
Artificial intelligence and automation in computer aided synthesis planning
Amol
Thakkar
 *ab,
Simon
Johansson
ac,
Kjell
Jorner
d,
David
Buttar
d,
Jean-Louis
Reymond
b and
Ola
Engkvist
*a
*ab,
Simon
Johansson
ac,
Kjell
Jorner
d,
David
Buttar
d,
Jean-Louis
Reymond
b and
Ola
Engkvist
*a
aHit Discovery, Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden. E-mail: amol.thakkar@dcb.unibe.ch; ola.engkvist@astrazeneca.com
bDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland
cDepartment of Computer Science and Engineering, Chalmers University of Technology, Gothenburg, Sweden
dEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Macclesfield, UK
First published on 5th November 2020
Abstract
In this perspective we deal with questions pertaining to the development of synthesis planning technologies over the course of recent years. We first answer the question: what is computer assisted synthesis planning (CASP) and why is it relevant to drug discovery and development? We draw a distinction between discovery and development, focusing on their differing requirements. We highlight the need for an automated synthesis platform which chemists can use to augment their workflows and what it entails. The interaction between experimental and computational scientists is emphasized as a key driver in the development of such technologies. Advances in the development and application of algorithms is then covered, drawing a distinction between physics based and statistical or data driven modelling paradigms, their use in, and how they contribute to augmented drug discovery and development. Finally, developments in the coupling of artificial intelligence and automation are discussed. Throughout, we emphasize the need for an inter-disciplinary approach, blurring the distinction between fields in the pursuit of artificial intelligence and automated platforms that can be integrated into chemical workflows.
Introduction
The use of artificial intelligence (AI) and automation to augment drug discovery and development has been the subject of several reviews in recent years and promises to accelerate both discovery and development in an effort to deliver medicines to patients faster.1–3 The subject has once again gained popularity, with key drivers being the accessibility of improved methods, increased computational power, and larger datasets. Artificially intelligent systems have the potential to transform drug discovery and development by conducting or assisting with tasks previously reserved for humans. In the brief history of the field, the definitions of what is deemed an ‘intelligent’ system have continued to change as technologies are outdated and new ones take their place.4–6 What once constituted artificial intelligence and automation no longer rouses interest among the chemical community as they have become routine tasks. For example, consider the collection of NMR spectra – a chemist is now able to submit samples for NMR analysis and await the result, with the machine carrying out automated sampling, recording of the spectra, and subsequent processing of the raw free induction decay data. This has recently been extended to the assignment of NMR spectra.7 As can be seen, these technologies have now become deeply embedded into chemical workflows and augment the ability of the chemists using them, allowing them to focus their time on analysis and the design of future experiments. Computer aided synthesis planning (CASP) has not yet reached the stage where it is an integral part of a chemist's workflow, but there has been much discussion about how best to integrate it, at which stage, what to expect, and what it will deliver.1 This ongoing debate signifies the beginning of a period of development by which members of several distinct research communities, ranging from biology, chemistry, mathematics, physics, robotics, and computer science must come together to build ‘intelligent’ and automated solutions that work for the chemist.Over the last 60 years, artificial intelligence has been used as a tool to find solutions to a plethora of chemical problems, from de novo design of compounds,8 and the reactions required to make them,9 to bioactivity prediction,10 and safety assessment.11 However, despite attempts to create platforms for CASP, none have experienced widespread adoption, with the exception of chemical search engines such as Reaxys and SciFinder. There are of course reasons other than their potential limitations and performance that contributed to lack of adoption during the early years, for instance the accessibility of computers, the internet, and barriers to entry in the form of steep learning curves. However, in the last few years, the tools have become more accessible, which in the context of the time taken for development of the underlying mathematical frameworks, is a relatively short period of time. Furthermore, there is a behavioral element that has limited adoption which is well-summarized by the late Carl Djerassi:12
“Symbolic manipulations by computers are in principle important in two areas of chemistry – synthesis and structure elucidation. It is the former where the use of computers has not been widely accepted because of the fear that thinking man will simply be reduced to an appendage to a machine. The synthetic chemist wishes to be both architect and building contractor – the former function being the intellectually and aesthetically more pleasing one – and it is precisely this architectural role that the computer is perceived partially to usurp”.
These behavioral aspects toward adoption have been discussed by Griffen et al. and provide a view of the problems the community and companies face, and have faced regarding the adoption of computational tools.13 At present, however, AI and automation cannot carry out the actions or higher-level reasoning required to run discovery and development cycles autonomously. Whilst technical improvements have been made toward this end, the behavioral aspect should not be overlooked.
As such, we believe that – in their current and future state – the algorithms presented henceforth, should be viewed as augmenting the ability of a human chemist to arrive at the desired solution. Thereby, they will act as tools to inspire and inform the decision maker rather than to replace or fully automate the design, make, analyze, and test (DMTA) cycle. In this regard the goal for the computational tools outlined herein is to improve the productivity of chemists, especially with regards to well-established practices, thus allowing more time to focus on novel or more difficult chemistries.
Whilst fully automated chemistry is one goal towards which AI and automation is being developed, this should be with the end goal of facilitating the work of a wet-lab chemist, rather than with the aim of replacing lab based chemists. The authors emphasize that synthetic chemistry is not necessarily the bottle neck in drug discovery, and is only one contributing factor in the process. Bender has discussed this in more detail with a view on efficacy and safety in drug development,14 and there are several ongoing works in the clinical phase to improve the whole process.15,16 Nevertheless, to facilitate development in any of the highlighted areas, an interdisciplinary approach bringing together experts from different fields is required. In addition, emphasis should be placed on ease of use and accessibility of the tools that are developed. Successful approaches may be characterized as those with a shallow learning curve for the experimentalist, a rich data source for the theoretician or data scientist, and tight-knit integration throughout the community from discovery to development. The approach should also be scalable, adaptable, reliable and most importantly, meet the needs of the end user.
To begin our foray into CASP we first define it as encompassing, but not limited to: (1) retrosynthetic analysis, the task of breaking a given compound down into simpler precursors; (2) reaction prediction, the task of predicting the product of a reaction given a set of precursors; (3) reaction condition prediction, the task of predicting a set of conditions (e.g. catalyst, temperature, solvent) under which a given reaction takes place (Fig. 1). In addition to these central tasks, we also consider the related task of reaction optimization, improving a pre-defined objective such as yield or purge of impurities by adjusting the conditions under which the reaction is carried out. We do not refer to reaction discovery explicitly as the definition of a new reaction is not well defined. For instance a novel reaction could be thought of as a new set of conditions for a known transformation, consider coupling reactions for example, for which there are a plethora of catalysts demanding specific substrate choices or reactions which are mechanistically different. Another crucial aspect of chemical synthesis we omit in this perspective is the role artificial intelligence can play in optimizing isolation and purification techniques. All of the highlighted tasks come together to form a system capable of predicting and optimizing synthetic pathways to target molecules. As such, CASP tools have many possible areas of application within drug discovery and development, as well as in parallel functions in the agrochemicals and specialty chemicals industries.
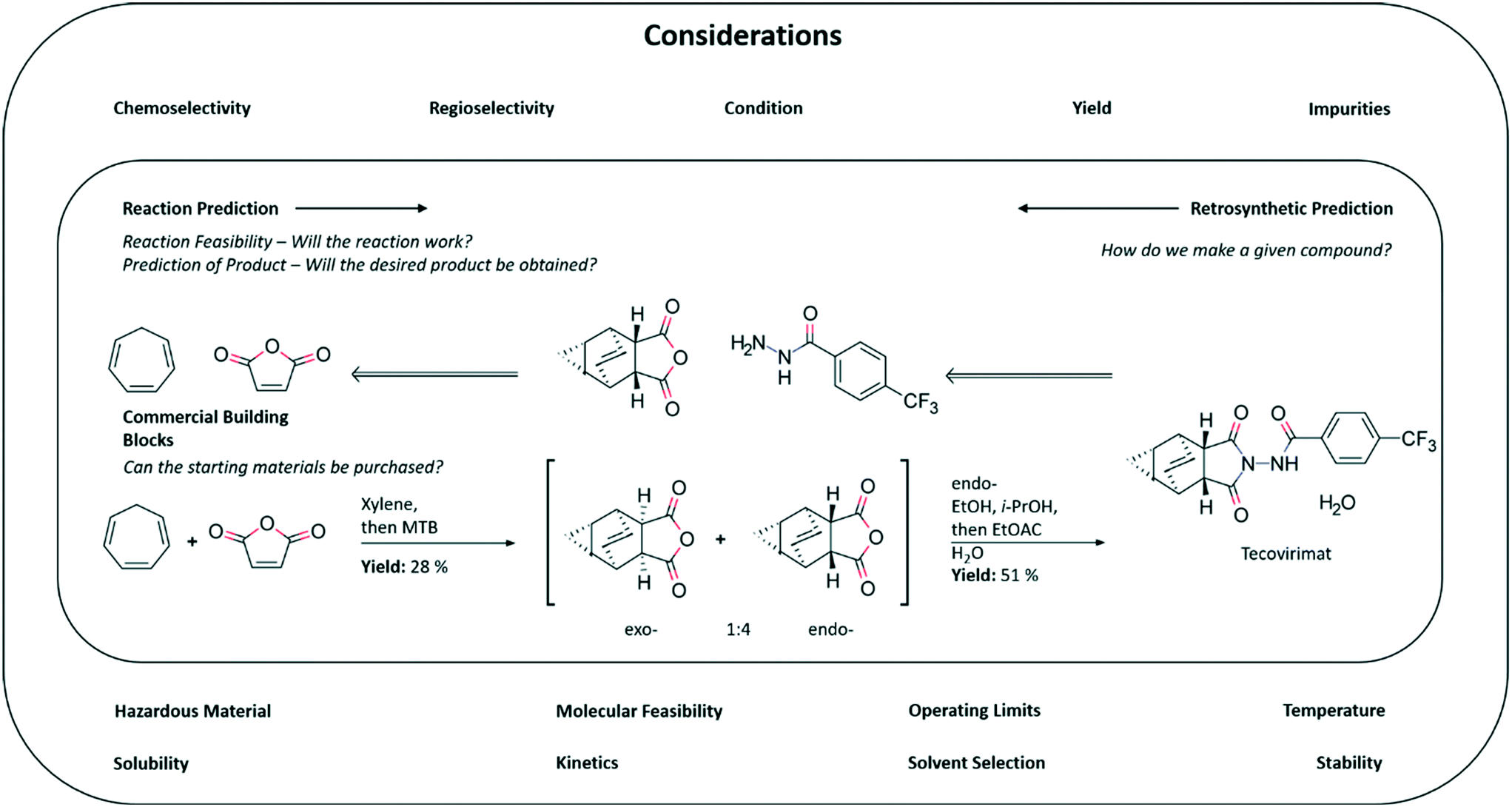 | ||
| Fig. 1 An efficient synthesis to tecovirimat annotated to exemplify potential application of CASP tools. There are several questions that CASP may be able to help answer using a mixture of statistical and physics based modelling from available datasets and from first principles. Models built with the aim of answering the questions outline in the figure, have the potential to augment the ability of the bench chemist. | ||
The role of CASP in drug discovery and development
It is important to draw a distinction between the discovery and development domains as requirements from the CASP tools differ. However, it is also important to note that whilst the domains differ in requirements there are also synergistic effects, thus they should not be considered in isolation. In the pharmaceutical industry, CASP tools have the potential for widespread use across discovery and development. For complex drug molecules, the number of synthetic possibilities available can be large, and CASP tools will require approaches to guide chemists to select the most appropriate route with respect to the goals of their project, patent restrictions, regulatory and environmental considerations, and probability that a certain route will succeed. The criteria behind what constitutes a successful synthesis differs for both discovery and development settings and will be discussed herein, in conjunction with how CASP tools can play a helping hand. Furthermore, with increasing focus on sustainable processes, CASP tools have the potential for providing an information rich environment to explore alternative processes that better meet the organizations sustainability goals across discovery and development.Drug discovery
In discovery chemistry, there is the potential for CASP to aid in the rapid identification of synthesis routes during hit finding campaigns and lead optimization. This can lower the barrier to synthesis, biological, and property screening, and ultimately project progression. The increasing interest in AI for de novo design, and it's use to find novel hits necessitates that some aspect of synthetic accessibility must be included to filter the vast number of virtually plausible compounds. In this sense, CASP can be used to filter compounds based on whether or not a synthetic route can be found starting from commercially available or in-house building blocks,17,18 although recent studies have also investigated how synthesizability can be incorporated already in the de novo design algorithm.19,20 Incorporation of CASP into de novo design could take the form of increasing the diversity of compounds obtained from a given scaffold.21 CASP can also be linked to automation to synthesize newly discovered hits, functionalize scaffolds for lead optimization, and for reaction screening and optimization. However, whilst incredibly useful in synthesizing libraries of compounds quickly, automated synthesis in the discovery setting can still be quite limited owing to both the number and types of transformations required in a multistep synthesis, which current automation platforms have not yet mastered.The success of any given synthesis is primarily governed by whether or not pure material can be obtained at the end of a synthetic route. There is no unanimous view regarding what constitutes a successful experiment or the yield one should obtain, and this differs between discovery and development. Often it is the case in discovery chemistry that there is either enough material to continue or there is not. Neither the yields nor the selectivity at each step in a synthetic sequence are necessarily optimized in the discovery setting, thus syntheses can be labor intensive and wasteful, although this can be deemed acceptable as the quantities of material handled at this stage are often below the gram scale. Often a given scaffold is synthesized in relatively large quantities (multi-gram) and functionalized to afford a series of analogues. Commercially available scaffolds whilst easy to access can prove to be difficult to patent, therefore CASP tools may be used to find syntheses to novel scaffolds which have been previously unexplored. In the pursuit of novel structures, CASP may be able to suggest known reactions but in a different context,22 or potentially invent new reactions towards this end.23,24 In both cases it remains to be seen whether the suggested reactions will work in the wet-lab and not just in silico consistently.
Drug development
Unlike discovery, drug development is focused on one or a few active pharmaceutical ingredients (API), and the ultimate goal of retrosynthesis and forward prediction tools is to aid in the identification of sustainable synthetic routes during the route design process.25 The selection of an optimal synthesis route is somewhat subjective, with scoring metrics such as yield, atom economy (AE), process mass intensity (PMI), and material costs being utilized to guide decision making.26,27 Whereas, in a discovery setting there is the potential to incorporate process-friendly synthetic steps early on in the life cycle of a drug candidate, the opportunity to do so is limited. This is because of the large number of compounds that are required to be synthesized in discovery compared to the few in development. The time and large resources dedicated to a given API, and the scale (multi-gram to kilograms) at which the chemistry is conducted is characteristic of the development process, where time can be spent optimizing, for example, yield, stereoselectivity, and number of steps. In addition, the scale on which the chemistry is conducted often requires a complete re-work of the medicinal chemistry synthetic route such that it may be compatible with manufacturing facilitates. Furthermore, sustainability becomes more important in development due to the scale on which the chemistry is conducted. Here, green chemistry principles are increasingly important to decrease both environmental footprint and overall production cost.25 For agrochemicals, cost of goods and environmental considerations are even greater, as products are made on multi-ton scale.28,29 These considerations could, e.g., completely rule out certain expensive reagents or energy-intensive steps. CASP could help to identify alternative greener reagents, catalysts, solvents, and less energy intensive processes, as well as reduce waste by predicting waste products in advance and minimizing their synthesis.25 In this context, the ACS Green Chemistry Institute Pharmaceutical Roundtable provide predictive tools for process chemistry.30 The importance of digitalization in chemical development is recognized with the concept of a digital twin31 built of modular components, entire plants or complete processes proposed to improve planning and productivity. The digital transformation of drug development will entail two aspects: digitization, the conversion all key information to a digital format; and digitalization, the leveraging of digitization to improve the drug development process. CASP tools tailored for development can be considered a component of the digital transformation.After an initial brainstorm, identifying several potential synthetic strategies, using both human and AI suggested routes, decisions must be made on which key steps to test and characterize experimentally. At this stage, quantitative rather than qualitative predictive models are key to guide the decision-making process, enabling chemists to test assumptions with respect to regio- or chemoselectivity, reaction rates, or other mechanistic assumptions. Quantum mechanical (QM) based models can achieve the necessary accuracy to facilitate such decisions and are increasingly utilized in drug development.32 Although QM methods have traditionally been used extensively, they are often categorized as computationally ‘expensive’. However, with the developments, increased accessibility, and ease of use of artificial intelligence (AI) methods, there is potential to speed up the predictive process, both in terms of the QM calculations themselves, and learning from datasets of QM-optimized structures.
Synergies between discovery and development
Whether in discovery or development, the end user is a chemist. The tools must be user friendly with a low barrier to entry and shallow learning curve, and offer an efficient way of literature searching, thereby complementing the functionalities of database providers such as SciFinder and Reaxys. Furthermore, routes must be presented with an associated probability of success or similar score such that comparisons can be made. These scores may take into account multiple objectives, such as yield, precedence, number of steps, cost and availability of starting materials, selectivity, presence of impurities, to name a few. The ability to rank routes with respect to both discovery and development may enable a blurring of the line between the two, as the information concerning development feasibility can be stored and learnt from to improve future discovery and development campaigns. There is a pressing need for improved data and knowledge management in this respect, to aid knowledge transfer from discovery teams to development, and also within the different stages within each of the two domains, i.e., from early to late chemical development. Giving discovery chemists access to ranking of routes from a development perspective could also lead to more development-friendly routes already at the discovery stage.The way in which the routes are ranked for both discovery and development can differ. In the discovery setting chemists may opt for divergent synthesis, favoring late stage diversification in order to obtain a series of analogues (Fig. 2). While the step count to any one compound may be longer, the total amount of steps to make all desired analogues may be lower. Whereas in development, there is a requirement for short efficient routes to obtain the target compound. Consider for example that discovery might target routes to make an aryl bromide for late-stage diversification through Suzuki couplings, whereas development may seek to carry out the Suzuki coupling much earlier in a convergent synthesis.
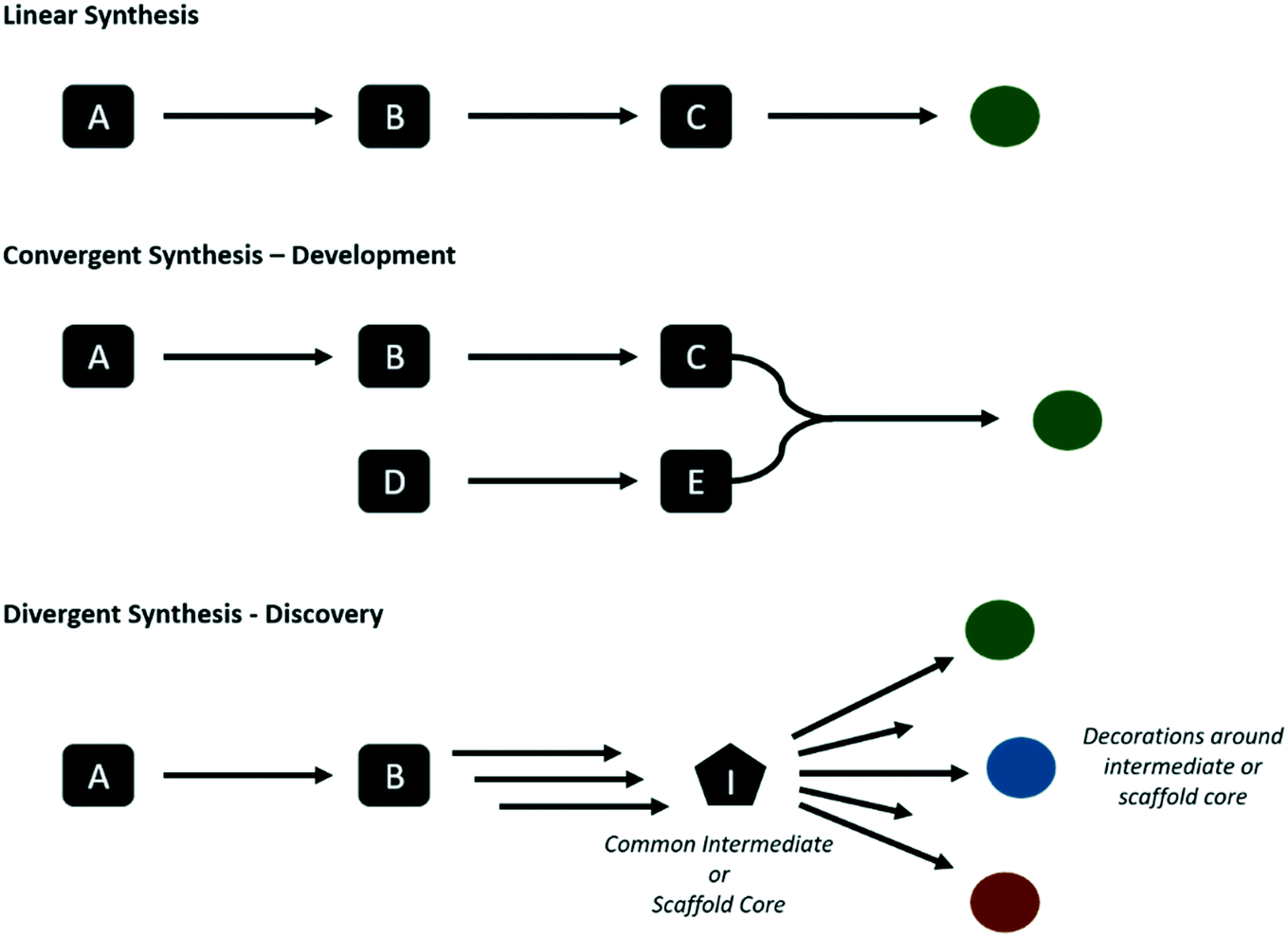 | ||
| Fig. 2 Illustration of linear, convergent, and divergent synthesis. Linear and convergent approaches can be used throughout the discovery and development process, however divergent synthesis are often used in discovery during and hit finding and lead optimization. | ||
The two domains, discovery and development lie on a continuum. The separation of the two varies between organization, and the point at which an active pharmaceutical ingredient (API) enters the development cycle can additionally vary depending on disease area and regulatory requirements. We do not aim to deal with this subject in detail and several reviews have been referred herein for the interested reader.29,33–35 Some problems to consider within both discovery and development are highlighted and we emphasise that there are several approaches that go towards addressing the various challenges. Broadly speaking the development domain is able to spend more time optimizing processes towards the optimal route. There is no universal definition for an optimal route and organisations may choose from a broad selection of metrics. This makes it difficult to handle algorithmically, as modelling techniques are dependent upon quantifiable metrics and as discussed with regards to data infrastructure later, data may not be available or there is no agreed upon measure. In cases where the optimal solution must be found the definition of optimal can vary between the two domains, and to complicate matters further depending on disease area also. What is optimal for discovery may not be optimal for development.
The infrastructure can be different between the two domains, and the techniques used in discovery may not scale to development. For instance consider the use of preparative high-performance liquid chromatography used to separate and purify crude reaction mixtures. The technique may not scale into the later large-scale development setting as is the case with many chromatographic techniques, thus other methods such as recrystallisation may be used to purge impurities in late stage chemical development. As highlighted the continuum between discovery and development means that the techniques used are less dependent on the domain, and rather the decisions that need to be made in a given project. For instance, non-selective routes may be favored in the discovery stage to generate stereoisomers that can then be separated later, giving rise to chemical diversity. Yet another approach may be to selectively synthesise a given isomer based on prior knowledge or experience with the orientation of compounds in the binding site. The stricter regulatory environment in development also demands that impurities are synthesized and assessed for ADMET properties. For this reason, it is important that alternate reaction pathways are well-understood so that they can be minimized. An understanding of impurities and alternative reaction pathways is desirable across both discovery and development, and CASP tools have the potential to enable chemists to explore these in more detail.
There are also synergies in the area of automation. CASP tools can benefit from the data generated though automation for reaction discovery, substrate scoping, and reaction condition optimization. In these situations, rather than finding routes, data for the predictions of regio-, chemo-, or stereo-selectivity, can be collected at scale in addition to the feasibility of reaction conditions, for a given set of substrates. This area of research is often associated with design of experiments (DOE),36,37 and the area of machine learning termed active learning.38
Availability of data – the foundations of predictive models
Datasets and availability
The Chemical Abstracts Services (CAS) is the largest provider of reaction data with 127 million reactions, recorded from 1840 to the present day.39 Other datasets include Reaxys (ca. 49 million reactions),40 SPRESI (ca. 4.6 million reactions),41 Pistachio (ca. 9.2 million reactions),42 and the United States Patent Office (USPTO) extracts which is the only publicly available dataset (ca. 3.3 million reactions).43,44 The USPTO dataset has additionally been sliced into several ‘benchmarking’ sets, such as the USPTO-50k dataset, to simplify the synthesis planning task with limited reaction classes.45 However, these are only a fraction of the available reaction data, and it remains to be seen how well newly published algorithms will scale to the size and noise present if all available reaction data were considered. To reduce the noise in reaction datasets, Toniato and co-workers have proposed methods by which a distinction is drawn between noise and signal based on the amount of times a deep learning model ‘forgets’ a sample during training, termed catastrophic forgetting.46 Furthermore, noise may be removed by using atom-mapping as only reactions that can be mapped will be considered as ‘correct’. The authors have followed this approach in their work,47 however, this is limited by the performance of the atom-mapping tool (Fig. 3).48,49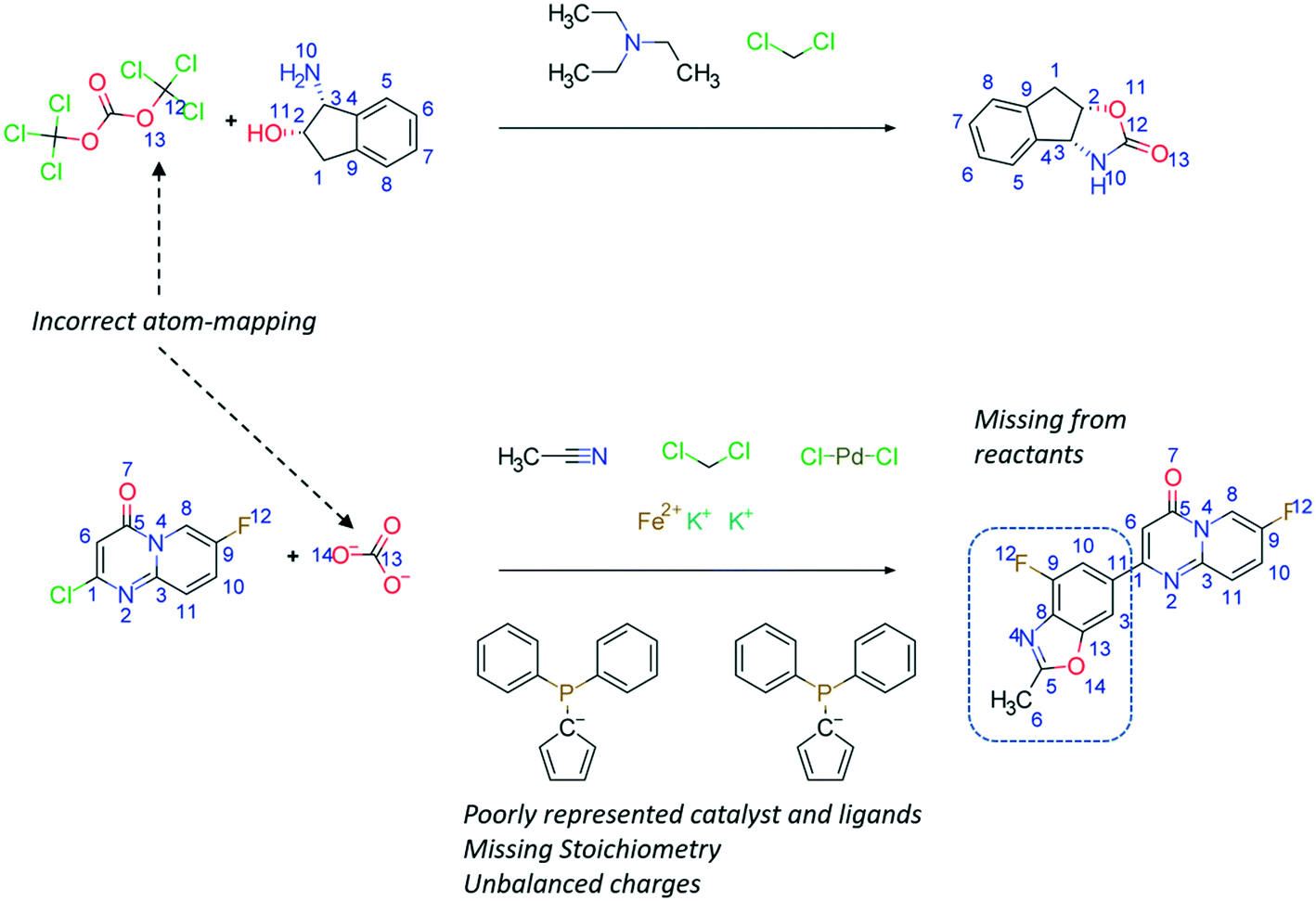 | ||
| Fig. 3 Examples of reactions taken directly from the public USPTO dataset without further modifications.43 The reactions exemplified here highlight limitations in the atom-mapping tools used, the representations of the catalyst and ligand, discrepancies in stoichiometry and charge balance, and missing reactants. These problems exist in all data sources examined by the authors in their studies. | ||
Datasets of computed structures and properties that can be used for reaction modelling have grown considerably in recent years. Green and co-workers recently created a database of transition states of more than 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reactions of small organic molecules,50 data which can be used to train deep learning models for reaction barrier prediction.51 In a similar way, von Lilienfeld and co-workers produced ca. 4500 transition states and 143000 reactant complex geometries and energies for SN2 and E2 reactions.52 St. John, Paton and Kim also generated data for 40000 organic molecules and the corresponding 200000 radicals species generated from hydrogen atom abstraction,53 which was used to train deep learning models to predict bond dissociation energies.54 Enormous datasets of quantum-chemical energies and structures are being generated for training machine learning models to reproduce quantum chemistry, such as the ANI-1 dataset with more than 20 million structures,55 and the QM9 dataset with structures, thermochemical data and electronic properties for 134000 molecules.56 Deposition of quantum-chemical calculations for individual articles as well as more comprehensive datasets to repositories such as QCArchive57 and ioChem-BD58 will increase data availability.
000 reactions of small organic molecules,50 data which can be used to train deep learning models for reaction barrier prediction.51 In a similar way, von Lilienfeld and co-workers produced ca. 4500 transition states and 143000 reactant complex geometries and energies for SN2 and E2 reactions.52 St. John, Paton and Kim also generated data for 40000 organic molecules and the corresponding 200000 radicals species generated from hydrogen atom abstraction,53 which was used to train deep learning models to predict bond dissociation energies.54 Enormous datasets of quantum-chemical energies and structures are being generated for training machine learning models to reproduce quantum chemistry, such as the ANI-1 dataset with more than 20 million structures,55 and the QM9 dataset with structures, thermochemical data and electronic properties for 134000 molecules.56 Deposition of quantum-chemical calculations for individual articles as well as more comprehensive datasets to repositories such as QCArchive57 and ioChem-BD58 will increase data availability.
For development of accurate reaction models, databases with kinetic data are of great importance. Green and West have collected kinetic data for numerous reactions in the RMG database.59,60 Many kinetic datasets compiled for individual studies61,62 and some databases are not readily available for download,63,64 and are therefore not accessible to the community. Clearly, establishing forms for sharing kinetic data is one hurdle that needs to be overcome to enable better reaction rate modelling. The situation is similar to conditions data from high-throughput experimentation, which is occasionally made available in machine readable format online,65 but most often shared in the supporting information as a hard-to-parse PDF file.66 The open reaction database is an initiative to collect publicly available reaction data to which academics and companies can contribute, in particular HTE data.67
Improvements in data infrastructure
Unfortunately, reaction data is not always consistent in annotations and is biased towards the most frequently used and positive outcome reactions.68 Notably, negative outcome reaction data are not often recorded, and their classification may be unreliable. This is because there can be multiple reasons for a failed or negative reaction, not least including the decision of the researcher to stop the reaction due to changing project demands, and cases where a researcher may accidently lose material, resulting in unreliable data entry. Whilst these problems are bound to occur, methods and standards for reporting chemical data through electronic laboratory notebooks should be improved. One such framework that could improve reporting methods are the FAIR principles for data management.69 Currently, electronic laboratory notebooks can be quite restrictive regarding the type of experiment conducted, often favoring single experiment entry over high throughput screening and successive design of experiments (DOE) for the identification of an optimal set of conditions. Furthermore, it is difficult for ELNs under their current implementation to capture multi-step synthesis. Therefore, it is not possible to easily extract full synthetic routes. This is also a problem in commercially available reaction datasets where single experiments are reported and the complete synthetic sequence is not easily known. Whilst modifications of ELNs can be made to facilitate high throughput data capture, additional data that is generated during the course of an experiment is difficult to capture, or is captured on an isolated platform. Consider the recording of heat and mass transfer, reaction quenching and work-up, or particle size during recrystallisation, which is often captured on proprietary software provided by the hardware vendor.In addition, reporting reaction data in ELNs is a mixture of free-text and specific fields for data entry. Increasing the number of fields imposes a rigid reporting structure at the expense of adaptability, whereas free-text is often not parsed during ELN export for use by informatics teams. Therefore, a degree of flexibility is also required in reaction reporting and data capture. To improve existing reporting schemes, methods for extracting and obtaining chemical data are being examined, ranging from natural language processing (NLP) which can tackle the free-text problem, to high throughput experimentation (HTE) and continuous processing which yield negative examples on which to train subsequent models.65,70,71 Whilst current ELNs may not necessarily be suited for HTE data, the role of HTE as a method of collecting large amounts of data, faster, has been recognized.38 Furthermore, efforts to create a public repository for reaction data are underway in the community, as well as discussions on schema and the redevelopment of laboratory notebooks to improve data capture.72–75
During early and late stage development there are several criteria to take into consideration aside from the feasibility of a particular reaction. These have been highlighted previously, so will only be briefly mentioned here. The cost of materials, the purge of impurities, knowledge transfer protocols for scale up, purification and crystallization, stability of APIs, and kinetic modelling to name a few examples may be stored in separate documents. The outcomes of route-finding campaigns are stored as individual entries in ELNs or documented in PowerPoint presentations and pdf documents. Thus, there is a need for a centralized repository for this information which is machine readable. While it may be the case that not all of the data will be used for modelling reasons, there is a need to improve infrastructure surrounding the storage and analysis of critical process data.
Improved publishing practices
Improvements in publishing practices could also prove beneficial to structured data repositories of reaction information. For instance, consider the compendium of synthetic routes published every year for approved drugs.76 These could be used as benchmarking sets for quality of synthetic routes, as well as providing a baseline to improve upon. Yet they are not available in a format which is easily machine-readable, despite being a source of valuable information, they remain locked in the literature. The same can be said for specialized areas of chemistry, in which there are several reviews outlining structure–reactivity relationships.77 These reviews can provide a wealth of information to which physics-based modelling can compare to experiment, and data-driven modelling learn relationships between structure and function. The subject of open source publishing and reproducibility has also been subject to ongoing debate.78–80 There have been several improvements in this regard during recent years, however there remain several opportunities to facilitate this process including the use of code sharing platforms.Data and AI driven CASP
There are two predominant approaches to CASP using a data-driven approach: 1) rule or template-based approaches, whether machine extracted or human-curated, and 2) rule or template-free approaches. These extremes lie on a continuous spectrum, with some studies combing the two. The main approaches used will be defined and outlined briefly in this section.Rule/template-based methods
One of the first attempts towards CASP was by Corey and co-workers, who attempted to codify and organize the rules of organic chemistry via a language called PATRAN (PAttern TRANslator), and while the language did not extend beyond the Logic and Heuristics Applied to Synthetic Analysis (LHASA) approach, it inspired the codification that is still in use today.81–83 The most well-known curated list of organic reaction rules took over 10 years of hand coding by expert chemists for incorporation into Synthia (formerly Chematica).84 the encoding of reaction rules is still ongoing as new chemistry is being discovered and older rules are refined. The approach has been validated in the laboratory on medicinally relevant targets.85 In addition to the transforms, functional group compatibilities and conditions under which the transformations where applicable where also encoded. Given the extent of the task, and the growing size of the chemical literature, another approach to encoding reaction rules was to automatically extract them from reaction SMILES in the form of SMIRKS patterns.86–89 These approaches may be faster but have been the subject of much debate concerning the accuracy with which they represent reactions and is discussed comprehensively by Molga et al.90 A recent study comparing a variety of proprietary and public databases found that approximately 2% of templates were common between the datasets.47 Whilst these are not necessarily different reactions, different structural variants are captured that artificially inflate the size of the rule set. To account for this Baylon et al. take a two-step approach. They first predict the reaction class or group, and subsequently a rule within the group which is used to enumerate the reactants from the given product.91Reaction rules or transformations are primarily used by expert system approaches to CASP,83,84,86,87,92,93 or more recently neural network classifiers for both the retrosynthesis and reaction prediction tasks.9,94,95 Neural network based systems are significantly faster than their predecessors such as the retrosynthesis tool ICSYNTH for finding full retrosynthetic pathways.87 However, because of the number of variables that must be accounted for when benchmarking one tool against another, including but not limited to: the reaction data underlying the tool, the scoring functions used, the availability of building blocks, and the implementation of the search algorithm, it is not immediately clear where one method is better than another. Rather each tool has the potential to excel in specific areas depending on the developers and end users priorities.
Segler and Waller use a neural network trained to predict which rule to apply in the retrosynthetic direction for a given compound from hundreds of thousands of possible rules.89 The network is employed as a ‘policy’ to enumerate potential synthetic routes represented as a tree to which Monte-Carlo tree search is applied (MCTS).9 The methodology inspired from game AI has been used to predict moves in games such as Go and Chess, as well as stock market prices.96 The approach combines historical ideas in CASP with developments in deep learning, resulting in the prediction of synthetic routes in seconds (Fig. 4). ASKCOS, developed by Coley and co-workers, takes inspiration from this approach for retrosynthetic route prediction, however they employ graph neural networks for predicting chemical reactivity and a NN classifier for selecting reaction conditions including catalyst, solvent, reagent and temperature.88,94,97,98 Furthermore, the neural networks used for prioritization fail to account for infrequently used reactions. They therefore do not prioritize templates (reaction rules) that could be used in silico but have not been used in the underlying reaction dataset. For example consider a Suzuki coupling that can be used to join two fragments together. If there are no examples of Suzuki couplings that have been used to join two fragments to form a ring, the model will be unable to predict such a reaction although it is possible. This has been partly addressed by domain specific modelling,99 and training NNs that account for template applicability.100,101
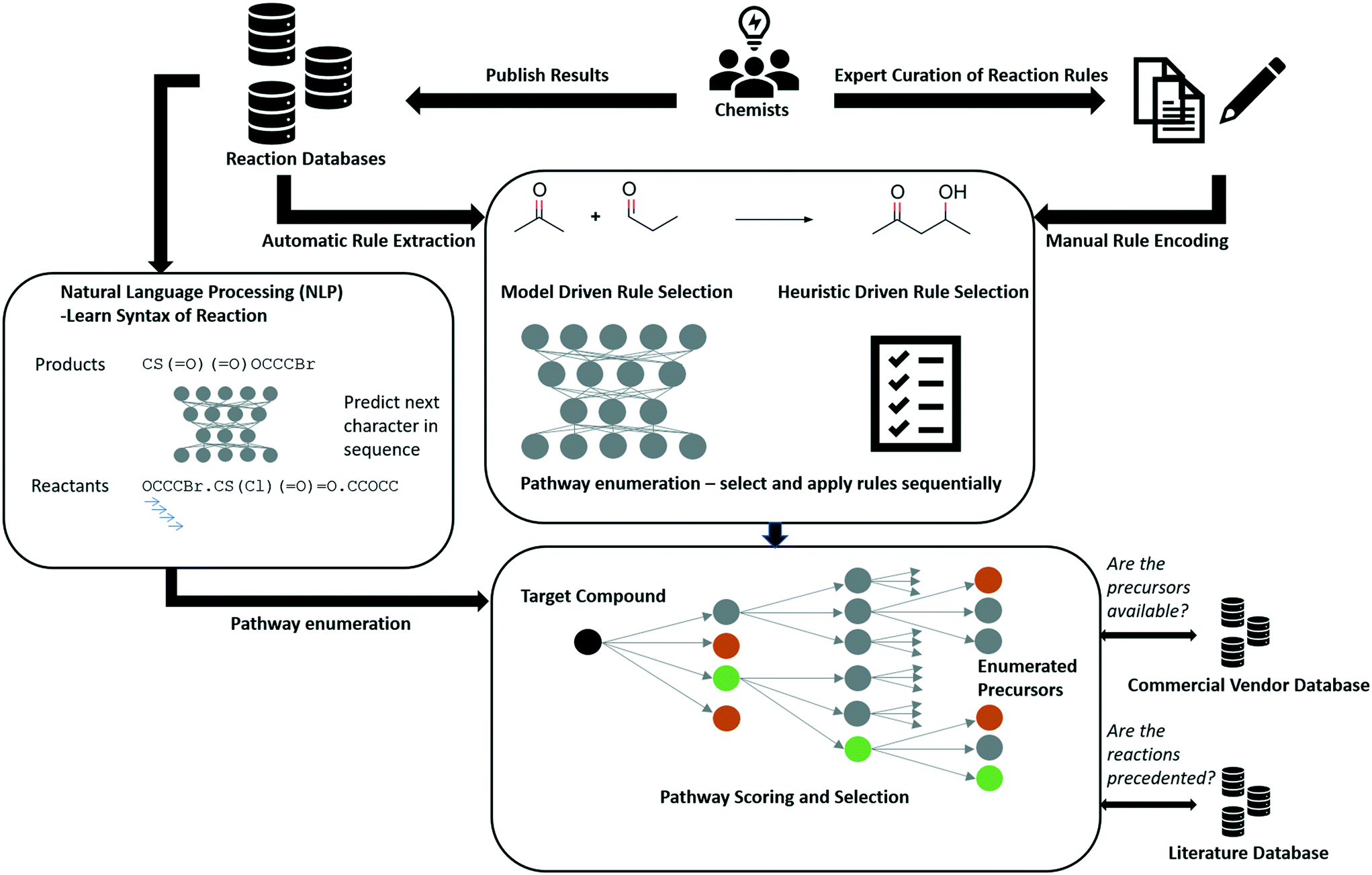 | ||
| Fig. 4 Schematic overview of rule-based approaches to CASP. Rules are either automatically extracted from the literature or manually encoded. The encoded rules represent chemical transformations which are either selected by heuristics or by models trained to learn the context in which a given rule can be applied. Model based approaches learn principles of chemical reactivity using statistical frameworks. The rules are used to enumerate synthetic pathways in the form of a tree which contains synthetic pathways selected by the scoring function. The enumerated precursors are checked to see if they are available from commercial vendors and the reactions are checked against the reaction datasets from which they were extracted to determine their precedence. Where rules are manually encoded a literature reference is associated with the rule. | ||
Notably, after the availability of datasets, the encoding or representation of chemical transformations is a bottle neck in predictive modelling. However, rules offer the advantage that predictions may be traced back to the underlying data, which is a feature that the end user wants.
Template free approaches – inspired by natural language processing (NLP)
The treatment of chemistry as a language has been explored both as a means of understanding chemical space, and codifying reaction transformations.45,102–104 The various encoding strategies are covered comprehensively by Öztürk et al.105 In contrast to rule-based approaches which predict a set of products or reactants by applying a transformation, NLP inspired approaches learn the syntax of the reactants or products depending on the task to be solved, most commonly from reaction SMILES. The problem is framed as a translation task, translating the reactants to products or vice versa. In one approach reaction SMILES are tokenized to give a vocabulary, much like a sentence may be split into its constituent words. The tokens are one-hot encoded into an n-dimensional binary vector, where the presence of a token is signified by a 1, where n is the size of the vocabulary. The vectors are fed to a neural network which learns to predict the next character/token in the sequence given a set of products or reactants, thereby reconstructing the original reaction or predicting a new one. Whilst these methods have shown promising results and improvements in line with developments in NLP, from sequence–sequence to transformer architectures within computer science,106,107 they lack the link back to the original data. However, they are potentially more interpretable than rule-based methods owing to the advent of attention, which can highlight areas of the reaction on which the algorithm focuses. This was recently demonstrated by Schwaller et al., whereby they were able to show that the algorithm implicitly learns atom-atom mapping.49 Thus the model is able to learn which atoms are changing during a reaction. Bort et al. employed similar approaches using an autoencoder and generative topographic mapping to sample novel reactions from reaction space learnt by the model.108Baldi and coworkers have taken an alternate approach to reaction prediction based on mechanistic information. They use an existing expert system to label their dataset with the required mechanistic information, thereby overcoming problems with poor data availability and annotation. Having defined a molecular orbital (MO) based reaction unit to model reactions as flows of electrons from sources to sinks, they use a two-stage machine learning approach to rank reactions that correspond to the most productive for a set of reactants and conditions.109 Recently this has been expanded to use NLP, specifically an architecture using long-short term memory (LSTM), which while less accurate includes more contextual information, and is able to predict reactive sites based solely on SMILES strings.110
Graph neural networks
Matrix representations of reactions were pioneered by Dugundji and Ugi, in the early 1970's where the reaction was described as an ‘R’ matrix, corresponding to the bond changes or changes of non-bonded valence electrons.111 In this respect, the ‘R’ matrix can be considered to be like a rule or template representing the transformation taking place. Similar ideas have now been extended and applied for both the retrosynthesis and reaction/condition prediction tasks using graph convolutional neural networks.88,112 More recently Shi et al. have used a graph to graphs (G2G) framework for the retrosynthesis task.113 The first step is reaction center identification which is common among rule-based methodologies, however rather than enumerate sets of precursors given a rule, the product is first broken into synthons (hypothetical units resembling reactants, in analogy to the formulation by Corey et al.).114 The reactants are then generated via a series of graph transformations from the synthons, thus taking into account that one synthon may correspond to multiple reactants. The graph transformations only affect small localized parts of the reactant/product as recognized by Somnath et al. who postulate that the graph topology is largely unaltered during the course of a reaction.115Reaction networks
Chemical reactions naturally lend themselves to representation as a graph or network, that is a set of vertices or nodes, molecules in this case, connected by directed edges, reactions. Typically, many studies concerned with route predictions deal with tree like structures, which can be considered sub-graphs of the overall reaction graph. However, several works have studied the statistics of reaction graphs at scale.84,116–118 Grzybowski et al. mapped the ‘Universe of Organic Chemistry’ and charted its evolution over time. In the process they identified a core set of organic compounds contributing to over 35% of known reactions.118 Furthermore, they frame the prediction of synthetic routes as a network optimization problem, whereby for a given set of products, they aim to find the set of substrates minimizing the cost. Similarly, Lapkin et al. use graph networks for the identification of strategic molecules in supply chains.117 They too have analyzed the statistics of the network of organic chemistry,116 and reach a consensus with the work of Grzybowski et al.118 Both found that on average six synthetic steps were required to synthesize any given compound from another in the network on average.Jacob and Lapkin additionally use a stochastic block model based on the network of organic chemistry to predict and discover new reaction pathways.119 Likewise, Segler and Waller identify complementary molecules in their graph. By doing so, they identify potential reaction partners for which the same reaction rules apply, thereby proposing reactions that appear to be novel.24
QM and AI driven synthesis (QM combined with AI for synthesis prediction)
QM based approaches are distinguished by requiring the generation of a three-dimensional structural representation for all reaction components being considered, from which molecular properties120 or complete reaction paths121 can be computed (Fig. 5). Such approaches are usually limited to the study of a single reaction class and the results can be qualitative or quantitative depending on the complexity of the reaction mechanism.122–124 QM approaches normally require significant user input and are often computationally intensive,125 making them currently unsuitable for routine exploitation with high-throughput CASP tools.126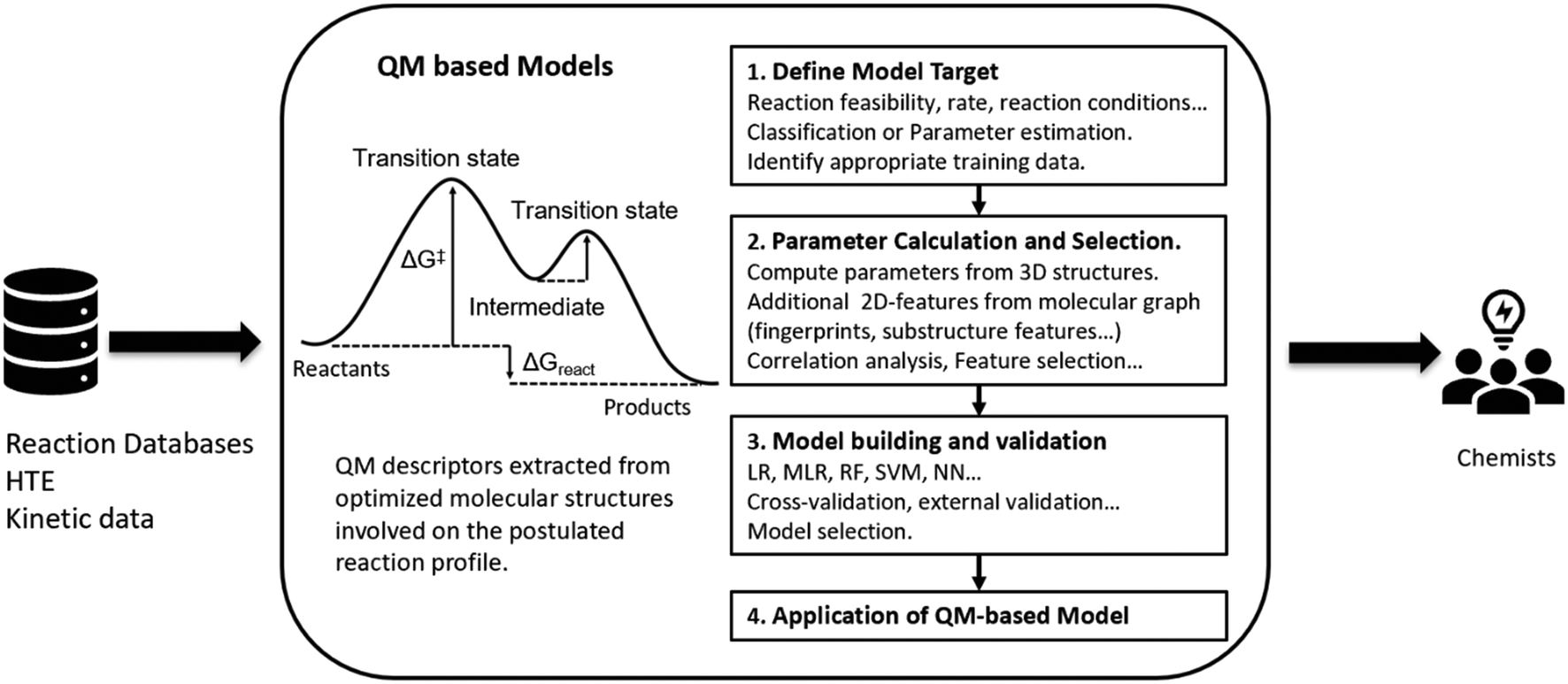 | ||
| Fig. 5 Schematic overview of QM-based approaches to synthesis prediction. Training data is extracted from existing sources and the model target selected for study. Quantum mechanical descriptors are computed from a three-dimensional structure involved on the reaction potential energy surface and can be augmented with two-dimensional descriptors based on the corresponding molecular graphs. The selected features are input into the model building process. A range of models and approaches are assessed using a suitable training set and the model performance is validated and the optimum model selected. The resultant model can then be utilised against the target of interest (reaction feasibility, selectivity, yield…) by chemists assessing proposed synthesis plans. | ||
QM approaches can be used to exhaustively explore reaction potential energy surfaces, enumerating all possible reaction outcomes. For example, Green and co-workers used their Reaction Mechanism Generator (RMG) software,59 coupled with QM-computed activation barriers, to explore feasible reactions for complex combustion processes.127 The global reaction route mapper (GRRM) from Maeda and co-workers systematically searches reaction surfaces to quantify reaction mechanisms and selectivity.128,129 Other strategies of this kind are the heuristic-aided quantum chemistry (HAQC)130,131 from the group of Aspuru-Guzik and the ReactionPredictor110,132 from the group of Baldi, which have been applied to more complex reaction mechanisms. In these implementations, the electronic re-organization of an organic reaction is encoded as chemical rules in the form of a SMIRKS pattern or as heuristic rules defining a basic reaction process. The full reaction surface for a chosen combination of reactants can then be explored and characterized with QM. Baldi et al. have also shown that a ML model for reaction success can be used to prioritize the requisite QM exploration.132 The inclusion of this QM reaction profiling enables the classification of the reactions obtained by applying the rules as feasible over a wide class of reaction mechanisms through ranking by thermodynamic and kinetic criteria. These approaches lay out how mechanistic based models can be combined with QM calculations for synthesis prediction. A related work by Nakai and co-workers identifies and ranks reactions by consideration of QM-derived descriptors such as orbital energies and Fukui functions for reactive atom pairs.133
In recent years, there has been increasing use of combining QM modelling with machine learning for quantitative chemical reactivity prediction.65,134 Doyle et al. used QM-calculated atomic, vibrational and electronic descriptors based only on the reactants and reagents coupled together with ML to build a quantitative model for the observed yield of Pd-catalyzed Buchwald–Hartwig reactions.65 The work also highlighted the synergies between high-throughput experimentation (HTE, vide infra) and QM-based machine learning to enable the prediction of novel substrate performance or optimal reaction conditions. Doyle et al. followed up this initial success with a similar approach for deoxyfluorination with sulfonyl fluorides, and developed a random forest model that could quantitatively predict reaction yield and optimum reaction conditions.135 Denmark and co-workers used models trained on average steric occupancy 3D grid descriptors together with substituent electronic descriptors to predict the enantioselectivity of chiral phosphoric acid-catalyzed thiol addition to N-acylimines.136 The work features the use of a universal training set, which is a representative subset of catalysts over chemical space that can be used to model a variety of reactions with different mechanisms, as long as they share the same catalyst scaffold.137
A hybrid approach combining 2D QSAR and modern 3D physical organic parameters derived from QM-based calculations is described by Sigman et al. to build a predictive model for enantioselectivity for an aza-Michael conjugate addition reaction.138 The work also highlights the ability of the approach for novel catalyst design and how quantitatively predictive reactivity models can be used in route optimization, which is a goal of CASP tools. Predictive models can also be derived from consideration of the product structures as described by Sigman et al.139 In this work a multivariate linear regression (MLR) model for a catalytically enantioselective Minisci reaction is reported. The model is built from computed features of the catalyst and product structures as these are most relevant to the enantioselective step. The mechanistic understanding encoded in the model is validated through successful enantioselectivity predictions to new reagents not included in the initial training set.
Experimentally, reactions are characterized by their reaction kinetics and it has been shown that quantitative models can be trained on such experimental data. Coelho et al. describe an approach to predict the second order rate constants for inverse-electron demand Diels–Alder (IEDDA) cycloadditions.140 A general MLR model for this reaction class based on 309 data points achieved a RMSE of 1.28 kcal mol−1 for the reaction barrier. As solvent polarity is a critical factor in reaction rates, the model includes a solvent term, enabling the prediction of reaction conditions from the resultant model. Buttar and co-workers studied the nucleophilic aromatic substitution (SNAr) reaction using a model which combined mechanistic modelling with machine learning to predict experimental activation free energies.141 By incorporating the computed activation energy as one of the features together with various steric and electronic features of the reactants and products, they achieved a mean absolute error of 0.8 kcal mol−1 on the external test set. The authors concluded that incorporating mechanistic information seems to be beneficial when limited data is available, as is most often the case for reaction kinetics. The obvious limitation of the method is that the reaction type needs to be mechanistically well-characterized so that the relevant transition states can be included in the model.
Late stage functionalization (LSF) is of increasing interest in drug development and deals with the ability to target specific C–H bonds in highly functionalized molecules. LSF exploitation in drug discovery makes regioselectivity prediction a key requirement for CASP tools. Predictive models to identify LSF opportunities are valuable tools for synthetic chemists. Jensen and co-workers developed the RegioSQM method for predicting site selectivity in electrophilic aromatic substitution (EAS) reactions to 81–96% accuracy, depending on cutoff values.142 The method uses the relative energies of the regioisomeric addition complexes with H+ as a model nucleophile. Norrby and co-workers report another predictive model for EAS that can predict the reaction site with 93% accuracy based on a random forest model built on QM descriptors and a simplified RegioSQM model.143 In a later study, they showed how a scale for directing group strength in LSF could be constructed from model QM calculations and validated it on a dataset of 150 C–H activation reactions, with 97% accuracy. Another example of coupling QM and ML for C–H functionalization is the work of Hong and co-workers, who report a model for predicting regioselectivity in radical C–H functionalization of heteroarenes.144 They trained a random forest (RF) model on DFT-calculated activation energies, and used the predicted barriers to infer site selectivity, achieving a 94.2% site accuracy (site for the major product) and an 89.9% selectivity accuracy (high, medium or low) in the out-of-sample test set. The study used 3406 radical functionalization reactions for training and found that models based on physically relevant QM based descriptors generalized better than models built on fingerprints, smooth overlap of atomic positions or bag of bonds features.
Quantitative reaction data is of limited availability in conventional reaction databases, but QM computational methods can be used to generate quantitative reaction data to aid training of synthesis prediction tools. Approaches to automate the exploration of reaction space using quantum mechanics are being developed,121,145,146 making the generation of such datasets achievable50,147 The generation of QM kinetic data sets will provide the opportunity for deep-learning CASP tools to train on quantitative end-points such as reaction rate. Transfer learning could then be used to fine-tune models pre-trained on QM data using a smaller amount of high-quality experimental data. QM-generated data could also be used to provide negative reaction data for model training, as such data is sparse in conventional reaction databases.
These recent examples highlight approaches to the development of data-driven chemical models and the increasing integration of QM and ML to build quantitative predictive models. AI-driven CASP tools will invariably at some point suggest key steps where regio-, chemo-selectivity or reaction success may be uncertain or hard to predict with simple models. Here, accurate QM-based models can be utilized to prioritize routes and reaction steps with higher accuracy.
The challenge for QM-based models will be to provide accurate and rapid results to CASP-generated route predictions. The development of low-cost quantum methods and the integration of computational modelling and cheminformatics with machine-learning approaches125 provide opportunities for speed improvements. The application of ML to develop transferable atomistic potentials for calculation of molecular energies has been demonstrated for conformational analysis where the RMSE versus benchmark DFT relative conformer energies was 0.6 kcal mol−1.55 This an exciting development that leads to rapid molecular energies at QM level accuracy. Additionally, detailed QM-based mechanistic studies of selected example reactions can still guide development of simpler predictive models, e.g., identifying if a transition state is stereoselectivity144 and guiding the selection of appropriate features. Along these lines, Grzybowski et al. high-lighted the importance of selecting physically meaningful descriptors in improving model performance and interpretability.148 Further studies will be required to understand how machine-learning approaches learn the principles of chemical reactivity,149 using QM-generated datasets to enable synthetic chemists to understand and interpret the decision making of models.150
Future challenges for quantitative QM reactivity models will be the extension of models to consider data sets with multiple possible mechanistic pathways, to rapidly identify unstable structures that could decompose via other processes and methods to assess whether data sets are representative for the model being considered.151 Future model development will be underpinned by high quality reaction data for different reaction classes and the exploitation of HTE for such data set generation will be an increasing trend. QM-based predictive models that are quantitatively predictive coupled with modern CASP tools will drive efficiency improvements in chemical synthesis, saving valuable time and resources for synthetic chemists.
Automation and autonomous discovery
The difference between automation and autonomy
There is a difference in definition between the terms of automation and autonomy. Whereas automation refers to any process in which a task, which is usually performed by a human, is instead conducted by machines, autonomy further includes an independence of the technological system and an ability to operate without human assistance. Coley et al. proposed seven questions to assess the autonomy of an experimental setup.126 The development of automated platforms for increasing throughput or attempts to perform autonomous experimentation has progressed over the past several decades. Early implementation of self-experimenting robots with machine learning integration was introduced in the area of genomics with the platforms “Adam” and “Eve”.152–154 The drive for building on this success for chemistry is ever growing and covers application areas from, e.g., renewable energy to kinetic model identification.155,156 In this section, we aim to cover the developments towards autonomous chemical discovery with pharmaceutical relevance.High-throughput experimentation: automation for generating data
Like other AI-based methods, autonomous systems are data-intensive and require good prior data to operate optimally. One idea to generate this data is using high-throughput experiments with automated dispensation, simultaneous reactions, and automated analytical tools. Scientists from Merck & Co. showed that it was possible to rapidly conduct a general nanomole-scale reaction on a 1536-well plate in a demonstration using the Buchwald–Hartwig amination, with verifiable results at the micromole-scale.157 With a reaction time of approximately 2 hours, the team examined the reaction of 3-bromopyridine with 16 different nucleophiles, together with 96 different catalyst and base combinations. The entire collection of samples was analyzed in 2.5 hours. It was done by collecting samples of different mass together in groups of four, so that they could be analyzed simultaneously with the LC-MS equipment. Doyle et al. used the same approach for reactions for three plates to conduct a total of 4608 reactions of Pd-catalyzed Buchwald–Hartwig aminations.65 Furthermore, this study also benchmarked several machine learning algorithms for predicting reaction yield. The root-mean square error (RMSE) on their validation set (30% of the data) was 7.2% with an R2 value of 0.92. Merck and Co. continued to improve their approach to reduce the analytical time, which was deemed to be the bottleneck of the experiments.158 In a later study, the LC-MS analysis was replaced with MALDI-TOF, which reduced the time of analysis for the entire 1536-sample to between 8 and 11 minutes. Unlike the first study, this project utilized glass plates instead of plastic plates, which increases the available chemical search space, as plastic plates limits the temperature range and suitable solvents. A group at Pfizer attempted to automate the data-generation process by using flow processing, using two UPLC-MS machines that were loaded alternately to continuously analyze micromolar scale samples.159 The system used a repurposed HPLC autosampler to freely sample the available reagents and yield all possible combinations within this space. This platform was demonstrated in a study on Suzuki–Miyaura coupling. The system conducted around 1500 reactions per day, for a total of 5740 reactions. It was noted by Merck that the flow processing format was limited to homogenous reactions of sufficiently high reaction kinetics to react faster than the UPLC-MS loading rate.158The data generation from HTE does not necessarily have to exhaustively test all configurations of combinatorial data to generate enough information to make inferences. Eyke et al. demonstrated that deep learning might also assist in the generation of new data, using active learning to steer the search.38 A neural network was trained to predict the yield and, using variational inference to assess the uncertainty of the model, an algorithm suggested the reaction combinations based on the current highest uncertainty of predicted reactions. This was demonstrated to give a significantly better prediction performance than a model trained by random selection, though one should note that the performance was identified to be affected by the batch size of data points to be added at each time step. The standard HTE plate sizes of 96 and 384 performed significantly worse than smaller batch sizes. Though this error rate was not detrimental to the predictiveness of the method, further development likely has to be made before it could be applied to predict the larger batch sizes, i.e., 1536-well plates. The study used using the Merck and Pfizer data as validation.157,159 The experiment started with a small subset of the respective experimental data, and the model was compared on the different methods for expanding the dataset.
Process chemistry and closed-loop optimization
For already known reactions, automated platforms for optimizing reaction conditions towards some optimization criteria have seen development. For example, the criteria could be optimizing towards, e.g., higher product yield, higher reaction rate, reduction of additives, or more environmentally friendly solvents. The integration of automated experimentation equipment with analytics and decision-making processes in an autonomous workflow is known as closed-loop optimization, or self-optimization. Houben et al. defined three necessary components for a closed-loop system:160• An automated reaction system: a controllable experimental setup to conduct a reaction with given parameters, coupled with in situ, (on site) chemical analysis tools to measure the current state.
• Cheminformatics: methods to mathematically describe existing data to determine the optimal outcome and direction of optimization.
• Automated design of experiment algorithms: algorithms for attributing the outcome of the observed data back to the controllable parameters for tuning the model. These should also suggest new configurations for the reaction system.
One of the early ways to implement an automated reaction system was with the use of microfluidics.161,162 This allowed for high-throughput experiments with high control and flexibility in addition to low consumption of chemicals. Jensen and co-workers studied microfluidic systems for the Suzuki–Miyaura reaction.163 By starting with two iterations of fractional factorial design, the platform then proceeded to optimize towards the G-optimum (minimizing the maximum variance in predicted values),164 while considering the discrete variables of reactants and the continuous variables of time and temperature simultaneously. This simultaneous optimization is more efficient than earlier approaches, which created redundant experiments as they varied one variable per experiment.165 However, as reactions on the microscale have different behavior with regards to heat and mass transfer, the results also have to be further verified in laboratories on a larger scale. Generally, conducting initial experiments on the micro-scale remains a popular and efficient approach. However, it should be noted that a separate development of automation has to be applied to the macro-scale. Poliakoff and co-workers pioneered platforms to tackle this challenge on the scale of 0.1–0.7 kg per day. This approach, used a modified simplex algorithm for optimization.166 This was also conducted with just having one parameter variable at each subsequent experiment.37,167 Using deep learning to steer the experimental design, Zhou et al. employed reinforcement learning to optimize a microdroplet reaction system and found optimal conditions for all four studied reactions within 30 minutes.168 They further trained the optimization model on prior training data of both similar and different underlying reaction mechanisms and found that the performance of the model improved compared to the baseline model.
The recent developments within closed-loop optimizations and a thorough review of modern monitoring techniques has been covered by Mateos et al.169 While closed-loop optimizing platforms have reached relative autonomy, one current problem is their specificity, as they often target only one problem reaction in their design. Modifying one platform to suit another study would require significant planning and would not be automated. Furthermore, most closed-loop platforms are designed to optimize towards a single response, e.g., yield, cost or flow rate.170 Multi-objective optimization can most simply be approached by performing a series of single-objective optimizations, subject to constraints for desirable values of the responses.171,172 An alternative is to consider a weighted sum of the different objectives, which oftentimes require prior knowledge of the system to impose weighting,173 although efforts have been made to perform automatic assignment of these weights according to an user-defined priority.174 In multi-objective optimization the set of optimal points, where one objective cannot be improved without costing performance towards another objective, is called the Pareto front. Lapkin and co-workers successfully identified the Pareto front between environmental and economic objectives for an example SNAr reaction and one N-benzylation reaction using the Thompson sampling efficient multi-objective (TS-EMO) algorithm.167 The same group then used the same type of algorithm on a Sonogashira reaction and a Claisen–Schmidt condensation reaction.37 Efficient Pareto front identification for pharmaceutical applications is motivated by competing objectives found in downstream processes, e.g., recovery and purification of products from natural sources. A framework for finding the Pareto front have been suggested by Galvanin and co-workers, however, it has only been demonstrated on simulated data.175 Another challenge is the use of different communication protocols in different laboratories and restrictive data formats and limited control accessibility for the monitoring equipment, which complicates the design of an interface for a central controlling computer. Some progress towards control frameworks has been made in settings outside of reaction optimization, such as controlling entire synthesis platforms using, e.g., a Raspberry Pi computer.176,177 For materials chemistry, one proposed platform by Li et al. might have answered several of these questions, connecting their product with a cloud server that would enable collaborations with other parties to centralize experimentation, rather than reproducing the platforms in other locations.155 The platform uses remote-controlled injections to a microfluidic reactor, with a robot arm transferring the sample to a CD spectrometer. The study incorporated an optimization algorithm assisted by reinforcement learning to create an autonomous laboratory.
Model based design of experiments
Generally, the structure of a closed-loop optimizing platform as we have described above, would have the objective of maximizing the yield as a function of the input parameters, e.g., temperatures, reactant concentrations and residence times. Experimental designs or design of experiments (DOE) instead conducts experiments with the focus on yielding the kinetic model that best describes the different outcomes of the reaction based on said parameters, optimizing for model accuracy and prioritizing experiments that would yield the most information rather than a single optimal configuration. Kinetic models such as these would primarily be applied during scale-up of laboratory reactions to full-scale industry processes.178 DOE was pioneered as a new area of statistics following the works of Fisher who introduced the concept of factorial design,179 to test multiple parameters at once, and the Latin square method for initializing the first trials for a factorial design to maximize the variation of parameters. The methods for DOE were further developed for models where one exploited underlying knowledge of the system. Franceschini et al. wrote an extensive review with regards to the history of this category of model based design of experiments (MBDOE).180 Here, MBDOE was defined using the following two criteria: the explicit use of model equations and current parameters to assess the ‘information content’ of the next experiment, and the application of an optimizing framework to solve the resulting numerical problem. The Jensen group made progress towards automating MBDOE by combining the framework with online HPLC analytics to estimate the kinetic parameters for a Dies–Alder reaction in a computer-controllable microreactor.181 Four kinetic rate expressions were suggested, and the model conducted sequential experiments to narrow down which expression that best described the observed results. A later study by the same group extended the framework for multi-step reactions by examining the SNAr reaction.182 The user of the model would input a kinetic model, and using maximum likelihood estimation or maximum a posteriori methods, optimal parameters would be estimated. Furthermore, the joint and total uncertainty of these two measures would be evaluated. The experiment which had the expectation of minimizing these uncertainties the most was selected to be conducted next.The MBDOE and self-optimization platforms do not necessarily have to be separate from each other. The Lapkin group conducted experiments on a C–H activation reaction by coupling the two techniques together; the MBDOE results were used as a prior to the optimization model.183 This yielded a working process design together with a kinetic model. They noted however, that for reactions of high complexity under a time constraint, where a physical model is infeasible, the use of ‘black-box’ optimization models which yield no kinetic understanding would be significantly faster and more efficient.
In the search of a platform that can be generalized towards a larger space of chemical reactions, Waldron et al. integrated Python code together with LabVIEW in order to operate the equipment of the platform.178 LabVIEW has been used in several studies due to the wide-spread support for instrument drivers by manufacturers.172,178,183–186 The MBDOE algorithms, which were implemented in Python, would send the reaction parameters to LabVIEW for execution. The LabVIEW software would then write the results in an excel output file, which the python software would read for further processing. The MBDOE model was demonstrated to achieve a lower parameter uncertainty than that of factorial design and operated on both steady-state and transient time-scales, concluding that the optimal setup depended on the relationship between analysis time and residence time. The same group later attempted to create a five-step generalized approach for setting up a MBDOE using their previously published software to analyse a benzoic acid esterification. The steps included were 1) a factorial screening to assess the experimental budget. 2) Test of candidate rate laws against the factorial design using a threshold to filter the models requiring the data to match the predictions in a 95% confidence interval. 3) A check for practical model identifiability, to see if parameters could be identified analytically. 4) Online MBDOE for model discrimination. More experimental data is added until only one candidate model still passes the threshold (95% χ2 test), 5) experiments for more precise model parameters. The order of the steps was designed to at each step automatically discard models that did not pass the requirements to reduce the number of practical experiments conducted. The framework could perform the individual experimental steps 1, 4 and 5 without supervision, but required manual input to switch between the steps. The demonstrated model conducted all experiments in 3 days.
Despite the growing popularity of AI and machine learning, the applications for DOE remain relatively few. It could be because current models already find solutions fast enough. There is, naturally, an exception. One study by Hartman and co-workers on the kinetic modelling of polymerization reactions used the classical Latin square for the design of the experiments, but chose to use neural networks for estimating the model to fit to the data generated.186 The purpose of the model was to estimate the reaction kinetics even without explicit mechanistic knowledge of the degree of dependence of the variables, i.e., if the rate expression dependence for a component is polynomial, exponential or of a more complex non-linear nature.
Discovery-oriented platforms
Several approaches have been explored towards a general platform of experimentation. Most of the recent developments use modular equipment that can be easily adapted to the reaction of interest. Coley et al. developed a reaction platform which used a robotic arm to change the modular units for a synthesis according to the recommendations made by the controlling computer.94 This was coupled with the ASKCOS reaction prediction platform to explore the feasibility of a test set of 15 small molecules. Chemists translated the synthesis routes suggested by ASKCOS to instructions that the robot arm could follow by adding parameters such as residence time and concentrations. A total of 15 small molecules of pharmacological relevance were successfully synthesized. A similar system was constructed by Cronin and co-workers,71 with the aim to automate bench chemistry with a modular approach that could perform the chemical operations of: reaction, extraction, filtration and evaporation. To control the system, they wrote an interfacing program called the Chempiler to standardize the instructions to the hardware platform. In a demonstration to synthesize the compounds sildenafil, rufinamide and nytol, they reported yields comparable to reference manual synthesis. Bédard et al. also favoured a modular approach, creating a versatile platform with five ‘bays’ where one could place any of six different modules from the choice of heated reactor, cooled reactor, LED-based photochemistry reactor, packed-bed reactor, membrane based liquid–liquid separator or a bypass. The components could be swapped according to specifications, and up 6 reagents could be used.187 This platform used continuous flow synthesis and allowed for either optimization or exploration of reaction conditions. More recently, Chatterjee et al. argued that previous approaches used a linear experimental configuration, which had disadvantages in the amount of equipment needed, and instead suggested a setup of modules arranged radially around a central station.184 The modules used 16-position valves to store different samples simultaneously, and included the modules of reagent dispensing, reaction, standby (for storage of intermediary compounds in multi-step reactions) and collection (endpoint).The setup modules consisted of commercially available hardware, with custom software. The capacity of this setup involved storage of 12 different reagents and operational settings of temperature up to 150 °C and pressure up to 9 bar.It is also possible to automate experiments using pure robotics, as in the case of the Automatic Synthesis Lab used by Eli Lilly.188 This system uses a total of four six-axis robots and three separate laboratory benches to reach an intended throughput of 120 reactions per day at the scale of 100 mg per reaction. It is reported to have been in development since 2008, conducting a total of 16349 reactions during 2011. More recently IBM have coupled their predictive tools to cloud and automation in order to deliver an autonomous synthesis platform. However, it is not yet clear what the full capabilities of the platform are, and upcoming publications from the group should reveal further details.189
While less noteworthy for flexibility, and more so for its practical applications, Adamo et al. proposed a portable chemical platform for manufacturing small-molecule chemicals in continuous end-to-end synthesis on demand, within the space of ∼1.3 m3, which was described as “drug manufacturing in a fridge-size box”.190 This platform was designed to address sudden increases in demand in select (and potentially remote) geographical regions. It was reported to reach a throughput of thousands of doses per day. It managed to produce four common pharmaceuticals with sufficient purity to meet U.S. Pharmacopeia standards. Another platform that has traded versatility and ability to swap components to focus on one particular use is the automated flow peptide synthesis (AFPS) developed by Pentelute and co-workers.191 By storing up to 27 bottles of amino acid at a time, the platform is designed to synthesize peptides at a rate of 40 seconds per addition. The platform has been demonstrated to synthesize peptides of up to a length of 164 amino acids.192 Platforms that specialize in one type of reaction can exploit similarities to a higher degree than generic platforms. Burke and co-workers noted for small molecule synthesis using building-blocks, that N-methyliminodiacetic acid (MIDA) boronates had a high affinity for silica gel, low mobility with MeOH:Et2O as an eluent and high mobility when eluted with tetrahydrofuran, which led to a generalized purification protocol.193 The result platform had the modules for deprotection, coupling and purification. Instead of a large storage of different building-blocks, customized cartridges containing the MIDA boronates and NaOH were designed.
Instead of putting the modularity and flexibility into focus, studies have also emphasized the decision-making algorithm and chemical exploration. Granda et al. modeled synthesis feasibility using a mix of machine learning models to assess whether a reaction was successful and to perform decision-making on which reaction to explore.194 This setup used batch chemistry to study a chemical space of 18 building block molecules in 969 combinations. The authors highlighted that it found four new reactions. In flow chemistry on the microfluid scale, Desai et al. used a random forest regression model for activity prediction to steer the synthesis.195 After 21 synthesized compounds, the setup found a novel template of Abl kinase inhibitors.
Automation without a platform
Unlike previous research which had focused on automating the laboratory equipment, connecting the different appliances into one interface, Cooper and co-workers instead built a robotic chemist.196 This robot was designed to move around a regular laboratory with standard equipment. The tools in the laboratory were each fitted with a cube used to enhance calibration. Touching the cube on different sides allowed the robot to reach a positional precision of 0.12 mm and an angular precision of 0.005°. The robot operated with batches of 16 experiments and autonomously conducted 688 experiments total over the course of 8 days. The decision-making algorithm for this robot used Bayesian optimization which used a function to optimize towards highest yield with a parameter for how ‘greedy’ or ‘optimistic’ the algorithm would be towards compounds of high uncertainty. Each batch varied this parameter to create a mix of greedy and conservative search.Which questions are suitable for autonomous chemical systems?
In their review, Zhong et al. discuss the applicability of automated (and by extension autonomous) systems.197 For drug discovery, later stages of the process, e.g., clinical trials quite naturally require human interaction. Furthermore, machine learning and AI-guided processes are often suitable for questions and tasks that have a quantitative nature due to their strength in optimization towards a goal. While a model might predict specified chemical properties, current popular machine learning models and neural networks have difficulties in questions that require answers on a higher level, e.g., “why is drug x more effective than drug y?” rather than “which drug is more effective?”Automated and autonomous platforms also have a common weakness, that the application domain of a constructed setup generally would be very specific. Small alterations to a setup could have big effects on the systems control or the monitoring, which hampers the generalizability of the system. As such, it might sometimes be inefficient to construct a fully automated system, rather than conducting the reaction manually. Furthermore, the approaches using LabVIEW, which have previously been mentioned in this review, while they might be easy to transfer between similar setups, require manual modification of code if different types of equipment has to be used, which is hardly desirable for the lab chemist. Cherkasov et al. attempted to solve this problem with a platform called OpenFlowChem, which had the goal of reducing the assembly time for programming down to the same time-scale of building the setup itself.198 In the end however, the authors conceded that at the current stage, LabVIEW based frameworks still have an implementation advantage, due to the existing support for instrument drivers, as the investment cost of competing methods would have to translate all measures into instructions that the instruments can perform, a trade-off cost that would be too steep for laboratory scale. Conclusively, autonomous systems cannot with the current progress be used for all chemistry but should be viewed as a tool that will be increasingly more prominent in the chemist's workflow as the apparatus becomes more sophisticated. One of the reasons that limit adoption and utility is the engineering barriers that must be overcome when transferring reactions from the bench into robotic friendly formats. A starting point is the transfer of reactions that are commonly used in pharmaceutical development.199 However, an additional caveat that must be considered is that not all reactions will be amenable to automation, and there are physical limitations to the variety of reactions a platform will be able to conduct, not least because of the number of building blocks, solvents, and reagents available to a platform at any given time.
Outlook
It is generally expected that ML and AI will be more and more important for drug discovery and drug development in the future, and discovery chemistry and process chemistry are no exceptions. While it is always difficult to predict the future there are several current trends that are expected to grow in importance. One is a tighter integration between ML/AI and automation, this integration will be fueled by the progress in chemistry automation generating more and more data that can be used to refine the ML models. Another area that is emerging is the combination of machine learning and quantum mechanics. Efforts are underway to both model the energy, wavefunction and electron density with ML. This exciting development might have a transformative impact of mechanistic understanding and prediction of the optimal synthetic route. It is also expected that deep learning based molecular design and synthesis prediction will be much tighter integrated in the future. The best generated molecules will be scored according to synthetic feasibility and prioritized either for synthesis on an automation platform or manual synthesis in the laboratory based on a suggested reaction protocol generated by the synthesis prediction software tool. Many algorithms also exist as open-source, the increased adaptation of open source is also expected to speed up the innovation of novel synthesis prediction algorithms.There are initiatives that aim to alleviate one of the current biggest bottlenecks, the lack of large reaction data sets in the public domain. The open reaction database is an initiative to collect publicly available reaction data to which academics and companies can contribute, in particular HTE data.67 The public access to high quality datasets of both successful and failed reactions will hopefully spur faster development of novel methods and make it possible to benchmark algorithms in a transparent way. The benchmarking of novel algorithms should be a priority for the community since novel algorithms are published almost weekly. To enhance data integration and exploitation it is also desirable that better chemical reaction ontologies will be developed and FAIR principles for the reaction data will be implemented.69 There is a chemical reaction ontology which has been developed, RXNO, however, it appears not to be maintained in the public domain, and whilst different approaches have been taken for reaction classification no maintained open source programs exist for automating the annotation.200,201 It is also desirable that it becomes easier to extract reaction data from publications. While pdf is a convenient format to read, it would be much easier if reaction data including supplementary information would be accessible in a markup language like XML. It is also important that the burden of annotating and recording reaction data is not increased for the synthetic chemists. If it would be too cumbersome or time-consuming to record the data in an ELN, the synthetic chemists might be reluctant to invest the necessary time and effort. It is important that the import of reaction data to an ELN is as smooth and easy as possible with automatic checks that all essential data have been captured.
There is also a trend to form private–public partnerships (PPP) to speed up the innovation in drug discovery. A PPP with a focus on improving CASP is the MLPDS (Machine Learning for Pharmaceutical Discovery and Synthesis Consortium) consortia.202 The consortia consist of researchers from MIT and 14 companies. Their goal stated on their homepage is to “facilitate the design of useful software for the automation of small molecule discovery and synthesis”. Another PPP is C-CAS (Center for Computer Assisted Synthesis).203 The consortium is led by University of Notre Dame and consists of five universities and five industrial partners. The mission statement for C-CAS is “Using quantitative, data-driven approaches to make synthetic chemistry more predictable”. These collaborations will facilitate the design of useful software for the automation of small molecule discovery and synthesis in the coming years. In addition, such collaborations involve working alongside experimentalists from both industry and academia. As such, it is hoped that students working in the field from an experimental background will improve their data literacy, and those working on the computational aspects will benefit from close collaboration with domain experts and the end user.204
Privacy preserving machine learning could in principle be applied to improve synthesis prediction for the participating partners. Thus, the synthesis prediction models of the participating partners would be improved with reaction data from other partners, however, the underlying reaction data wouldn't be shared. No such existing initiative exists currently. There is a PPP for improving bioactivity predictions, MELLODDY.205 MELLODDY (Machine Learning Ledger Orchestration For Drug Discovery) consists of seven public partners and ten pharmaceutical partners and their vision statement on their homepage is “MELLODDY aims to leverage the world's largest collection of small molecules with known biochemical or cellular activity to enable more accurate predictive models and increase efficiencies in drug discovery”. Thus, there exists an example that might be transferable to privacy preserving machine learning for synthesis prediction.
With the advances in automation and data analysis, it also becomes very important that synthetic chemists are trained in automation and data analysis. The general need for training scientist in working with “Big Data” has been highlighted.206 Training in data science for the next generation of chemists is for instance also an integral part of C-CAS consortia, with the explicit aim to train a “new generation of data chemists”. Training in data science is also part of the needed cultural change to being able to reap the benefits of the progress in faster data generation. The potential conflicts have recently been highlighted between more traditional chemistry views and AI driven methods.13 However, in the end no chain is stronger than its weakest link so it is crucial to get everyone on-board and working in a more data centric culture.
Conclusions
Synthesis prediction and chemistry automation has seen renewed interest in the last few years and the progress has been remarkable. We aim in this review to cover the most important current trends in synthesis prediction for both discovery- and process chemistry. Historical rule-based systems for route prediction have been complemented by software based on ML and large datasets of reactions as training sets. In addition, there is an increased uptake of QM based methods to support CASP.32 Progress in chemistry automation like plate-based chemistry has transformed productivity and facilitated high-throughput chemistry, which is key for generating the large and consistent reaction data sets needed for accurate model building. It is also expected with further progress in chemistry automation, reaction data generation, computer hardware and algorithm development for synthesis prediction that the speed of innovation will be maintained and that the coming decade will see significant productivity gains in discovery- and process-chemistry.Lastly, we reiterate that the task of coupling automation with predictive modelling is by no means complete and there is considerable work to be done, both in terms of technological advancement and in terms of integration into chemists workflows. The authors emphasize that synthetic chemistry is not necessarily the bottle neck in drug discovery, and is only one contributing factor in the process. Nevertheless, to facilitate development in any of the highlighted areas, an interdisciplinary approach bringing together experts from different fields is required. In addition, emphasis should be placed on ease of use and accessibility of the tools that are developed. Successful approaches may be characterized as those with a shallow learning curve for the experimentalist, a rich data source for the theoretician or data scientist, and tight-knit integration throughout the community from discovery to development. The approach should also be scalable, adaptable, reliable and most importantly, meet the needs of the end user.
Author contributions
AT wrote the ‘Introduction’, ‘Availability of Data’, and ‘Data and AI Driven CASP’. AT, KJ, and DB wrote ‘The Role of CASP in Drug Discovery and Development’. KJ and DB wrote ‘QM and AI driven Synthesis’. SJ wrote ‘Automation and Autonomous Discovery’. OE wrote the Outlook and Conclusions. All authors improved and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AT was funded by AstraZeneca and the Swiss National Science Foundation (SNF). SJ was supported by the Wallenberg Artificial Intelligence, Autonomous Systems, and Software Program (WASP), funded by the Knut and Alice Wallenberg Foundation. KJ is a fellow of the AstraZeneca PostDoc program. The authors would like to thank Jeffrey Johannes and Simon Hardy for their comments.References
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis, J. Med. Chem., 2020, 63(16), 8667–8682 CrossRef CAS.
- P. Schneider, W. P. Walters, A. T. Plowright, N. Sieroka, J. Listgarten, R. A. Goodnow, J. Fisher, J. M. Jansen, J. S. Duca, T. S. Rush, M. Zentgraf, J. E. Hill, E. Krutoholow, M. Kohler, J. Blaney, K. Funatsu, C. Luebkemann and G. Schneider, Rethinking Drug Design in the Artificial Intelligence Era, Nat. Rev. Drug Discovery, 2020, 19(5), 353–364 CrossRef CAS.
- A. M. Jordan, Artificial Intelligence in Drug Design—the Storm before the Calm?, ACS Med. Chem. Lett., 2018, 9(12), 1150–1152 CrossRef CAS.
- E. A. Feigenbaum, Some Challenges and Grand Challenges for Computational Intelligence, J. Assoc. Comput. Mach., 2003, 50(1), 32–40 CrossRef.
- A. M. Turing, I.—Computing Machinery and Intelligence, Mind, 1950, LIX, 236, pp. 433–460 Search PubMed.
- P. W. Battaglia, J. B. Hamrick, V. Bapst, A. Sanchez-Gonzalez, V. Zambaldi, M. Malinowski, A. Tacchetti, D. Raposo, A. Santoro, R. Faulkner, C. Gulcehre, F. Song, A. Ballard, J. Gilmer, G. Dahl, A. Vaswani, K. Allen, C. Nash, V. Langston, C. Dyer, N. Heess, D. Wierstra, P. Kohli, M. Botvinick, O. Vinyals, Y. Li and R. Pascanu, Relational Inductive Biases, Deep Learning, and Graph Networks, 2018, arXiv:1806.01261, https://ui.adsabs.harvard.edu/abs/2018arXiv180601261B (accessed June 01, 2018) Search PubMed.
- A. Howarth, K. Ermanis and J. M. Goodman, Dp4-Ai Automated Nmr Data Analysis: Straight from Spectrometer to Structure, Chem. Sci., 2020, 11(17), 4351–4359 RSC.
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, The Rise of Deep Learning in Drug Discovery, Drug Discovery Today, 2018, 23(6), 1241–1250 CrossRef.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning Chemical Syntheses with Deep Neural Networks and Symbolic Ai, Nature, 2018, 555, 604 CrossRef CAS.
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkasov and A. Tropsha, Qsar without Borders, Chem. Soc. Rev., 2020, 49(11), 3525–3564 RSC.
- T. Unterthiner, A. Mayr, G. Klambauer and S. Hochreiter Toxicity Prediction Using Deep Learning, 2015, arXiv e-prints, arXiv:1503.01445, https://ui.adsabs.harvard.edu/abs/2015arXiv150301445U, (accessed March 01, 2015).
- R. K. Lindsay, B. G. Buchanan, E. A. Feigenbaum and J. Lederberg, Applications of Artificial Intelligence for Organic Chemistry: The Dendral Project, McGraw-Hill Book Company, 1980 Search PubMed.
- E. J. Griffen, A. G. Dossetter and A. G. Leach, Chemists: Ai Is Here; Unite to Get the Benefits, J. Med. Chem., 2020, 63(16), 8695–8704 CrossRef CAS.
- A. Bender, Will Robotics, Ai and Cloud Computing in Chemical Synthesis Save Drug Discovery? A Closer Look, http://www.drugdiscovery.net/2020/09/09/will-robotics-ai-and-cloud-computing-in-chemical-synthesis-save-drug-discovery-a-closer-look/.
- K.-K. Mak and M. R. Pichika, Artificial Intelligence in Drug Development: Present Status and Future Prospects, Drug Discovery Today, 2019, 24(3), 773–780 CrossRef.
- S. M. McKinney, M. Sieniek, V. Godbole, J. Godwin, N. Antropova, H. Ashrafian, T. Back, M. Chesus, G. C. Corrado, A. Darzi, M. Etemadi, F. Garcia-Vicente, F. J. Gilbert, M. Halling-Brown, D. Hassabis, S. Jansen, A. Karthikesalingam, C. J. Kelly, D. King, J. R. Ledsam, D. Melnick, H. Mostofi, L. Peng, J. J. Reicher, B. Romera-Paredes, R. Sidebottom, M. Suleyman, D. Tse, K. C. Young, J. De Fauw and S. Shetty, International Evaluation of an Ai System for Breast Cancer Screening, Nature, 2020, 577(7788), 89–94 CrossRef CAS.
- P. Ertl and A. Schuffenhauer, Estimation of Synthetic Accessibility Score of Drug-Like Molecules Based on Molecular Complexity and Fragment Contributions, J. Cheminf., 2009, 1(1), 8 Search PubMed.
- W. Gao and C. W. Coley, The Synthesizability of Molecules Proposed by Generative Models, J. Chem. Inf. Model., 2020 DOI:10.1021/acs.jcim.0c00174.
- S. Krishna Gottipati, B. Sattarov, S. Niu, Y. Pathak, H. Wei, S. Liu, K. M. J. Thomas, S. Blackburn, C. W. Coley, J. Tang, S. Chandar and Y. Bengio, Learning to Navigate the Synthetically Accessible Chemical Space Using Reinforcement Learning, 2020, arXiv, arXiv:2004.12485, https://ui.adsabs.harvard.edu/abs/2020arXiv200412485K, (accessed April 01, 2020).
- J. Bradshaw, B. Paige, M. J. Kusner, M. H. S. Segler and J. M. Hernández-Lobato, A Model to Search for Synthesizable Molecules, 2019, arXiv e-prints arXiv:1906.05221, https://ui.adsabs.harvard.edu/abs/2019arXiv190605221B, (accessed June 01, 2019).
- F. Chevillard and P. Kolb, Scubidoo: A Large yet Screenable and Easily Searchable Database of Computationally Created Chemical Compounds Optimized toward High Likelihood of Synthetic Tractability, J. Chem. Inf. Model., 2015, 55(9), 1824–1835 CrossRef CAS.
- A. Thakkar, N. Selmi, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, “Ring Breaker”: Neural Network Driven Synthesis Prediction of the Ring System Chemical Space, J. Med. Chem., 2020, 63(16), 8791–8808 CrossRef CAS.
- R. Herges and C. Hoock, Reaction Planning: Computer-Aided Discovery of a Novel Elimination Reaction, Science, 1992, 255(5045), 711 CrossRef CAS.
- M. H. S. Segler and M. P. Waller, Modelling Chemical Reasoning to Predict and Invent Reactions, Chem. – Eur. J., 2017, 23(25), 6118–6128 CrossRef CAS.
- J. Li and M. D. Eastgate, Making Better Decisions During Synthetic Route Design: Leveraging Prediction to Achieve Greenness-by-Design, React. Chem. Eng., 2019, 4(9), 1595–1607 RSC.
- R. Dach, J. J. Song, F. Roschangar, W. Samstag and C. H. Senanayake, The Eight Criteria Defining a Good Chemical Manufacturing Process, Org. Process Res. Dev., 2012, 16(11), 1697–1706 CrossRef CAS.
- M. D. Eastgate, M. A. Schmidt and K. R. Fandrick, On the Design of Complex Drug Candidate Syntheses in the Pharmaceutical Industry, Nat. Rev. Chem., 2017, 1(2), 0016 CrossRef CAS.
- G. T. Whiteker, Applications of the 12 Principles of Green Chemistry in the Crop Protection Industry, Org. Process Res. Dev., 2019, 23(10), 2109–2121 CrossRef CAS.
- R. B. Leng, M. V. M. Emonds, C. T. Hamilton and J. W. Ringer, Holistic Route Selection, Org. Process Res. Dev., 2012, 16(3), 415–424 CrossRef CAS.
- Institute, A. G. C. Tools for Innovation in Chemistry, https://www.acsgcipr.org/tools-for-innovation-in-chemistry/.
- N. Kockmann, Digital Methods and Tools for Chemical Equipment and Plants, React. Chem. Eng., 2019, 4(9), 1522–1529 RSC.
- Y.-H. Lam, Y. A. Abramov, R. Ananthula, J. Elward, L. Hilden, S. Nilsson-Lill, P.-O. Norrby, A. Ramirez, E. C. Sherer, J. Mustakis and G. J. Tanoury, Applications of Quantum Chemistry in Pharmaceutical Process Development: Current State and Opportunities, Org. Process Res. Dev., 2020, 24(8), 1496–1507 CrossRef CAS.
- H.-J. Federsel, Chemical Process Research and Development in the 21st Century: Challenges, Strategies, and Solutions from a Pharmaceutical Industry Perspective, Acc. Chem. Res., 2009, 42(5), 671–680 CrossRef CAS.
- M. K. O'Brien, M. Kolb, T. J. Connolly, J. C. McWilliams and K. Sutherland, Early Chemical Development at Legacy Wyeth Research, Drug Discovery Today, 2011, 16(1), 81–88 CrossRef.
- H.-J. Federsel, Process R&D under the Magnifying Glass: Organization, Business Model, Challenges, and Scientific Context, Bioorg. Med. Chem., 2010, 18(16), 5775–5794 CrossRef CAS.
- V. Fath, N. Kockmann, J. Otto and T. Röder, Self-Optimising Processes and Real-Time-Optimisation of Organic Syntheses in a Microreactor System Using Nelder–Mead and Design of Experiments, React. Chem. Eng., 2020, 5(7), 1281–1299 RSC.
- A. D. Clayton, A. M. Schweidtmann, G. Clemens, J. A. Manson, C. J. Taylor, C. G. Niño, T. W. Chamberlain, N. Kapur, A. J. Blacker, A. A. Lapkin and R. A. Bourne, Automated Self-Optimisation of Multi-Step Reaction and Separation Processes Using Machine Learning, Chem. Eng. J., 2020, 384, 123340 CrossRef CAS.
- N. S. Eyke, W. H. Green and K. F. Jensen, Iterative Experimental Design Based on Active Machine Learning Reduces the Experimental Burden Associated with Reaction Screening, ChemRxiv, 2020.
- Cas Content, https://www.cas.org/about/cas-content (accessed 13 Feb) Search PubMed.
- Elsevier Fact Sheet, https://www.elsevier.com/__data/assets/pdf_file/0005/91616/Reaxys-Fact-Sheet-2019-web.pdf, (accessed 29-Apr).
- InfoChem Spresi, https://www.infochem.de/about/spresi, (accessed 29-Apr).
- Pistachio, https://www.nextmovesoftware.com/pistachio.html, (accessed 30-Jan).
- D. Lowe, Chemical Reactions from Us Patents (1976-Sep2016), https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873, (accessed Apr 31, 2018).
- D. Lowe, Extraction of Chemical Structures and Reactions from the Literature, Doctoral thesis, University of Cambridge, 2012 Search PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models, ACS Cent. Sci., 2017, 3(10), 1103–1113 CrossRef CAS.
- A. Toniato, P. Schwaller, A. Cardinale, J. Geluykens and T. Laino, Unassisted Noise-Reduction of Chemical Reactions Data Sets, ChemRxiv, 2020.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, Datasets and Their Influence on the Development of Computer Assisted Synthesis Planning Tools in the Pharmaceutical Domain, Chem. Sci., 2020, 11(1), 154–168 RSC.
- W. Jaworski, S. Szymkuć, B. Mikulak-Klucznik, K. Piecuch, T. Klucznik, M. Kaźmierowski, J. Rydzewski, A. Gambin and B. A. Grzybowski, Automatic Mapping of Atoms across Both Simple and Complex Chemical Reactions, Nat. Commun., 2019, 10(1), 1434 CrossRef.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Unsupervised Attention-Guided Atom-Mapping, ChemRxiv, 2020.
- C. A. Grambow, L. Pattanaik and W. H. Green, Reactants, Products, and Transition States of Elementary Chemical Reactions Based on Quantum Chemistry, Sci. Data, 2020, 7(1), 137 CrossRef CAS.
- C. A. Grambow, L. Pattanaik and W. H. Green, Deep Learning of Activation Energies, J. Phys. Chem. Lett., 2020, 11(8), 2992–2997 CrossRef CAS.
- R. G. Falk, S. N. Heinen, M. Bragato and O. Anatole von Lilienfeld, Thousands of Reactants and Transition States for Competing E2 and SN2 Reactions, 2020, arXiv:2006.00504, https://ui.adsabs.harvard.edu/abs/2020arXiv200600504F, (accessed May 01, 2020) Search PubMed.
- P. C. St. John, Y. Guan, Y. Kim, B. D. Etz, S. Kim and R. S. Paton, Quantum Chemical Calculations for over 200,000 Organic Radical Species and 40,000 Associated Closed-Shell Molecules, Sci. Data, 2020, 7(1), 244 CrossRef CAS.
- P. C. St. John, Y. Guan, Y. Kim, S. Kim and R. S. Paton, Prediction of Organic Homolytic Bond Dissociation Enthalpies at near Chemical Accuracy with Sub-Second Computational Cost, Nat. Commun., 2020, 11(1), 2328 CrossRef CAS.
- J. S. Smith, O. Isayev and A. E. Roitberg, Ani-1, a Data Set of 20 Million Calculated Off-Equilibrium Conformations for Organic Molecules, Sci. Data, 2017, 4(1), 170193 CrossRef CAS.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Quantum Chemistry Structures and Properties of 134 Kilo Molecules, Sci. Data, 2014, 1(1), 140022 CrossRef CAS.
- D. G. A. Smith, D. Altarawy, L. A. Burns, M. Welborn, L. N. Naden, L. Ward, S. Ellis, B. P. Pritchard and T. D. Crawford, The Molssi Qcarchive Project: An Open-Source Platform to Compute, Organize, and Share Quantum Chemistry Data, WIREs Comput. Mol. Sci., 2020, e1491 Search PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The Iochem-Bd Platform, J. Chem. Inf. Model., 2014, 55(1), 95–103 CrossRef.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Reaction Mechanism Generator: Automatic Construction of Chemical Kinetic Mechanisms, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- https://rmg.mit.edu/database/ .
- M. Glavatskikh, T. Madzhidov, D. Horvath, R. Nugmanov, T. Gimadiev, D. Malakhova, G. Marcou and A. Varnek, Predictive Models for Kinetic Parameters of Cycloaddition Reactions, Mol. Inf., 2019, 38, 1800077 CrossRef CAS.
- T. Gimadiev, T. Madzhidov, I. Tetko, R. Nugmanov, I. Casciuc, O. Klimchuk, A. Bodrov, P. Polishchuk, I. Antipin and A. Varnek, Bimolecular Nucleophilic Substitution Reactions: Predictive Models for Rate Constants and Molecular Reaction Pairs Analysis, Mol. Inf., 2019, 38, 1800104 CrossRef.
- https://kinetics.nist.gov/solution/ .
- https://kinetics.nist.gov/kinetics/ .
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Predicting Reaction Performance in C–N Cross-Coupling Using Machine Learning, Science, 2018, 360(6385), 186–190 CrossRef CAS.
- Z. Jaman, D. L. Logsdon, B. Szilágyi, T. J. P. Sobreira, D. Aremu, L. Avramova, R. G. Cooks and D. H. Thompson, High-Throughput Experimentation and Continuous Flow Evaluation of Nucleophilic Aromatic Substitution Reactions, ACS Comb. Sci., 2020, 22(4), 184–196 CrossRef CAS.
- Open Reaction Database, https://ord-schema.readthedocs.io/en/latest/.
- X. Jia, A. Lynch, Y. Huang, M. Danielson, I. Lang'at, A. Milder, A. E. Ruby, H. Wang, S. A. Friedler, A. J. Norquist and J. Schrier, Anthropogenic Biases in Chemical Reaction Data Hinder Exploratory Inorganic Synthesis, Nature, 2019, 573(7773), 251–255 CrossRef CAS.
- M. D. Wilkinson, M. Dumontier, I. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. da Silva Santos, P. E. Bourne, J. Bouwman, A. J. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. T. Evelo, R. Finkers, A. Gonzalez-Beltran, A. J. G. Gray, P. Groth, C. Goble, J. S. Grethe, J. Heringa, P. A. C. ‘t Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. J. Lusher, M. E. Martone, A. Mons, A. L. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. A. Swertz, M. Thompson, J. van der Lei, E. van Mulligen, J. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao and B. Mons, The Fair Guiding Principles for Scientific Data Management and Stewardship, Sci. Data, 2016, 3(1), 160018 CrossRef.
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Automated Extraction of Chemical Synthesis Actions from Experimental Procedures, Nat. Commun., 2020, 11(1), 3601 CrossRef.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Organic Synthesis in a Modular Robotic System Driven by a Chemical Programming Language, Science, 2019, 363(6423), eaav2211 CrossRef CAS.
- C. W. Coley, W. H. Green and K. F. Jensen, Rdchiral: An Rdkit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application, J. Chem. Inf. Model., 2019, 59(6), 2529–2537 CrossRef CAS.
- Rmg Database, https://rmg.mit.edu/database/.
- Iochem, https://www.iochem-bd.org/.
- The Molssi Quantum Chemistry Archive.
- A. C. Flick, C. A. Leverett, H. X. Ding, E. McInturff, S. J. Fink, C. J. Helal, J. C. DeForest, P. D. Morse, S. Mahapatra and C. J. O'Donnell, Synthetic Approaches to New Drugs Approved During 2018, J. Med. Chem., 2020, 63(19), 10652–10704 CrossRef CAS.
- S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Concerted Nucleophilic Aromatic Substitution Reactions, Angew. Chem., Int. Ed., 2019, 58(46), 16368–16388 CrossRef CAS.
- W. P. Walters, Modeling, Informatics, and the Quest for Reproducibility, J. Chem. Inf. Model., 2013, 53(7), 1529–1530 CrossRef CAS.
- G. A. Landrum, Reproducibility in Cheminformatics and Computational Chemistry Research: Certainly We Can Do Better Than This, J. Cheminf., 2013, 5(1), O4 Search PubMed.
- R. D. Clark, A Path to Next-Generation Reproducibility in Cheminformatics, J. Cheminf., 2019, 11(1), 62 Search PubMed.
- E. Corey, A. Long and S. Rubenstein, Computer-Assisted Analysis in Organic Synthesis, Science, 1985, 228(4698), 408–418 CrossRef CAS.
- E. J. Corey and W. T. Wipke, Computer-Assisted Design of Complex Organic Syntheses, Science, 1969, 166(3902), 178 CrossRef CAS.
- D. A. Pensak and E. J. Corey, Lhasa—Logic and Heuristics Applied to Synthetic Analysis, in Computer-Assisted Organic Synthesis, American Chemical Society, 1977, vol. 61, pp. 1–32 Search PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Computer-Assisted Synthetic Planning: The End of the Beginning, Angew. Chem., Int. Ed., 2016, 55(20), 5904–5937 CrossRef.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 2018, 4(3), 522–532 CAS.
- J. Law, Z. Zsoldos, A. Simon, D. Reid, Y. Liu, S. Y. Khew, A. P. Johnson, S. Major, R. A. Wade and H. Y. Ando, Route Designer: A Retrosynthetic Analysis Tool Utilizing Automated Retrosynthetic Rule Generation, J. Chem. Inf. Model., 2009, 49(3), 593–602 CrossRef CAS.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Route Design in the 21st Century: The Icsynth Software Tool as an Idea Generator for Synthesis Prediction, Org. Process Res. Dev., 2015, 19(2), 357–368 CrossRef.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Prediction of Organic Reaction Outcomes Using Machine Learning, ACS Cent. Sci., 2017, 3(5), 434–443 CrossRef CAS.
- M. H. S. Segler and M. P. Waller, Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction, Chem. – Eur. J., 2017, 23(25), 5966–5971 CrossRef CAS.
- K. Molga, E. P. Gajewska, S. Szymkuć and B. A. Grzybowski, The Logic of Translating Chemical Knowledge into Machine-Processable Forms: A Modern Playground for Physical-Organic Chemistry, React. Chem. Eng., 2019, 4(9), 1506–1521 RSC.
- J. L. Baylon, N. A. Cilfone, J. R. Gulcher and T. W. Chittenden, Enhancing Retrosynthetic Reaction Prediction with Deep Learning Using Multiscale Reaction Classification, J. Chem. Inf. Model., 2019, 59(2), 673–688 CrossRef CAS.
- H. Satoh and K. Funatsu, Sophia, a Knowledge Base-Guided Reaction Prediction System - Utilization of a Knowledge Base Derived from a Reaction Database, J. Chem. Inf. Comput. Sci., 1995, 35(1), 34–44 CrossRef CAS.
- W. L. Jorgensen, E. R. Laird, A. J. Gushurst, J. M. Fleischer, S. A. Gothe, H. E. Helson, G. D. Paderes and S. Sinclair, Cameo: A Program for the Logical Prediction of the Products of Organic Reactions, Pure Appl. Chem., 1990, 62(10), 1921 CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A Robotic Platform for Flow Synthesis of Organic Compounds Informed by Ai Planning, Science, 2019, 365(6453), eaax1566 CrossRef CAS.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, Neural Networks for the Prediction of Organic Chemistry Reactions, ACS Cent. Sci., 2016, 2(10), 725–732 CrossRef CAS.
- D. Silver, A. Huang, C. J. Maddison, A. Guez, L. Sifre, G. van den Driessche, J. Schrittwieser, I. Antonoglou, V. Panneershelvam, M. Lanctot, S. Dieleman, D. Grewe, J. Nham, N. Kalchbrenner, I. Sutskever, T. Lillicrap, M. Leach, K. Kavukcuoglu, T. Graepel and D. Hassabis, Mastering the Game of Go with Deep Neural Networks and Tree Search, Nature, 2016, 529, 484 CrossRef CAS.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, Using Machine Learning to Predict Suitable Conditions for Organic Reactions, ACS Cent. Sci., 2018, 4(11), 1465–1476 CrossRef CAS.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, A Graph-Convolutional Neural Network Model for the Prediction of Chemical Reactivity, Chem. Sci., 2019, 10(2), 370–377 RSC.
- A. Thakkar, N. Selmi, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, ‘Ring Breaker': Neural Network Driven Synthesis Prediction of the Ring System Chemical Space, ChemRxiv, 2020.
- E. J. Bjerrum, A. Thakkar and O. Engkvist, Artificial Applicability Labels for Improving Policies in Retrosynthesis Prediction, ChemRxiv, 2020.
- M. E. Fortunato, C. W. Coley, B. C. Barnes and K. F. Jensen, Data Augmentation and Pretraining for Template-Based Retrosynthetic Prediction in Computer-Aided Synthesis Planning, J. Chem. Inf. Model., 2020, 60(7), 3398–3407 CrossRef CAS.
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Organic Chemistry as a Language and the Implications of Chemical Linguistics for Structural and Retrosynthetic Analyses, Angew. Chem., Int. Ed., 2014, 53(31), 8108–8112 CrossRef CAS.
- J. Nam and J. Kim, Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions, 2016, arXiv:1612.09529.
- P. Schwaller, T. Gaudin, D. Lányi, C. Bekas and T. Laino, “Found in Translation”: Predicting Outcomes of Complex Organic Chemistry Reactions Using Neural Sequence-to-Sequence Models, Chem. Sci., 2018, 9(28), 6091–6098 RSC.
- H. Öztürk, A. Özgür, P. Schwaller, T. Laino and E. Ozkirimli, Exploring Chemical Space Using Natural Language Processing Methodologies for Drug Discovery, Drug Discovery Today, 2020, 25(4), 689–705 CrossRef.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS.
- P. Karpov, G. Godin and I. V. Tetko, in A Transformer Model for Retrosynthesis, Artificial Neural Networks and Machine Learning – ICANN 2019: Workshop and Special Sessions, ed. I. V. Tetko, V. Kůrková, P. Karpov and F. Theis, Springer International Publishing, Cham, 2019, pp. 817–830 Search PubMed.
- W. Bort, I. I. Baskin, P. Sidorov, G. Marcou, D. Horvath, T. Madzhidov, A. Varnek, T. Gimadiev, R. Nugmanov and A. Mukanov, Discovery of Novel Chemical Reactions by Deep Generative Recurrent Neural Network, ChemRxiv, 2020.
- M. A. Kayala, C.-A. Azencott, J. H. Chen and P. Baldi, Learning to Predict Chemical Reactions, J. Chem. Inf. Model., 2011, 51(9), 2209–2222 CrossRef CAS.
- D. Fooshee, A. Mood, E. Gutman, M. Tavakoli, G. Urban, F. Liu, N. Huynh, D. Van Vranken and P. Baldi, Deep Learning for Chemical Reaction Prediction, Mol. Syst. Des. Eng., 2018, 3(3), 442–452 RSC.
- J. Dugundji and I. Ugi, in An Algebraic Model of Constitutional Chemistry as a Basis for Chemical Computer Programs, Computers in Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1973, pp. 19–64.
- S. Ishida, K. Terayama, R. Kojima, K. Takasu and Y. Okuno, Prediction and Interpretable Visualization of Retrosynthetic Reactions Using Graph Convolutional Networks, J. Chem. Inf. Model., 2019, 59(12), 5026–5033 CrossRef CAS.
- C. Shi, M. Xu, H. Guo, M. Zhang and J. Tang, A Graph to Graphs Framework for Retrosynthesis Prediction, 2020, arXiv e-prints, arXiv:2003.12725, https://ui.adsabs.harvard.edu/abs/2020arXiv200312725S, (accessed March 01, 2020).
- E. J. Corey, General Methods for the Construction of Complex Molecules, in The Chemistry of Natural Products, Butterworth-Heinemann, 1967, pp. 19–37 Search PubMed.
- V. R. Somnath, C. Bunne, C. W. Coley, A. Krause and R. Barzilay, Learning Graph Models for Template-Free Retrosynthesis, 2020, arXiv e-prints, arXiv:2006.07038, https://ui.adsabs.harvard.edu/abs/2020arXiv200607038S, (accessed June 01, 2020).
- P.-M. Jacob and A. Lapkin, Statistics of the Network of Organic Chemistry, React. Chem. Eng., 2018, 3(1), 102–118 RSC.
- J. M. Weber, P. Lió and A. A. Lapkin, Identification of Strategic Molecules for Future Circular Supply Chains Using Large Reaction Networks, React. Chem. Eng., 2019, 4(11), 1969–1981 RSC.
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, The Wired Universe of Organic Chemistry, Nat. Chem., 2009, 1, 31 CrossRef CAS.
- P.-M. Jacob and A. Lapkin, Prediction of Chemical Reactions Using Statistical Models of Chemical Knowledge, ChemRxiv, 2018 DOI:10.26434/chemrxiv.6954908.v1.
- M. Karelson, V. S. Lobanov and A. R. Katritzky, Quantum-Chemical Descriptors in Qsar/Qspr Studies, Chem. Rev., 1996, 96(3), 1027–1044 CrossRef CAS.
- A. L. Dewyer and P. M. Zimmerman, Finding Reaction Mechanisms, Intuitive or Otherwise, Org. Biomol. Chem., 2017, 15(3), 501–504 RSC.
- Q. Peng, F. Duarte and R. S. Paton, Computing Organic Stereoselectivity – from Concepts to Quantitative Calculations and Predictions, Chem. Soc. Rev., 2016, 45(22), 6093–6107 RSC.
- Z. Liu, C. Patel, J. N. Harvey and R. B. Sunoj, Mechanism and Reactivity in the Morita–Baylis–Hillman Reaction: The Challenge of Accurate Computations, Phys. Chem. Chem. Phys., 2017, 19(45), 30647–30657 RSC.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis, ACS Catal., 2019, 9(8), 6803–6813 CrossRef CAS.
- S. Grimme and P. R. Schreiner, Computational Chemistry: The Fate of Current Methods and Future Challenges, Angew. Chem., Int. Ed., 2018, 57(16), 4170–4176 CrossRef CAS.
- K. F. Jensen, C. W. Coley and N. S. Eyke, Autonomous Discovery in the Chemical Sciences Part I: Progress, Angew. Chem., Int. Ed., 2020, 59, 2–38 CrossRef.
- M. R. Harper, K. M. Van Geem, S. P. Pyl, G. B. Marin and W. H. Green, Comprehensive Reaction Mechanism for N-Butanol Pyrolysis and Combustion, Combust. Flame, 2011, 158(1), 16–41 CrossRef CAS.
- W. M. C. Sameera, S. Maeda and K. Morokuma, Computational Catalysis Using the Artificial Force Induced Reaction Method, Acc. Chem. Res., 2016, 49(4), 763–773 CrossRef CAS.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, Implementation and Performance of the Artificial Force Induced Reaction Method in the Grrm17 Program, J. Comput. Chem., 2018, 39(4), 233–251 CrossRef CAS.
- D. Rappoport and A. Aspuru-Guzik, Predicting Feasible Organic Reaction Pathways Using Heuristically Aided Quantum Chemistry, J. Chem. Theory Comput., 2019, 15(7), 4099–4112 CrossRef CAS.
- D. Rappoport, C. J. Galvin, D. Y. Zubarev and A. Aspuru-Guzik, Complex Chemical Reaction Networks from Heuristics-Aided Quantum Chemistry, J. Chem. Theory Comput., 2014, 10(3), 897–907 CrossRef CAS.
- P. Sadowski, D. Fooshee, N. Subrahmanya and P. Baldi, Synergies between Quantum Mechanics and Machine Learning in Reaction Prediction, J. Chem. Inf. Model., 2016, 56(11), 2125–2128 CrossRef CAS.
- M. Fujinami, J. Seino and H. Nakai, Quantum Chemical Reaction Prediction Method Based on Machine Learning, Bull. Chem. Soc. Jpn., 2020, 93(5), 685–693 CrossRef CAS.
- J. P. Reid and M. S. Sigman, Holistic Prediction of Enantioselectivity in Asymmetric Catalysis, Nature, 2019, 571(7765), 343–348 CrossRef CAS.
- M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, Deoxyfluorination with Sulfonyl Fluorides: Navigating Reaction Space with Machine Learning, J. Am. Chem. Soc., 2018, 140(15), 5004–5008 CrossRef CAS.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of Higher-Selectivity Catalysts by Computer-Driven Workflow and Machine Learning, Science, 2019, 363(6424), eaau5631 CrossRef CAS.
- J. J. Henle, A. F. Zahrt, B. T. Rose, W. T. Darrow, Y. Wang and S. E. Denmark, Development of a Computer-Guided Workflow for Catalyst Optimization. Descriptor Validation, Subset Selection, and Training Set Analysis, J. Am. Chem. Soc., 2020, 142(26), 11578–11592 CrossRef CAS.
- T. T. Metsänen, K. W. Lexa, C. B. Santiago, C. K. Chung, Y. Xu, Z. Liu, G. R. Humphrey, R. T. Ruck, E. C. Sherer and M. S. Sigman, Combining Traditional 2d and Modern Physical Organic-Derived Descriptors to Predict Enhanced Enantioselectivity for the Key Aza-Michael Conjugate Addition in the Synthesis of Prevymis™ (Letermovir), Chem. Sci., 2018, 9(34), 6922–6927 RSC.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, Predictive Multivariate Linear Regression Analysis Guides Successful Catalytic Enantioselective Minisci Reactions of Diazines, J. Am. Chem. Soc., 2019, 141(48), 19178–19185 CrossRef CAS.
- J. M. J. M. Ravasco and J. A. S. Coelho, Predictive Multivariate Models for Bioorthogonal Inverse-Electron Demand Diels–Alder Reactions, J. Am. Chem. Soc., 2020, 142(9), 4235–4241 CrossRef CAS.
- K. Jorner, T. Brinck, P.-O. Norrby and D. Buttar, Machine Learning Meets Mechanistic Modelling for Accurate Prediction of Experimental Activation Energies, Chem. Sci., 2020 10.1039/D0SC04896H.
- J. C. Kromann, J. H. Jensen, M. Kruszyk, M. Jessing and M. Jørgensen, Fast and Accurate Prediction of the Regioselectivity of Electrophilic Aromatic Substitution Reactions, Chem. Sci., 2018, 9(3), 660–665 RSC.
- A. Tomberg, M. J. Johansson and P.-O. Norrby, A Predictive Tool for Electrophilic Aromatic Substitutions Using Machine Learning, J. Org. Chem., 2018, 84(8), 4695–4703 CrossRef.
- X. Li, S. Q. Zhang, L. C. Xu and X. Hong, Predicting Regioselectivity in Radical C−H Functionalization of Heterocycles through Machine Learning, Angew. Chem., Int. Ed., 2020, 59, 13253–13259 CrossRef CAS.
- G. N. Simm, A. C. Vaucher and M. Reiher, Exploration of Reaction Pathways and Chemical Transformation Networks, J. Phys. Chem. A, 2019, 123(2), 385–399 CrossRef CAS.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15(5), 2847–2862 CrossRef CAS.
- C. A. Grambow, A. Jamal, Y.-P. Li, W. H. Green, J. Zádor and Y. V. Suleimanov, Unimolecular Reaction Pathways of a Γ-Ketohydroperoxide from Combined Application of Automated Reaction Discovery Methods, J. Am. Chem. Soc., 2018, 140(3), 1035–1048 CrossRef CAS.
- W. Beker, E. P. Gajewska, T. Badowski and B. A. Grzybowski, Prediction of Major Regio-, Site-, and Diastereoisomers in Diels–Alder Reactions by Using Machine-Learning: The Importance of Physically Meaningful Descriptors, Angew. Chem., Int. Ed., 2019, 58(14), 4515–4519 CrossRef CAS.
- J. A. Kammeraad, J. Goetz, E. A. Walker, A. Tewari and P. M. Zimmerman, What Does the Machine Learn? Knowledge Representations of Chemical Reactivity, J. Chem. Inf. Model., 2020, 60(3), 1290–1301 CrossRef CAS.
- T. Hanser, C. Barber, S. Guesné, J. F. Marchaland and S. Werner, Applicability Domain: Towards a More Formal Framework to Express the Applicability of a Model and the Confidence in Individual Predictions, in Advances in Computational Toxicology, Springer, 2019, vol. 30, pp. 215–232 Search PubMed.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Predictive and Mechanistic Multivariate Linear Regression Models for Reaction Development, Chem. Sci., 2018, 9(9), 2398–2412 RSC.
- R. D. King, K. E. Whelan, F. M. Jones, P. G. K. Reiser, C. H. Bryant, S. H. Muggleton, D. B. Kell and S. G. Oliver, Functional Genomic Hypothesis Generation and Experimentation by a Robot Scientist, Nature, 2004, 427(6971), 247–252 CrossRef CAS.
- R. D. King, J. Rowland, S. G. Oliver, M. Young, W. Aubrey, E. Byrne, M. Liakata, M. Markham, P. Pir, L. N. Soldatova, A. Sparkes, K. E. Whelan and A. Clare, The Automation of Science, Science, 2009, 324(5923), 85 CrossRef CAS.
- K. Williams, E. Bilsland, A. Sparkes, W. Aubrey, M. Young, L. N. Soldatova, K. De Grave, J. Ramon, M. de Clare, W. Sirawaraporn, S. G. Oliver and R. D. King, Cheaper Faster Drug Development Validated by the Repositioning of Drugs against Neglected Tropical Diseases, J. R. Soc., Interface, 2015, 12(104), 20141289 CrossRef.
- J. Li, J. Li, R. Liu, Y. Tu, Y. Li, J. Cheng, T. He and X. Zhu, Autonomous Discovery of Optically Active Chiral Inorganic Perovskite Nanocrystals through an Intelligent Cloud Lab, Nat. Commun., 2020, 11(1), 2046 CrossRef CAS.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, Closed-Loop Model-Based Design of Experiments for Kinetic Model Discrimination and Parameter Estimation: Benzoic Acid Esterification on a Heterogeneous Catalyst, Ind. Eng. Chem. Res., 2019, 58(49), 22165–22177 CrossRef CAS.
- A. Buitrago Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules, Science, 2015, 347(6217), 49–53 CrossRef CAS.
- S. Lin, S. Dikler, W. D. Blincoe, R. D. Ferguson, R. P. Sheridan, Z. Peng, D. V. Conway, K. Zawatzky, H. Wang, T. Cernak, I. W. Davies, D. A. DiRocco, H. Sheng, C. J. Welch and S. D. Dreher, Mapping the Dark Space of Chemical Reactions with Extended Nanomole Synthesis and Maldi-Tof Ms, Science, 2018, 361(6402), eaar6236 CrossRef.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, A Platform for Automated Nanomole-Scale Reaction Screening and Micromole-Scale Synthesis in Flow, Science, 2018, 359(6374), 429 CrossRef CAS.
- C. Houben and A. A. Lapkin, Automatic Discovery and Optimization of Chemical Processes, Curr. Opin. Chem. Eng., 2015, 9, 1–7 CrossRef.
- S. Krishnadasan, R. J. C. Brown, A. J. deMello and J. C. deMello, Intelligent Routes to the Controlled Synthesis of Nanoparticles, Lab Chip, 2007, 7(11), 1434–1441 RSC.
- J. P. McMullen, M. T. Stone, S. L. Buchwald and K. F. Jensen, An Integrated Microreactor System for Self-Optimization of a Heck Reaction: From Micro- to Mesoscale Flow Systems, Angew. Chem., Int. Ed., 2010, 49(39), 7076–7080 CrossRef CAS.
- B. J. Reizman, Y.-M. Wang, S. L. Buchwald and K. F. Jensen, Suzuki–Miyaura Cross-Coupling Optimization Enabled by Automated Feedback, React. Chem. Eng., 2016, 1(6), 658–666 RSC.
- A. C. Atkinson and A. N. Donev, Optimum Experimental Designs, Clarendon Press, 1992 Search PubMed.
- J. P. McMullen and K. F. Jensen, Integrated Microreactors for Reaction Automation: New Approaches to Reaction Development, Annu. Rev. Anal. Chem., 2010, 3(1), 19–42 CrossRef CAS.
- A. J. Parrott, R. A. Bourne, G. R. Akien, D. J. Irvine and M. Poliakoff, Self-Optimizing Continuous Reactions in Supercritical Carbon Dioxide, Angew. Chem., Int. Ed., 2011, 50(16), 3788–3792 CrossRef CAS.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Machine Learning Meets Continuous Flow Chemistry: Automated Optimization Towards the Pareto Front of Multiple Objectives, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- Z. Zhou, X. Li and R. N. Zare, Optimizing Chemical Reactions with Deep Reinforcement Learning, ACS Cent. Sci., 2017, 3(12), 1337–1344 CrossRef CAS.
- C. Mateos, M. J. Nieves-Remacha and J. A. Rincón, Automated Platforms for Reaction Self-Optimization in Flow, React. Chem. Eng., 2019, 4(9), 1536–1544 RSC.
- D. N. Jumbam, R. A. Skilton, A. J. Parrott, R. A. Bourne and M. Poliakoff, The Effect of Self-Optimisation Targets on the Methylation of Alcohols Using Dimethyl Carbonate in Supercritical Co2, J. Flow Chem., 2012, 2(1), 24 CrossRef CAS.
- D. Cortés-Borda, K. V. Kutonova, C. Jamet, M. E. Trusova, F. Zammattio, C. Truchet, M. Rodriguez-Zubiri and F.-X. Felpin, Optimizing the Heck–Matsuda Reaction in Flow with a Constraint-Adapted Direct Search Algorithm, Org. Process Res. Dev., 2016, 20(11), 1979–1987 CrossRef.
- J. S. Moore and K. F. Jensen, Automated Multitrajectory Method for Reaction Optimization in a Microfluidic System Using Online Ir Analysis, Org. Process Res. Dev., 2012, 16(8), 1409–1415 CrossRef CAS.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, A Novel Internet-Based Reaction Monitoring, Control and Autonomous Self-Optimization Platform for Chemical Synthesis, Org. Process Res. Dev., 2016, 20(2), 386–394 CrossRef CAS.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Chimera: Enabling Hierarchy Based Multi-Objective Optimization for Self-Driving Laboratories, Chem. Sci., 2018, 9(39), 7642–7655 RSC.
- A. Pankajakshan, C. Waldron, M. Quaglio, A. Gavriilidis and F. Galvanin, A Multi-Objective Optimal Experimental Design Framework for Enhancing the Efficiency of Online Model Identification Platforms, Engineering, 2019, 5(6), 1049–1059 CrossRef CAS.
- M. O'Brien, A. Hall, J. Schrauwen and J. van der Made, An Open-Source Approach to Automation in Organic Synthesis: The Flow Chemical Formation of Benzamides Using an Inline Liquid-Liquid Extraction System and a Homemade 3-Axis Autosampling/Product-Collection Device, Tetrahedron, 2018, 74(25), 3152–3157 CrossRef.
- R. J. Ingham, C. Battilocchio, J. M. Hawkins and S. V. Ley, Integration of Enabling Methods for the Automated Flow Preparation of Piperazine-2-Carboxamide, Beilstein J. Org. Chem., 2014, 10, 641–652 CrossRef.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, An Autonomous Microreactor Platform for the Rapid Identification of Kinetic Models, React. Chem. Eng., 2019, 4(9), 1623–1636 RSC.
- R. A. Fisher, Design of Experiments, Br. Med. J., 1936, 1(3923), 554 CrossRef.
- G. Franceschini and S. Macchietto, Model-Based Design of Experiments for Parameter Precision: State of the Art, Chem. Eng. Sci., 2008, 63(19), 4846–4872 CrossRef CAS.
- J. P. McMullen and K. F. Jensen, Rapid Determination of Reaction Kinetics with an Automated Microfluidic System, Org. Process Res. Dev., 2011, 15(2), 398–407 CrossRef CAS.
- B. J. Reizman and K. F. Jensen, An Automated Continuous-Flow Platform for the Estimation of Multistep Reaction Kinetics, Org. Process Res. Dev., 2012, 16(11), 1770–1782 CrossRef CAS.
- A. Echtermeyer, Y. Amar, J. Zakrzewski and A. Lapkin, Self-Optimisation and Model-Based Design of Experiments for Developing a C–H Activation Flow Process, Beilstein J. Org. Chem., 2017, 13, 150–163 CrossRef CAS.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Automated Radial Synthesis of Organic Molecules, Nature, 2020, 579(7799), 379–384 CrossRef CAS.
- B. J. Reizman and K. F. Jensen, Feedback in Flow for Accelerated Reaction Development, Acc. Chem. Res., 2016, 49(9), 1786–1796 CrossRef CAS.
- B. A. Rizkin, A. S. Shkolnik, N. J. Ferraro and R. L. Hartman, Combining Automated Microfluidic Experimentation with Machine Learning for Efficient Polymerization Design, Nat. Mach. Intell., 2020, 2(4), 200–209 CrossRef.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Reconfigurable System for Automated Optimization of Diverse Chemical Reactions, Science, 2018, 361(6408), 1220 CrossRef.
- A. G. Godfrey, T. Masquelin and H. Hemmerle, A Remote-Controlled Adaptive Medchem Lab: An Innovative Approach to Enable Drug Discovery in the 21st Century, Drug Discovery Today, 2013, 18(17), 795–802 CrossRef CAS.
- T. Laino, Roborxn: Automating Chemical Synthesis, https://www.ibm.com/blogs/research/2020/08/roborxn-automating-chemical-synthesis/.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, On-Demand Continuous-Flow Production of Pharmaceuticals in a Compact, Reconfigurable System, Science, 2016, 352(6281), 61 CrossRef CAS.
- A. J. Mijalis, D. A. Thomas, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis, Nat. Chem. Biol., 2017, 13(5), 464–466 CrossRef CAS.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Synthesis of Proteins by Automated Flow Chemistry, Science, 2020, 368(6494), 980–987 CrossRef CAS.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Synthesis of Many Different Types of Organic Small Molecules Using One Automated Process, Science, 2015, 347(6227), 1221–1226 CrossRef CAS.
- J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Controlling an Organic Synthesis Robot with Machine Learning to Search for New Reactivity, Nature, 2018, 559(7714), 377–381 CrossRef CAS.
- B. Desai, K. Dixon, E. Farrant, Q. Feng, K. R. Gibson, W. P. van Hoorn, J. Mills, T. Morgan, D. M. Parry, M. K. Ramjee, C. N. Selway, G. J. Tarver, G. Whitlock and A. G. Wright, Rapid Discovery of a Novel Series of Abl Kinase Inhibitors by Application of an Integrated Microfluidic Synthesis and Screening Platform, J. Med. Chem., 2013, 56(7), 3033–3047 CrossRef CAS.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, A Mobile Robotic Chemist, Nature, 2020, 583(7815), 237–241 CrossRef CAS.
- J. Zhong, J. Riordon, T. C. Wu, H. Edwards, A. R. Wheeler, K. Pardee, A. Aspuru-Guzik and D. Sinton, When Robotics Met Fluidics, Lab Chip, 2020, 20(4), 709–716 RSC.
- N. Cherkasov, Y. Bai, A. J. Expósito and E. V. Rebrov, Openflowchem – a Platform for Quick, Robust and Flexible Automation and Self-Optimisation of Flow Chemistry, React. Chem. Eng., 2018, 3(5), 769–780 RSC.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Expanding the Medicinal Chemistry Synthetic Toolbox, Nat. Rev. Drug Discovery, 2018, 17, 709 CrossRef.
- G. M. Ghiandoni, M. J. Bodkin, B. Chen, D. Hristozov, J. E. A. Wallace, J. Webster and V. J. Gillet, Development and Application of a Data-Driven Reaction Classification Model: Comparison of an Electronic Lab Notebook and Medicinal Chemistry Literature, J. Chem. Inf. Model., 2019, 59(10), 4167–4187 CrossRef CAS.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules, Org. Biomol. Chem., 2006, 4(12), 2337–2347 RSC.
- Mit Mlpds, https://mlpds.mit.edu/.
- Ccas, https://ccas.nd.edu/.
- https://cen.acs.org/synthesis/Automation-people-Training-new-generation/97/i42 .
- Melloddy, https://www.melloddy.eu/.
- I. V. Tetko, O. Engkvist, U. Koch, J.-L. Reymond and H. Chen, Bigchem: Challenges and Opportunities for Big Data Analysis in Chemistry, Mol. Inf., 2016, 35(11–12), 615–621 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |