
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of substituted triphenylenes via nickel-mediated Yamamoto coupling†
Zachary W. Schroeder,
Joshua LeDrew,
Vanessa M. Selmani and
Kenneth E. Maly *
*
Department of Chemistry & Biochemistry, Wilfrid Laurier University, Waterloo, Ontario N2L 3C5, Canada. E-mail: kmaly@wlu.ca
First published on 13th December 2021
Abstract
Substituted triphenylenes show promise as organic semiconductors because of their ability to form columnar liquid crystalline phases featuring extended π-stacked arrays. While there are several methods for preparing triphenylenes, including oxidative cyclization reactions such as the Scholl reaction, as well as transition metal-catalyzed aryne cyclotrimerization, these methods are not effective for electron deficient triphenylenes. Here we demonstrate that the nickel-mediated Yamamoto coupling of o-dibromoarenes is a concise and efficient way to prepare substituted triphenylenes, including electron-deficient systems that are otherwise challenging to prepare. We also demonstrate the application of this approach to prepare electron deficient discotic mesogens composed of triphenylenes bearing imide and thioimide groups.
Introduction
A key component in the design of organic semiconducting materials is the ability to tune properties such as the frontier molecular orbital energies and molecular packing. Promoting π-stacking favours molecular orbital overlap that is important for charge transport in these systems. One strategy to promote effective π-stacking is to design disk-shaped molecules that form columnar mesophases, where molecules self-organize into π-stacked arrays, making them attractive candidates as organic semiconducting materials.1–3Substituted triphenylenes have been extensively explored and are well-established discotic mesogens.4–6 These triphenylenes are usually prepared via oxidative aryl–aryl bond forming reactions such as the Scholl reaction,7,8 which works very well for electron-rich systems but is not effective for electron-poor compounds. Other common approaches for the preparation of triphenylenes include cross-coupling followed by oxidative cyclization, as well as palladium-catalyzed aryne cyclotrimerization.9 Despite these methods, access to electron-deficient triphenylenes remains a challenge.10
The design and synthesis of electron deficient triphenylenes would provide access to potential n-type materials with low LUMO energies. Furthermore, several studies have shown that electron-withdrawing groups on discotic mesogens promote the formation of stable columnar liquid crystalline phases, likely through improved π-stacking interactions.11–14 These observations suggest that the preparation of electron-deficient triphenylenes may lead to discotic liquid crystals with broad mesophase ranges. Therefore, there is impetus to develop new approaches for the preparation of electron-deficient triphenylene derivatives.
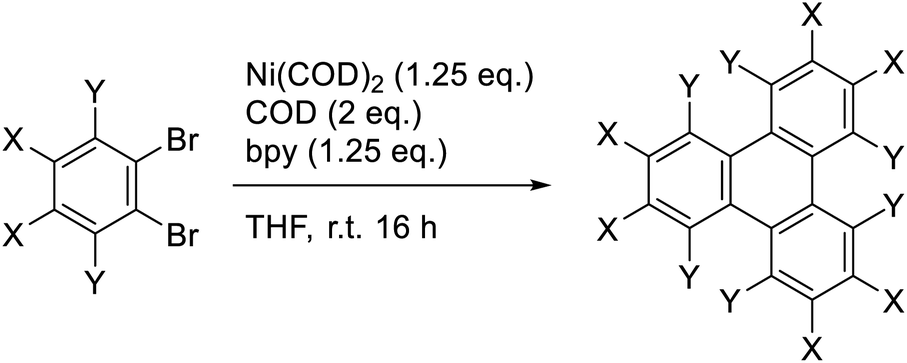
The Yamamoto coupling consists of a homocoupling of aryl bromides in the presence of a Ni(0) complex to form a new aryl–aryl bond. This reaction has been used for the synthesis of biaryls,15,16 as well as the preparation of conjugated polymers17–19 and extended two- and three-dimensional macromolecular frameworks.20 When ortho-dibromobenzene is subjected to Yamamoto coupling conditions, cyclotrimerization occurs to form the corresponding 3-fold symmetric triphenylene (Scheme 1).21 Recently, this approach has been used to prepare 3-fold symmetric polycyclic aromatic hydrocarbons such as trinaphthylenes and larger starphene derivatives.22–24 The use of the Yamamoto coupling to prepare substituted triphenylenes, however, has received little attention. This reaction should be amenable to the preparation of triphenylenes, including electron deficient triphenylenes that are not readily prepared by other methods.
 | ||
| Scheme 1 Cyclotrimerization of o-dibromobenzene using the Yamamoto coupling.21 | ||
Here we report the use of the nickel-mediated Yamamoto coupling to prepare a series of triphenylenes 2a–f and compare this approach to other methods for preparing the same compounds. In particular, we focus on electron-deficient triphenylenes, which are otherwise challenging to prepare using established methods. We demonstrate that this method is viable for the preparation of triphenylenes and in several cases offers the advantage of a more concise and high yielding synthesis as compared to other approaches. We also demonstrate that this approach can be used to prepare electron-deficient discotic compounds such as triphenylene trisimide (3) and its thionated analog (4) in order to compare their mesomorphic properties.
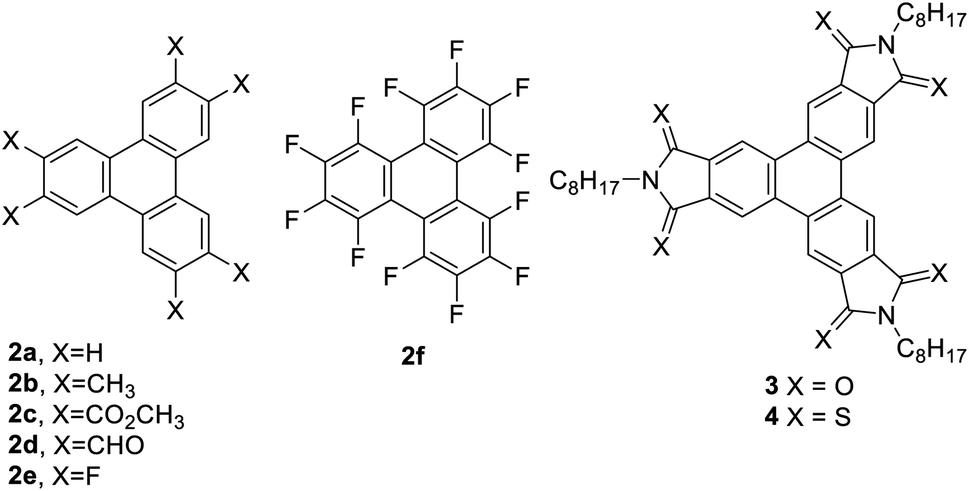
Results and discusison
To explore the potential of the Yamamoto coupling, a series of substituted o-dibromobenzenes were used. o-Dibromobenzene (1a), 1,2-dibromo-4,5-difluorobenzene (1e), and 1,2-dibromo-3,4,5,6-tetrafluorobenzene (1f) were obtained from commercial sources. 4,5-Dibromo-o-xylene25 (1b) and dimethyl 4,5-dibromophthalate26,27 (1c) were prepared according to literature procedures, and 4,5-dibromophthalaldehyde (1d) was prepared in two steps from 4,5-dibromophthalic acid (refer to ESI† for details).Triphenylenes 2a–f were chosen as targets because they have all been prepared previously by different methods, permitting a comparison of the Yamamoto coupling with previously reported approaches. They were also selected because they included a range of electron-donating and electron-withdrawing groups. Compounds 1a–f were each subjected to reaction with stoichiometric Ni(COD)2 (1.25 eq.), COD (2 eq.), and 2,2′-bipyridine (1.25 eq.) in THF at room temperature overnight to afford the corresponding triphenylenes in modest to good yields (Table 1).
|
||
|---|---|---|
| Substrate | Product | Yield (%) |
| 1a (X = H, Y = H) | 2a | 59 |
| 1b (X = CH3, Y = H) | 2b | 58 |
| 1c (X = CO2CH3, Y = H) | 2c | 74 |
| 1d (X = CHO, Y = H) | 2d | 0 |
| 1e (X = F, Y = H) | 2e | 44 |
| 1f (X = Y = F) | 2f | 20 |
Triphenylene (2a) has been prepared by a number of different methods, including the Yamamoto coupling. The previous report of the formation of triphenylene in 60% yield using the Yamamoto coupling used Ni(COD)2 in the presence of PPh3 in DMF at 70 °C.21 It should be noted that the reported yield was not an isolated yield but was measured by gas chromatography. We employed conditions adapted from Rüdiger et al.,22 who used Ni(COD)2 in the presence of COD and 2,2′-bipyridine (bpy) in THF at room temperature, thereby avoiding the use of DMF as a solvent and the need for elevated temperatures. Gratifyingly, we obtained triphenylene 2a in 59% isolated yield, comparable to that reported previously.21
Using the same conditions, hexamethyltriphenylene 2b was prepared in a 58% yield. To evaluate the utility of the Yamamoto, we compared this synthesis with the previously reported synthetic approaches to 2b. There are two previously described synthetic approaches to 2b. The first involves palladium-catalyzed aryne cyclotrimerization using the corresponding trimethylsilyl aryl triflate,28 While the second approach involves oxidative cyclization of the corresponding terphenyl, which was prepared by Suzuki coupling (Scheme 2).29 The aryne cyclotrimerization reaction is reported to proceed in 34% yield,28 and also requires several steps to prepare the aryne precursor, requiring cryogenic conditions.28,30 Alternatively, Rathore and coworkers showed that oxidative cyclization of the o-terphenyl derivative proceeded in quantitative yield.29 However, the preparation of the o-terphenyl precursor via Suzuki-coupling proceeded in a modest 32% over two steps. Clearly, each of these approaches has its disadvantages and the Yamamoto coupling represents a much more concise synthesis and improved yield as compared to both methods.
 | ||
| Scheme 2 Previously reported synthetic approaches to compound 2b. | ||
Having demonstrated that the Yamamoto coupling offers and improved synthesis of electron-rich triphenylenes, we turned our attention to electron-deficient triphenylenes (2c–f). Compound 2c, which bears ester groups on the triphenylene, has been synthesized previously.31 Since oxidative cyclizations are not generally effective for the synthesis of electron-deficient triphenylenes, the previous synthesis by Osawa et al. made use of a Cava reaction as a key step to form the triphenylene (Scheme 3). Specifically, the synthesis began by benzylic bromination of the relatively expensive hexamethylbenzene to form hexakis(bromomethyl)benzene in quantitative yield. The Cava reaction of hexakis(bromomethyl)benzene with dimethyl acetylene-dicarboxylate (DMAD) in the presence of sodium iodide gave the hexahydrotriphenylene in 14% yield, which was oxidized with MnO2 in 95% yield to give 2c. This synthesis has the drawbacks of starting with the relatively expensive hexamethylbenzene and the low yield of the Cava reaction. In contrast, Yamamoto coupling of 1c, which is readily prepared in two steps from 1b,26 proceeds in a yield of 74%. This reaction demonstrates that electron-poor triphenylenes can be prepared using this approach, providing an alternative to existing methods.
 | ||
| Scheme 3 Synthesis of compound 2c reported by Osawa et al.31 | ||
In contrast to the preparation of hexaester 2c, efforts to prepare hexaaldehyde 2d were unsuccessful, resulting in a complex mixture with no evidence of the desired product. One potential explanation for the failure of the Yamamoto coupling of 1d is that aldehydes are known to react directly with zero-valent nickel complexes, undergoing reactions such as homodimerization to form esters.32 This observation highlights a limitation in the scope of the Yamamoto coupling when other reactive functional groups are present.
Fluorinated triphenylenes 2e and 2f are of interest in the context of crystal engineering and supramolecular chemistry because of their ability to engage in arene–perfluoroarene interactions in the solid state.33–38 These two compounds could be prepared via the Yamamoto coupling of the corresponding dibromoarenes 1e and 1f in modest yields. Compound 2e has been prepared previously by palladium catalyzed aryne cyclotrimerization reactions.9,39,40 The initial report of the aryne cyclotrimerization used the corresponding trimethylsilyl aryl triflate as the aryne precursor and gave compound 2e in 52% yield. However, the synthesis of the aryne precursor required three steps from commercially available 2-bromo-4,5-difluorophenol, with some of these steps having modest yields.9,39 More recently, Greaney and coworkers reported an aryne cyclotrimerization method using the appropriate bromoaryl boronate ester and showed that 2e could be prepared in 33% yield using this approach.40 While the yield of the final step was lower, the aryne precursor was prepared in one step from commercially available starting material. In contrast, the Yamamoto coupling produced 2e in 44% yield directly from commercially available 1e, suggesting that this approach may represent a slight improvement over the method of Greaney and coworkers. Perfluorotriphenylene 2f has been prepared by Ullmann coupling of 1,2-diiodotetrafluorobenzene using copper bronze at high temperature, furnishing 2f in only a 5% yield.41–43 Using the Yamamoto coupling, the yields are slightly improved, providing 2f in a 20% yield. Thus, even though the yields of the Yamamoto coupling are modest for the preparation of fluorinated triphenylenes, this approach still represents an improved approach as compared to previous syntheses.
Having shown that the Yamamoto coupling is a concise approach for the preparation of a variety of triphenylenes, including electron-deficient systems, we sought to demonstrate that it can be used for the preparation of electron-deficient discotic liquid crystalline materials. We sought to prepare imide (3) which was previously shown to exhibit a columnar liquid crystalline phase.28 The previous synthesis followed a similar approach to that of hexaester 2c: benzylic bromination of hexamethylbenzene, followed by a Cava reaction with the alkyl maleimide in the presence of NaI in DMF.28 The crude product of the reaction was then treated with bromine and finally trimethylamine to provide compound 3 in a yield only 18% over three steps. Given the low yield and use of expensive reactants, we sought an alternative synthesis that made use of the Yamamoto coupling and used o-xylene as an inexpensive starting material.
Our alternative synthesis of triphenylene imide 3 is outlined in Scheme 4. 4,5-Dibromo-o-xylene25 was oxidized to the corresponding phthalic acid in moderate yield as described previously and then converted into the anhydride 5 in quantitative yield.44 Anhydride 5 was then reacted with octyl amine in the presence of Ni(OAc)2 and Ac2O in a modification of a method described by Branda and coworkers45 to give imide 6. Compound 6 was then subjected to the Yamamoto coupling conditions using Ni(COD)2, COD, and 2,2′bipyridine (bpy) in THF to furnish the desired compound 3 in 65% yield.
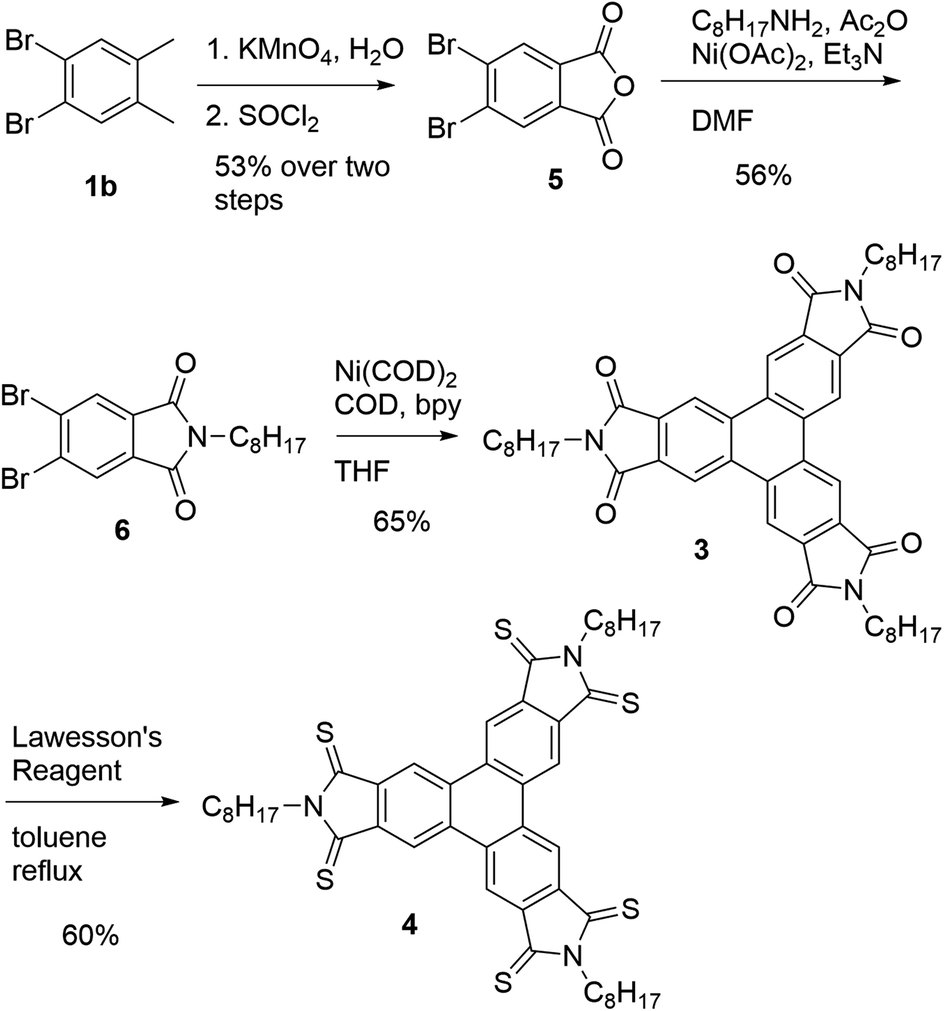 | ||
| Scheme 4 Synthesis of compounds 3 and 4. | ||
An investigation of the mesomorphic properties of 3 by polarized optical microscopy and differential scanning calorimetry showed that the compound indeed exhibited an ordered columnar mesophase as described previously by Wu and coworkers.28 The observed mesomorphic properties are consistent with those previously reported, showing a clearing point at ca. 228 °C.
Previous studies on imide materials have shown that thionation can be used to improve electron-accepting ability of imides by lowering the LUMO energy, producing materials with lower HOMO–LUMO gaps.27,46–51 We have also shown that thionation of imide containing discotic liquid crystalline compounds has only a small impact on liquid crystallinity while altering the electronic properties significantly.27,51 We therefore sought to explore the effect of thionation on the mesomorphic properties of compound 4. Compound 4 was readily accessed by 6-fold thionation of 3 giving the desired product in 60% yield.
Thioimide 4 shows a red shift in the absorption compared to the corresponding imide (3) as shown by UV-visible spectroscopy (Fig. 1). This red shift is consistent with previously reported thionated imides, which show a lowering of the HOMO–LUMO gap primarily through a lowering of the LUMO energy.
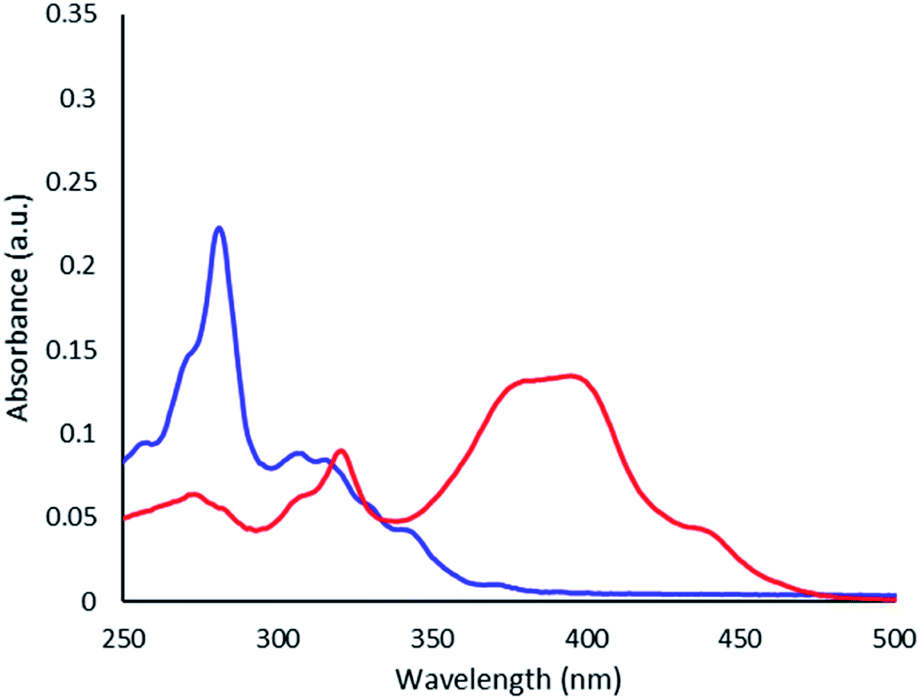 | ||
| Fig. 1 UV-visible spectra of 3 (blue line) and 4 (red line) in CH2Cl2 (2 × 10−6 M). | ||
Preliminary investigation of the mesomorphic properties of compound 4 by polarized optical microscopy showed compound 4 underwent a transition to the isotropic phase at approximately 320 °C. Upon cooling, a phase transition to a columnar mesophase was observed. Polarized optical microscopy revealed dendritic textures consistent with a columnar hexagonal phase (Fig. 2). This texture was preserved to room temperature, suggesting a glass formation. Unfortunately, heating above 300 °C also appeared to result in at least partial decomposition. Consequently, the phase transition temperatures were reduced with each heating and cooling cycle. As such, the phase transitions were not readily observed by differential scanning calorimetry, nor is it possible to make meaningful characterization of the mesophase, or comparison of the mesophase characteristics with compound 3. That said, in this system, thionation has the effect of dramatically increasing the clearing point of these compounds to the point of decomposition. These observations are in contrast to previous studies on the thionation of imide liquid crystals, where thionation had only a small impact on the mesophase range.27,51 In this case, where all six imide oxygen atoms are replaced by sulfur has a dramatic effect on the phase transition temperatures.
 | ||
| Fig. 2 Polarized optical micrograph of thioimide 4 upon cooling from isotropic liquid. | ||
Conclusions
In summary, we have shown that the nickel-mediated Yamamoto coupling of o-dibromoarenes can be used for the preparation of substituted triphenylenes. This approach is more concise and high yielding than several existing synthetic approaches, in particular for electron-deficient systems that are not readily prepared by oxidative cyclization methods. One drawback of this approach is the use of stoichiometric amounts of Ni(COD)2. We applied this method to the preparation of electron-deficient discotic mesogen 3 bearing imide groups, and converted it to the corresponding thioimide (4). Compound 4 shows evidence for a columnar mesophase with a high clearing point, but decomposition near the clearing point prevents a clear comparison with compound 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Natural Sciences and Engineering Council of Canada (NSERC), the Canada Foundation for Innovation, the Ontario Research Fund, and Wilfrid Laurier University for support. This work was supported by the Research Support Fund. Z. W. S. thanks NSERC for an Undergraduate Student Research Award and J. L. thanks NSERC for a Postgraduate Scholarship Award and the Ontario Government for an Ontario Graduate Scholarship. We thank Dmitry D. Gusev for the generous use of his glove box. We also thank Dr A. Furtos at the Université de Montréal for mass spectrometric analyses.Notes and references
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed.
- S. Kumar, Chemistry of Discotic Liquid Crystals: From Monomers to Polymers, CRC Press, Boca Raton, 2011 Search PubMed.
- S. Kumar, Liq. Cryst., 2004, 31, 1037–1059 CrossRef CAS.
- A. N. Cammidge and R. J. Bushby, in Handbook of Liquid Crystals, ed. D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, 1998, vol. 2B Search PubMed.
- R. C. Borner, R. J. Bushby and A. N. Cammidge, Liq. Cryst., 2006, 33, 1439–1448 CrossRef CAS.
- D. Pérez and E. Guitián, Chem. Soc. Rev., 2004, 33, 274–283 RSC.
- D. Peña, S. Escudero, D. Pérez, E. Guitián and L. Castedo, Angew. Chem., Int. Ed., 1998, 37, 2659–2661 CrossRef.
- H. Bock, M. Rajaoarivelo, S. Clavaguera and É. Grelet, Eur. J. Org. Chem., 2006, 2889–2893 CrossRef CAS.
- E. J. Foster, R. B. Jones, C. Lavigueur and V. E. Williams, J. Am. Chem. Soc., 2006, 128, 8569–8574 CrossRef CAS PubMed.
- J. A. Paquette, C. J. Yardley, K. M. Psutka, M. a. Cochran, O. Calderon, V. E. Williams and K. E. Maly, Chem. Commun., 2012, 48, 8210–8212 RSC.
- K. M. Psutka, J. Williams, J. A. Paquette, O. Calderon, K. J. A. Bozek, V. E. Williams and K. E. Maly, Eur. J. Org. Chem., 2015, 1456–1463 CrossRef CAS.
- J. A. Paquette, K. M. Psutka, C. J. Yardley and K. E. Maly, Can. J. Chem., 2017, 95, 399–409 CrossRef CAS.
- M. F. Semmelhack, P. Helquist, L. D. Jones, L. Keller, L. Mendelson, L. S. Ryono, J. G. Smith and R. D. Stauffer, J. Am. Chem. Soc., 1981, 103, 6460–6471 CrossRef CAS.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 7547–7560 CrossRef CAS.
- T. Yamamoto, K. Sanechika and A. Yamamoto, J. Polym. Sci., Part C: Polym. Lett., 1980, 18, 9–12 CAS.
- T. Yamamoto, K. Sanechika and A. Yamamoto, Bull. Chem. Soc. Jpn., 1983, 56, 1497–1502 CrossRef CAS.
- M. Kobayashi, J. Chen, T. C. Chung, F. Moraes, A. J. Heeger and F. Wudl, Synth. Met., 1984, 9, 77–86 CrossRef CAS.
- T. Ben, H. Ren, M. Shengqian, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- Z. h. Zhou and T. Yamamoto, J. Organomet. Chem., 1991, 414, 119–127 CrossRef CAS.
- E. C. Rüdiger, F. Rominger, L. Steuer and U. H. F. Bunz, J. Org. Chem., 2016, 81, 193–196 CrossRef PubMed.
- V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J. V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, J. Am. Chem. Soc., 2017, 139, 18508–18511 CrossRef CAS PubMed.
- E. C. Rüdiger, M. Porz, M. Schaffroth, F. Rominger and U. H. F. Bunz, Chem.–Eur. J., 2014, 20, 12725–12728 CrossRef PubMed.
- C. Structures, P. R. Ashton, U. Girreser, D. Giuffrida, F. H. Kohnke, J. P. Mathias, F. M. Raymo, A. Z. Slawin, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1993, 115, 5422–5429 CrossRef.
- Z. Chen, S. Dong, C. Zhong, Z. Zhang, L. Niu, Z. Li and F. Zhang, J. Photochem. Photobiol., A, 2009, 206, 213–219 CrossRef CAS.
- K. M. Psutka, J. LeDrew, H. Taing, S. H. Eichhorn and K. E. Maly, J. Org. Chem., 2019, 84, 10796–10804 CrossRef CAS PubMed.
- J. Yin, H. Qu, K. Zhang, J. Luo, X. Zhang, C. Chi and J. Wu, Org. Lett., 2009, 11, 3028–3031 CrossRef CAS PubMed.
- L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, J. Org. Chem., 2010, 75, 4748–4760 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211–1214 CrossRef.
- T. Osawa, T. Kajitani, D. Hashizume, H. Ohsumi, S. Sasaki, M. Takata, Y. Koizumi, A. Saeki, S. Seki, T. Fukushima and T. Aida, Angew. Chem., Int. Ed., 2012, 51, 7990–7993 CrossRef CAS PubMed.
- Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res., 2015, 48, 1746–1755 CrossRef CAS PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- F. Cozzi, S. Bacchi, G. Filippini, T. Pilati and A. Gavezzotti, Chem.–Eur. J., 2007, 13, 7177–7184 CrossRef CAS PubMed.
- C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W. Clegg and C. Viney, Chem. Commun., 1999, 2493–2494 RSC.
- G. W. Coates, A. R. Dunn, L. M. Henling, D. A. Dougherty and R. H. Grubbs, Angew. Chem., Int. Ed., 1997, 36, 248–251 CrossRef CAS.
- C. E. Smith, P. S. Smith, R. L. Thomas, E. G. Robins, J. C. Collings, C. Dai, A. J. Scott, S. Borwick, A. S. Batsanov, S. W. Watt, S. J. Clark, C. Viney, J. a. K. Howard, W. Clegg and T. B. Marder, J. Mater. Chem., 2004, 14, 413 RSC.
- M. Weck, A. R. Dunn, K. Matsumoto, G. W. Coates, E. B. Lobkovsky and R. H. Grubbs, Angew. Chem., Int. Ed., 1999, 38, 2741–2745 CrossRef CAS PubMed.
- D. Peña, D. Pérez, E. Guitián and L. Castedo, J. Am. Chem. Soc., 1999, 121, 5827–5828 CrossRef.
- J. A. García-López and M. F. Greaney, Org. Lett., 2014, 16, 2338–2341 CrossRef PubMed.
- V. B. Smith and A. G. Massey, Tetrahedron, 1969, 25, 5495–5501 CrossRef CAS.
- M. B. Hursthouse, V. B. Smith and A. G. Massey, J. Fluorine Chem., 1977, 10, 145–155 CrossRef CAS.
- C. S. Frampton, D. D. MacNicol and S. J. Rowan, J. Mol. Struct., 1997, 405, 169–178 CrossRef CAS.
- D. Mo, H. Chen, Y. Zhu, H. H. Huang, P. Chao and F. He, ACS Appl. Mater. Interfaces, 2021, 13, 6147–6155 CrossRef CAS PubMed.
- A. M. Asadirad, S. Boutault, Z. Erno and N. R. Branda, J. Am. Chem. Soc., 2014, 136, 3024–3027 CrossRef CAS PubMed.
- B. A. Llewellyn, E. S. Davies, C. R. Pfeiffer, M. Cooper, W. Lewis and N. R. Champness, Chem. Commun., 2016, 52, 2099–2102 RSC.
- N. Pearce, E. S. Davies, W. Lewis and N. R. Champness, ACS Omega, 2018, 3, 14236–14244 CrossRef CAS PubMed.
- F. S. Etheridge, R. Fernando, J. A. Golen, A. L. Rheingold and G. Sauve, RSC Adv., 2015, 5, 46534–46539 RSC.
- T.-F. Yang, S.-H. Huang, Y.-P. Chiu, B.-H. Chen, Y.-W. Shih, Y.-C. Chang, J.-Y. Yao, Y.-J. Lee and M.-Y. Kuo, Chem. Commun., 2015, 51, 13772–13775 RSC.
- A. J. Tilley, C. Guo, M. B. Miltenburg, T. B. Schon, H. Yan, Y. Li and D. S. Seferos, Adv. Funct. Mater., 2015, 25, 3321–3329 CrossRef CAS.
- K. M. Psutka and K. E. Maly, RSC Adv., 2016, 6, 78784–78790 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental details, and NMR spectra. See DOI: 10.1039/d1ra07931j |
| This journal is © The Royal Society of Chemistry 2021 |