entry of Al3+ into the crystal lattice. H+ was highly related to the formation of ZHS in the electrolyte, and the good reversibility was due to the enhancement of H+ diffusion in the host material after ion doping and lignin coating.52 At the same time, the oxygen vacancies generated with Al3+ substitution in L + Al@α-MnO2 further reduced the resistance of H+ insertion/extraction.35 Therefore, almost no ZHS was attached to the electrode surface after charging, further protecting the electrode structure and improves the cycle stability.
4. Conclusions
In summary, L + Al@α-MnO2 was successfully fabricated as high-performance cathode materials through Al3+ doping and lignin coating via hydrothermal route. The resulting material presented a morphology of short nanorod, which had a larger specific surface area and could provide more active sites. The lignin coating could allow Al3+ to be fully doped into the MnO2 lattice during the synthesis process. The doping of Al3+ improved the insertion/extraction of H+ to reduce the residue of zinc hydroxide sulfate in the electrode during the charge–discharge process. The related spectroscopic analysis was consistent with the results of local structural changes in the material and theoretical assumptions. The results showed that L + Al@α-MnO2 by Al3+ doping and lignin coating effectively alleviated the structural collapse caused by Mn dissolution, thus stabilizing the electrochemical performance of the electrode. These positive effects made the Zn/MnO2 battery operate in high specific capacity (66.7% higher than α-MnO2 after 3000 cycles). The synergistic effect of lignin and Al3+ could provide a reference for the synthesis of next-generation electrode materials with using the modification method of polymers and metal ions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21908205 and 22078308), Henan Provincial Key Research and Development Program (No. 202102210312 and 202102310019), Innovation Leadership Program in Sciences and Technologies for Central Plains Talent Plan (No. 214200510009), Henan Postdoctoral Foundation (No. 201901010), Young Elite Scientists Sponsorship Program by Henan Association for Science and Technology (No. 2021HYTP022), China Postdoctoral Science Foundation (No. 2019M662535), Innovation Leadership Program in Sciences and Technologies for Zhengzhou Talent Gathering Plan, Local Outstanding Contribution Talent Project in Sciences and Technologies for Zhengzhou Talent Gathering Plan (No. 20180400042), Jiangsu Province Natural Science Foundation for Distinguished Young Scholars (No. BK20190035).References
- J. Wang, X. Sun, H. Zhao, L. Xu, J. Xia, M. Luo, Y. Yang and Y. Du, J. Phys. Chem. C, 2019, 123, 22735–22741 CrossRef CAS.
- C. Zhang, X. Yu, H. Chen, L. Li, D. Sun, X. Chen and X. Hao, J. Alloys Compd., 2021, 864, 158685 CrossRef CAS.
- W. Xiong, T. K. Hoang, D. Yang, Y. Liu, M. Ahmed, J. Xu, X. Q. Qiu and P. Chen, J. Energy Storage, 2019, 26, 100920 CrossRef.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- X. Liu, J. Yi, K. Wu, Y. Jiang, Y. Liu, B. Zhao, W. Li and J. Zhang, Nanotechnology, 2020, 31, 122001 CrossRef CAS PubMed.
- D. Bin, F. Wang, A. G. Tamirat, L. Suo, Y. Wang, C. Wang and Y. Xia, Adv. Energy Mater., 2018, 8, 1703008 CrossRef.
- L. Shen, Y. Jiang, Y. Liu, J. Ma, T. Sun and N. Zhu, Chem. Eng. J., 2020, 388, 124228 CrossRef CAS.
- W. Xu and Y. Wang, Nano-Micro Lett., 2019, 11, 1–30 CrossRef PubMed.
- H. Chen, X. Zhang, L. Chen, Z. Yang, X. Zeng and J. Meng, J. Energy Storage, 2020, 27, 101139 CrossRef.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- J. Xin, C. Liu, Z. Qiu, J. Zhou, Q. Wang, Y. Liu and B. Guo, RSC Adv., 2018, 8, 26906–26909 RSC.
- J. Wang, J. G. Wang, H. Liu, C. Wei and F. Kang, J. Mater. Chem. A, 2019, 7, 13727–13735 RSC.
- H. Shen, B. Liu, Z. Nie, Z. Li, S. Jin, Y. Huang and H. Zhou, RSC Adv., 2021, 11, 14408–14414 RSC.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang and Y. Shao, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- L. Xu, Y. Zhang, J. Zheng, H. Jiang, T. Hu and C. Meng, Mater. Today Energy, 2020, 18, 100509 CrossRef.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158–12164 CrossRef CAS PubMed.
- L. Zhang, C. Liang, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- J. J. Huang, Y. Y. Lia, R. K. Xie, J. W. li, Z. H. Tian, G. L. Chai, Y. W. Zhang, F. L. Lai, G. J. He, C. T. Liu, T. X. Liu and D. J. Brett, J. Energy Chem., 2021, 58, 147–155 CrossRef.
- L. Lin, W. Lei, S. Zhang, Y. Liu, G. G. Wallace and J. Chen, Energy Storage Mater., 2019, 19, 408–423 CrossRef.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- F. Raza, X. Ni, J. Wang, S. Liu, Z. Jiang, C. Liu, H. F. Chen, F. Amjad and A. Ju, J. Energy Storage, 2020, 30, 101467 CrossRef.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
- L. Li, T. K. Hoang, J. Zhi, M. Han, S. Li and P. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 12834–12846 CrossRef CAS PubMed.
- B. Lee, S. Y. Chong, H. R. Lee, K. Y. Chung and H. O. Si, Sci. Rep., 2014, 4, 6066 CrossRef CAS PubMed.
- C. Li, X. Zhang, W. He, G. Xu and R. Sun, J. Power Sources, 2020, 449, 227596 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- S. Lian, C. Sun, W. Xu, W. Huo, Y. Luo, K. Zhao, G. Yao, W. Xu, Y. Zhang and Z. Li, Nano Energy, 2019, 62, 79–84 CrossRef CAS.
- J. Wang, J. G. Wang, H. Liu, Z. You, C. Wei and F. Kang, J. Power Sources, 2019, 438, 226951 CrossRef CAS.
- J. Ming, J. Guo, C. Xia, W. Wang and H. N. Alshareef, Mater. Sci. Eng. R Rep., 2019, 135, 58–84 CrossRef.
- H. Peng, H. Fan, C. Yang, Y. Tian, C. Wang and J. Sui, RSC Adv., 2020, 10, 17702–17712 RSC.
- Y. D. Jiao, L. Q. Kang, J. B. Gair, K. M. Coll, J. W. Li, H. B. Dong, H. Jiang, R. Wang, F. Corà, D. J. Brett, G. J. He and I. P. Parkin, J. Mater. Chem. A, 2020, 8, 22075–22082 RSC.
- Z. Hu, X. Xu, C. Chi, T. Li and Y. Zhang, Nano Energy, 2015, 11, 226–234 CrossRef CAS.
- Y. Liu, G. He, H. Jiang, I. P. Parkin, P. R. Shearing and D. J. Brett, Adv. Funct. Mater., 2021, 31, 2010445 CrossRef CAS.
- Y. Zhang, L. Tao, C. Xie, D. Wang, Y. Zou, R. Chen, Y. Wang, C. Jia and S. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed.
- J. Cao, D. Zhang, X. Zhang, S. Wang, J. Han, Y. Zhao, Y. Huang and J. Qin, Appl. Surf. Sci., 2020, 534, 147630 CrossRef CAS.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- J. W. Xu, Q. L. Gao, Y. M. Xia, X. S. Lin, W. L. Liu, M. M. Ren, F. G. Kong, S. J. Wang and C. Lin, J. Colloid Interface Sci., 2021, 598, 419–429 CrossRef CAS PubMed.
- J. Wang, Y. Qian, Y. Deng, D. Liu, H. Li and X. Qiu, Appl. Surf. Sci., 2016, 390, 617–622 CrossRef CAS.
- Y. Ge and Z. Li, ACS Sustainable Chem. Eng., 2018, 6, 7181–7192 CrossRef CAS.
- Z. Ma, F. Jing, L. Hou, L. Fan, Y. Zhao, Y. Fan and X. Qin, Nano, 2020, 15, 2050014 CrossRef CAS.
- Z. Jian, S. Wang, J. Yu, C. Hong, H. Tan, Q. Liu and J. Wu, J. Solid State Electrochem., 2014, 18, 1585–1591 CrossRef.
- Y. Zhang, Y. Liu, Z. Liu, X. Wu, Y. Wen, H. Chen, X. Ni, G. Liu, J. Huang and S. Peng, J. Energy Chem., 2022, 64, 23–32 CrossRef.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y. W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- R. A. Davoglio, G. Cabello, J. F. Marco and S. R. Biaggio, Electrochim. Acta, 2018, 261, 428–435 CrossRef CAS.
- T. H. Wu and W. Y. Liang, ACS Appl. Mater. Interfaces, 2021, 13, 23822–23832 CrossRef CAS PubMed.
- F. Kataoka, T. Ishida, K. Nagita, V. Kumbhar, K. Yamabuki and M. Nakayama, ACS Appl. Energy Mater., 2020, 3, 4720–4726 CrossRef CAS.
- Y. Li, X. Wang, Y. Gao, Q. Zhang, G. Tan, Q. Kong, S. Bak, G. Lu, X. Q. Yang and L. Gu, Adv. Energy Mater., 2019, 9, 1803087 CrossRef.
- Q. Gao, J. Wang, B. Ke, J. Wang and Y. Li, Ceram. Int., 2018, 44, 18770–18775 CrossRef CAS.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- P. Mulvaney, R. Cooper, F. Grieser and D. Meisel, J. Phys. Chem., 1990, 94, 8339–8345 CrossRef CAS.
- T. H. Wu and W. S. Lin, Electrochim. Acta, 2021, 394, 139134 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06808c |
| This journal is © The Royal Society of Chemistry 2021 |