
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew 1H-benzimidazole-2-yl hydrazones with combined antiparasitic and antioxidant activity†
Maria A. Argirovaa,
Miglena K. Georgievab,
Nadya G. Hristova-Avakumovac,
Dimitar I. Vuchevd,
Galya V. Popova-Daskalovad,
Kameliya K. Anichinae and
Denitsa Y. Yancheva *a
*a
aInstitute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., build. 9, 1113 Sofia, Bulgaria. E-mail: denitsa.pantaleeva@orgchm.bas.bg
bLAQV-REQUIMTE, Department of Chemistry, NOVA School of Science and Technology, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal
cDepartment of Medical Physics and Biophysics, Faculty of Medicine, Medical University of Sofia, 2 Zdrave Str., 1431 Sofia, Bulgaria
dDepartment of Infectious Diseases, Parasitology and Tropical Medicine, Medical University, Plovdiv, Bulgaria
eUniversity of Chemical Technology and Metallurgy, 8 Kliment Ohridski Blvd., 1756 Sofia, Bulgaria
First published on 14th December 2021
Abstract
Parasitic infections, caused mainly by the species Trichinella spiralis (T. spiralis), are widespread around the world and lead to morbidity and mortality in the population. Meanwhile, some studies have showed that these parasites induce oxidative stress in the infected host. With the aim of developing a class of compounds combining anthelmintic with antioxidant properties, a series of new benzimidazolyl-2-hydrazones 5a-l, bearing hydroxyl- and methoxy-groups, were synthesized. The anthelmintic activity on encapsulated T. spiralis was studied in vitro thus indicating that all hydrazones were more active than the clinically used anthelmintic drugs albendazole and ivermectin. 5b and 5d killed the total parasitic larvae (100% effectiveness) after 24 hours incubation period at 37 °C in both concentrations (50 and 100 μg ml−1). The antioxidant activity of the target compounds was elucidated in vitro against stable free radicals DPPH and ABTS as well as iron induced oxidative damage in model systems containing biologically relevant molecules lecithin and deoxyribose. The two 2,3- and 3,4-dihydroxy hydrazones 5b and 5d were the most effective radical scavengers in all studied systems. DFT calculations were applied to calculate the reaction enthalpies in polar and nonpolar medium and estimate the preferred mechanism of antioxidant activity. The relative radical scavenging ability of compounds 5a-l showed a good correlation to the experimentally observed trends. It was found that the studied compounds are capable to react with various free radicals – ˙OCH3, ˙OOH and ˙OOCH3, through several possible reaction pathways – HAT in nonpolar medium, SPLET in polar medium and RAF in both media.
1. Introduction
Trichinellosis is a natural and synanthropic focal zooanthroponosis, which is caused by parasites of the genus Trichinella.1 Human trichinellosis is usually associated with the species Trichinella spiralis (T. spiralis) which has a cosmopolitan distribution, but is most often found in the areas with temperate climate where swine are raised.2–4The complex interactions between the Trichinella parasites and the host lead to different clinical manifestations of the disease, which can be grouped into two phases related to the life cycle of the parasite – gastrointestinal (enteral) phase5 and muscular (parenteral) phase.6 Conventional treatment of the human trichinellosis is complex and includes the use of the benzimidazole anthelmintic drugs albendazole or mebendazole (Fig. 1) in combination with antipyretics, anti-inflammatory drugs and vitamins to control the temperature and intense inflammatory reactions accompanying the clinical picture of the disease.7
 | ||
| Fig. 1 Designed prototype scaffold based on antioxidant and anthelmintic active pharmacophores. | ||
The tissue damages and immune system dysfunction which arise during trichinellosis as well as toxic manifestation caused from the massive destruction of parasites during the treatment with anthelmintic drugs can be in many cases conditioned by the oxidative stress state that accompanies this infection.8 Studies have shown that changes in the levels of some antioxidant enzymes such as glutathione peroxidase (GPx), superoxide dismutase (SOD), glutathione-S-transferase (GST) and catalase (CAT) in the host's muscle tissues during trichinellosis were observed.9–11 Therefore, antioxidants are expected to help protecting the hosts against these injurious factors. Gabrashanska et al.8 examined the effect of dietary selenium in the form of organic compound Sel-Plex on the oxidative and antioxidant status, mortality, and body weight in rats experimentally infected with the nemataode T. spiralis. Their results confirmed that selenium at low doses contributes to antioxidant response and improvement of the antioxidant status in host has been observed. Analogously, resveratrol, orally applied at a dose of 20 mg kg−1 once daily for two weeks, significantly improved the redox status of the small intestine and muscles during the intestinal and muscular phases of T. spiralis infection in mice.12
In view of the above, we present here the design and synthesis of new benzimidazole derivatives as potential anti-trichinella compounds that combine anthelmintic and antioxidant actions in one molecule. Thus, in addition to their ability to kill parasites, these biologically active substances may lead to an improvement of the antioxidant status in the host.
The benzimidazoles are a large chemical family widely used in veterinary and human medicine to treat helminthic infections13 and an appropriate structural platform for antioxidant discovery efforts.14 In examining the recent literature, certain fragments were observed in the structure of benzimidazole-based antioxidants – a benzimidazole core, phenolic hydroxyl and/or methoxyl groups and hydrazone moiety (Fig. 1).15–17
On the other hand, in our previous paper,18 we reported the synthesis of a series of benzimidazolyl-2-hydrazones of fluoro-, hydroxy- and methoxy-substituted benzaldehydes and 1,3-benzodioxole-5-carbaldehyde (Fig. 1). The benzimidazoles showed potent anthelmintic activity against isolated muscle larvae of T. spiralis in an in vitro experiment as well as moderate antiproliferative activity against MCF-7 breast cancer cells.18 The antiparasitic and anticancer effects were correlated to the ability of the compounds to interfere with tubulin polymerization, as demonstrated by in vitro testing. As a next step in our research18 and on the basis of the above mentioned facts, the present study reports the synthesis of a new series of 1H-benzimidazol-2-yl hydrazones containing hydroxyphenyl and methoxyphenyl moieties, their anthelmintic activity as well as antioxidant and radical scavenging properties determined using in vitro assays – DPPH, ABTS, and iron induced oxidative damage in model systems containing lecithin and deoxyribose. In order to rationalize the observed effects, a detailed computational study of the radical scavenging properties using density functional theory (DFT) was carried out. Thermodynamic parameters related to the main antioxidant mechanisms were computed. Feasibility of the reactions with ˙OCH3, ˙OOH and ˙OOCH3 were estimated by thermodynamic calculations as well. The polarity of the medium was accounted in the study by incorporation of water and benzene solvent.
2. Results and discussion
2.1. Chemistry
According to the literature benzimidazole-2-thiols could be synthesized thorough various methods such as reaction of benzene-1,2-diamine with carbon disulfide and potassium hydroxide in the presence of ethyl alcohol and water; with potassium dithiocarbamate (potassium ethyl xanthate);19 reaction of benzene-1,2-diamine with thiophosgene in chloroform20 as well as refluxing solution with thiocyanate at 120–130 °C.21 In this study the new benzimidazole-2-yl hydrazones 5a-l were obtained according to the following reaction scheme (Scheme 1).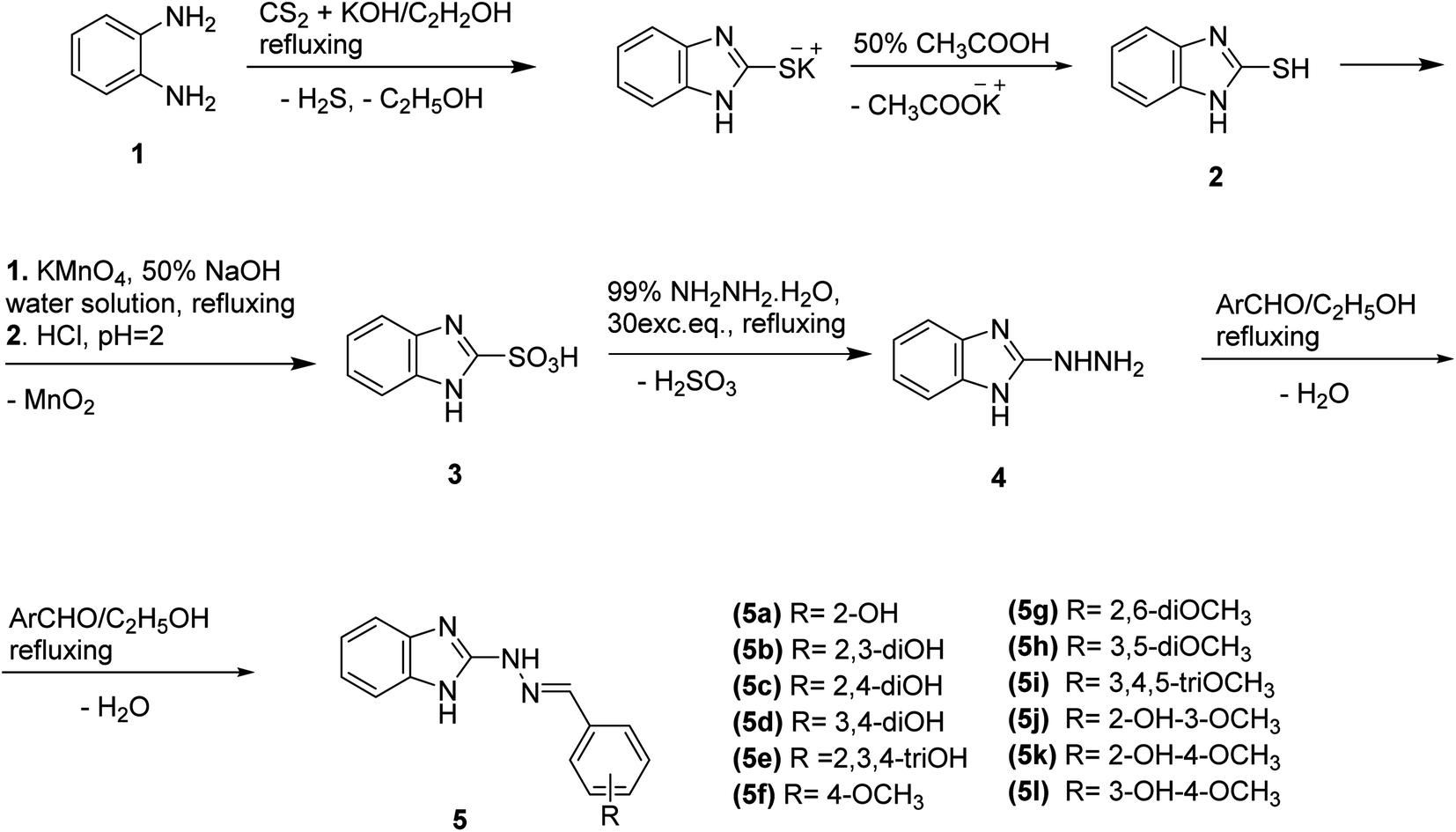 | ||
| Scheme 1 Synthesis of 1H-benzimidazole-2-yl hydrazone derivatives 5a-l. | ||
The synthesis of the 1H-benzimidazol-2-yl-thiol 2 was carried out by refluxing ethanol–water solution of potassium hydroxide, carbon disulphide and o-phenylenediamine according to the procedure described by us earlier.22 1H-Benzimidazol-2-yl-sulfonic acid 3 was obtained by oxidation of 1H-benzimidazole-2-thiol in a 50% sodium hydroxide solution of potassium permanganate for 1 h. The filtrate was acidified with hydrochloric acid to pH = 1 and the resulting precipitate of the sulfonic acid was filtered off and washed with water.23 The 1H-benzimidazol-2-yl-sulfonic acid 3 was converted in hydrazine 4 by refluxing it for 3 hours in excess of 99% hydrazine hydrate according to the described protocol.24
The target compounds 5a-l were synthesized through reaction of condensation between 1H-benzimidazole-2-yl-hydrazine 4 and various hydroxyl- and methoxy-benzaldehydes in molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and using ethanol, 99%, as a solvent.
:1 and using ethanol, 99%, as a solvent.
Identification of the 1H-benzimidazole-2-yl hydrazones was achieved by spectroscopic techniques FT-IR, 1H NMR and 13C NMR. The spectral data are given in the Experimental section.
The formation of the azomethine bond was identified by disappearance of the strong doublet for N–H stretching vibration, characteristic for the hydrazine 4, and appearance of a bands for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching in the region 1600–1620 cm−1 of the IR spectra of the 1H-benzimidazole-2-yl hydrazone products. The N–H bonds in the benzimidazolyl fragment and hydrazone moiety gave rise to strong peaks varying within the region 3370–3160 cm−1. The C–O stretching vibrations of the methoxyl groups, were found as intense broad bands around 1265 and 1135 cm−1. In the IR spectra of the compounds containing hydroxyl groups, another band for C–O stretching was observed around 1200 cm−1, while the bands of the corresponding O–H stretching vibrations appeared from 3332 to 3468 cm−1.
N stretching in the region 1600–1620 cm−1 of the IR spectra of the 1H-benzimidazole-2-yl hydrazone products. The N–H bonds in the benzimidazolyl fragment and hydrazone moiety gave rise to strong peaks varying within the region 3370–3160 cm−1. The C–O stretching vibrations of the methoxyl groups, were found as intense broad bands around 1265 and 1135 cm−1. In the IR spectra of the compounds containing hydroxyl groups, another band for C–O stretching was observed around 1200 cm−1, while the bands of the corresponding O–H stretching vibrations appeared from 3332 to 3468 cm−1.
In the 1H-NMR spectra of 5a-l the signals for the protons of the azomethine group were found at 7.9–8.0 ppm as singlets. The benzimidazole protons produced multiplet signals within the interval 6.9–7.3 ppm in DMSO solvent, while the phenyl protons resonated in the interval 6.9–7.8 ppm. The chemical shift values for the protons for the labile NH and OH protons varied in a broader range – 11.26–11.55 and 9.87–10.95 ppm.
Generation of nitrosamine impurities during the synthetic process, degradation or biotransformation is considered a severe toxicity issue due to the mutagenic and carcinogenic properties of nitrosamines.25–30 In the past few years, nitrosamine impurities (above the maximum acceptable limit that is in the range from 26.5 to 96 ng per day) were detected in various drugs and lead even to recalls from the market of several sartans used as angiotensin II receptor blockers, the diuretic drug hydrochlorothiazide, the anti-ulcer agents ranitidine and nizatidine, the antidiabetic drug metformin, and other medicines.25,26,31
The key step in thе synthesis of the drug valsartan is the formation of the tetrazole ring by the reaction of nitrile intermediate with tributyltin azide. Sodium nitrite is being used to expel remaining sodium azide reagent and under acidic condition, the nitrite ion could form nitrous acid, which could react with products from the degradation process of the solvent dimethylformamide (DMF) used in the route of synthesis.28,32 The tetrazole ring formation in the drug losartan involved using the solvent DMF and NaN3, which were precursors for obtaining of the nitrosamines. The synthetic pathways of irbesartan33,34 and candesartan35 also contained azide–nitrile coupling to form tetrazole ring, which were risk factors for nitrosamine contamination. Ranitidine, that is a H2-blocker for control of the acidity in the stomach, has been reported as the most reactive precursor of N-Nitrosodimethylamine (NDMA).26,36 Metformin in reaction of oxidation could form NDMA under high-pressure oxidative conditions or heat.37 As well as, dimethylamine is the main precursor in the synthesis of ranitidine and metformin, thus obtaining NDMA impurities.38 Last year FDA has found new nitrosamine impurities in the life-saving drugs rifampin and rifapentine (1-methyl-4-nitrosopiperazine (MNP) and 1-cyclopentyl-4-nitrosopiperazine (CPNP)) but they were not recalled from the market due to their importance.39
The nitrosamine derivatives could be obtained in presence of primary, secondary, tertiary amines or quaternary ammonium salts along with nitrosating agents such as sodium nitrite, nitrous acid (HNO2) or nitrite (NO2) under acidic conditions. Some carbamates, N-alkyl amides, cyanamides, guanidines, amidines, hydroxylamines, hydrazines, hydrazones, and hydrazides might form nitrosamine impurities.26,40 In presence of O2 could be formed other nitrosating agents – nitrosyl chloride (NOCl), nitrogen oxides (N2O3, N2O4), nitrosonium tetrafluoroborate (NOBF4)40 and nitric oxide (NO)41 that could produce nitrosamines.26 In addition, decomposition of solvents used in the route of synthesis of the drugs, such as dimethylformamide (DMF), dimethylacetamide (DMAc) or diethylacetamide (DEA), could lead to producing the mutagenic nitrosamines impurities.27,28
In this context, it should be mentioned that the synthetic route suggested by us does not include the use of nitric acid, nitrite or solvents containing dialkylamino fragment and the structure of 5a-l does not comprise tetrazole ring, dimethyl or piperazine fragments. In effort to assess the potential formation of nitrosamine impurities in the synthetic route, compound 5a was studied by UV and 15N-NMR spectroscopy. The UV spectra of nitrosamines in ethanol are characterized by two absorption bands – one of high intensity around 230 nm (similar to the band characterizing a N-NO2 group), and another one of lower intensity within the region 345–374 nm (attributed to resonance structures close to the NN band).42 However hydrazone 5a showed strong own absorption around 230 nm in ethanol which hampers the detection of potential nitrosamine impurities (Fig. S6, cf. ESI†). In the 15N-NMR spectrum of 5a in DMSO-d6 one signal was registered at 320 ppm downfield of ammonia, taken as 0.0 ppm (Fig. S7, cf. ESI†) corresponding to the N-atom from the azomethine bond, and no other signals were found that might be attributed to nitrosamines. Reported chemical shifts for nitrosamine compounds are above 500 ppm on the ammonia scale.43 Other studies reported for simple symmetrically-substituted nitrosamines NMR shifts from −107.2 to −146.7 ppm for N1(–NO) and from +154.6 to +175.9 ppm for N2(O), referenced to 15NO3Na.44 In unsymmetrically substituted nitrosamines N1 resonates respectively from −103.7 to −153.6 ppm, while N2 – from +157.0 to +176.9 ppm.44
The nitrosation and formation of the nitrosamines could be also derived endogenous in the stomach. Тhe conversion of L-arginine to citrulline in poly-morphonuclear leukocytes, is catalyzed by nitric oxide synthases (NOS) that lead to generation of nitric oxide (NO) which could react with oxygen thus producing nitrosating agents, such as N2O3 and N2O4. Under certain conditions, these nitrosating agents could react with secondary amines to form nitrosamines.45 On the other hand, it was shown that simultaneous presence of antioxidants such as ascorbate is able to block the intragastric nitrosation of drugs.40
The low risk of nitrosamine formation from benzimidazole fragment is supported by the data available for the benzimidazole-containing drug albendazole whose principal route for primary metabolism is the rapid oxidation of its sulphide group to sulphoxide, followed by further oxidation to sulphone, and deacetylation of the carbamate group to an amine.46
2.2. Anthelmintic activity
The larvicidal effects of the newly synthesized compounds 5a-l on encapsulated T. spiralis ML were studied in vitro. Biological assay results, shown in Table 1, indicate that all benzimidazoles in the tested concentrations were more active than the clinically used anthelmintic drugs albendazole and ivermectin.| Comp. | Concentrations 50 (100) μg ml−1 in μM | Efficacyb (%) | Efficacyb (%) | ||
|---|---|---|---|---|---|
| After 24 h | After 48 h | ||||
| 50 μg ml−1 | 100 μg ml−1 | 50 μg ml−1 | 100 μg ml−1 | ||
| a Control – 100 parasites.b p < 0.05. | |||||
| 5a | 0.198 (0.396) | 72.45 | 90.05 | 80.07 | 95.44 |
| 5b | 0.186 (0.372) | 100.00 | 100.00 | 100.00 | 100.00 |
| 5c | 0.186 (0.372) | 95.03 | 95.78 | 98.05 | 98.87 |
| 5d | 0.186 (0.372) | 100.00 | 100.00 | 100.00 | 100.00 |
| 5e | 0.176 (0.352) | 90.03 | 100.00 | 94.87 | 100.00 |
| 5f | 0.188 (0.376) | 5.74 | 15.34 | 24.60 | 40.25 |
| 5g | 0.169 (0.337) | 95.45 | 95.67 | 98.58 | 100.00 |
| 5h | 0.169 (0.337) | 10.33 | 95.20 | 28.17 | 97.98 |
| 5i | 0.153 (0.306) | 90.46 | 95.00 | 94.95 | 98.06 |
| 5j | 0.177 (0.354) | 40.15 | 100.00 | 61.47 | 100.00 |
| 5k | 0.177 (0.354) | 90.09 | 95.87 | 95.00 | 98.79 |
| 5l | 0.177 (0.354) | 85.43 | 100.00 | 91.03 | 100.00 |
| Albendazole | 0.188 (0.377) | 10.68 | 10.80 | 14.63 | 15.61 |
| Ivermectin | 0.057 (0.114) | 45.39 | 49.10 | 62.17 | 78.83 |
The evaluation of the anthelmintic properties of the benzimidazole derivatives indicated that the hydrazones bearing only hydroxyl groups on the phenyl ring (compounds 5a–5e) showed remarkable activity. 5a with a single hydroxyl group in 2-position of the phenyl ring exhibited 90% larvicidal effect at concentration 100 μg ml−1 after 24 h. The introduction of a second hydroxyl group at 4-position of hydrazone 5a to give compound 5c produced 5% enhancement in activity. The shift of OH group from 4- to 3-position to give hydrazone 5b led to further increase in activity (100% efficacy at a concentration of 50 μg ml−1). In accordance with these and our earlier results,18 it can be assumed that the presence of a OH group in the 3-position of the phenyl moiety of the molecule accounts for a larger share of the anti-Trichinella spiralis activity. The benzimidazolyl-2-hydrazone of 2,3-dihydroxybenzaldehyde (compound 5b) and of 3,4-dihydroxybenzaldehyde (compound 5d), respectively, killed the total parasitic larvae (100% effectiveness after 24 hour incubation period at 37 °C) in concentrations of 50 μg ml−1. A comparison of the biological effects revealed that the presence of two or three hydroxyl groups in the phenyl moiety led to benzimidazole derivatives 5b, 5c, 5d and 5e endowed with a much better anthelmintic activity against T. spiralis as compared with that of the naturally occurring stilbenoid polyphenol – resveratrol, which in study in vitro has showed a lethal effect on muscle Trichinella larvae only upon exposure to very higher concentrations (440 and 880 μM) for 72 h.47 On the other hand, there are numerous reports of natural phenolic compounds (including flavonoids) with anthelmintic effects against parasitic nematodes other than T. Spiralis48–50 and their anthelmintic activity increases with the number of hydroxyl groups.50
However, the presence of a hydroxyl group at 3-position in combination with a 4-methoxyl moiety (compound 5l) led to slight reduction in activity (85% larvicidal effect after 24 h in concentration of 50 μg ml−1) as compared to hydrazone 5d. Methoxy-analogues (5f and 5h) showed less pronounced activities against the parasites, comparable to that of the anthelmintic albendazole, after 24 h of exposure in 50 μg ml−1 concentration. The larvicidal effects of the tested compounds (excluding 5f, 5h, 5j and 5l) were dose-independent i.e. the viability of T. spiralis larvae was almost unchanged with 2-fold increase in the dose (100 μg ml−1, Table 1).
Based on the in vitro activity of hydrazones 5a-l on T. spiralis ML as well as that of other recently investigated 1H-benzimidazolyl-2-hydrazones,18 it was outlined that the combination of benzimidazole and hydrazone pharmacophores in addition with hydroxyl and/or methoxyl groups on the arylidene moiety favorably modulates the anti-Trichinella spiralis activity. Indeed, the studied benzimidazole hydrazones showed remarkable larvicidal effect superior to the benzimidazole-containing derivatives earlier synthesized by us.22,51–53 Thus, compounds 5a-l deserve more detailed pharmacological and toxicological investigations for the development of potential new anti-Trichinella spiralis agents.
2.3. Experimental estimation of the radical scavenging activity
The observed radical scavenging effects of the new 1H-benzimidazole-2-yl hydrazones against stable free radicals ABTS and DPPH are illustrated in Fig. 2A and B. The performed experiments denoted that the absorbance values of the working solutions of both radicals decreased as the sample concentrations of the tested compounds increased. The curves for the relationship between the concentration and the radical scavenging activity (RSA), obtained on the basis of these results, were with well-expressed linear dependences (R2 < 0.95). They were used to estimate the C-50 values of the compounds and to compare their scavenging activity. Two known antioxidants – quercetin and melatonin, were used as reference compounds in the experiments in order to judge better on the relative radical scavenging properties of the 1H-benzimidazole-2-yl hydrazones 5a-l. Melatonin contains an indolyl heterocycle, structurally close to the benzimidazole one, while quercetin is a flavonoid compound including a catechol fragment which is also a structural unit relevant the hydrozones studied by us.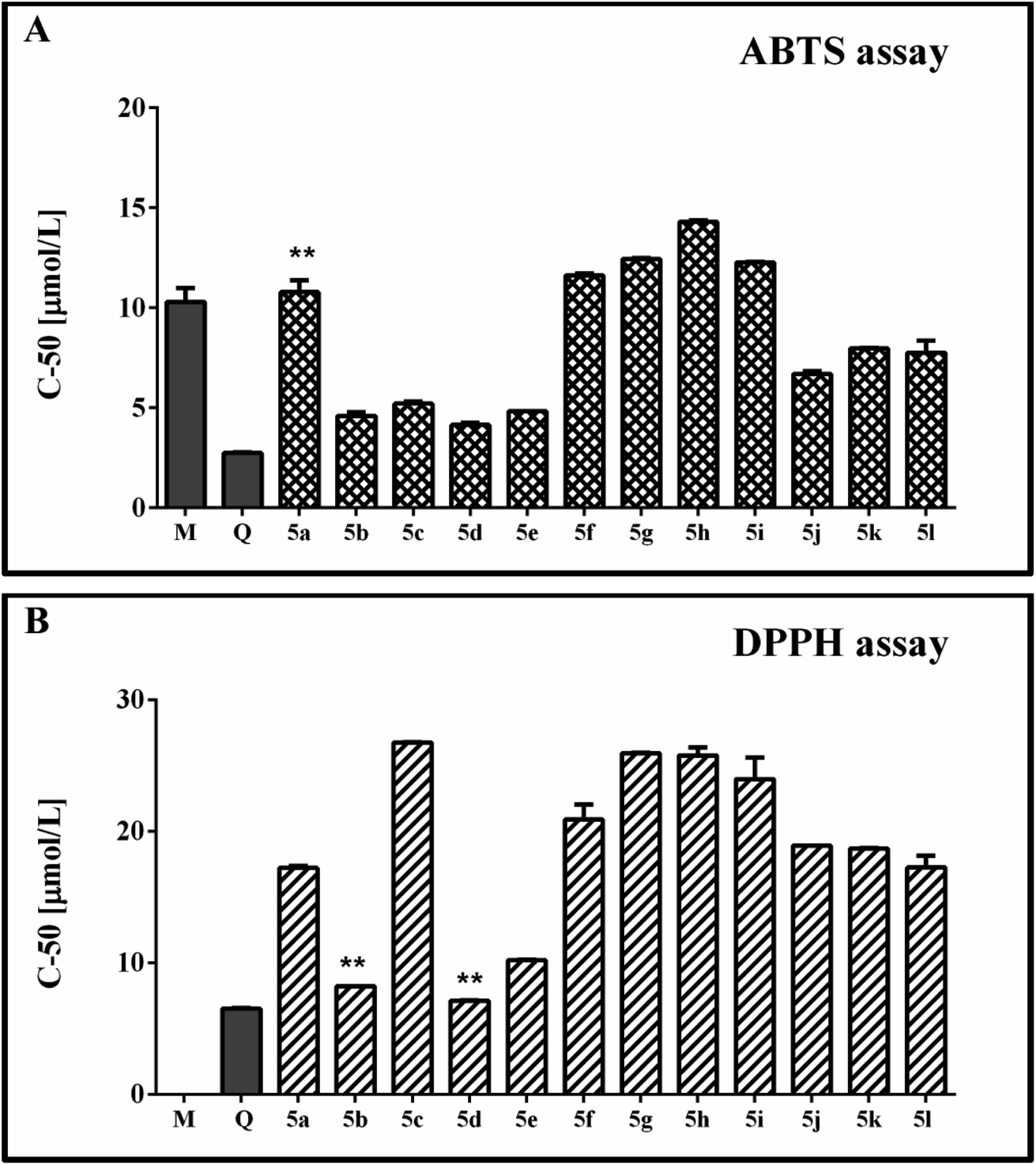 | ||
| Fig. 2 Anti-radical properties of compounds 5a-l in spectrophotometric model systems containing stable free radicals. Results have been presented as C-50 values calculated on the base of the concentration–RSA% relationship. (A) Data from the ABTS cation radical containing model systems; (B) data obtained using the DPPH assay. Melatonin (M) and quercetin (Q) have been used as reference compounds. Data are expressed as the mean ± SD. Groups were compared by one-way ANOVA with Bonferroni post hoc test. The results were considered statistically significant when p < 0.05. ** (p > 0.05) compared with Melatonin; ● (p > 0.05) compared with quercetin. | ||
The data obtained from the ABTS model system denoted C-50 values of the studied 1H-benzimidazole-2-yl hydrazones close or lower than that of the reference melatonin. The hydrazones bearing only methoxy groups in the phenyl ring (mono-, di- and tri-methoxy) had higher C-50 values compared to the references, suggesting necessity of higher amount of these compounds compared to melatonin and quercetin in order to achieve 50% RSA. Within this group of compounds, 5h was the least active – with the highest observed C-50 value – 14.30 μmol l−1. 5f, 5g and 5i had statically identical C-50 values. They were lower compared to 5h suggesting better scavenging activity. 5f had an equivalent C-50 value with the mono-hydroxyphenyl hydrazone 5a.
The presence of both methoxy and hydroxy groups in the tested 1H-benzimidazole-2-yl hydrazones induced significant decrease in the C-50 value compared to 5f and 5a. The C-50 values of this subgroup comprising 5j–5l were lower than 8 μmol l−1. They demonstrated necessity of lower concentration of the active substance to induced 50% decrease of the RSA% compared to melatonin, but did not reach the C-50 concentration of quercetin.
The di- and trihydroxy substituted 1H-benzimidazole-2-yl hydrazones 5b–5e demonstrated the best RSA within the whole series. They had very similar and in some cases even identical C-50 values ranging from 4.15 to 5.20 μmol l−1. The observed effect was much higher compared to melatonin, but still did not reach the quercetin's effectiveness.
In the DPPH model system, the reference melatonin did not exert capability to decrease the absorbance value of the radical solution. As observed in the ABTS system, among the group of the di- and trihydroxy substituted benzimidazoles were the most potent scavengers. The C-50 value of 5e was slightly higher than quercetin (with a statistically significant difference). 5b and 5d denoted identical concentration as the mentioned reference. The hydrazone with the mono-hydroxyphenyl moiety 5a and the hydrazones with mixed methoxy/hydroxy substitution i.e. 5j–5l showed equivalent C-50 values. The compounds with di- and trimethoxyphenyl moiety, similarly to the ABTS system, exerted the highest C-50 values together with the dihydroxy substituted benzimidazoles 5c.
The effect of the tested new 1H-benzimidazole-2-yl hydrazones on iron induced lecithin oxidative damage is presented in Fig. 3A. In the samples containing the reference compounds and all hydrazones, except for 5k, administrated at concentration 90 μM, the absorbance values at 532 nm were lower compared to the controls. This indicates decrease of the extent of molecular damage and well expressed protection effect against ferrous induced peroxidation. The estimated extent of molecular damage in the presence of quercetin was around 20%. In the samples containing mono-, di- and trimethoxyphenyl substituted hydrazones the observed extent of molecular damage was higher than 45%. The effect of the mono-methoxy hydrazone 5f was statistically identical to those of the dimethoxy substituted derivatives 5g and 5h. The modulation effect of 5f was also comparable to the results for the hydrazone with mono hydroxyphenyl moiety 5a. The trimethoxy substituted hydrazone 5i denoted more substantial protection compared to 5f but it was still equivalent to the one of 5g as well to that of 5j and 5l. The di- and trihydroxy substituted hydrazones 5b, 5d and 5e showed excellent protective effect – for 5d and 5e equal to the effect of quercetin.
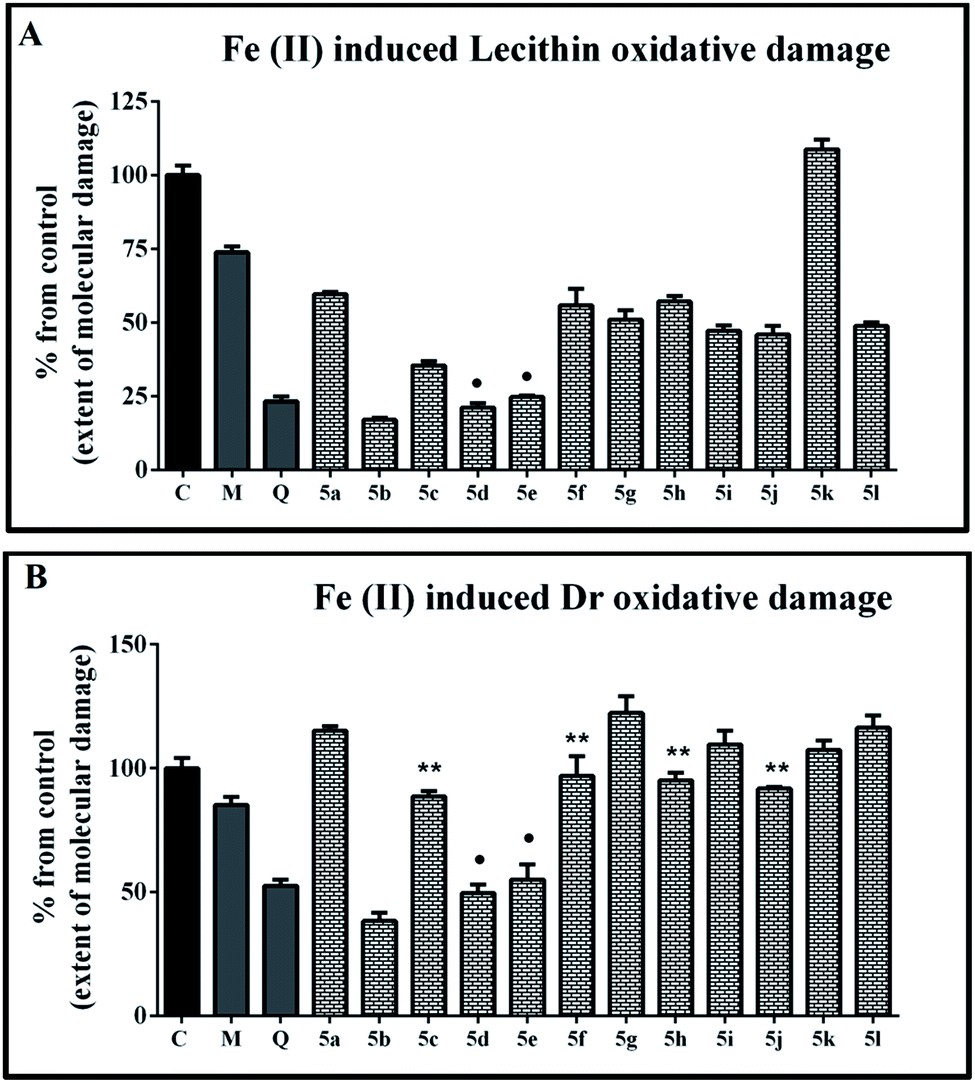 | ||
| Fig. 3 Protection effects of compounds 5a-l on iron induced oxidative damage of biologically relevant molecules: (A) extent of molecular oxidative damage observed in lecithin containing model system (1 mg ml−1) in the presence of compounds 5a-l (90 μM); (B) extent of molecular oxidative damage observed in deoxyribose containing model system (0.5 mmol l−1) in the presence of compounds 5a-l (80 μM). Melatonin (M) and quercetin (Q) at the same concentration have been used as reference compounds in both systems. Data were compared by one-way ANOVA with Bonferroni post hoc test. Results are presented as “% of molecular damage” means ± SD. The results were considered statistically significant when p < 0.05. ** (p > 0.05) compared with melatonin; ● (p > 0.05) compared with quercetin. | ||
The ability of the compounds to protect the deoxyribose molecules from iron induced molecular degradation is shown in Fig. 3B. In this system the tested benzimidazoles displayed both prooxidant and antioxidant effects depending on the molecular structure. The extent of the observed prooxidant effect was negligible to modest. The mono-, di- and trimethoxy substituted hydrazones either increased the absorbance values compared to the controls or did not exert any modulation effect. More substantial deoxyribose degradation and enhancement of the induced by iron molecular damage has been observed in the samples containing 5a, 5g and 5l. For all of these samples the estimated parameter was over 115%.
As in the lecithin containing system, the derivatives possessing hydroxyl groups in vicinal positions in the phenyl ring were better molecular protectors under the conditions of ferrous iron induced peroxidation. Compounds 5d and 5e had an effect equivalent to that of the reference quercetin and denoted extent of molecular damage close to 50%. The strongest protection effect was observed with the samples containing 5b which exerted extent of molecular damage lower than 40% – considerably better protection effect compared to both reference compounds.
Comparing the structures and the exerted modulation effects of the studied compounds it is inferred that the presence of a more than one hydroxyl group in the molecular structure of the derivatives is a crucial factor affecting their capability to protect the biologically important molecules in both chosen model systems. This relationship follows the trends found in the anthelmintic study.
The performed assays denoted a higher radical scavenging ability of the hydroxyl substituted 1H-benzimidazole-2-yl hydrazones than other earlier studied benzimidazole derivatives as for example benzimidazoles containing N-methyl-1,3,4-thiadiazol-2-amine and 4-methyl-2H-1,2,4-triazole-3(4H)-thione moieties,54 2-amino- and 5-aryl-1,3,4-oxadiazole moieties55 and N,N-disubstituted benzimidazole-2-thione hydrazone derivatives.16 On the other side, taking into account the comparison to the reference compounds used in the experiments, it could be concluded that the observed effects of the hydroxyl substituted 1H-benzimidazole-2-yl hydrazones are close to those of catechin and flavonol compounds56 and superior to melatonin derivatives.57
2.4. Molecular structure optimization
In order to rationalize the observed effects, the molecular structure and the radical scavenging properties of compounds 5a-l were characterized by DFT methods. In consideration of the fact that the studied compounds can exist in two tautomeric forms – amino and imino (Fig. 4), their geometries were optimized fully using (U)B3LYP density functional theory (DFT) method and 6-311++G** basis set in gas phase and solvents: water and benzene. Solvent effects were included in the optimizations by using the Polarizable Continuum Model (PCM). Single-point (SP) energy calculations were performed additionally at the M062X/6-311+G** level of theory, on the optimized B3LYP geometries. The calculated geometries included also Z and E configurations of the double CN (azomethine) bond, s-cis and s-trans configuration of the N–N bond, and different orientations of the substituents in the phenyl ring for both tautomeric forms (Fig. 4). The calculated Gibbs free energies G of the possible isomers of all studied compounds in amino and imino form are listed in the ESI.†
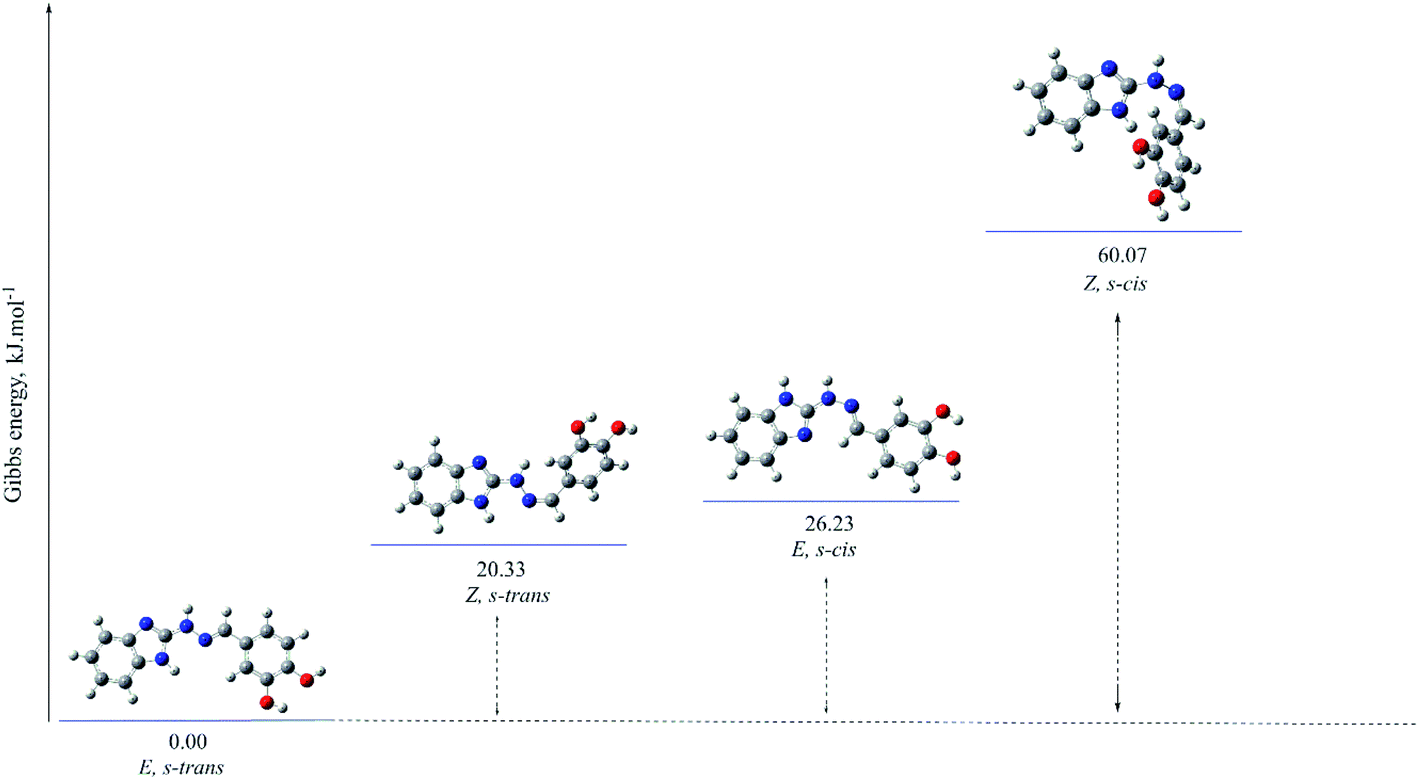 | ||
| Fig. 4 Optimized molecular structure and relative Gibbs energies (in kJ mol−1) of the possible isomers of compound 5d in amino tautomeric form, obtained at B3LYP/6-311++G(d,p) level of theory in gas phase. | ||
Summarizing the computations, the imino form strongly prevails in all compounds containing hydroxyl group in ortho position of the phenyl ring, otherwise the amino form is the favourable one. The energy difference between them is about 2.06–12.88 kJ mol−1 (cf. ESI†).
For compound 5d containing two hydroxyl groups at positions 3 and 4 in the phenyl ring and showing the most potent radical scavenging effect towards free stable radicals ABTS and DPPH, the amino:imino ratio is: 81.66:18.34 (gas phase); 91.97:8.03 (water); 96.18:3.82 (benzene) according to the B3LYP calculations. The results obtained with SP M06-2X functional indicate even greater prevalence of the amino form in all kind of media: 90.60:9.40 (gas phase); 96.87:3.13 (water); 98.37:1.63 (benzene) respectively.
On the other hands, for compound 5b containing also two hydroxyl groups, but at positions 2 and 3 in the phenyl ring and showing the highest protection effect towards lecithin and deoxyribose, the amino:imino ratio is 3.48:96.52 (gas phase); 9.98:90.02 (water); 4.60:95.40 (benzene) according to the B3LYP calculations.
The optimized molecular structures of the most stable isomers of compounds 5b (in imino tautomeric form) and 5d (in amino tautomeric form) are presented for illustration in Fig. 4 and 5. The computational results showed that E configuration of the double azomethine bond is more stable for both amino or imino form, as well as s-trans – for the arrangement around the N–N bond. The molecular geometry of the preferred amino and imino isomers is essentially flat.
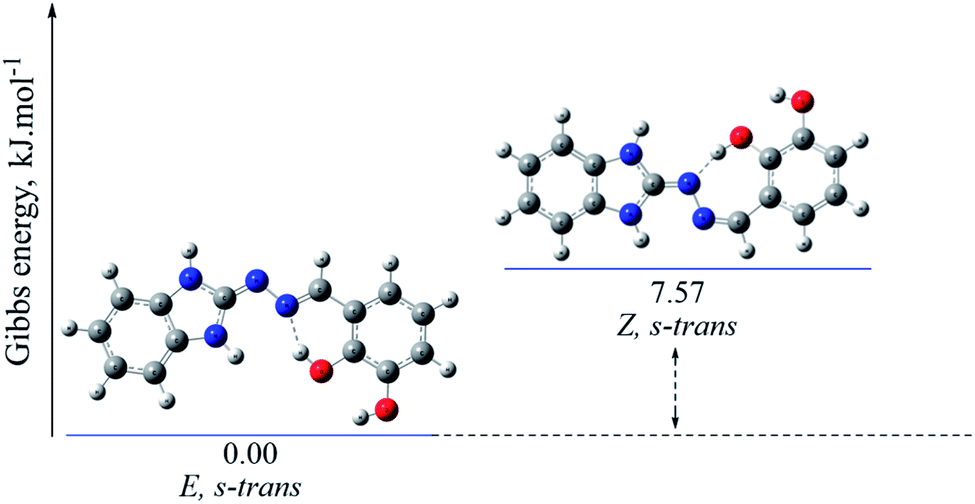 | ||
| Fig. 5 Optimized molecular structure and relative Gibbs energies (in kJ mol−1) of the possible isomers of compound 5b in imino tautomeric form, obtained at B3LYP/6-311++G(d,p) level of theory in gas phase. | ||
The gas-phase energy differences between the different isomers of the compounds with preferred amino forms are within the following ranges: E, s-trans → E, s-cis (ΔG 16.90–47.29 kJ mol−1) → Z, s-trans (ΔG 16.91–34.60 kJ mol−1) (cf. ESI†).
The theoretically estimated geometry of the amino E, s-trans forms is in a significant correlation with earlier reported X-ray experimental data.58 The calculated bond lengths of compound 5d are close to the values of the benzimidazolyl-2-hydrazone of pyridine-2-carbaldehyde examined by Pervova et al. (Fig. 6).58 The E configuration of the double CN bond of the target hydrazones is compatible with the reported structures of hydrazones containing benzothiazole59 as well as benzimidazole fragment.58
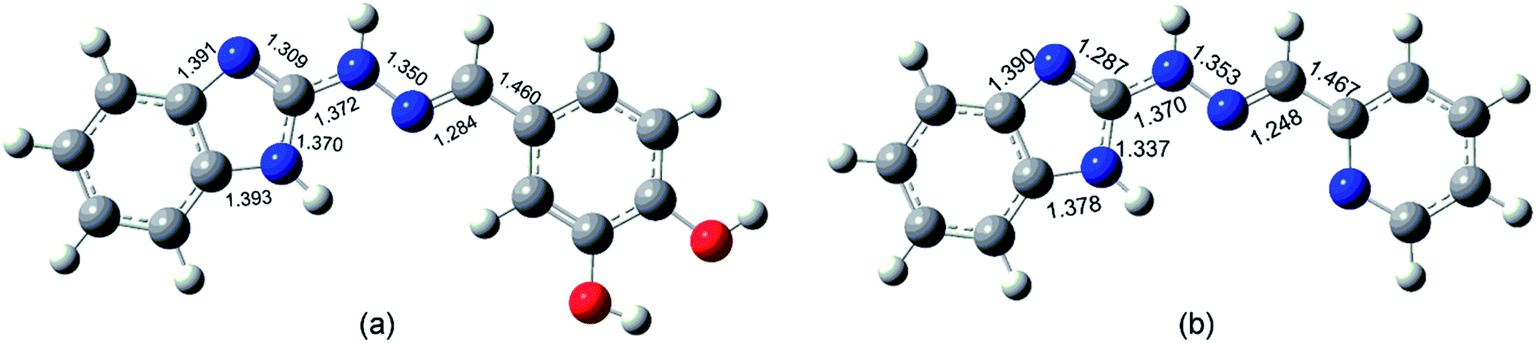 | ||
| Fig. 6 General view of the molecule of compound 5d (a) and benzimidazolyl-2-hydrazone of pyridine-2-carbaldehyde (b,58) with selected bond lengths. | ||
The gas-phase energy differences between the different isomers of the compounds with preferred imino forms vary as follows: E, s-trans → E, s-cis (ΔG 0.88–35.34 kJ mol−1) → Z, s-trans (ΔG 4.17–28.34 kJ mol−1) (cf. ESI†).
2.5. Computational study of the radical scavenging activity
At molecular level, the studied hydrazones 5a-l can exert their radical scavenging activity through several probable mechanisms (Scheme 2). The first one of them comprises direct hydrogen atom transfer from the antioxidant to the free radical (HAT mechanism). Other probable mechanisms are the two-steps reaction pathways where the electron and proton are transferred in separate steps from the antioxidant to the free radical: single electron transfer followed by proton transfer (SET-PT mechanism) and sequential proton loss electron transfer (SPLET mechanism). A fourth possible way for deactivation of the free radicals is radical adduct formation (RAF mechanism) where the antioxidant covalently binds to the free radical.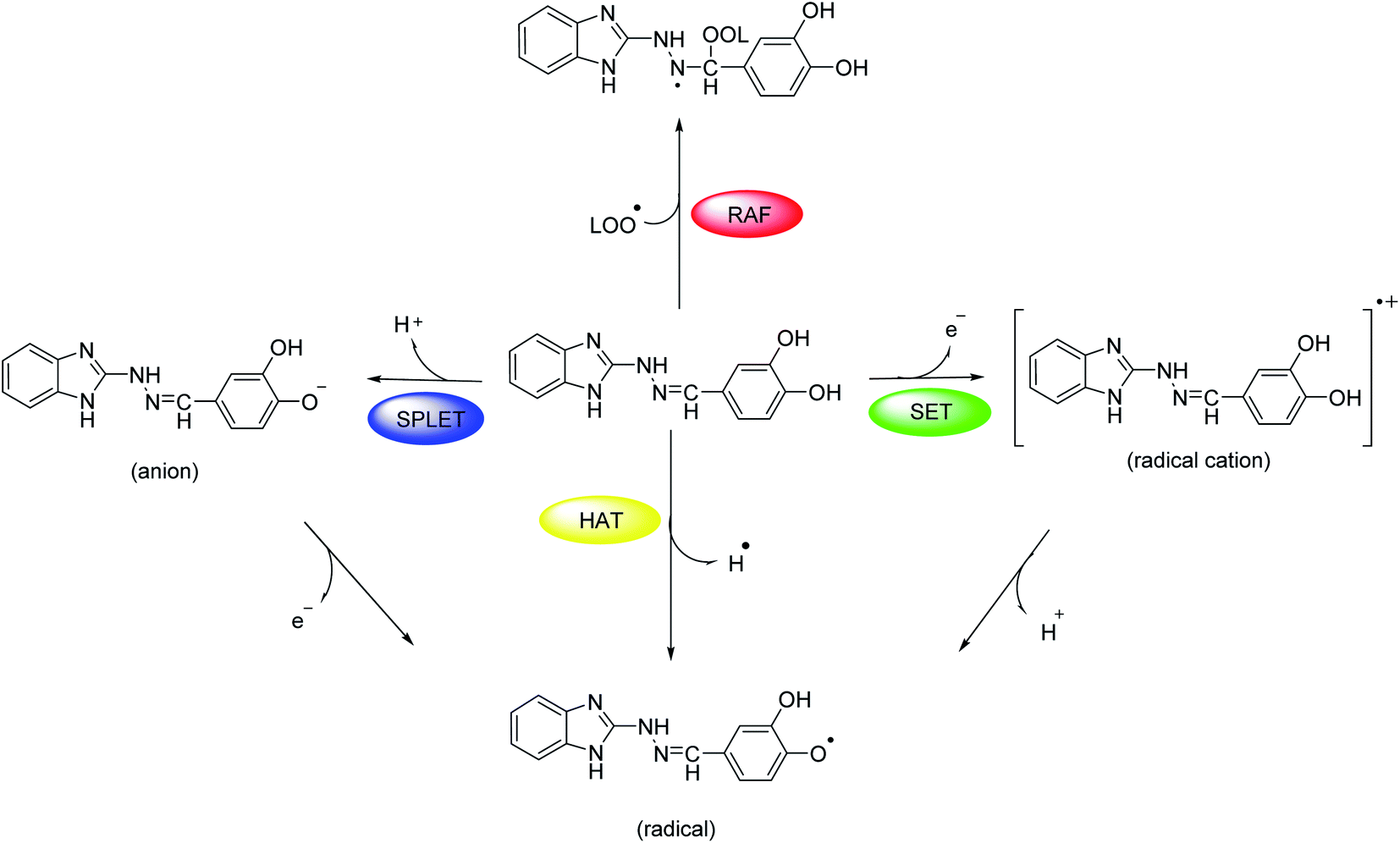 | ||
| Scheme 2 Probable mechanisms of radical-scavenging activity. | ||
In the same way, the molecular structure of the studied 1H-benzimidazole-2-yl hydrazones suggest several possible sites for hydrogen atom transfer, deprotonation and radical adduct formation. For instance, compound 5d might cleave the bonds N5–H10, N11–H24, O20–H22, O21–H23 and C13–H25 (for numbering cf. Scheme 3) in order to transfer a hydrogen atom or a proton as shown in blue in Scheme 3. Radical attack for adduct formation is also possible at several reaction sites: C1, C2, C4, C6 etc. (shown in red in Scheme 3). The reactivity for HAT of the above mentioned sites in compound 5d was estimated by calculating the respective bond dissociation enthalpies (BDE) resulting in the formation of radicals R1–R5 (Scheme 3). The most reactive site is denoted by the lowest enthalpy. It was previously demonstrated that DFT/B3LYP calculations are a good choice for estimation of the reaction enthalpies as they can provide a fairly accurate agreement between the calculated values and the available experimental data as well as describe reliably the structure–activity relationship for antioxidants with diverse chemical structure at reasonable computational time.60–64 On the other hand, the use of at least double-zeta basis set, including polarization and diffuse functions, is recommended,65 therefore the B3LYP//6-311++G(d,p) combination was applied in the present calculations. The results obtained at this computational level allow a useful comparison with earlier studied antioxidants.
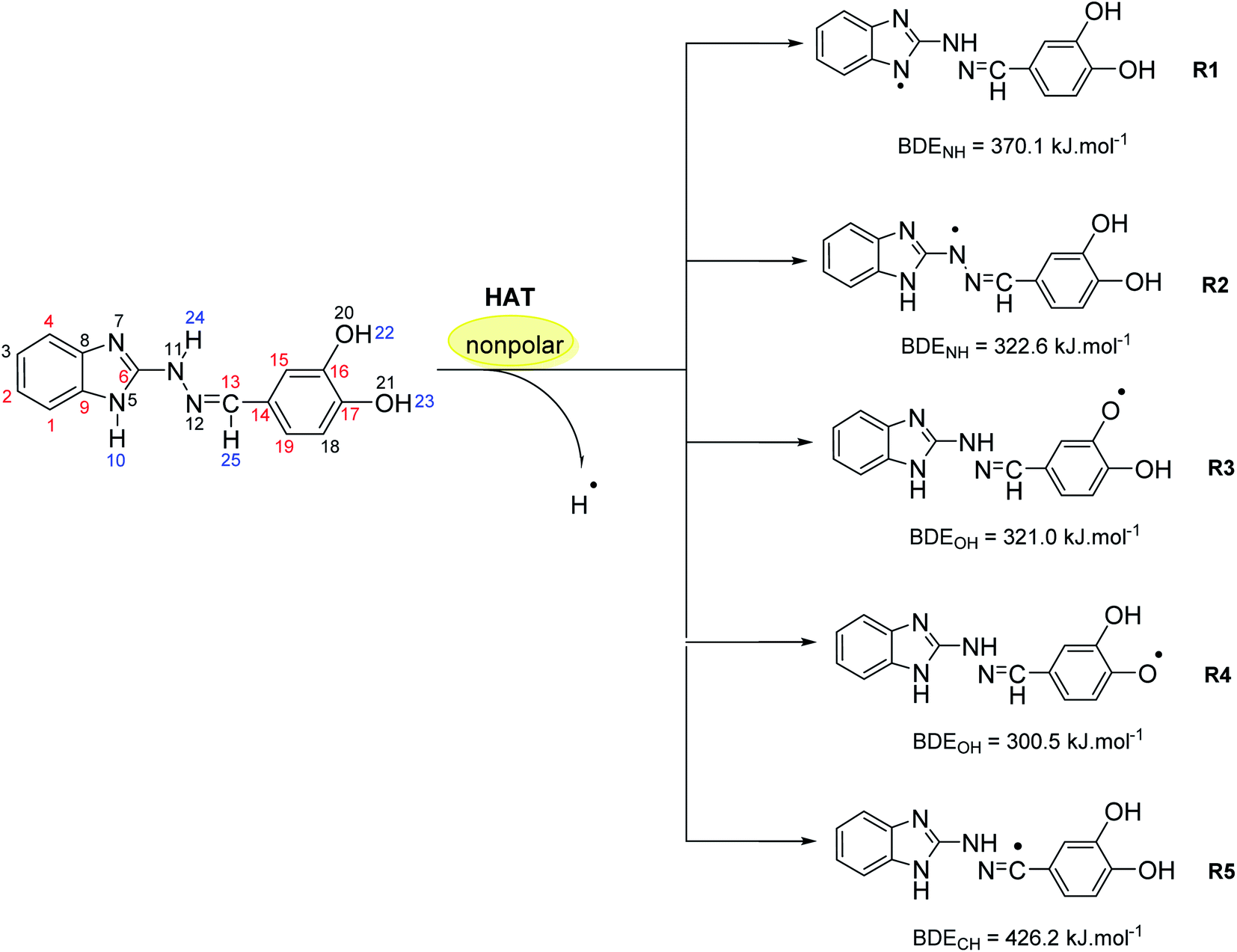 | ||
| Scheme 3 Possible sites for hydrogen atom transfer (in blue) and radical adduct formation (in red) for compound 5d along with the radicals formed by HAT and the respective bond dissociation enthalpies, calculated at B3LYP/6-311++G(d,p) level of theory in benzene. | ||
The calculations in gas phase and benzene medium outlined the hydroxyl group at para position in the phenyl ring as the most active one (BDE in benzene 300.5 kJ mol−1; BDE in gas phase 293.2 kJ mol−1), followed by the hydroxyl group at meta position in the phenyl ring (BDE in benzene 321.0 kJ mol−1; BDE in gas phase 313.4 kJ mol−1) and the amino group in the hydrazine chain (BDE in benzene 322.6 kJ mol−1; BDE in gas phase 316.5 kJ mol−1). Moreover, the calculated BDE values are close to the calculated BDE values of known phenolic antioxidants such as α-tocopherol (BDE in gas phase 297 kJ mol−1, calculated at the same level of theory60) and quercetin (with lowest O–H BDE value estimated to 305 kJ mol−1 at the same level of theory66).
The amino group in the benzimidazolyl fragment and the C–H bond from the azomethine group of compound 5d are characterized by a much lower reactivity – with BDE values in benzene 370.1 kJ mol−1 (BDE in gas phase 366.7 kJ mol−1) and 426.2 kJ mol−1 (BDE in gas phase 418.7 kJ mol−1), thus not expected to contribute significantly to the radical scavenging properties of the compound. For comparison, the BDE of the indolyl N–H bond in melatonin and the methylene group next to the indolyl fragment were also calculated. The first one is 355 kJ mol−1, whereas the latter – 428 kJ mol−1, respectively.
The radical scavenging ability of the antioxidants is related to the stability of the formed radicals, which on turn is affected by the delocalization of the unpaired electron over the conjugated system.67 In order to estimate the stability of R4, R3 and R2 radicals of 5d, the Natural Bond Orbital (NBO) spin density distribution was examined and compared (Fig. 7).
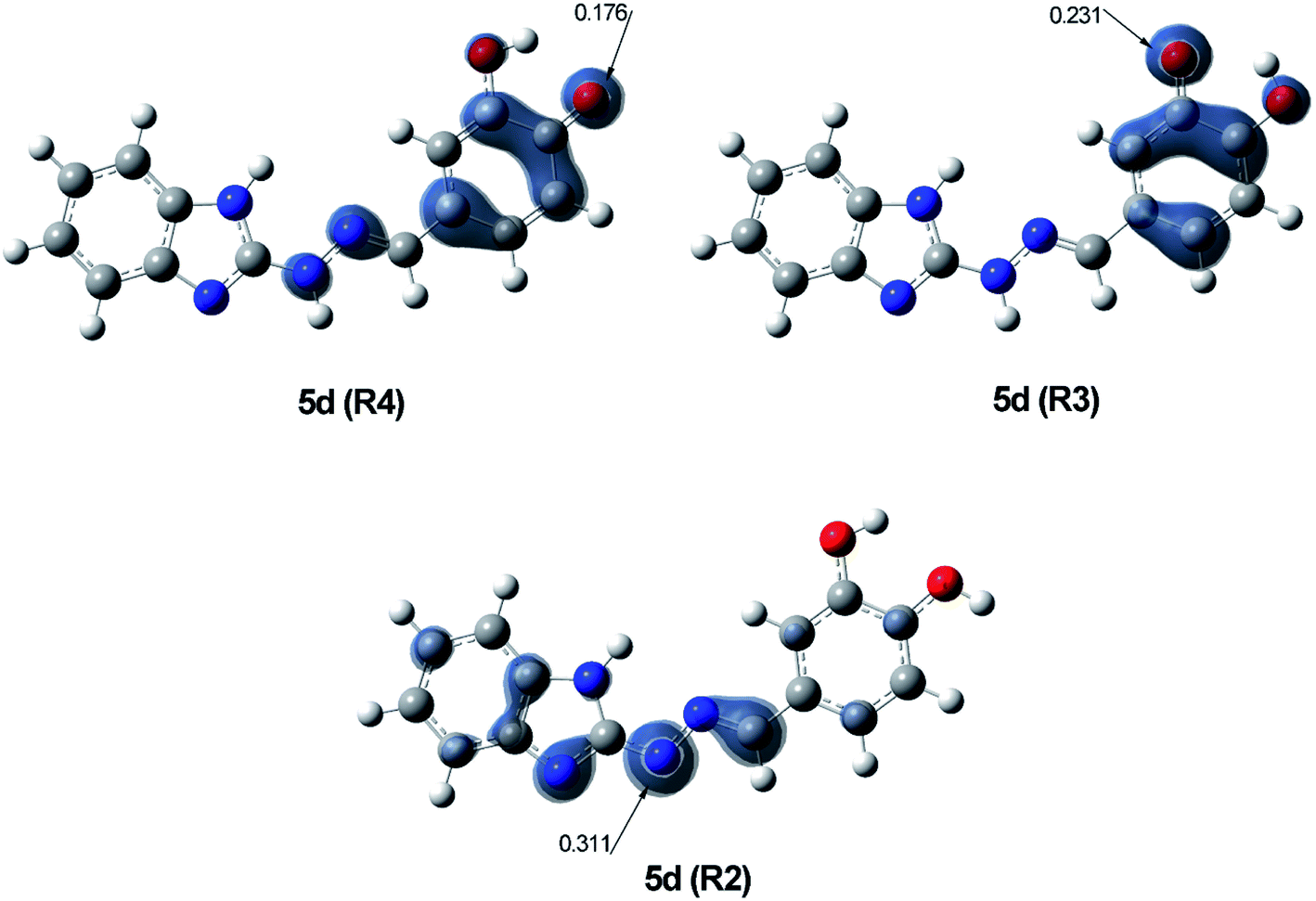 | ||
| Fig. 7 Visualisation of spin density of the three most stable radicals R4, R3 and R2 of 5d, calculated at B3LYP/6-311++G(d,p) level of theory in benzene. | ||
Radical R4 showed the highest delocalization of spin density – only 0.176 of the unpaired electron are found on O21, while the greater part of it is spread over the phenyl ring and the hydrazone chain. In radical R3 the spin density is delocalized only over the phenyl ring with 0.231 e− localized on O20, while in radical R2 it is limited within the hydrazone chain and part of the benzimidazole ring, with 0.311 e− localized on N11. These data indicate R4 as the most stable radical, followed by R3 and R2. The order of stability of radicals R4, R3 and R2, correspond to the order of reactivity at the different reaction sites established by the calculated BDE values. Such correlation between the two parameters has been previously discussed for other phenolic antioxidants.68–71
Further the reaction enthalpies for HAT, SET-PT and SPLET mechanisms of all hydrazones 5a-l were computed in gas phase and water. The ability to deactivate free radical via HAT mechanism was described by the BDE values. The ionization potentials (IP) were used to describe the ability of the compounds to transfer a single electron via SET-PT mechanism, while the proton affinities (PA) were used to estimate the ability to transfer a proton via SPLET. Based on the comparison of the BDE values to IPs and PAs, it is possible to judge which mechanism is expected to govern the antioxidant action of the studied compounds. The lower the reaction enthalpy, the more easily would be followed the particular reaction pathway. On the other hand, the comparison of the reaction enthalpies of the different compounds might clarify the relationship between the molecular structure and the experimentally determined radical scavenging activity.
The reaction enthalpies for each compound were modelled at all possible reaction sites. Fig. 8A presents the lowest BDEs, IPs and PAs found for all compounds in gas phase. As it could be seen the IP and PA values lay far above the respective BDE values for all studied compounds (Fig. 8A), pointing out that the SET-PT and SPLET mechanism require significantly more energy than HAT and therefore are not likely to occur in nonpolar phase.
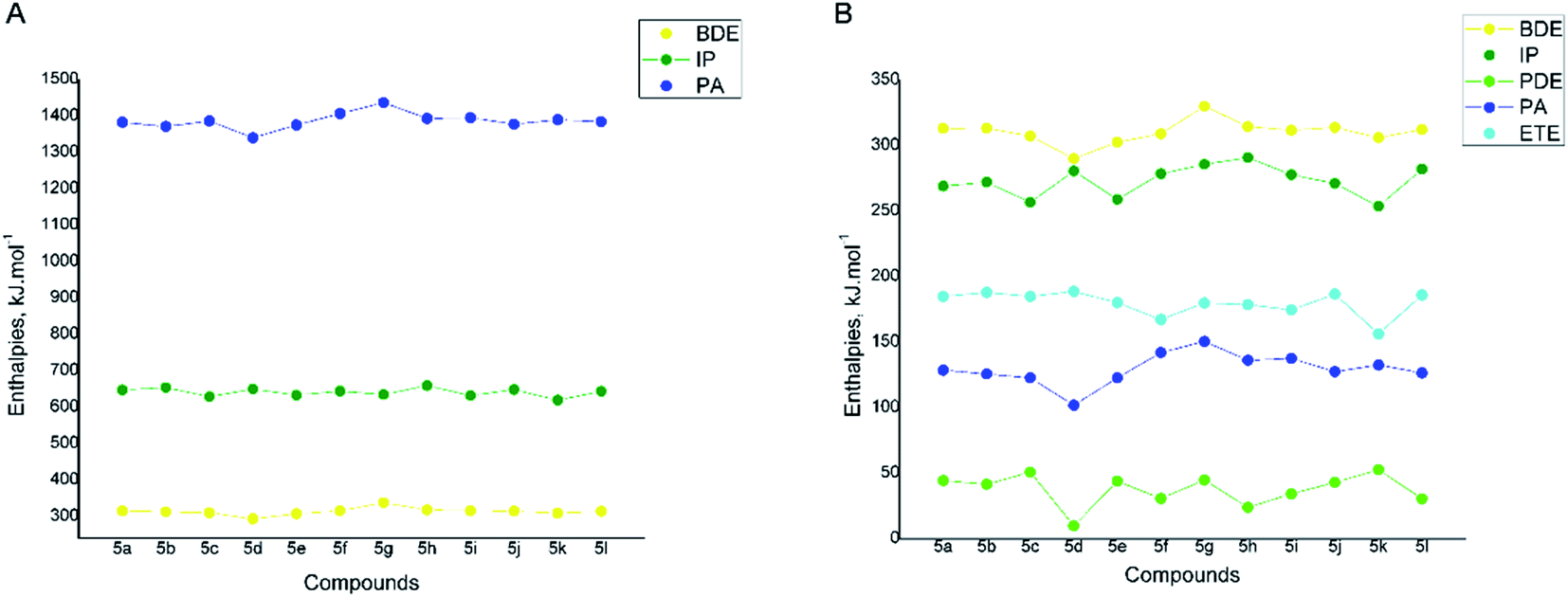 | ||
| Fig. 8 Reaction enthalpies for compounds 5a-l in gas phase (A) and water (B): dissociation enthalpy (BDE), ionization potential (IP), proton dissociation enthalpy (PDE), proton affinity (PA), and electron transfer enthalpy (ETE), calculated at B3LYP/6-311++G** level of theory. | ||
It was established that for compounds 5d and 5l, bearing hydroxyl groups in 3 and 4 position of the phenyl ring, the lowest BDE corresponds to the cleavage of one of the O–H bonds and are 293.21 and 314.49 kJ mol−1, respectively. For compounds 5f–i, where only methoxyl groups are present in the phenyl ring, the lowest BDEs are connected to the cleavage of the N5–H10 bond from the benzimidazole fragment. Correspondingly they are higher than the O–H BDE values of 5c–e and range from 315.24 to 373.81 kJ mol−1. For all derivatives containing hydroxyl groups in 2-position of the phenyl ring the lowest BDE are connected to the cleavage of the N5–H10 bond similarly to 5f–i. It should be noted that the compounds bearing vicinal hydroxyl groups i.e. 5b–e possess the lowest BDEs within the whole studied series and the 3,4-dihydroxyl derivative 5d is outlined as the most active one with BDE of 293.21 kJ mol−1. Based on that, it is expected to be the most active via HAT mechanism in nonpolar medium.
In water the BDE values of compounds 5a-l are slightly lowered in comparison to the gas phase ones (they vary in the range 291.75–330.01 kJ mol−1), but follow the same trend (Fig. 8B).
On the other hand, the calculated O–H and N–H proton affinities of 5a-l in water are much lower than the respective BDEs showing that in polar medium the first step (deprotonation) of SPLET mechanism is favored over HAT. The PAs are connected to the same functional groups as in the HAT process. PAs are between 108.30 and 150.47 kJ mol−1 – the lowest values were found again for the compounds bearing vicinal hydroxyl groups 5b–5e, and particularly for the 3,4-dihydroxyl derivative 5d. The second step (electron transfer) of the SPLET mechanism, described by the electron transfer enthalpy (ETE), also requires lower energy than the homolytic cleavage of the O–H or N–H bond for all studied compounds.
The ionization potentials of 5a-l in water are lower than the BDEs, but still higher than the PA values which indicate that the SET-PT mechanism is less probable than the SPLET in polar medium.
Summarizing the data in polar medium, it could be concluded that in this case SPLET is regarded as the most probable mechanism of antioxidant action and the derivatives with vicinal hydroxyl groups are expected again to show the highest activity. These results are in good agreement with the experimentally determined radical scavenging effects in DPPH, lecithin and deoxyribose assays, where the hydroxyl substituted hydrazones demonstrated the most potent radical scavenging effect. The presence of a phenyl fragment with vicinal hydroxyl groups has been associated to improved radical scavenging properties due to its conversion to ortho-quinone intermediates.72,73
The ability of the studied compounds to deactivate different free radicals was modeled by transition states involving attack by ˙OCH3, ˙OOH and ˙OOCH3, at all reactive sites identified in the preceding computational study. The calculations were carried out with compound 5d at SP M06-2X/6-311++G** level of theory. M06-2X/6-311++G** was selected for the computations taking into account the reliability of this method for transition state studies.69,74,75 The activation energies (ΔG‡) for all studied transition states in gas phase and solvent (water and benzene) for all three attacking radicals are presented in Table 2.
| TSs | ΔG‡((U)M06-2X/6-311++G**//(U)B3LYP/6-311++G**) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ˙OCH3 | ˙OOH | ˙OOCH3 | |||||||
| Gas phase | Water | Benzene | Gas phase | Water | Benzene | Gas phase | Water | Benzene | |
| TS H10 | 67.64 | 69.23 | 77.44 | 90.83 | 81.24 | 90.59 | 95.07 | 86.47 | 97.32 |
| TS H24 | 33.15 | 89.83 | — | 64.81 | 77.91 | 74.32 | 74.46 | 82.93 | 84.91 |
| TS H22 | — | 18.08 | — | 75.71 | 81.95 | 86.09 | 80.93 | 81.31 | 90.32 |
| TS H23 | — | 18.30 | — | 68.97 | 83.31 | 74.56 | 71.73 | 83.13 | 67.47 |
| TS H25 | 71.01 | 83.94 | 79.93 | 115.15 | 125.92 | 122.74 | 119.82 | 132.74 | 128.83 |
The most stable transition states TS H24, TS H22 and TS H23 correspond to the formation of the most stable radicals R2, R3 and R4 for all attacking species. Unfortunately, we could not locate transition states between 5d and ˙OCH3 for TS H24 in benzene and TS H22 and TS H23 in gas phase and benzene. The scans made (5d)-O⋯H⋯O–CH3 for TS H22's and TS H23's and (5d)-N⋯H⋯O–CH3 for TS H24 do not indicate maximum in the energies corresponding to transition state (cf. ESI, Fig. S1†). Having in mind the activation energies for TS H22 (˙OCH3) and TS H23 (˙OCH3) in water (around 18 kJ mol−1 only) and the activation energies of the TS's for the other attacking radicals (values in gas phase and benzene lower than in water), it seems that the not detected TS's should have lower than 18 kJ mol−1 activation energy.
The optimized structures of the most stable TS's (Fig. 9) show similarity in regard to the bond distances (in Å) for the breaking of O⋯H and N⋯H and forming new H⋯O bonds with the different free radicals. The distances in TS H24's are longer compared to the other two transition states (TS H23 and TS H22) for the same attacking free radical.
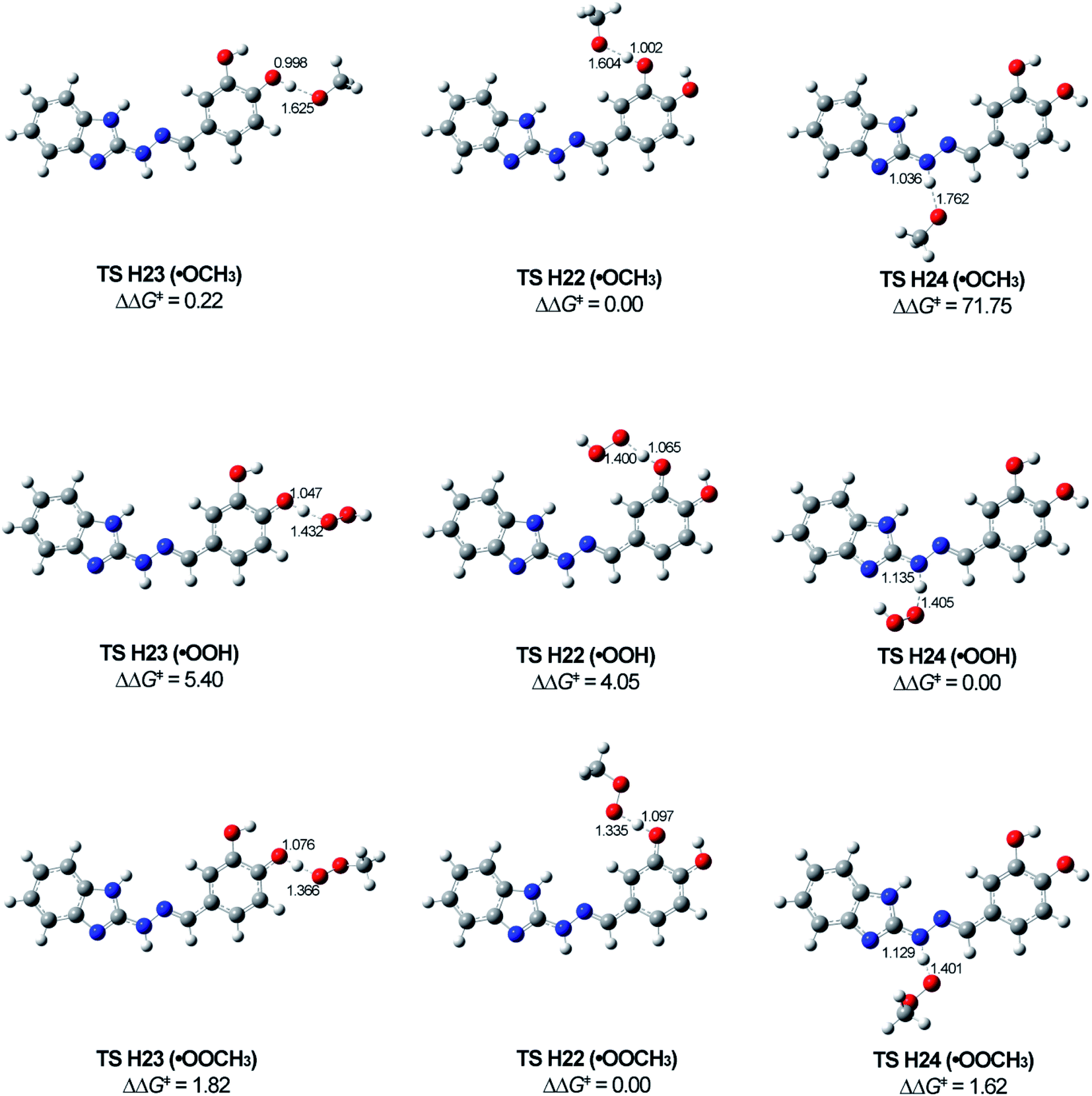 | ||
| Fig. 9 Optimized structures of the TSs for the most stable radicals R2, R3 and R4 corresponding to the HAT mechanism. ΔΔG‡ shows the difference in the activation energies between the TSs for every one of the studied radicals. Calculations are done on SP M06-2X/6-311++G** method in solvent water. | ||
According to our theoretical study, the radical scavenging can go through RAF mechanism as well. The calculations showed that the most stable transition states (TS) between the studied radicals and 5d are at C13 position (Table 3).
| TSs | ΔG‡((U)M06-2X/6-311++G**//(U)B3LYP/6-311++G**) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ˙OCH3 | ˙OOH | ˙OOCH3 | |||||||
| Gas phase | Water | Benzene | Gas phase | Water | Benzene | Gas phase | Water | Benzene | |
| TS C1 | 68.15 | 74.93 | 75.89 | 102.16 | 104.87 | 107.80 | 115.53 | 119.58 | 122.07 |
| TS C2 | 70.23 | 72.73 | 74.54 | 101.96 | 100.35 | 107.29 | 115.26 | 114.57 | 119.53 |
| TS C4 | 72.23 | 71.69 | 75.85 | 102.99 | 100.31 | 107.88 | 117.04 | 116.04 | 121.04 |
| TS C6 | 83.64 | 92.37 | 91.53 | 101.62 | 109.66 | 109.84 | 107.90 | 118.34 | 116.40 |
| TS C9 | 73.18 | 78.53 | 80.13 | 107.77 | 107.56 | 112.57 | 121.37 | 121.36 | 126.40 |
| TS C13 | 52.87 | 60.20 | 59.55 | 73.13 | 82.25 | 80.70 | 80.72 | 91.13 | 88.93 |
| TS C14 | 82.30 | 83.24 | 87.81 | 113.28 | 114.10 | 117.98 | 125.51 | 127.23 | 130.16 |
| TS C15 | 70.08 | 70.10 | 76.12 | 102.25 | 101.91 | 107.36 | 112.21 | 113.95 | 117.81 |
| TS C16 | 70.29 | 72.34 | 82.25 | 107.55 | 101.44 | 108.84 | 117.23 | 115.83 | 121.77 |
| TS C17 | 47.72 | 61.16 | 57.31 | 83.81 | 91.66 | 91.33 | 95.10 | 104.29 | 102.26 |
| TS C19 | 65.02 | 70.42 | 71.00 | 94.11 | 97.89 | 98.95 | 106.31 | 112.12 | 112.56 |
The optimized geometries of the possible transition states involved in the RAF mechanism are shown in Fig. 10. The most relevant parameter is the C⋯O distance corresponding to the forming new bond and it was found to be around 2 Å for all calculated TS's.
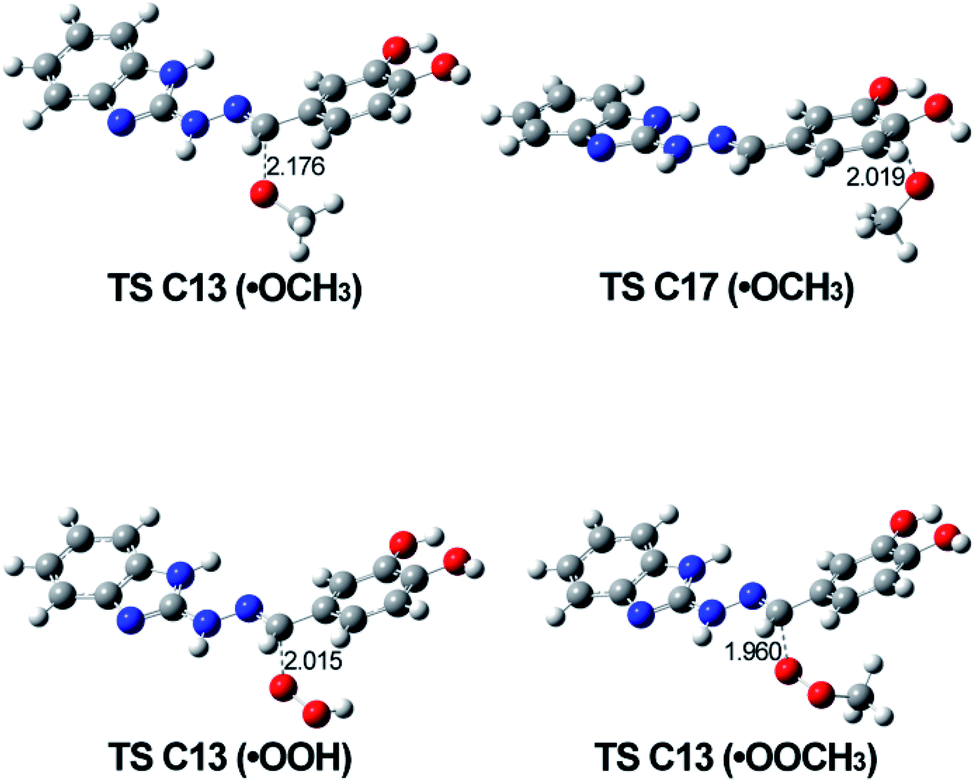 | ||
| Fig. 10 Optimized structures of the most probable TSs corresponding to the RAF mechanism for every one of the studied radicals. Calculations are done in solvent water. | ||
The values of ΔGreaction in Table 4 for this mechanism showed that concerning the ˙OCH3, the reaction can proceed at C13 and C17 position in 5d in water and benzene; when the radical is ˙OOH, the reaction still will be possible at C13 (water and benzene), while when the reacting species is ˙OOCH3, the reaction will follow this mechanism with difficulty.
| Reaction site | ΔGreaction((U)M06-2X/6-311++G**//(U)B3LYP/6-311++G**) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ˙OCH3 | ˙OOH | ˙OOCH3 | |||||||
| Gas phase | Water | Benzene | Gas phase | Water | Benzene | Gas phase | Water | Benzene | |
| a Having in mind the values of ΔGreaction and ΔG‡ for the TSs of HAT and RAF mechanisms, the reaction between ˙OCH3 and 5d will go mainly through HAT mechanism at H23, H22 and H24 position. | |||||||||
| HAT | |||||||||
| H10 | −36.36 | −46.88 | −37.65 | 34.50 | 23.15 | 32.96 | 40.00 | 30.75 | 39.62 |
| H24 | −84.27 | −87.42 | −82.32 | −13.41 | −17.39 | −11.71 | −7.91 | −9.79 | −5.05 |
| H22 | −84.32 | −84.46 | −83.50 | −13.46 | −14.43 | −12.89 | −7.96 | −6.83 | −6.23 |
| H23 | −99.63 | −99.29 | −98.46 | −28.77 | −29.26 | −27.85 | −23.27 | −21.66 | −21.20 |
| H25 | 3.36 | 2.21 | 5.44 | 74.22 | 72.24 | 76.05 | 79.71 | 79.84 | 82.70 |
|
|||||||||
| RAF | |||||||||
| C13 | −72.93 | −59.30 | −63.33 | −12.69 | 2.89 | −1.05 | −1.74 | 14.23 | 7.63 |
| C17 | −33.50 | −20.52 | −24.00 | 33.22 | 43.65 | 41.08 | 43.15 | 54.25 | 51.38 |
| C19 | −7.15 | 3.53 | 0.05 | 49.64 | 57.48 | 56.23 | 61.73 | 69.46 | 67.33 |
| C6 | −7.44 | 6.19 | 2.97 | 46.14 | 59.22 | 55.08 | 59.45 | 73.11 | 69.60 |
| C15 | −0.32 | 6.64 | 6.26 | 55.90 | 61.67 | 62.62 | 66.13 | 71.97 | 72.00 |
| C1 | 5.92 | 16.26 | 13.97 | 63.08 | 70.87 | 70.68 | 76.25 | 84.87 | 83.93 |
| C2 | 12.27 | 20.53 | 19.17 | 67.01 | 70.94 | 72.33 | 80.20 | 85.44 | 87.28 |
| C4 | 3.91 | 10.93 | 11.13 | 61.61 | 63.63 | 66.99 | 72.86 | 75.68 | 79.21 |
| C9 | 32.02 | 42.70 | 40.60 | 91.60 | 96.73 | 98.32 | 106.09 | 110.98 | 112.45 |
| C14 | 31.47 | 37.22 | 37.58 | 87.01 | 92.49 | 91.45 | 98.65 | 103.71 | 106.64 |
| C16 | 4.70 | 8.28 | 11.17 | 68.99 | 69.92 | 73.65 | 80.22 | 81.57 | 86.95 |
Hypothetically, the RAF mechanism can occur at N12 (Scheme 3) as well, but any attempt to locate the corresponding products and transition states brought us failure.
When the reacting species is ˙OOH both mechanisms (HAT and RAF) can be followed: HAT – at H23, H22 and H24, and RAF – at C13 position. The most stable TSs for HAT and RAF mechanism have similar values of ΔG‡ (HAT: H24–77.91 (water) and 74.32 (benzene) kJ mol−1 and RAF: C13–82.25 (water) and 80.70 (benzene) kJ mol−1).
The ˙OOCH3 could be deactivated only by HAT mechanism. The calculations showed a lower reactivity of the radical compared with the other two studied radicals. According to the ΔGreaction, the reactions are slightly exothermic and in the same time the ΔG‡ of the most stable TSs are higher than the ΔG‡ of the other two radicals at the respective positions.
All gathered computational data demonstrated that the newly synthesized 1H-benzimidazole-2-yl hydrazones possess very versatile radical scavenging properties – several reaction sites characterized by relatively low reaction enthalpies and possibility to act simultaneously through several possible reaction pathways. It was also found that the predicted activities of compounds 5a-l are in good correlation with the experimentally observed antiradical activity.
3. Conclusions
To conclude, several 1H-benzimidazole-2-yl hydrazones as potent multi target drugs, combining antioxidant and anthelmintic activity, were synthesized using o-phenylendiamine as precursor and variety of methoxy- and hydroxyl-substituted benzaldehydes. The newly synthesized 1H-benzimidazole-2-yl hydrazones exhibited higher anthelmintic activity against isolated Trichinella spiralis muscle larvae in vitro than the clinically used anthelmintic drugs albendazole and ivermectin in the tested concentrations. The 1H-benzimidazole-2-yl hydrazones containing more than one hydroxyl groups in the phenyl moiety (compounds 5b–e) showed a potent anthelmintic activity – above 90% larvicidal effect at concentration 50 μg ml−1 after 24 h, while the methoxy derivatives showed less pronounced activities against the parasites. Compounds 5b and 5d were the most active ones with 100% effectiveness after the 24 hour incubation period at 37 °C in concentrations of 50 μg ml−1 and 100 μg ml−1.The in vitro evaluation of the antioxidant properties of the new 1H-benzimidazole-2-yl hydrazones based on the stable free radicals ABTS and DPPH as well as ferrous iron induced oxidative molecular damage in lecithin and deoxyribose, revealed that the di- and trihydroxy substituted 1H-benzimidazole-2-yl hydrazones 5b–5e were the most potent scavengers within the whole series. The strongest protection effect was observed with compounds 5d and 5e – denoting extent of molecular damage close or lower than 50%, respectively, and compound 5b – having similar or even ameliorated protection effect compared to the reference quercetin. In this way, it was established that the presence of more than one hydroxyl group especially in the case when occupying positions next to each other in the phenyl moiety of the studied 1H-benzimidazole-2-yl hydrazones is beneficial for the manifestation of both anthelmintic and radical scavenging activity.
The molecular geometry of the synthesized hydrazones was studied by computational methods, suggesting that the E, s-trans isomer is the favorable one. Regarding the possible tautomeric forms (amino and imino forms), it was found that the imino form is strongly preferred in all compounds containing hydroxyl group in ortho position of the phenyl ring, while the amino form is the favourable one for all others. In order to clarify the radical-scavenging activity of the 1H-benzimidazole-2-yl hydrazones, the reaction enthalpies for HAT, SET-PT and SPLET mechanisms of all compounds 5a-l were computed in gas phase, benzene and water at B3LYP/6-311++G** level of theory. In nonpolar medium, it could be concluded that the most probable mechanism is HAT. However, in polar medium due to the lower proton affinities of the compounds than the BDEs, SPLET mechanism is favored over HAT. Based on the gained computational data the derivatives possessing vicinal hydroxyl groups are expected to show the highest activity which could explain the results obtained from the in vitro assays. It was also shown that the studied compounds are capable to react with various free radical through several possible reaction pathways, including RAF, with C13 position being the most active for radical adduct formation.
4. Experimental
4.1. Reagents and materials
The o-phenylenediamine, 98% and the benzaldehydes (2-hydroxybenzaldehyde, 97%, 2,3-dihydroxybenzaldehyde, 98%, 2,4-dihydroxybenzaldehyde, 98%, 3,4-dihydroxybenzaldehyde, 98%, 2,3,4-trihydroxybenzaldehyde, 98%, 4-methoxybenzaldehyde, 98%, 2,6-dimethoxybenzaldehyde, 98%, 3,5-dimethoxybenzaldehyde, 98%, 3,4,5-trimethoxybenzaldehyde, 98%, 2-hydroxy-3-methoxybenzaldehyde, 99%, 2-hydroxy-4-methoxybenzaldehyde, 99%, 3-hydroxy-4-methoxybenzaldehyde, 99%) used for the preparation of the target benzimidazolyl hydrazones were obtained from Alfa-Aesar (United Kingdom) and Sigma Aldrich.The progress of the reaction was monitored by thin layer chromatography (TLC) on standard silica gel pre-coated plates (60 F254, 0.25 mm, Merck KGaA, Darmstadt, Germany) and eluted by benzene–methanol (3:1, v/v).
The melting points (mp) were measured by Büchi B-540 instrument (Büchi Labortechnik AG, Flawil, Switzerland) and were uncorrected. The IR spectra of the synthesized compounds in solid state were recorded on a Bruker Tensor 27 FT spectrometer in ATR (attenuated total reflectance) mode with a diamond crystal accessory. The spectra were referenced to air as a background by accumulating 64 scans, at a resolution of 2 cm−1. 1H and 13C NMR spectra were measured on a Bruker Avance II+ 600 MHz NMR instrument in deuterated dimethyl sulfoxide-d6 (DMSO-d6) as a solvent, at room temperature. Chemical shifts (δ) are reported in parts per million (ppm), coupling constants (J) – in Hz, and splitting patterns are denoted as follows: s (singlet), d (doublet), t (triplet), m (multiplet).
4.2. Synthesis
Yield: 75%; Rf = 0.65 (benzene:methanol = 3:1, v/v); IR (νmax/cm−1): 3153 (νN–H); 3114 (νAr-H); 2559 (νS–H); 1623, 1511, 1466 (νC–C–Ar); 1178 (νC–S); 743 (δAr).
Yield: 60%; Rf = 0.20 (benzene:methanol = 3:1, v/v); IR (νmax/cm−1): 3080, 3059, 3014 (νAr-H), 1631 (δN–H), 1619 (νCN), 1192 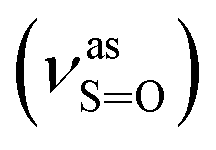 , 1057
, 1057 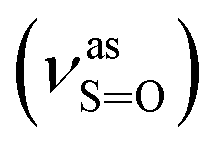 , 653 (νS–O).
, 653 (νS–O).
Yield: 76%; mp. 221.8–223.9 °C; Rf = 0.29 (benzene:methanol = 3:1, v/v); IR (νmax/cm−1): 3314, 3273 (νN–H), 3091, 2993 (νAr-H), 1664 (δN–H), 1620 (νCN), 1587, 1556 (νC–C–Ar).
:1). The solid product was filtered off and recrystallized from ethanol.
4.2.4.1. 2-(2-Hydroxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5a). The mixture was refluxed for 3 hours. Yield: 71%; mp. 275.1–277.7 °C; Rf = 0.69 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3351 (νO–H), 3285 (νN–H), 3063, 3046, 3019 (νCHarom), 1627 (δNH), 1608 (νCN), 1245 (νAr–O); 727 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.50 (bs, 1H, NH), 11.30 (bs, 1H, NH), 10.19 (s, 1H, OH), 8.31 (s, 1H, NCH), 7.79–7.77 (d, J = 7.42 Hz, 1H, Ar), 7.21–7.17 (m, 3H, Ar-bz, Ar), 6.98–6.92 (m, 2H, Ar), 6.89–6.86 (m, 2H, Ar-bz); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 156.28, 130.37, 127.73, 121.07, 119.85, 119.69, 116.40, 109.85.
4.2.4.2. 2-(2,3-Dihydroxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5b). The mixture was refluxed for 3 hours. Yield: 70%; mp. 298.0–300.4 °C; Rf = 0.25 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3403 (νO–H), 3382 (νN–H), 3060 (νCHarom), 1628 (δNH), 1614 (νCN), 1262, 1212 (νAr–O); 720, 710 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.50 (bs, 1H, NH), 9.33 (s, 1H, OH), 8.30 (s, 1H, NCH), 7.21–7.18 (m, 3H, Ar-bz; Ar), 6.95 (s, 2H, Ar-bz), 6.78–6.76 (dd, J = 7.75 Hz, 1.52 Hz, 1H, Ar), 6.70–6.67 (t, J = 15.54, 7.72 Hz, 1H, Ar); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 145.92, 145.01, 121.50, 119.42, 118.31, 116.15.
4.2.4.3. 2-(2,4-Dihydroxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5c). The mixture was refluxed for 4 hours. Yield: 40%; mp. 284.5–286.8 °C; Rf = 0.66 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3401 (νO–H), 3338 (νN–H), 3056 (νCHarom), 1626 (δNH), 1610 (νCN), 1219, 1261 (νAr–O), 732 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.39 (s, 1H, NH), 10.95 (s, 1H, NH), 10.19 (s, 1H, OH), 9.70 (s, 1H, OH), 8.19 (s, 1H, NCH), 7.52–7.51 (d, J = 8.98 Hz, 1H, Ar), 7.18–7.15 (m, 2H, Ar), 6.95–6.89 (m, 2H, Ar-bz), 6.32–6.30 (m, 2H, Ar); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 159.94, 157.95, 129.38, 112.63, 107.92, 102.81.
4.2.4.4. 2-(3,4-Dihydroxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5d). The mixture was refluxed for 4 hours and 30 minutes. Yield: 50%; mp. 215.1–217.3 °C; Rf = 0.20 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3407 (νO–H), 3257 (νN–H), 3057 (νCHarom), 1635 (δNH), 1606 (νCN), 1268 (νAr–O), 744, 751 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.46 (bs, 1H, NH), 9.28 (bs, 2H, OH), 7.85 (s, 1H, NCH), 7.24 (d, J = 2.09 Hz, 1H, Ar), 7.23–7.21 (d, J = 8.37 Hz, 2H, Ar-bz), 7.02–7.01 (dd, J = 8.16 Hz, 2.02 Hz, 1H, Ar), 6.93 (s, 2H, Ar-bz), 6.78–6.76 (d, J = 8.10 Hz, 1H, Ar); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 154.00, 147.22, 145.98, 141.88, 127.13, 119.46, 115.87, 113.78.
4.2.4.5. 2-(2,3,4-Trihydroxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5e). The mixture was refluxed for 4 hours. Yield: 56%; mp. 248.5–250.5 °C; Rf = 0.27 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3468, 3332 (νO–H), 3252 (νN–H), 3062 (νCHarom), 1631 (δNH), 1612 (νCN), 1212, 1266 (νAr–O), 730 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.46 (s, 1H, NH), 9.87 (bs, 1H, OH), 9.42 (bs, 1H, OH), 8.14 (s, 1H, NCH), 7.17–7.16 (m, 2H, Ar-bz), 6.96–6.92 (m, 3H, Ar-bz, Ar), 6.37–6.36 (d, J = 8.47 Hz, 1H, Ar); 3C NMR (150 MHz, DMSO-d6) δ (ppm): 153.76, 147.92, 146.55, 133.13, 119.30, 113.14, 107.87.
4.2.4.6. 2-(4-Methoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5f). The mixture was refluxed for 3 hours. Yield: 63%; mp. 241.7–243.8 °C; Rf = 0.46 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3388 (νN–H), 3060, 3030 (νCHarom), 2927 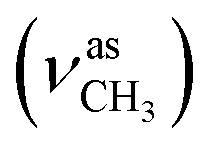 , 2835
, 2835 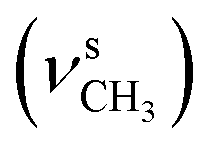 , 1651 (δNH), 1609 (νCN), 1458
, 1651 (δNH), 1609 (νCN), 1458 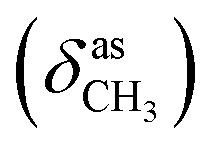 , 1378
, 1378 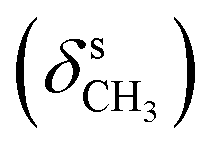 , 1244, 1136 (νC–O–C), 728, 714 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.43 (bs, 2H, NH), 7.97 (s, 1H, NCH), 7.76–7.73 (m, 2H, Ar), 7.24–7.20 (m, 2H, Ar), 7.00–6.96 (m, 2H, Ar-bz), 6.97–6.92 (m, 2H, Ar-bz), 3.80 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 160.38, 153.98, 140.99, 128.37, 114.55, 55.70.
, 1244, 1136 (νC–O–C), 728, 714 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.43 (bs, 2H, NH), 7.97 (s, 1H, NCH), 7.76–7.73 (m, 2H, Ar), 7.24–7.20 (m, 2H, Ar), 7.00–6.96 (m, 2H, Ar-bz), 6.97–6.92 (m, 2H, Ar-bz), 3.80 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 160.38, 153.98, 140.99, 128.37, 114.55, 55.70.
4.2.4.7. 2-(2,6-Dimethoxybenzylidene)-1-(1H-benzimidazol-2-yl)-hydrazine (5g). The mixture was refluxed for 3 hours. Yield: 75%; mp. 227.5–229.7 °C; Rf = 0.72 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3321 (νN–H), 3105, 3054, 3019 (νCHarom), 2946 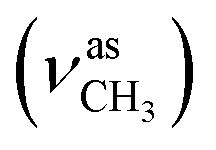 , 2847
, 2847  , 1640 (δNH), 1605 (νCN), 1459
, 1640 (δNH), 1605 (νCN), 1459  , 1384
, 1384  , 1253, 1086 (νC–O–C), 743, 727 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.26 (bs, 1H, NH), 11.15 (bs, 1H, NH), 8.16 (s, 1H, NCH), 7.32–7.28 (t, J = 16.75 Hz, 8.38 Hz, 1H, Ar), 7.23–7.20 (dd, J = 7.71 Hz, 2.85 Hz, 2H, Ar-bz), 6.97–6.94 (m, 1H, Ar-bz), 6.91–6.87 (m, 1H, Ar-bz), 6.72–6.69 (d, J = 8.44 Hz, 2H, Ar), 3.81 (s, 6H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 158.80, 153.64, 136.83, 133.76, 130.76, 120.91, 119.33, 115.62, 111.98, 110.14, 104.66, 56.35.
, 1253, 1086 (νC–O–C), 743, 727 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.26 (bs, 1H, NH), 11.15 (bs, 1H, NH), 8.16 (s, 1H, NCH), 7.32–7.28 (t, J = 16.75 Hz, 8.38 Hz, 1H, Ar), 7.23–7.20 (dd, J = 7.71 Hz, 2.85 Hz, 2H, Ar-bz), 6.97–6.94 (m, 1H, Ar-bz), 6.91–6.87 (m, 1H, Ar-bz), 6.72–6.69 (d, J = 8.44 Hz, 2H, Ar), 3.81 (s, 6H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 158.80, 153.64, 136.83, 133.76, 130.76, 120.91, 119.33, 115.62, 111.98, 110.14, 104.66, 56.35.
4.2.4.8. 2-(3,5-Dimethoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5h). The mixture was refluxed for 3 hours. Yield: 66%; mp. 251.7–253.8 °C; Rf = 0.73 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3389 (νN–H), 3060 (νCHarom), 2960  2838
2838 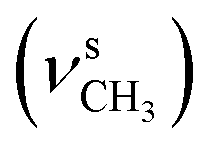 , 1647 (δNH), 1638 (νCN), 1458
, 1647 (δNH), 1638 (νCN), 1458 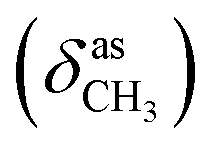 , 1357
, 1357 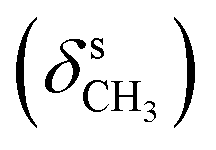 , 1266, 1127 (νC–O–C), 733 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.56 (bs, 1H, NH), 11.48 (bs, 1H, NH), 7.94 (s, 1H, NCH), 7.27–7.22 (m, 2H, Ar-bz), 6.99–6.92 (m, 4H, Ar-bz, Ar), 6.50–6.49 (m, 1H, Ar), 3.80 (s, 6H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 161.11, 153.83, 141.01, 137.56, 133.62, 121.03, 119.73, 115.52, 109.81, 104.92, 101.30, 55.81.
, 1266, 1127 (νC–O–C), 733 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.56 (bs, 1H, NH), 11.48 (bs, 1H, NH), 7.94 (s, 1H, NCH), 7.27–7.22 (m, 2H, Ar-bz), 6.99–6.92 (m, 4H, Ar-bz, Ar), 6.50–6.49 (m, 1H, Ar), 3.80 (s, 6H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 161.11, 153.83, 141.01, 137.56, 133.62, 121.03, 119.73, 115.52, 109.81, 104.92, 101.30, 55.81.
4.2.4.9. 2-(3,4,5-Trimethoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5i). The mixture was refluxed for 3 hours. Yield: 60%; mp. 216.1–218.5 °C; Rf = 0.4 (benzene
:methanol = 4:1, v/v); IR (νmax/cm−1): 3380 (νN–H), 3060, 3005 (νCHarom), 2940 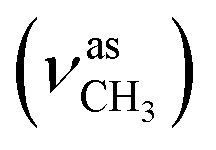 , 2834
, 2834 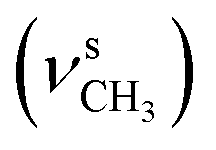 , 1655 (δNH, νCN), 1612 (νCN), 1462
, 1655 (δNH, νCN), 1612 (νCN), 1462 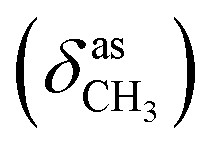 , 1359
, 1359 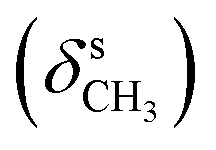 , 1228, 1121 (νC–O–C), 728 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.55 (bs, 2H, NH), 7.95 (s, 1H, NCH), 7.27–7.25 (m, 2H, Ar-bz), 7.11 (s, 2H, Ar), 6.99–6.95 (m, 2H, Ar-bz), 3.86 (s, 6H, OCH3), 3.69 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.76, 141.38, 138.76, 131.08, 104.35, 60.58, 56.52.
, 1228, 1121 (νC–O–C), 728 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.55 (bs, 2H, NH), 7.95 (s, 1H, NCH), 7.27–7.25 (m, 2H, Ar-bz), 7.11 (s, 2H, Ar), 6.99–6.95 (m, 2H, Ar-bz), 3.86 (s, 6H, OCH3), 3.69 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.76, 141.38, 138.76, 131.08, 104.35, 60.58, 56.52.
4.2.4.10. 2-(2-Hydroxy-3-methoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5j). The mixture was refluxed for 2 hours and 30 minutes. Yield: 74%; mp. 252.7–254.4 °C; Rf = 0.74 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3363 (νO–H), 3305 (νN–H), 3061, 2997 (νCHarom), 2965 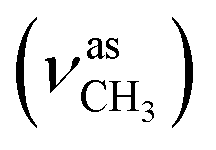 , 2836
, 2836 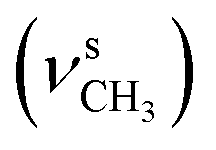 , 1626 (δNH), 1606 (νCN), 1479
, 1626 (δNH), 1606 (νCN), 1479 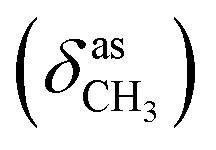 , 1369
, 1369 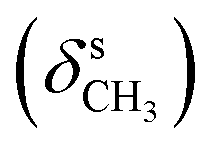 , 1262 (νAr–O); 1240, 1065 (νC–O–C), 738, 718 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.52 (bs, 1H, NH), 8.32 (s, 1H, NCH), 7.41–7.40 (d, J = 7.70 Hz, 1H, Ar), 7.20–7.19 (m, 2H, Ar-bz), 6.95–6.93 (m, 3H, Ar-bz, Ar), 6.83–6.81 (t, J = 15.78 Hz, 7.85 Hz, 1H, Ar), 3.82 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.92, 148.32, 145.82, 121.45, 119.34, 112.55, 56.32.
, 1262 (νAr–O); 1240, 1065 (νC–O–C), 738, 718 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.52 (bs, 1H, NH), 8.32 (s, 1H, NCH), 7.41–7.40 (d, J = 7.70 Hz, 1H, Ar), 7.20–7.19 (m, 2H, Ar-bz), 6.95–6.93 (m, 3H, Ar-bz, Ar), 6.83–6.81 (t, J = 15.78 Hz, 7.85 Hz, 1H, Ar), 3.82 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.92, 148.32, 145.82, 121.45, 119.34, 112.55, 56.32.
4.2.4.11. 2-(2-Hydroxy-4-methoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5k). The mixture was refluxed for 4 hours. Yield: 53%; mp. 251.0–253.1 °C; Rf = 0.76 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3402 (νO–H), 3236 (νN–H), 3068, 3054 (νCHarom), 2963 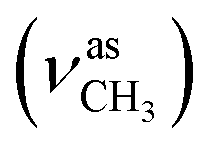 , 2838
, 2838 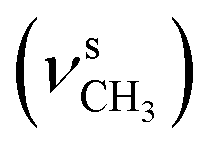 , 1656 (δNH), 1625 (νCN), 1460
, 1656 (δNH), 1625 (νCN), 1460 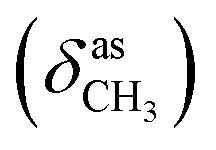 , 1378
, 1378 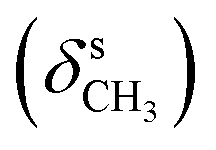 , 1267 (νAr–O); 1222, 1027 (νC–O–C), 730, 713 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.44 (bs, 1H, NH), 11.15 (bs, 1H, NH), 10.38 (bs, 1H, OH), 8.23 (s, 1H, NCH), 7.64–7.63 (d, J = 8.49 Hz, 1H, Ar), 7.17 (s, 2H, Ar-bz), 6.94–6.93 (d, J = 14.77 Hz, 2H, Ar-bz), 6.49–6.48 (d, J = 8.66 Hz, 1H, Ar), 6.45 (s, 1H, Ar), 3.79 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 161.42, 157.88, 153.94, 129.27, 121.04, 114.06, 106.54, 101.34, 55.62.
, 1267 (νAr–O); 1222, 1027 (νC–O–C), 730, 713 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.44 (bs, 1H, NH), 11.15 (bs, 1H, NH), 10.38 (bs, 1H, OH), 8.23 (s, 1H, NCH), 7.64–7.63 (d, J = 8.49 Hz, 1H, Ar), 7.17 (s, 2H, Ar-bz), 6.94–6.93 (d, J = 14.77 Hz, 2H, Ar-bz), 6.49–6.48 (d, J = 8.66 Hz, 1H, Ar), 6.45 (s, 1H, Ar), 3.79 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 161.42, 157.88, 153.94, 129.27, 121.04, 114.06, 106.54, 101.34, 55.62.
4.2.4.12. 2-(3-Hydroxy-4-methoxybenzylidene)-1-(1H-benzimidazol-2-yl)hydrazine (5l). The mixture was refluxed for 3 hours. Yield: 55%; mp. 267.1–269.5 °C; Rf = 0.5 (benzene
:methanol = 3:1, v/v); IR (νmax/cm−1): 3382 (νO–H), 3265 (νN–H), 3058, 3007 (νCHarom), 2954 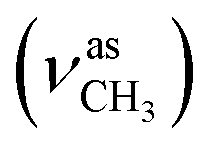 , 2834
, 2834  , 1629 (δNH), 1602 (νCN), 1461
, 1629 (δNH), 1602 (νCN), 1461  , 1375
, 1375  , 1274 (νAr–O); 1233, 1094 (νC–O–C), 733 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.51 (bs, 1H, NH), 7.89 (s, 1H, NCH), 7.33–7.32 (d, J = 1.93 Hz, 1H, Ar), 7.22 (s, 2H, Ar-bz), 7.11–7.09 (dd, J = 8.24 Hz, 1.99 Hz, 1H, Ar), 6.96–6.92 (m, 3H, Ar-bz, Ar), 3.80 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.95, 149.19, 147.05, 141.53, 133.73, 128.54, 120.95, 119.45, 115.50, 113.13, 112.15, 109.79, 56.10.
, 1274 (νAr–O); 1233, 1094 (νC–O–C), 733 (γC–H); 1H NMR (600 MHz, DMSO-d6) δ (ppm): 11.51 (bs, 1H, NH), 7.89 (s, 1H, NCH), 7.33–7.32 (d, J = 1.93 Hz, 1H, Ar), 7.22 (s, 2H, Ar-bz), 7.11–7.09 (dd, J = 8.24 Hz, 1.99 Hz, 1H, Ar), 6.96–6.92 (m, 3H, Ar-bz, Ar), 3.80 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6) δ (ppm): 153.95, 149.19, 147.05, 141.53, 133.73, 128.54, 120.95, 119.45, 115.50, 113.13, 112.15, 109.79, 56.10.
4.3. In vitro anthelmintic assay
Benzimidazole derivatives 5a-l were tested for anthelmintic activity against T. spiralis muscle larvae (ML) provided from The National Centre for Infectious and Parasitic Diseases, Department of Parasitology and Tropical medicine, Sofia, Bulgaria.In brief, freshly decapsulated larvae of the parasite (100 specimens for 1 ml physiological solution) were exposed to 50 and 100 μg ml−1 concentrations of experimental compounds dissolved in dimethyl sulfoxide (DMSO), and were incubated at 37 °C in “humid” chamber with a thermostat. Albendazole and ivermectin were used in this test as standard anthelmintics. The microscopic observations for vitality of T. spiralis ML were carried out after 24 h and 48 h, respectively. The efficacy (%) of newly synthesized 1H-benzimidazol-2-yl hydrazones on the viability of the parasites was calculated as the average value of three experiments as previously described:18
| Efficacy% = (the number of dead parasite larvae/total parasite larvae) × 100% |
4.4. Estimation of the radical-scavenging activity and protective effect
ABTS assay – the experiments were performed as described by Re et al.76 As first step ABTS˙+ stock solution was prepared by mixing 14 mM ABTS and potassium persulfate 2.45 mM (final concentration). The resulting mixture was allowed to stay in the dark for 12–16 h (until a primary solution of the radical with stable absorbance has been obtained). The suspension was then diluted until obtaining final working solution with absorbance 0.70 ± 0.01 units at 734 nm. The reading of the absorbance values has been done exactly 1 hour after mixing the ABTS radical working solution with different concentrations of the tested compounds.
DPPH assay – the experimental procedure was performed according to Grupy et al.,77 a standard purple colored working solution with absorbance of 1 at 517 nm (characteristic band for DPPH˙) was prepared in ethanol. Two milliliters of the DPPH radical solution were allowed to react with different concentrations of the tested hydrazones for 1 hour. After the incubation the relative decrease in the absorbance at 517 nm was registered and the radical scavenging activity was calculated.
On the base of the obtained data has been estimated the radical scavenging activity (RSA %) calculated in the following way:
| RSA% = ((Acontrol − Asample)/Acontrol) × 100 |
4.5. Computational methods
Full geometry optimizations have been performed with the Gaussian 09 software package79 employing density functional theory (DFT) with the hybrid functional B3LYP80 and the 6-311++G** basis set.81,82 Solvent effects in water and benzene were included in the optimizations by using the Polarizable Continuum Model (PCM).83 Harmonic vibrational frequencies have been calculated for all located stationary structures to verify whether they are minima or transition states. Zero-point energies and thermal corrections have been taken from unscaled vibrational frequencies. Single-point energy calculations in gas phase and single-point PCM energy calculations, in water and benzene, were performed at the M06-2X/6-311++G**84 levels of theory. All energies are in kJ mol−1 and were calculated at 25 °C. All bond lengths are reported in Å.Dissociation enthalpy (BDE), ionization potential (IP), proton dissociation enthalpy (PDE), proton affinity (PA), and electron transfer enthalpy (ETE) of the most stable conformers will be calculated according eqn (1)–(5).60
| BDE = H(A˙) + H(H˙) − H(A–H) | (1) |
| IP = H(A˙+) + H(e−) − H(A–H) | (2) |
| PDE = H(A˙) + H(H+) − H (A˙+) | (3) |
| PA = H(A−) + H(H+) − H(A–H) | (4) |
| ETE = H(A˙) + H(e−) − H(A−) | (5) |
The enthalpy of hydrogen atom, H(H), for each solvent are obtained by the same method and basis set. All reaction enthalpies will be calculated for 25 °C. The enthalpies of proton H(H+), and electron, H(e−), in gas phase: 6.197 kJ mol−1 and 3.145 kJ mol−1, were applied in accordance with previously estimated values.60 The respective enthalpies of hydrated proton, H(H+), and hydrated electron, H(e−), were used also as reported in the literature:60 H(H+) = −1083.5 kJ mol−1 and H(e−) = −232.8 kJ mol−1.
Author contributions
M. A., D. Y. and K. A. conceived and designed the study, M. A. performed the chemical synthesis, N. A. performed radical-scavenging activity experiments, D. V. and G. P.-D. performed parasitological experiments, M. A., M. G. and D. Y. performed theoretical calculations. All the authors contributed to the paper writing.Conflicts of interest
The authors declare that there is no conflict of interests related to this study.Acknowledgements
This work has been financially supported by the National Science Fund of Bulgaria, Contract KII-06-H39/4. Research equipment of Distributed Research Infrastructure INFRAMAT, part of Bulgarian National Roadmap for Research Infrastructures, supported by Bulgarian Ministry of Education and Science was used in a part of this investigation.Notes and references
- F. Bruschi and K. D. Murrell, New aspects of human trichinellosis: the impact of new Trichinella species, review, Postgrad. Med. J., 2002, 78, 15 CrossRef CAS PubMed.
- X. Bai, X. Hu, X. Liu, B. Tang and M. Liu, Current Research of Trichinellosis in China, Front. Microbiol., 2017, 8, 1472 CrossRef PubMed.
- G. Kim, M. H. Choi, J. H. Kim, Y. M. Kang, H. J. Jeon, Y. Jung, M. J. Lee and M. D. Oh, An outbreak of trichinellosis with detection of Trichinella larvae in leftover wild boar meat, J. Korean Med. Sci., 2011, 26(12), 1630 CrossRef PubMed.
- Trichinellosis, Annual Epidemiological Report for 2017, https://www.ecdc.europa.eu/en/publications-data/trichinellosis-annual-epidemiological-report-2017 Search PubMed.
- W. I. Khan, Physiological changes in the gastrointestinal tract and host protective immunity: learning from the mouse-T. spiralis model, Parasitology, 2008, 135, 671 CrossRef CAS PubMed.
- J. Dupouy-Camet, W. Kociecka, F. Bruschi, F. Bolas-Fernandez and E. Pozio, Opinion on the diagnosis and treatment of human trichinellosis, Expert Opin. Pharmacother., 2002, 3(8), 1117 CrossRef PubMed.
- J. H. Diaz, R. J. Warren and M. J. Oster, The Disease Ecology, Epidemiology, Clinical Manifestations, and Management of Trichinellosis Linked to Consumption of Wild Animal Meat, Wilderness Environ. Med., 2020, 31(2), 235 CrossRef PubMed.
- M. Gabrashanska, S. Petkova and S. E. Teodorova, The antioxidant status in Trichinella spiralis-infected rats, improved by Selenium supplementation, Open Chem., 2019, 5(1), 001 Search PubMed.
- M. Derda, E. Wandurska-Nowak and E. Hadas, Changes in the level of antioxidants in the blood from mice infected with Trichinella spiralis, Parasitol. Res., 2004, 93, 207 CrossRef PubMed.
- K. Boczon, E. Hadas, E. Wandurska-Nowak and M. Derda, A stimulation of antioxidants in muscles of Trichinella spiralis infected rats, Acta Parasitol., 1996, 41, 136 CAS.
- M. Derda and E. Hadas, Antioxidants and proteolytic enzymes in experimental trichinellosis, Acta Parasitol., 2000, 45, 356 CAS.
- D. I. Elgendy, A. A. Othman, M. A. Hasby Saad, N. A. Soliman and S. E. Mwafy, Resveratrol reduces oxidative damage and inflammation in mice infected with Trichinella spiralis, J. Helminthol., 2020, 94(140), 1 Search PubMed.
- S. Sharma and N. Anand, Approaches to Design and Synthesis of Antiparasitic Drugs, Chapter 8 – Benzimidazoles, Pharmacochem. Libr., 1997, 25, 195 Search PubMed.
- V. A. Kosolapov, A. A. Spasov, V. A. Anisimova and O. N. Zhukovskaya, Condensed Benzimidazoles Are a Novel Scaffold for Antioxidant Agents' Search and Development, Antioxidants, 2019, 1 CAS.
- A. Baldisserotto, M. Demurtas, I. Lampronti, M. Tacchini, D. Moi, G. Balboni, S. Vertuani, S. Manfredini and V. Onnis, In-Vitro Evaluation of Antioxidant, Antiproliferative and Photo-Protective Activities of Benzimidazolehydrazone Derivatives, Pharmaceuticals, 2020, 13, 68 CrossRef CAS PubMed.
- N. Anastassova, D. Yancheva, A. Mavrova, M. Kondeva-Burdina, V. Tzankova, N. Hristova-Avakumova and V. Hadjimitova, Design, synthesis, antioxidant properties and mechanism of action of new N,N'-disubstituted benzimidazole-2-thione hydrazone derivatives, J. Mol. Struct., 2018, 1165, 162 CrossRef CAS.
- M. Ali, S. Ali, M. Khan, U. Rashid, M. Ahmad, A. Khan, A. Sulaiman Al-Harrasi, F. Ullah and A. Latif, Synthesis, biological activities, and molecular docking studies of 2-mercaptobenzimidazole based derivatives, Bioorg. Chem., 2018, 80, 472 CrossRef CAS PubMed.
- K. Anichina, M. Argirova, R. Tzoneva, V. Uzunovac, A. Mavrova, D. Vuchev, G. Popova-Daskalova, F. Fratev, M. Guncheva and D. Yancheva, 1H-Benzimidazole-2-yl Hydrazones as Tubulin-targeting Agents: Synthesis, Structural Characterization, Anthelmintic activity and Antiproliferative activity against MCF-7 breast carcinoma cells and Molecular docking studies, Chem.-Biol. Interact., 2021, 345, 109540 CrossRef CAS PubMed.
- J. F. Van Allan and B. D. Deason, 2-Mercaptobenzimidazole, Org. Synth., 1950, 30, 56 CrossRef CAS.
- O. Billeter and A. Steiner, Ber., 1887, 20, 231 Search PubMed.
- E. Lellmann, Ann., 1883, 221, 9 Search PubMed.
- A. Ts. Mavrova, K. K. Anichina, D. I. Vuchev, J. A. Tsenov, P. S. Denkova, M. S. Kondeva and M. K. Micheva, Antihelminthic activity of some newly synthesized 5(6)-(un)substituted-1H-benzimidazol-2-ylthioacetylpiperazine derivatives, Eur. J. Med. Chem., 2006, 41, 1412 CrossRef CAS PubMed.
- A. Ts. Mavrova, D. Wesselinova, N. Vassilev and J. A. Tsenov, Design, synthesis and antiproliferative properties of some new 5-substituted-2-iminobenzimidazole derivatives, Eur. J. Med. Chem., 2013, 63, 696 CrossRef CAS PubMed.
- G. A. Mokrushina, S. K. Kotovskaya, G. N. Tyurenkova, V. I. Il'enko, V. G. Platonov and I. V. Kiseleva, Synthesis of 2-hydrazinobenzimidazoles and their antiinfluenza activity, Pharm. Chem. J., 1988, 22, 146 CrossRef.
- S. S. Bharate, Critical Analysis of Drug Product Recalls due to Nitrosamine Impurities, J. Med. Chem., 2021, 64, 2923 CrossRef CAS PubMed.
- B. Tuesuwan and V. Vongsutilers, Nitrosamine Contamination in Pharmaceuticals: Threat, Impact, and Control, J. Pharm. Sci., 2021, 110, 3118 CrossRef CAS PubMed.
- J. C. Beard and T. M. Swager, An Organic Chemist's Guide to N-Nitrosamines: Their Structure, Reactivity, and Role as Contaminants, J. Org. Chem., 2021, 86, 2037 CrossRef CAS PubMed.
- T. Shaikh, A. Gosar and H. Sayyed, Nitrosamine Impurities in Drug Substances and Drug Products, J. Adv. Pharm. Pract., 2020, 2(1), 48 Search PubMed.
- J. L. Caulfield, The chemistry of nitric oxide-induced deamination and cross-linking of DNA, Doctoral dissertation, Massachusetts Institute of Technology, 1997, vol. 1.
- R. Nilsson, The molecular basis for induction of human cancers by tobacco specific nitrosamines, Regul. Toxicol. Pharmacol., 2011, 60(2), 268 CrossRef CAS PubMed.
- Control of nitrosamine impurities in human drugs: Guidance for industry, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), 2020, https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/control-nitrosamineimpurities-human-drugs Search PubMed.
- P. Buhlmayer, F. Ostermayer and T. Schmidlin, Acyl compounds, US. Pat., 5399578A, Novartis Pharmaceuticals, 1995 Search PubMed.
- C. A. Bernhart, P. M. Perreaut and B. P. Ferrari, A new series of imidazolones: highly specific and potent nonpeptide AT1 angiotensin II receptor antagonists, J. Med. Chem., 1993, 36(22), 3371 CrossRef CAS PubMed.
- C. Bernhart, J.-C. Breliere and J. Clement, N-substituted heterocyclic derivatives, their preparation and the pharmaceutical compositions in which they are present, U.S. Pat., 5270317, U.S. Patent and Trademark Office, Washington, DC, 1993 Search PubMed.
- T. Naka, K. Nishikawa and T. Kato, Benzimidazole derivatives, their production and use, European Patent, 0459136B1, European Patent Office, Munich, Germany, 1996.
- R. Shen and S. A. Andrews, Demonstration of 20 pharmaceuticals and personal care products (PPCPs) as nitrosamine precursors during chloramine disinfection, Water Res., 2011, 45(2), 944 CrossRef CAS PubMed.
- I. Badran, A. D. Manasrah and N. N. Nassar, A combined experimental and density functional theory study of metformin oxy-cracking for pharmaceutical wastewater treatment, RSC Adv., 2019, 9(24), 13403 RSC.
- J. Yang, T. A. Marzan, W. Ye, C. D. Sommers, J. D. Rodriguez and D. A. Keire, A cautionary tale: quantitative LC-HRMS analytical procedures for the analysis of N-Nitrosodimethylamine in metformin, AAPS J., 2020, 22, 89 CrossRef CAS PubMed.
- Liquid Chromatography-High Resolution Mass Spectrometry (LC-ESI-HRMS) Method for the Determination of MNP in Rifampin and CPNP in Rifapentine Drug Substance and Drug Product, U. S. Food and Drug Administration (FDA), https://www.fda.gov/media/142092/download Search PubMed.
- S. S. Mirvish, Formation of N-nitroso compounds: chemistry, kinetics, and in vivo occurrence, Toxicol. Appl. Pharmacol., 1975, 31(3), 325 CrossRef CAS PubMed.
- T. Itoh, Y. Matsuya, H. Maeta, M. Miyazaki, K. Nagata and A. Ohsawa, Reaction of secondary and tertiary amines with nitric oxide in the presence of oxygen, Chem. Pharm. Bull., 1999, 47(6), 819 CrossRef CAS.
- M. Piskorz and T. Urbanski, Ultraviolet and Infrared Spectra of Some Nitrosamines, Bull. Acad. Pol. Sci., Ser. Sci. Chim., 1963, 11, 607 CAS.
- K. A. Thorn and L. G. Cox, Nitrosation and Nitration of Fulvic Acid, Peat and Coal with Nitric Acid, PLoS One, 2016, 11, e0154981 CrossRef PubMed.
- J. P. Gouesd and G. J. Martin, Application of 13C and 15N Spectroscopy to the Study of Electronic Delocalization in N-N Bonds: Nitrosamines, Hydrazones, Triazenes and Related Protonated Species, Org. Magn. Reson., 1979, 12(5), 263 CrossRef.
- I. T. Vermeer, L. Y. Henderson, E. J. Moonen, L. G. Engels, J. W. Dallinga, J. M. van Maanen and J. C. Kleinjans, Neutrophil-mediated formation of carcinogenic N-nitroso compounds in an in vitro model for intestinal inflammation, Toxicol. Lett., 2004, 154, 175 CrossRef CAS PubMed.
- Albendazole – summary report, committee for medicinal products for veterinary use, European Medicines Agency Veterinary Medicines and Inspections, EMEA/MRL/865/03-final, June 2004, https://www.ema.europa.eu/en/documents/mrl-report/albendazole-summary-report-3-committee-veterinary-medicinal-products_en.pdf Search PubMed.
- S. Ozkoc, S. Tuncay, S. B. Delibas and C. Akisu, In vitro effects of resveratrol on Trichinella spiralis, Parasitol. Res., 2009, 105(4), 1139 CrossRef PubMed.
- N. Mukherjee, S. Mukherjee, P. Saini, P. Roy and S. P. S. Babu, Phenolics and Terpenoids; the promising new search for anthelmintics: a critical review, Mini-Rev. Med. Chem., 2016, 16, 1415 CrossRef CAS PubMed.
- V. Spiegler, E. Liebau and A. Hensel, Medicinal plant extracts and plant-derived polyphenols with anthelmintic activity against intestinal nematodes, Nat. Prod. Rep., 2017, 34, 627 RSC.
- M. Liu, S. K. Panda and W. Luyten, Plant-Based Natural Products for the Discovery and Development of Novel Anthelmintics against Nematodes, Biomolecules, 2020, 10, 426 CrossRef CAS PubMed.
- A. Ts. Mavrova, K. K. Anichina, D. I. Vuchev, J. A. Tsenov, M. S. Kondeva and M. K. Micheva, Synthesis and antitrichinellosis activity of some 2-substituted-[1,3]thiazolo[3,2-a]benzimidazol-3(2H)-ones, Bioorg. Med. Chem., 2005, 13, 5550 CrossRef CAS PubMed.
- A. Ts. Mavrova, P. Denkova, Y. A. Tsenov, K. K. Anichina and D. I. Vutchev, Synthesis and antitrichinellosis activity of some bis(benzimidazol-2-yl)amines, Bioorg. Med. Chem., 2007, 15, 6291 CrossRef CAS PubMed.
- A. Mavrova, K. Anichina, O. Izevbekhai, D. Vutchev, G. Popova-Daskalova, D. Yancheva and S. Stoyanov, New 1,3-Disubstituted benzimidazole-2-ones as promising scaffold for the antitrihinellosis development, J. Chem. Technol. Metall., 2021, 56(1), 3 CAS.
- C. Kuş, G. Ayhan-Kilcigil, S. Ozbey, F. B. Kaynak, M. Kaya, T. Coban and B. Can-Eke, Synthesis and antioxidant properties of novel N-methyl-1,3,4-thiadiazol-2-amine and 4-methyl-2H-1,2,4-triazole-3(4H)-thione derivatives of benzimidazole class, Bioorg. Med. Chem., 2008, 16, 4294 CrossRef PubMed.
- İ. Kerimov, G. Ayhan-Kılcıgil, E. D. Özdamar, B. Can-Eke, T. Çoban, S. Özbey and C. Kazak, Design and One-Pot and Microwave-Assisted Synthesis of 2-Amino/5-Aryl-1,3,4-oxadiazoles Bearing a Benzimidazole Moiety as Antioxidants, Arch. Pharm., 2012, 345, 549 CrossRef PubMed.
- R. Hirano, W. Sasamoto, A. Matsumoto, H. Itakura, O. Igarashi and K. Kondo, Antioxidant Ability Of Various Flavonoids Against Dpph Radicals And Ldl Oxidation, J. Nutr. Sci. Vitaminol., 2001, 47, 357 CrossRef CAS PubMed.
- C. Phiphatwatcharaded, P. Puthongking, P. Chaiyarit, N. P. Johns, S. Sakolchai and P. Mahakunakorn, The anti-oxidant effects of melatonin derivatives on human gingival fibroblasts, Arch. Oral Biol., 2017, 79, 55 CrossRef CAS PubMed.
- I. G. Pervova, S. A. Melkozerov and P. A. Slepukhin, Synthesis, Structure, and Photochemical Properties of Hetarylaldehydes Benzimidazolyl-2-hydrazones, Russ. J. Gen. Chem., 2010, 80(5), 987 CrossRef CAS.
- E. B. Lindgren, J. D. Yoneda and K. Z. Leal, J. Mol. Struct., 2012, 1036, 19 CrossRef.
- E. Klein, V. Lukes and M. Ilcin, DFT/B3LYP study of tocopherols and chromans antioxidant action energetics, Chem. Phys., 2007, 336, 51 CrossRef CAS.
- G. Mazzone, N. Malaj, A. Galano, N. Russoa and M. Toscano, Antioxidant properties of several coumarin–chalcone hybrids from theoretical insights, RSC Adv., 2015, 5, 565 RSC.
- J. Wang, H. Tang, B. Hou, P. Zhang, Q. Wang, B.-L. Zhang, Y.-W. Huang, Y. Wang, Z.-M. Xiang, C.-T. Zi, X.-J. Wang and J. Sheng, Synthesis, antioxidant activity, and density functional theory study of catechin derivatives, RSC Adv., 2017, 7, 54136 RSC.
- K. Perez-Cruz, M. Moncada-Basualto, J. Morales-Valenzuela, G. Barriga-Gonzalez, J. A. Squella, P. Navarrete-Encina and C. Olea-Azar, Synthesis and antioxidant study of new polyphenolic hybrid-coumarins, Arabian J. Chem., 2017, 11, 525 CrossRef.
- Zh. Velkov, M. Traykov, I. Trenchev, L. Saso and A. Tadjer, Topology-Dependent Dissociation Mode of the O–H Bond in Monohydroxycoumarins, J. Phys. Chem. A, 2019, 123, 5106 CrossRef CAS PubMed.
- A. Galano, Free Radicals Induced Oxidative Stress at a Molecular Level: The Current Status, Challenges and Perspectives of Computational Chemistry Based Protocols, J. Mex. Chem. Soc., 2015, 59(4), 231 Search PubMed.
- A. Vaganek, J. Rimarcık, V. Lukes and E. Klein, On the energetics of homolytic and heterolytic O–H bond cleavage in flavonoids, Comput. Theor. Chem., 2012, 991, 192 CrossRef CAS.
- C. J. Parkinson, P. M. Mayer and L. Radom, An assessment of theoretical procedures for the calculation of reliable radical stabilization energies, J. Chem. Soc., Perkin Trans. 1, 1999, 2, 2305 RSC.
- E. T. Denisov and T. G. Denisova, The reactivity of natural phenols, Russ. Chem. Rev., 2009, 78(11), 1047 CrossRef CAS.
- P. Alov, I. Tsakovska and I. Pajeva, Computational Studies of Free Radical-Scavenging Properties of Phenolic Compounds, Curr. Top. Med. Chem., 2015, 15, 85 CrossRef CAS PubMed.
- Y. Z. Zheng, G. Deng, Q. Liang, D. F. Chen, R. Guo and R. C. Lai, Antioxidant Activity of Quercetin and Its Glucosides from Propolis: A Theoretical Study, Sci. Rep., 2017, 7, 7543 CrossRef PubMed.
- L. Messaadiaa, Y. Bekkar, M. Benamira and H. Lahmar, Predicting the antioxidant activity of some flavonoids of Arbutus plant: a theoretical approach, Chem. Phys. Impact, 2020, 1, 100007 CrossRef.
- B. Zhou and Z. L. Liu, Bioantioxidants: From chemistry to biology, Pure Appl. Chem., 2005, 77, 1887 CAS.
- J.-G. Fang, M. Lu, Z.-H. Chen, H.-H. Zhu, L. Yang, L.-M. Wu and Z.-L. Liu, Chem.–Eur. J., 2002, 8, 4191 CrossRef CAS.
- A. Galano and J. R. Alvarez-Idaboy, Kinetics of Radical-Molecule Reactions in Aqueous Solution: A Benchmark Study of the Performance of Density Functional Methods, J. Comput. Chem., 2014, 35, 2019 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, How Well Can New-Generation Density Functionals Describe the Energetics of Bond-Dissociation Reactions Producing Radicals?, J. Phys. Chem. A, 2008, 112, 1095 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evance, Antioxidant activity applying an improved ABTS radical cation decolorization assay, Free Radical Biol. Med., 1999, 1231 CrossRef CAS.
- P. Goupy, C. Dufour, M. Loonis and O. Dangles, Quantitative kinetic analysis of hydrogen transfer reactions from dietary polyphenols to the DPPH radical, J. Agric. Food Chem., 2003, 615 CrossRef CAS PubMed.
- T. Asakawa and S. Matsushita, Coloring conditions of thiobarbituric acid test for detecting lipid hydroperoxides, Lipids, 1980, 15, 137 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. D. McLean and G. S. Chandler, Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11-18, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions, J. Chem. Phys., 1980, 72, 650 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07419a |
| This journal is © The Royal Society of Chemistry 2021 |