
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable synthesis of favipiravir via conventional and continuous flow chemistry†
Thanat Tiyasakulchai‡
a,
Netnapa Charoensetakul‡a,
Thitiphong Khamkhenshorngphanuchb,
Chawanee Thongpanchanga,
Onsiri Srikunc,
Yongyuth Yuthavonga and
Nitipol Srimongkolpithak *a
*a
aNational Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani, Thailand. E-mail: nitipol.sri@biotec.or.th
bDepartment of General Education, Faculty of Science and Health Technology, Navamindradhiraj University, Bangkok, Thailand
cGovernment Pharmaceutical Organization (GPO), Bangkok, Thailand
First published on 1st December 2021
Abstract
Decagram scale synthesis of favipiravir was performed in 9 steps using diethyl malonate as cheap starting material. Hydrogenation and bromination steps were achieved by employing a continuous flow reactor. The synthetic process provided a total of 16% yield and it is suitable for larger-scale synthesis and production.
Introduction
Favipiravir (1, sold under the brand name Avigan, 6-fluoro-3-hydroxy-2-pyrazinecarboxamide) is an antiviral drug that has hitherto been used to treat influenza. Since the onset of the SARS-COV-2 (Severe Acute Respiratory Syndrome-Coronavirus-2) pandemic, favipiravir has been re-purposed to treat COVID-19 patients under emergency provision in several countries. Its efficacy against COVID-19 has been extensively investigated in clinical trials1,2 (see https://clinicaltrials.gov). Favipiravir's safety has already been demonstrated by its extensive use as an anti-influenza drug in Japan. Pharmacologically, favipiravir bears a pyrazine scaffold and works as a prodrug that undergoes biotransformation for the inhibition of virus RNA-dependent RNA polymerase.3 The favourable safety profile and known mechanism of action make favipiravir one of the promising re-purposing drugs against COVID-19.The synthesis routes of favipiravir have been reported by academic groups and companies, including an innovator Toyama Chemicals.3–5 However, most of the reported synthetic routes were carried out on a small scale and some required costly column chromatography. Their starting materials have been disclosed, including 2-aminomalonamide (2),6 3-hydroxypyridine-2-carboxamide (3),7,8 3-aminopyrazine-2-carboxylic acid (4),9 pyrazine-2-carbonitrile (5),10 2-aminopyrazine (6),11 synthesising favipiravir via the key intermediate 3,6-dichloropyrazine-2-carbonitrile (7). Toyama Chemicals' patent also indicated that it was possible to synthesize favipiravir from the starting material 8 via another key intermediate 9 (ref. 12) (Scheme 1). However, during the pandemic, Toyama Chemicals have indicated that diethyl malonate (10) was sourced as starting material for commercial production,13 but have not reported synthetic methods. We and other workers believe that diethyl malonate could be used for the synthesis of 2-aminomalonamide (2).3 Diethyl malonate is cheap and easy to source during the pandemic. However, the total synthetic route for the synthesis of favipiravir from diethyl malonate has not been reported and may need to be altered for scaled production.
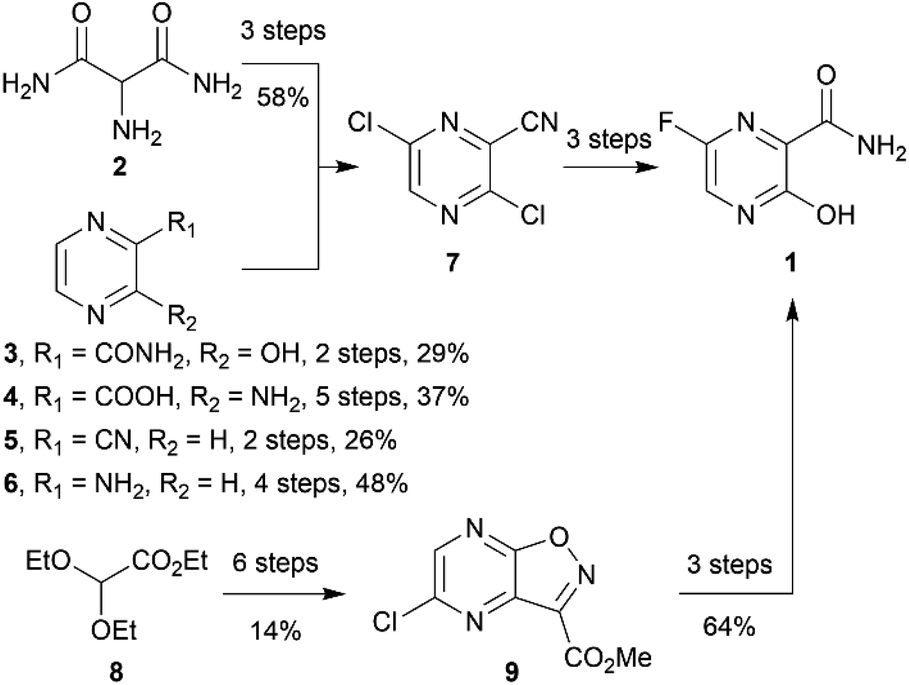 | ||
| Scheme 1 Reported starting materials for favipiravir synthesis. | ||
In this project, our group aimed to develop a scalable synthetic route of favipiravir from diethyl malonate. Each reaction was optimized and modified from the reported protocol. Moreover, continuous flow chemistry was also introduced for assistance in hydrogenation and bromination reactions, resulting in a 16% total yield via 9 steps synthesis (Scheme 2).
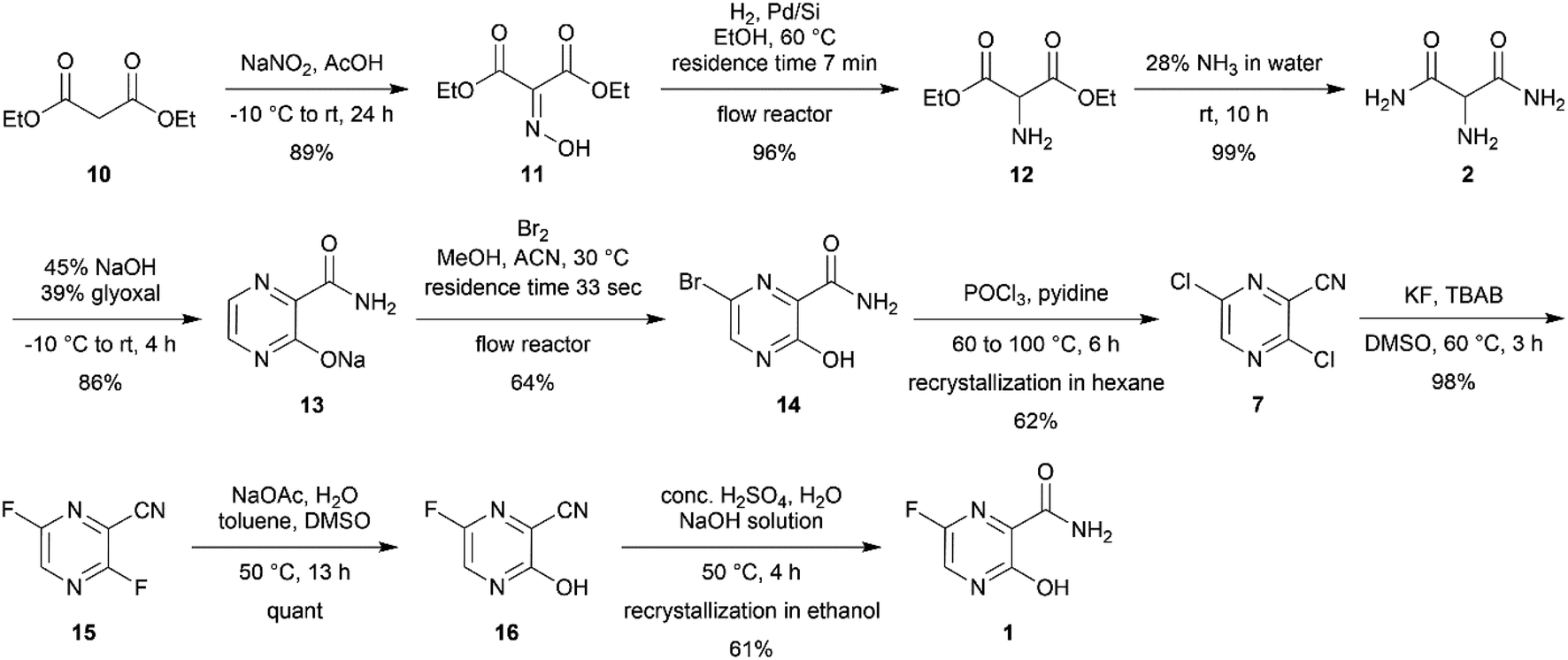 | ||
| Scheme 2 Total synthesis of favipiravir from diethyl malonate 10. | ||
Results and discussion
Our study was initiated by treating 100 g of 10 in glacial acetic acid with sodium nitrile in water slowly at −10 °C to obtain diethyl oximinomalonate (11)14 (Table S1†). Notably, oximinomalonate was not stable for long-term storage, whereas the HCl salt of diethyl 2-aminomalonate (12) was more stable and commercially available. The compound 11 underwent hydrogenation reaction in batch and continuous flow manners. While the batch reaction can not be completed in 24 hours under hydrogen at atmospheric pressure, the continuous flow reaction using Pd/Si in a packed bed column achieved a resident time in the column of only 7 minutes at 60 °C (Table 1 and Fig. 1). We found that crude 12 could be used without further purification
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Catalyst | Time | Temp. (°C) | Conversion (%) |
| a In a batch process.b Isolation yield ∼80% or able to use without further purification. | |||||
| 1a | H2 (excess) | Pd/C, 5% wt | 24 h | 30 | 66 |
| 2 | H2 (1 eq.) | Pd/Si | 7 min | 30 | 86 |
| 3 | H2 (4 eq.) | Pd/Si | 7 min | 30 | 96 |
| 4 | H2 (6 eq.) | Pd/Si | 7 min | 30 | 97 |
| 5 | H2 (6 eq.) | Pd/Si | 7 min | 60 | 100b |
| 6 | H2 (6 eq.) | Pd/Si | 14 min | 30 | 94 |
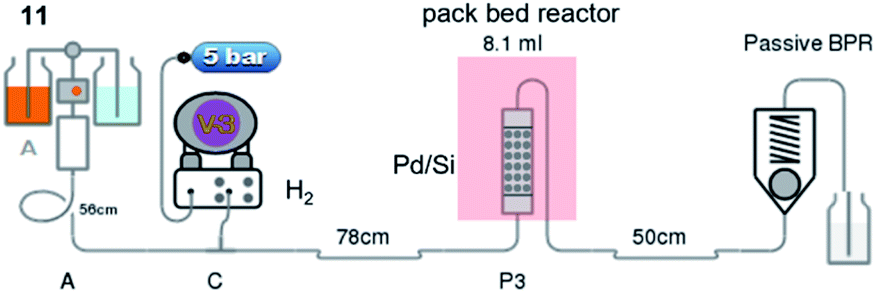 | ||
| Fig. 1 Hydrogenation of diethyl oximinomalonate (11) in a continuous flow reactor. | ||
Subsequently, 2-aminomalonamide 2 was obtained in high yield (99%) by treating crude 12 with 28% ammonia in water whereas ammonia in methanol, as indicated in the patent,6 resulted in several unidentified side products (Table S2†). 2 and glyoxal were then used for pyrazinamide cyclization in basic condition to provide pyrazinamide salt 13 in a good yield (81%) (Table S3†).
Subsequently, the bromination of 13 by batches provided a low to moderate yield (36%) after several optimizations were carried out (Table S4†). However, using continuous flow the brominating process of pyrazinamide salt 13 was successful with moderate yield (64%) using only short period of time (Table 2).
|
|||
|---|---|---|---|
| Entry | Condition | Time (min) | Yielda (%) |
| a Isolated yield.b In a batch process. | |||
| 1b | Br2 (1 eq.), AcOH (1.2 eq.), ACN, rt | 60 | 36 |
| 2 | Br2 (3 eq.), ACH, 30 °C | 2 | 48 |
| 3 | Br2 (4.5 eq.), ACN, 30 °C | 2 | 22 |
| 4 | Br2 (3 eq.), ACN, 60 °C | 2 | 32 |
| 5 | Br2 (3 eq.), ACN, 30 °C | 4 | 28 |
| 6 | Br2 (3 eq.), MeOH, 30 °C | 2 | 44 |
| 7 | Br2 (3 eq.), ACN, 30 °C | 0.55 | 64 |
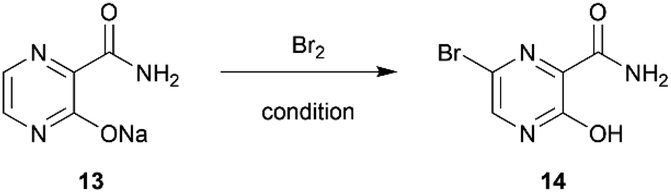
Notably, the reaction needed to be protected from light as the flow reactor was more transparent than the conventional batch synthesis. Also, the process still required a mixing tube as an auxiliary part to overcome the mass transfer issue (Fig. 2). After obtaining bromopyrazinamide 14 from extraction, it was chlorinated using POCl3 in pyridine to provide dichloro 7 and recrystallized in hexane in a good yield (62%) (Table S5†).
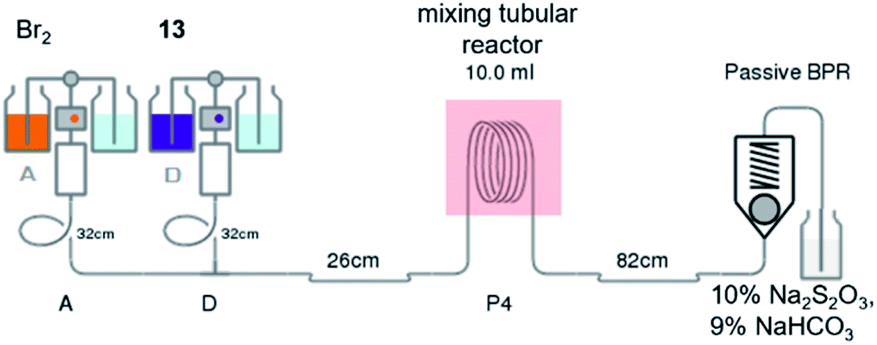 | ||
| Fig. 2 Bromination of sodium 2-carbamoylpyrazine-3-hydroxylate in a continuous flow reactor. | ||
The last three steps were modified from previous publications and patents.6,11 The reported syntheses revealed only milligram to multigram scale of production.6,7,9–11. Therefore, we optimized the following three steps: first, the fluorination of dichloro 7 was performed using the halex process to provide difluoro 15 in an excellent yield (Table S6†). The completion was monitored using TLC and NMR. The completion of difluorination was determined by the disappearance of starting material and monofluorination (Fig. 3). The 1H NMR monitoring indicated that the singlet signal at 8.58 ppm of starting material 7, doublet signal at 8.49 ppm (J = 8.1 Hz) of mono-fluorinated intermediate 15a, doublet signal at 8.46 ppm (J = 1.2 Hz) of mono-fluorinated intermediate 15b and doublet of doublet signal at 8.33 ppm (J = 8.1, 1.4 Hz) of 15. Whereas the 19F NMR monitoring also indicated the singlet signal at −75.34 ppm mono-fluorinated intermediate 15b, singlet signal at −79.39 ppm of mono-fluorinated intermediate 15a and two doublet signals at −77.23 and −81.12 ppm of 15. These NMR signals were useful for optimization and monitoring reaction. In order to complete the reaction, the halex process must be performed in perfluoroalkoxy (PFA) or hastelloy vessel due to the incompatibility of the reaction and glassware.
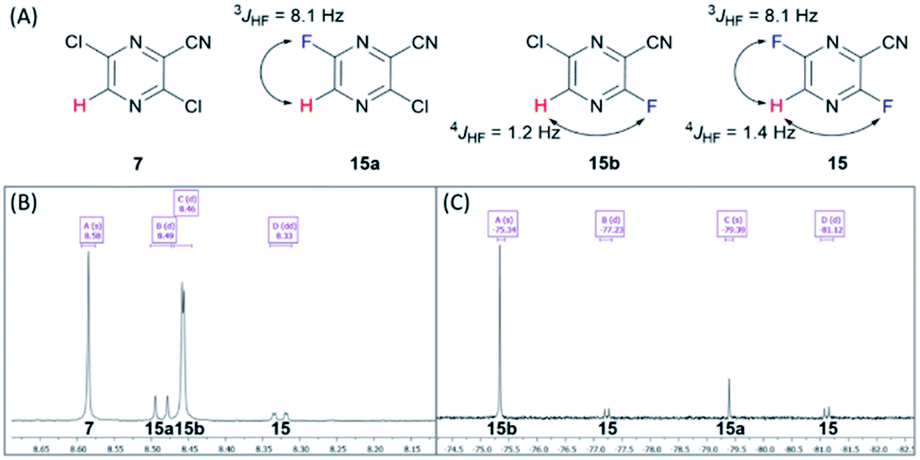 | ||
| Fig. 3 (A) Structure of 7, 15a, 15b and 15. (B) 1H NMR (CDCl3) spectra of 7, 15a, 15b and 15. (C) 19F NMR (CDCl3) spectra of 7, 15a, 15b and 15. | ||
Subsequently, the difluoro 15 was converted into monofluorinated compound 16 via substitution reaction in excellent yield (Table S7†). Finally, amide hydrolysis was carried out to convert compound 21 into favipiravir in moderate yield (61%) (Table S8†). The decolourization using activated charcoal and recrystallization in ethanol provided the crystalline (orthorhombic unite, (CCDC: 2106319†) off-white favipiravir. The following characterization revealed more than 98% purity (Fig. S1†), 0.13% water content (Table S9†), and total heavy metal was lower than 2 ppm (Table S10†).
Conclusions
We have demonstrated a total scalable synthesis of favipiravir. The continuous flow chemistry applied in hydrogenation and bromination steps considerably enhances yields. Our overall method provides a satisfactory yield and purity of favipiravir.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Center For Genetic Engineering and Biotechnology (BIOTEC) and Government Pharmaceutical Organization (GPO), grant numbers P2050576 and P2050882, respectively. We are grateful for the access to equipment at GPO, Innovation Institute PTT, BECTHAI, Phenomenex Thailand. We also thank Mr Sutichai Nithithanasilp and Miss Surisa Kongthong for HRMS and NMR data collection. We also had a great discussion with Dr Chaipat Lapinee and Dr Julian Mark Eyears. We are also grateful to Miss Waraporn Pinyo for collecting XRD data. Last, we are thankful for administration work from Ms. Linda Aree.Notes and references
- T. Manabe, D. Kambayashi, H. Akatsu and K. Kudo, BMC Infect. Dis., 2021, 21, 489–501 CrossRef CAS PubMed.
- C. C. Lai, C. M. Chao and P. R. Hsueh, J. Microbiol., Immunol. Infect., 2021, 54, 767–775 CrossRef CAS PubMed.
- C. De Savi, D. L. Hughes and L. Kvaerno, Org. Process Res. Dev., 2020, 24, 940–976 CrossRef CAS.
- S. V. Beldar and U. Jordis, Synthesis, 2020, 52, 3735–3750 CrossRef.
- Y. A. Titova and O. V. Fedorova, Chem. Heterocycl. Compd., 2020, 56, 659–662 CrossRef CAS PubMed.
- Nippon Soda Co., Ltd, US Pat., 20110275817A1, 2011 Search PubMed.
- S. V. Beldar and U. Jordis, Presented in part at the 13th Electronic Conference on Synthetic Organic Chemistry, Institute of Applied Synthetic Chemistry, 2009 Search PubMed.
- F. Shi, Z. Li, L. Kong, Y. Xie, T. Zhang and W. Xu, Drug Discoveries Ther., 2014, 8, 117–120 CrossRef PubMed.
- F.-L. Liu, C.-Q. Li, H.-Y. Xiang and S. Feng, Chem. Pap., 2017, 71, 2153–2158 CrossRef CAS.
- M. Li, CN Pat., 107226794A, 2017 Search PubMed.
- Q. Guo, M. Xu, S. Guo, F. Zhu, Y. Xie and J. Shen, Chem. Pap., 2019, 73, 1043–1051 CrossRef CAS.
- Toyamachemical Co. Ltd, US Pat., 20130245264A1, FUJIFILM Corporation, 2013 Search PubMed.
- NIKKEI Asia, accessed April 2021, https://asia.nikkei.com/Spotlight/Coronavirus/Japan-aims-to-make-Avigan-for-coronavirus-minus-imported-material.
- E. V. Antina, G. B. Guseva, A. E. Loginova, A. S. Semeikin and A. I. V’yugin, Russ. J. Gen. Chem., 2010, 80, 2374–2381 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2106318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06963b |
| ‡ TT and NC contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2021 |