
Production of jet-fuel-range molecules from biomass-derived mixed acids†
Elnaz
Jamalzade
abd,
Koorosh
Kashkooli
ad,
Liam
Griffin
ad,
G. Peter
van Walsum
 ad and
Thomas J.
Schwartz
*acd
ad and
Thomas J.
Schwartz
*acd
aDepartment of Chemical & Biomedical Engineering, University of Maine, Orono, ME 04469, USA. E-mail: thomas.schwartz@maine.edu
bDepartment of Chemistry, University of Maine, Orono, ME 04469, USA
cFrontier Institute for Research in Sensor Technology, University of Maine, Orono, ME 04469, USA
dForest Bioproducts Research Institute, University of Maine, Orono, ME 04469, USA
First published on 21st January 2021
Abstract
Biomass has received considerable attention as a feedstock for the replacement of crude oil for producing both energy and high-value chemicals. In this work, we use a combination of chemical and biological processing to produce long-chain linear and branched ketones with low oxygen content. A mixture of medium-chain-length carboxylic acids was obtained by methane-inhibited, open-culture anaerobic fermentation of lignocellulosic biomass, and this mixture was further oligomerized using heterogeneous chemical catalysis. The products fall in the range of C10–C20 molecules that can potentially be blended with existing hydrocarbon jet fuels. We used a Pd/CeZrOx catalyst to achieve >90% yield to C11+ ketones starting from C2–C4 mixed acids. The acids are first recovered from the fermentation broth as ethyl esters by reactive distillation using Amberlyst-45 as a catalyst. We evaluated the activity of several bifunctional catalysts for upgrading these ethyl esters into long-chain ketones, finding that 0.25 wt% Pd/CeZrOx was most active. Using a combination of experimental reaction kinetics measurements and gas-phase thermodynamics calculations, we postulate a reaction network that explains the production of the most abundant products via a combination of direct ester ketonization, dehydration, and hydrogenation.
Introduction
Aviation is essentially a fossil fuel industry, one which uses about 8 million barrels of oil every day.1 The worldwide demand for jet fuel has been steadily increasing since 1980, with the consumption rate rising from 2.1m barrels per day in 1980 to 4.1m barrels per day in 2018.2 In 2016 the International Civil Aviation Organization (ICAO) reported that 2% of global CO2 emissions can be attributed to the aviation sector.3 The average annual air traffic rate is projected to further increase by 4.1% per year over the period from 2015–2025, which could make the aviation industry a significant fossil-based CO2 emitter by 2050.3 Unlike other transportation sectors, where there might be an alternative source of energy such as renewable electricity, there is currently no way to fly nearly 8 million people every day without using the chemical potential energy stored in jet fuel molecules. Electric airplanes would require the use of heavy batteries, leading to substantial modifications to airport infrastructure and updating of existing designs or, worse, requiring the development of entirely new airframes. So, it is crucial to find another carbon-neutral energy source that works with the current engine technology and fuel supply infrastructure. In this regard, biomass-based jet fuel is an attractive option because it provides a feasible and relatively short-term solution to decrease carbon emissions.4,5There are several pathways that upgrade biomass feedstocks to jet fuel additives, many of which are available at commercial or pre-commercial scales, including the alcohol-to-jet, oil-to-jet, gas-to-jet, and sugar-to-jet processes.6–10 The fuel closest to commercialization is hydroprocessed esters and fatty acids (HEFA).11–13 However, the use of vegetable oils to produce HEFA fuels has led to increases in food costs in some parts of the world, and HEFA fuels are difficult to produce sustainably with high yields on a per acre basis.14 In contrast, the sugar-to-jet pathway in particular is a flexible chemical upgrading strategy that has attracted significant attention in the last decade due to the global abundance of cellulosic biomass and the availability of several technologies to convert it to water-soluble sugars.6,15,16 Transportation fuels must meet rigorous standards to smoothly drive engines while also maintaining engine life and suppressing harmful exhaust gas. The main components in standard of jet fuel should be C10–C12 branched or cyclic alkanes and C13–C16 multiply branched alkanes.17
In this work we apply a three-step process to convert lignocellulosic biomass to long-chain, low-oxygen species containing 7–19 carbons, which are in the correct carbon-chain range to be used as jet fuel blendstocks. The first step uses open (i.e., mixed) culture fermentation (OCF) for digestion of lignocellulosic biomass to produce fatty acids with chains between 4 and 8 carbons long. OCF has many advantages, including the ability to digest a variety of different organic materials including carbohydrates, proteins, and fats using enzymes made during fermentation, which adds competitive flexibility of the types of biomass feedstocks that can be upgraded.10,18 In addition, OCF is a single-pot method, allowing for a multistep fermentation to occur in a single vessel without needing to sterilize the system, which is a significant advantage over monoculture fermentations, including ethanol production and the acetone–butanol–ethanol (ABE) process. In the second step of our process, the carboxylic-acid-rich fermentation broth undergoes acid-catalyzed esterification, where reactive distillation is used to recover and volatilize the acids while avoiding equilibrium limitations.
Because these esters are relatively short by jet-fuel standards (e.g., they contain 8 or fewer carbons), the mixture of esterification products is subsequently subjected to catalytic C–C coupling to produce the long-chain species required for jet fuel applications. These reactions, shown in Scheme 1, include (a) direct ester ketonization, in which two esters react to form a linear ketone, with CO2 and H2O released as byproducts, (b) Claisen Dieckmann condensation/hydrogenation, in which two esters react to form a heavier, branched ketone, (c) the retro-Tischenko reaction followed by aldol condensation to produce ethanol and short-chain ketones, (d) ester hydrolysis followed by acid condensation/decarbonylation to produce a linear ketone, and (e) sequential aldol condensation/Michael addition of the linear ketones produced by (a–d) to form branched diones. The combination of reactions (a–e) yields a mixture of C7–C19 linear and branched ketones and alkanes.10 Such materials can be readily upgraded to hydrocarbon fuel blendstocks using mild hydrodeoxygenation (HDO).19
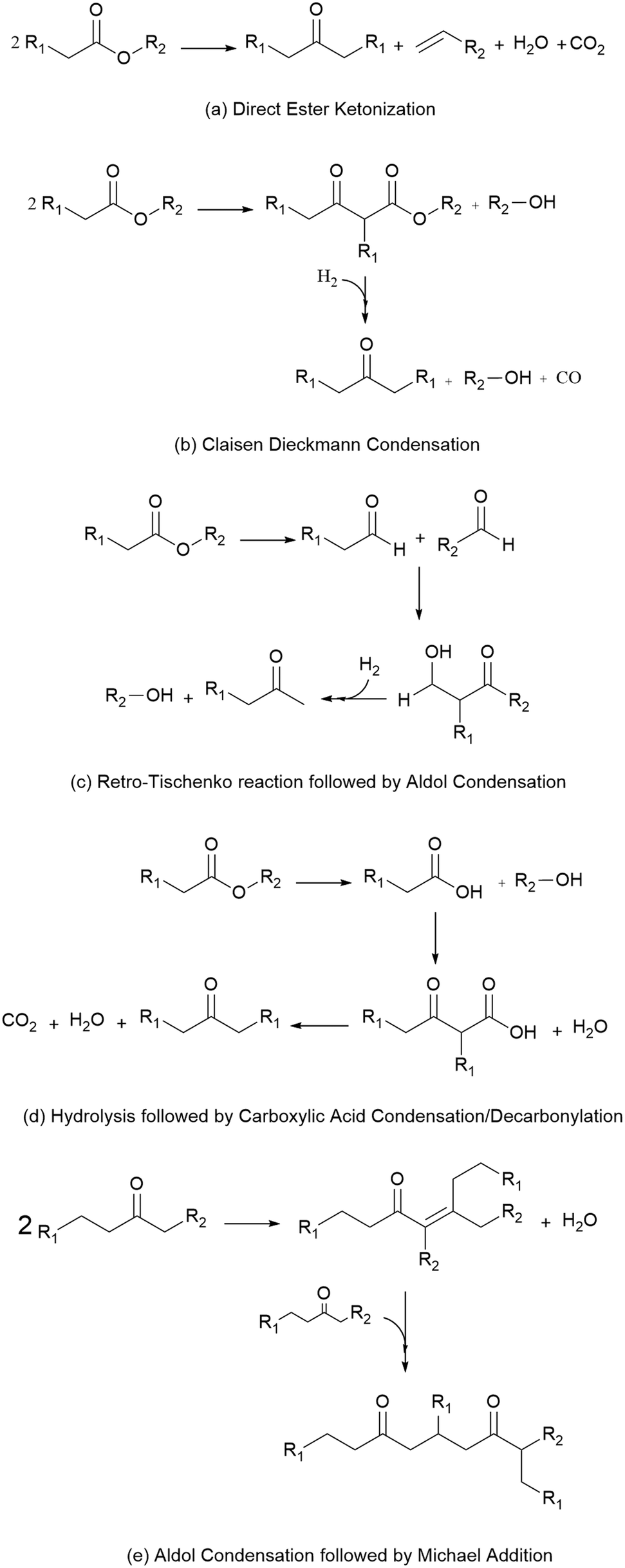 | ||
| Scheme 1 Catalytic reactions carried out in this work. | ||
The reactions shown in Scheme 1 require a catalyst containing a combination of acidic sites (reactions b, d and e), basic sites (reactions a–c and e), and reduced metal sites (reactions b and c). As described in the work of Gaertner et al.,20 CeZrOx contains a combination of acidic and basic functionalities that, when used as a catalyst support for a metal such as Pd, can facilitate ketonization and condensation reactions, with the Pd sites saturating C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and some CO bonds. Consequently, in this work we evaluate whether C–C coupling of short-chain organic esters can be carried out in a single reactor system containing a Pd/CeZrOx catalyst. We show that whole biomass can be converted to compounds suitable for blending with jet fuel (following HDO) using a combination of biocatalytic processes for biomass deconstruction and defunctionalization (i.e., OCF) and chemical catalytic processes for reactive separation, carbon chain elongation, and deoxygenation.
C bonds and some CO bonds. Consequently, in this work we evaluate whether C–C coupling of short-chain organic esters can be carried out in a single reactor system containing a Pd/CeZrOx catalyst. We show that whole biomass can be converted to compounds suitable for blending with jet fuel (following HDO) using a combination of biocatalytic processes for biomass deconstruction and defunctionalization (i.e., OCF) and chemical catalytic processes for reactive separation, carbon chain elongation, and deoxygenation.
Experimental
Catalyst preparation
CeZrOx with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ce:Zr molar ratio was prepared by co-precipitation of nitrate precursors following the procedure described by Kunkes et al.21 Briefly, aqueous solutions of Ce(NO3)3·6H2O (Aldrich, 99.9%) and ZrO(NO3)2 (Alfa Aesar, 99.9%) were mixed, after which concentrated (28%) aqueous NH4OH was added dropwise to raise the pH to 10, at which point a mixed carbonate material precipitated from solution. This mixed carbonate was aged at pH 10 for 3 days, after which the solids were separated by filtration, washed with ultra-pure water and ethanol, dried overnight at 373 K, and calcined in air at 623 K for 2 h to form the CeZrOx mixed oxide. The Pd/CeZrOx catalyst was prepared via incipient wetness impregnation (IWI) of CeZrOx with an aqueous solution of Pd(NO3)2 (Aldrich, 99.99%). The impregnated solid was dried overnight at 373 K and calcined in air at 623 K for 4 h. The catalyst was packed into a flow reactor and reduced at 623 K for 3 h in 50 sccm H2 gas immediately prior to reaction. Synthetic hydrotalcite (Aldrich) was calcined at 773 K for 4 h to obtain a MgO–Al2O3 mixed oxide. The Cu/MgO–Al2O3 and Pd/MgO–Al2O3 catalysts were prepared via IWI of MgO–Al2O3 with an aqueous solution of Cu(NO3)2 (Aldrich, 99.99%) or Pd(NO3)2 (Aldrich, 99.99%), respectively. The impregnated solid was dried overnight at 373 K and calcined in air at 773 K for 4 h.22 Prior to use, Amberlyst-45™ (Dow) was washed with 1 L deionized water (18 MΩ), dried, crushed, and sieved to less than 150 microns.
:1 Ce:Zr molar ratio was prepared by co-precipitation of nitrate precursors following the procedure described by Kunkes et al.21 Briefly, aqueous solutions of Ce(NO3)3·6H2O (Aldrich, 99.9%) and ZrO(NO3)2 (Alfa Aesar, 99.9%) were mixed, after which concentrated (28%) aqueous NH4OH was added dropwise to raise the pH to 10, at which point a mixed carbonate material precipitated from solution. This mixed carbonate was aged at pH 10 for 3 days, after which the solids were separated by filtration, washed with ultra-pure water and ethanol, dried overnight at 373 K, and calcined in air at 623 K for 2 h to form the CeZrOx mixed oxide. The Pd/CeZrOx catalyst was prepared via incipient wetness impregnation (IWI) of CeZrOx with an aqueous solution of Pd(NO3)2 (Aldrich, 99.99%). The impregnated solid was dried overnight at 373 K and calcined in air at 623 K for 4 h. The catalyst was packed into a flow reactor and reduced at 623 K for 3 h in 50 sccm H2 gas immediately prior to reaction. Synthetic hydrotalcite (Aldrich) was calcined at 773 K for 4 h to obtain a MgO–Al2O3 mixed oxide. The Cu/MgO–Al2O3 and Pd/MgO–Al2O3 catalysts were prepared via IWI of MgO–Al2O3 with an aqueous solution of Cu(NO3)2 (Aldrich, 99.99%) or Pd(NO3)2 (Aldrich, 99.99%), respectively. The impregnated solid was dried overnight at 373 K and calcined in air at 773 K for 4 h.22 Prior to use, Amberlyst-45™ (Dow) was washed with 1 L deionized water (18 MΩ), dried, crushed, and sieved to less than 150 microns.
Catalyst characterization
The structure of the CeZrOx supports was verified by X-ray diffraction (XRD). A PANalytical X'PertPro diffractometer with a monochromated CuKα X-ray source was used for the diffraction studies. Diffractograms were collected over a 2θ range from 15 to 80 deg, with 0.02 deg intervals and a dwell time of 12 s.The surface area, total pore volume, and pore size distribution of each catalyst was determined by nitrogen porosimetry measurements that were made on a Micromeritics ASAP 2020 instrument. The samples were degassed under vacuum at 473 K for 8 h to ensure the samples were thoroughly dried and free of adsorbed impurities.23 Isotherms were measured at 77 K from P/P0 < 2 × 10−5 to 0.995, spanning the micro- and meso-pore range. The surface areas were calculated according to the Brunauer–Emmett–Teller (BET) equation24 in the relative pressure range of 0.05–0.25 (P/P0) and the total pore volume was calculated using the Barrett–Joyner–Halenda (BJH) equation25 corrected as described by Kruk, Jaroniec, and Sayari (KJS)26 using the adsorption branch of the N2 isotherm.
Fermentation
Open culture fermentation (OCF) was carried out on lime-pretreated yellow birch wood cultured in a roller bottle apparatus held at 310 K, as described elsewhere.27 Briefly, the pretreatment conditions for the yellow birch wood chips consisted of cooking at 10% dry solids/liquor at 423 K for 6 h with a 10% lime-to-wood ratio followed by solids particle-size-reduction achieved by grinding. The resulting solid material was supplemented with 4% chicken manure and 2% corn steep liquor as nutrient supplements, and inoculated with 4% marine organic sediment, compost, and prior fermentation broth (all wet basis), and fermented anaerobically for up to 30 days. Ethanol and/or lactic acid were added to the fermentation periodically to enhance chain elongation of the carboxylic acids and enhance the titer of medium chain-length fatty acids (MCFAs), which consisted primarily of hexanoic acid.For the purpose of material balances, we define the digestible compounds (DC) as all the digestible carbohydrates and organic acids originating from the pretreated wood and nutrient supplements. DC does not include any supplemented ethanol and/or lactic acid added to the fermentation, nor does DC include lignin, since lignin is generally not digestible in anaerobic conditions. Digestible solids (DS) refers to the fraction of DC that is derived from the solid carbohydrates (cellulose and hemicellulose) in the feed. The material balances are done on a carbon basis, with conversions (Xi) and the MCFA selectivity (SMCFA) defined as:
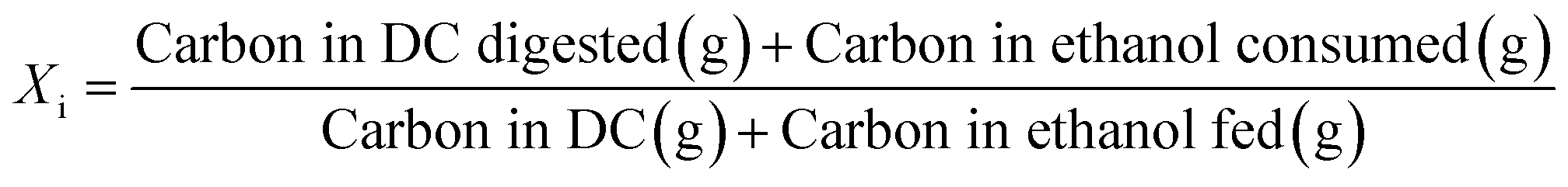 | (1) |
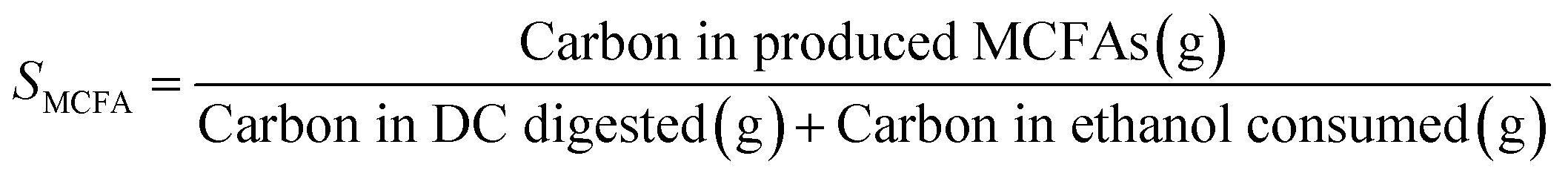 | (2) |
Reactive distillation
The C–C coupling reactions studied here occur in the vapor phase, which necessitates recovery of the acids produced by OCF. Reactive distillation to form ethyl esters was used to accomplish both separation and volatilization, performed using a Dean–Stark apparatus (Chemglass). Hexanoic acid was chosen as a model compound to represent the mixture of acids produced by OCF. In a typical experiment, 20 mL of 30 g L−1 hexanoic acid (Acros Organics, 99%) in 6-undecanone solvent (TCI America, 98%), was added to a 100 mL three-necked reactor flask containing a magnetic stir bar and 0.05 g of Amberlyst 45™. The temperature of the reaction mixture was first ramped to 333 K at a rate of 33 K min−1, then to 428 K at a rate of 3 K min−1. The reaction mixture was stirred at 300 rpm, which was continued while a 2x molar excess of ethanol (Acros Organics, Absolute, 99.5%) was added dropwise to the reactor from an addition funnel. The reaction mixture was then held at 428 K for three hours, at which point condensation into the Dean–Stark receiver slowed significantly and the heating mantle was turned off. Once cooled, both the reaction mixture and organic product were decanted into collection bottles for analysis by gas chromatography.Ester oligomerization reactions
Ethyl hexanoate (Sigma-Aldrich, >99%), hydrogen (Matheson, 99.999%), and helium (Matheson, 99.999%) were all used as-purchased. The conversion of ethyl hexanoate over CeZrOx, MgO–Al2O3, Pd/CeZrOx, Pd/MgO–Al2O3, and Cu/MgO–Al2O3, was carried out at in a fixed bed, U-shaped reactor consisting of a quarter-inch stainless steel tube, shown schematically in Fig. 1. The catalyst was packed between two plugs of quartz wool. The reactor was heated by a well-insulated furnace. K-type thermocouples (Omega) were used to measure the reactor temperature, which was controlled by a PID controller (Automation Direct, Solo-4848) connected to a variable transformer (Staco, 3PN series). Mass flow controllers (MKS, Type 247) were used to regulate the flow of H2 and He during the experiments. The liquid feed was pumped by a syringe pump (New Era Pump Systems, NE-1000) to a length of 1/8 inch tubing used as an evaporator, which in turn fed the reactor. A back-pressure regulator (Swagelok) was used to control the total pressure, which was measured by two gauges at the inlet and outlet of the reactor. A gas–liquid separator was submerged in an ice bath and used to collect the liquid products for analysis. The catalyst was reduced in situ at 623 K (ramp rate of 274 K min−1) for 2 h in flowing H2 (22 sccm). After the reduction was completed, the temperature and pressure were adjusted and the flow of H2 was started. The weight hourly space velocity (WHSV) was calculated as the mass flow rate of ethyl hexanoate feed normalized by the mass of catalyst.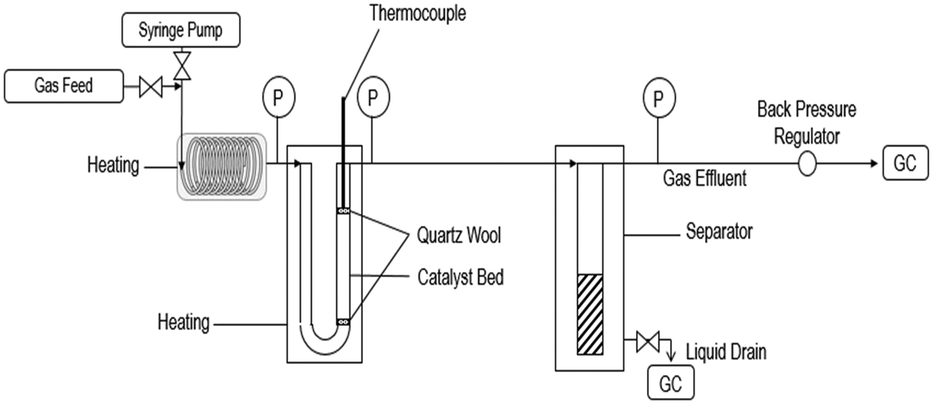 | ||
| Fig. 1 Schematic of the flow reactor system used for these studies. | ||
Analytical methods
Analysis of the fermentation products was performed using a Shimadzu high performance liquid chromatograph (HPLC) equipped with a refractive index detector (RID). Separation was achieved at 333 K using an Aminex HPX-87H column (Biorad) with an aqueous mobile phase (0.6 mL min−1) buffered with 0.005 M H2SO4. Concentrations were determined by comparison with external standards. Carbon analysis of the inputs and outputs of the fermentation was done using a CHNO analyser (PerkinElmer).Analysis of the liquid products of the catalytic reactions was performed with an Agilent gas chromatograph (model 7820A), equipped with an HP-5 column (30 m × 0.32 mm × 0.25 μm) and a flame ionization detector (FID). Unknown species were identified with an Agilent 7820A GC coupled to an Agilent 5975 mass selective detector (MSD). Mass spectra of unknown species were compared against standard spectra from NIST, confirmed based on the atomic mass of the molecular ion fragment and estimated retention indices based on an alkane scale. Liquid samples were collected every 24 hours. The GC program (used for both the GC/FID and GC/MSD) was as follows: helium was used as a carrier gas with a flow rate of 25 mL min−1, and the inlet temperature set point was 483 K; the oven temperature was held at 373 K for 2 min then increased to 383 K at a heating rate of 275 K min−1, held at 383 K for 3 min, increased to 393 K at a heating rate of 278 K min−1, held at 393 K for 2 min, increased to 523 K at a heating rate of 280 K min−1 and held at 523 K for 3 min; the detector temperature was set to 523 K. Quantification of each compound was determined based on a standard curve using n-butanol as an internal standard.
Fractional conversions were calculated according to eqn (3), where X represents the conversion of species i, Ff,i represents the feed flowrate of species i, and Fi represents the molar flow rate of species i. The carbon selectivities toward reaction products, i, were calculated according to eqn (4), where S represents the selectivity and Fi represents the molar flow rate of species i, and nC,i represents the number of carbon atoms in species i. Mass balances typically closed to greater than 90 wt%, and carbon balances typically closed to greater than 70 mol%. Cases where the carbon balance did not fully close were due to the formation of low-concentration, minority products that could not be readily quantified. Initial selectivities were obtained by extrapolation to zero-time assuming first-order deactivation. Steady-state selectivities were obtained by averaging the first several points obtained after the reaction was observed to reach steady state, typically after approx. 48 h.
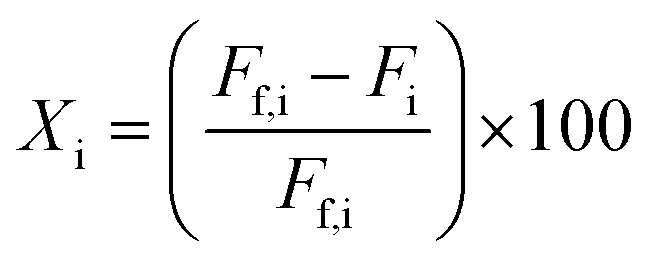 | (3) |
 | (4) |
Results and discussion
Fermentation
The conversion of lignocellulosic biomass to MCFAs is shown in Fig. 2. The open culture fermentation converted lime-pretreated yellow birch wood chips supplemented with chicken manure to a mixture of carboxylic acids. Fermentations were carried out in batch culture at 310 K. Intermediate metabolites included ethanol and lactic acid, which were subsequently consumed by chain elongation that yielded primarily butyric and hexanoic acids. The highest yields of MCFAs were achieved when the fermentations were supplemented with ethanol and/or lactic acid. When supplemental ethanol was fed in two increments of 10 g L−1 to the fermenter, production of 15 g L−1 hexanoic acid contributed to a total MCFA titer greater than 17 g L−1, achieved with 35% feedstock conversion and 41% MCFA selectivity. These fermentations also generated small amounts octanoic acid, but these were not accurately quantified by the aqueous-phase HPLC. The maximum concentration of aliphatic acids produced with lactic acid supplementation included more than 40 g L−1 butyric acid, up to 12 g L−1 propionic acid, but less than 4 g L−1 of hexanoic acid. Combining ethanol and lactic acid supplementation led to an elevated combination of hexanoic and butyric acids, at 14 and 7 g L−1 respectively. The fermentation conditions applied to generate organic acids for the esterification experiment included two ethanol supplements of 10 g L−1 each. To capture all the MCFAs prior to upgrading, the fermentation broth was extracted with 6-undecanone (the major primary product of C–C coupling, vide infra).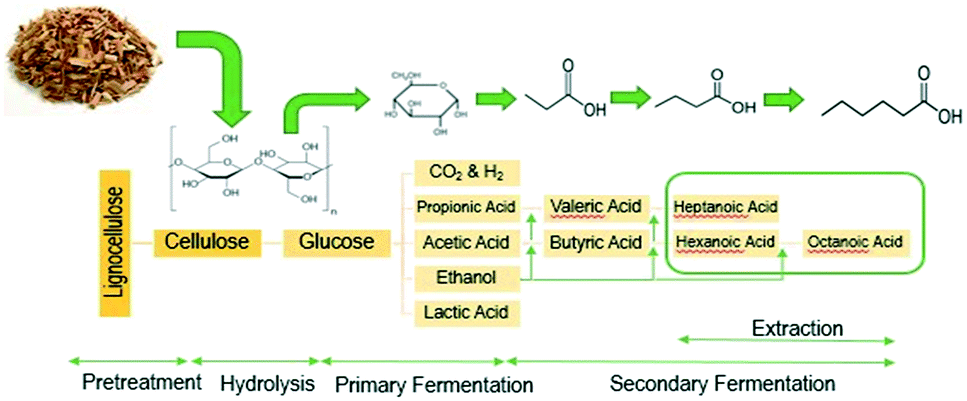 | ||
| Fig. 2 Schematic of the pretreatment and two-stage open culture fermentation process. | ||
Material balances on the fermentation were performed on a carbon basis. Table 1 presents the carbon balance for a fermentation supplemented with ethanol doses adding up to 20 g L−1.
| Carbon source or product | Carbon (g) | |||
|---|---|---|---|---|
| Batch 3 | Batch 4 | Batch 5 | Batch 6 | |
| Batches 3 and 5 added ethanol in three doses: 10 + 5 + 5 g L−1. Batches 4 and 6 added ethanol in 2 doses: 10 + 10 g L−1. | ||||
| Fermentation inputs | ||||
| Pretreated wood | 19.85 | 19.85 | 19.85 | 19.85 |
| Pretreatment liquor | 1.6 | 1.6 | 1.6 | 1.6 |
| Marine sediment and CSL | 0.02 | 0.02 | 0.02 | 0.02 |
| Manure | 2.75 | 2.75 | 2.75 | 2.75 |
| Fermentation inoculum | 0.29 | 0.29 | 0.29 | 0.29 |
| Ethanol | 5.15 | 4.17 | 5.15 | 4.17 |
| Total C into fermentation | 29.65 | 28.72 | 29.66 | 28.68 |
| Fermentation outputs | ||||
| Biotic carbon dioxide (gas) | 0.51 | 0.46 | 0.78 | 0.86 |
| Residual solids | 22.01 | 22.3 | 21.79 | 20.28 |
| Residual liquids | 6.88 | 6.06 | 7.18 | 5.95 |
| Liquid samples removed | 0.51 | 0.49 | 0.51 | 0.5 |
| Total C out of fermentation | 29.91 | 29.3 | 30.25 | 27.59 |
| Difference | 0.26 | 0.58 | 0.59 | −1.09 |
| % error | 0.88 | 2.02 | 1.99 | −3.8 |
Esterification
To identify reactive distillation conditions that lead to high yields of ethyl hexanoate from ethanol and hexanoic acid, we evaluated the effect on the ethyl hexanoate yield of the molar ratio of hexanoic acid-to-ethanol, the reactor space–time (τ), and the reaction temperature. The effect of space–time was studied by varying the catalyst loading, on a per-gram-of-feed basis, from 0.037 to 0.201 g:g of hexanoic acid, as shown in Fig. 3. As expected, increasing the space–time results in increased fractional conversion of hexanoic acid, reaching complete conversion above τ = 0.3 h. The selectivity to ethyl hexanoate was 100% in all cases. Notably, when carrying out esterification of MCFAs extracted from the spent fermentation broth, some amount of catalyst inhibition was observed, indicated by the triangle (▲) in Fig. 3. While Fig. 3 reports only the conversion of hexanoic acid, the other MCFAs present in the fermentation broth were also converted to esters with 100% selectivity. The fractional conversions were as follows: XC4 = 8%, and XC6 = 58%. Catalyst inhibition by biogenic impurities is known to be an important problem in the catalytic conversion of biologically derived molecules.28 For solid acid catalysts as used here, residual proteins from the fermentation can lead to deactivation by partial plugging of catalyst pores.29 Similarly, there may also be inhibition of the acidic sites of the esterification catalyst due to amino acids that are co-extracted with the MCFAs.30 Accordingly, we suggest that the likely reason for the lower fractional conversion of hexanoic acid here is the presence of contaminants, likely amino acids and denatured proteins, in the feed that lead to catalyst deactivation, a problem which has been previously addressed by the inclusion of polymer-derived microenvironments surrounding the active sites.31
 | ||
| Fig. 3 Influence of reactor spacetime on fractional conversion of hexanoic acid. Spacetime was defined based on the mass of hexanoic acid in the reactor, and selectivity to ethyl hexanoate was 100% in all cases. Reaction conditions: Amberlyst 45™, 428 K, 300 rpm, 3 h, ethanol:hexanoic acid = 2:1 g:g. Hexanoic acid feed (●), fermentation broth feed (▲). | ||
Ester oligomerization reactions
Ethyl hexanoate (1 in Table 2) was used as a representative esterification product and was upgraded at 623 K over a Pd/CeZrOx catalyst in the presence of H2. A wide range of liquid products was observed, and these are listed along with their selectivities at complete conversion in Table 3. Analysis of the gas phase revealed the presence of small amounts of ethanol, ethylene, and acetaldehyde but not of any significant other gas-phase organics. The behavior of the major products as a function of time-on-stream is shown in Fig. 4. Continuous reaction was performed for 30 days, with the catalyst deactivating over the first several days before stabilizing. Notably, the selectivity to 2 gradually increased during the reaction, ultimately reaching 55% after 20 days. The selectivity toward 6 decreased from 7% to 3% over the first 15 days of time-on-stream, after which it stabilized. The other major products, 17 and 3, maintained nearly constant selectivities of 8% and 13%, respectively over the entire 30 day experiment. The remaining products are lumped by carbon-number range, and the distributions are shown in Fig. 5. The selectivity to deoxygenation products (i.e., alkanes) decreases while the selectivity to C–C coupling products (i.e., ketones, alcohols, etc.) increases over the same time range, which suggests that the Pd sites undergo deactivation during the first several days of time-on-stream, while the C–C bond-forming reactions occur mostly over sites on the CeZrOx support, which remains active.| Compound | IUPAC name | #Ca |
|---|---|---|
| a Number of carbon atoms. | ||
| 1 | Ethyl hexanoate | 8 |
| 2 | 6-Undecanone | 11 |
| 3 | 6-Undecanol | 11 |
| 4 | 5-Undecene | 11 |
| 5 | 2-Undecene | 11 |
| 6 | Undecane | 11 |
| 7 | Ethylene | 2 |
| 8 | 1-Butene | 4 |
| 9 | 1-Hexene | 6 |
| 10 | 5-Ethyl-6-undecanone | 13 |
| 11 | Ethyl-2-butyl-3-oxooctanoate | 14 |
| 12 | Hexanoic acid | 6 |
| 13 | 2-Butyl-3-oxooctanic acid | 12 |
| 14 | Hexanal | 6 |
| 15 | Acetaldehyde | 2 |
| 16 | 3-(Hydroxymethyl)-2-heptanone | 8 |
| 17 | 2-Heptanone | 7 |
| 18 | 6-Tridecanone | 13 |
| 19 | 6-Dodecanone | 12 |
| 20 | 4-Nonanone | 9 |
| 21 | 8-Methyl-7-tridecene-6-one | 14 |
| 22 | 8-Methyl-6-tridecanone | 14 |
| 23 | 12-Methyl-8-propyl-610-heptadecadione | 21 |
| 24 | 4-Methyl-2-nonanone | 10 |
| 25 | 2-Nonanone | 9 |
| 26 | 8-pentadecanone | 15 |
| 27 | Hexadecane | 16 |
| 28 | 4-Decanone | 10 |
| Cmpd. | Product | #C | Initial Sel. (%) | Steady-state Sel. (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.25 wt% Pd/CeZrOx, 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, conversion 99%, and WHSV = 0.046 h−1. | ||||
| 17 | 2-Heptanone | 7 | 8 | 8 |
| — | 4-Methyl 3-heptanone | 8 | 1 | <1 |
| 20 | 4-Nonanone | 9 | 3 | 1 |
| 25 | 2-Nonanone | 9 | 3 | 2 |
| 6 | Undecane | 11 | 7 | 3 |
| — | 3-Ethyl 2-nonanone | 11 | 1 | 1 |
| 2 | 6-Undecanone | 11 | 47 | 55 |
| 3 | 6-Undecanol | 11 | 12 | 13 |
| — | Tridecane | 13 | <1 | <1 |
| — | 2-Methyl 1-dodecanol | 13 | 7 | 7 |
| 10 | 5-Ethyl 6-undecanone | 13 | 1 | 1 |
| 18 | 6-Tridecanone | 13 | 1 | <1 |
| — | 6-Methyl tridecane | 14 | 1 | <1 |
| — | 6-Methyl pentadecane | 16 | 2 | 2 |
| — | 6-Propyl tridecane | 16 | 3 | 3 |
| — | Nonadecane | 19 | 1 | 1 |
| — | 5-Methyl octadecane | 19 | 1 | <1 |
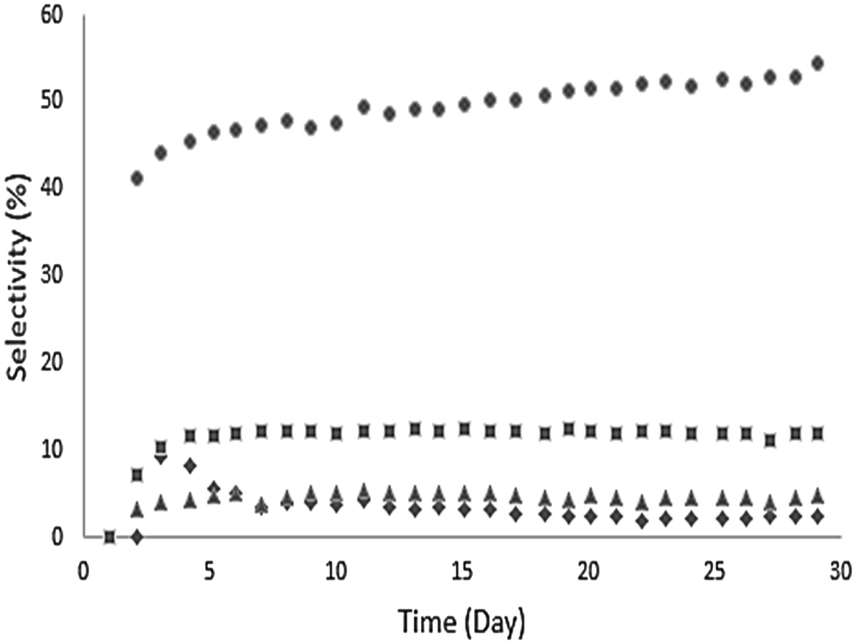 | ||
| Fig. 4 Time course of selectivity to the major products of the conversion of ethyl hexanoate over 0.25 wt% Pd/CeZrOx at 623 K, 134.9 kPas H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. Ethyl hexanoate conversion >99%. 6-Undecanone (●), 6-undecanol (■), 2-heptanone (▲), undecane (◆). | ||
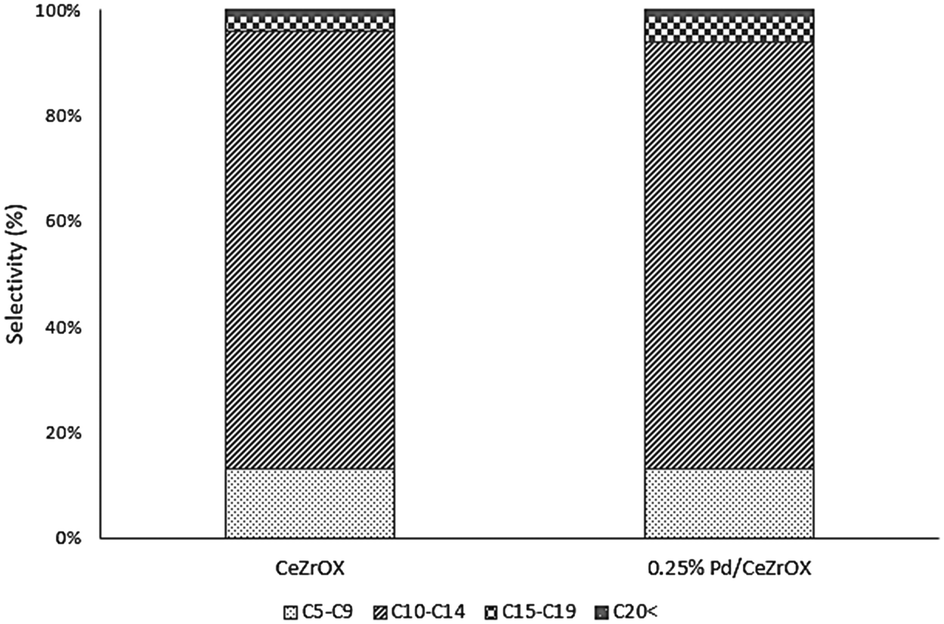 | ||
| Fig. 5 Comparison of steady-state product carbon number distributions from the conversion of ethyl hexanoate over CeZrOx and 0.25 wt% Pd/CeZrOx at 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. | ||
Given that much of the reactivity described above appears to be catalyzed only by the CeZrOx support, we obtained results at the same reaction conditions (reaction temperature of 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1) for bulk CeZrOx without any Pd nanoparticles, the products of which are presented in Table 4. We observed 60% selectivity to 2 at steady state. The other major products were 6 and 17, although the selectivity of 6 decreased during the course of 6 days of time-on-stream, from 18% to 8%. The selectivity to 17 was constant at ca. 11%. The carbon-number distributions for all products are shown in Fig. 5. Notably, the product distributions obtained both with and without Pd nanoparticles after 5 days of time-on-stream are nearly identical, confirming our hypothesis that deactivation of the Pd/CeZrOx catalyst is primarily due to loss of Pd activity.
| Cmpd. | Product | #C | Initial Sel. (%) | S.S. Sel. (%) |
|---|---|---|---|---|
| a Reaction conditions: CeZrOx at 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. | ||||
| 17 | 2-Heptanone | 7 | 10 | 11 |
| 20 | 4-Nonanone | 9 | 1 | 2 |
| — | Decane | 10 | 1 | <1 |
| 6 | Undecane | 11 | 18 | 8 |
| — | 3-Undecene | 11 | 1 | <1 |
| 4 | 5-Undecene | 11 | 4 | 3 |
| 2 | 6-Undecanone | 11 | 56 | 60 |
| 3 | 6-Undecanol | 11 | 1 | 2 |
| — | 3-Dodecene | 12 | <1 | 1 |
| 19 | 6-Dodecanone | 12 | 2 | 1 |
| 18 | 6-Tridecanone | 13 | 1 | 1 |
| — | Tridecanal | 13 | <1 | <1 |
| — | 1-Dodecanol-2-methyl | 13 | <1 | 2 |
| — | 6-Tetradecanone | 14 | 3 | 4 |
| — | 6-pentadecanone | 15 | 5 | 1 |
| 15 | 8-pentadecanone | 15 | <1 | <1 |
| — | 1-Hexadecene | 16 | <1 | <1 |
| — | Tetradecanoic acid, 2-oxo-, ethyl ester | 16 | <1 | <1 |
| — | 2-Methyl hexadecane | 17 | <1 | <1 |
| — | 7-Heptadecanone | 17 | <1 | 1 |
| — | 1-Heptadecen-7,10-dione | 17 | <1 | <1 |
| — | 2(3H)-Furanone, dihydro-5-tetradecyl | 18 | <1 | <1 |
| — | Cyclopentane, 2-hexyloctyl | 19 | <1 | <1 |
| — | 10-Nonadecanone | 19 | <1 | <1 |
| — | Androstene-3á,17á-diol | 19 | <1 | <1 |
| — | Androstan-17-one, 3-hydroxy | 19 | <1 | <1 |
| — | Heptadecane, 2,6,10,15-tetramethyl | 21 | 2 | 1 |
| — | 5-Methyl-6-heneicosen-11-one | 22 | <1 | <1 |
If carbon deposition is the primary mode responsible for deactivation of the Pd sites, then decreasing the temperature (in the presence of H2) may lead to an improvement in activity by favoring hydrogenation reactions vs. dehydrogenation. As shown in Fig. 6, the conversion unsurprisingly decreases at lower temperatures, as does the selectivity to long-chain products, possibly due to high activation barriers for C–C bond forming reactions, although we note this effect could also be due to a change in selectivity caused by decreasing the extent of reaction (i.e., decreasing the conversion leads to fewer secondary and tertiary products in the series reactions). That at least some of the decrease in selectivity at low temperature is due to a decrease in the extent of reaction is supported by an observation of increased selectivity to C15–C19 species when 6-undecanone is fed to this catalyst at 623 K, as shown in Fig. 7, and we speculate that the decrease shown in Fig. 6 likely originates from an amalgamation of both effects. Importantly, though, the origin of the decrease in selectivity is ultimately not significant here because the lower reaction temperatures do not substantially influence the deactivation behavior of the catalyst, suggesting that desired operation is at 623 K.
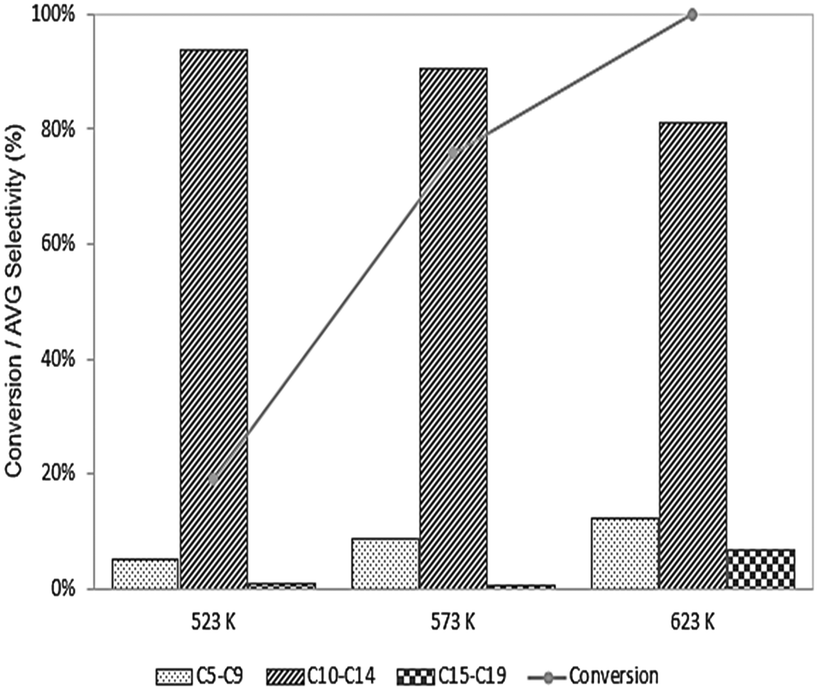 | ||
| Fig. 6 The effect of the reaction temperature on the products selectivity and conversion of ethyl hexanoate over 0.25 wt% Pd/CeZrOx, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. | ||
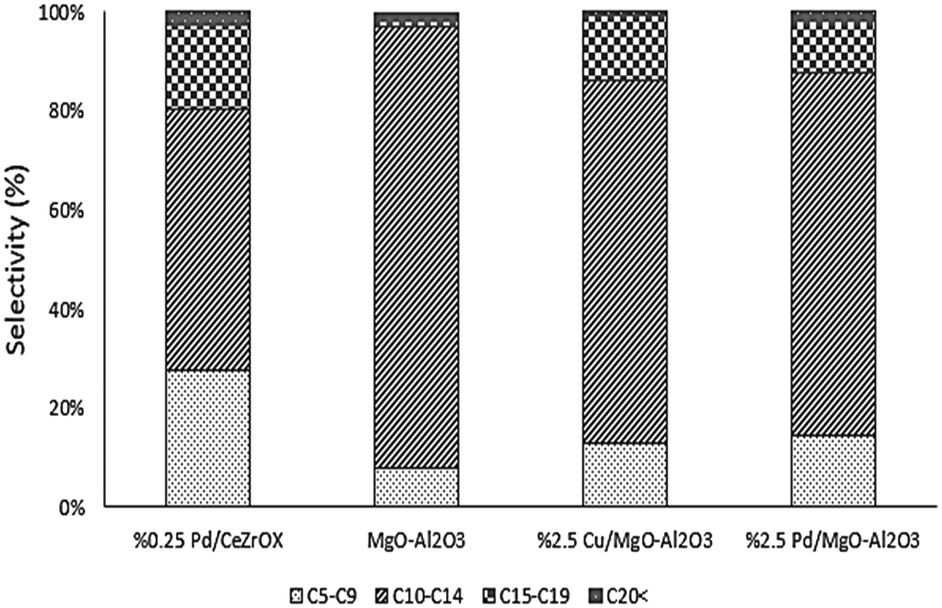 | ||
| Fig. 7 Comparison of steady-state product carbon number distributions based on their carbon number from conversion of 6-undecanone over 0.25 wt% Pd/CeZrOx, MgO–Al2O3, Cu/MgO–Al2O3 and Pd/MgO–Al2O3 at 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. | ||
In an effort to increase the carbon numbers of the products by favoring sequential reactions, we evaluated subsequent reaction of 6-undecannone (2), which is the major product of ethyl hexanoate upgrading. To favor aldol-type condensation reactions, MgO–Al2O3 was selected as a catalyst because it is reported to possess not only strongly basic sites needed for C–C bond formation32,33 but also acid–base site pairs with appropriate strength to achieve high turnover frequencies for aldol condensation (due to insertion of Al into the MgO framework).34 Temperature-programmed desorption of CO2 confirms the presence of strong base sites on this catalyst (see Fig. S3†). Using the same reaction conditions as in our previous experiments (i.e., 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1), we again achieved complete conversion, in this case of 2. The main product was 19, with 58% selectivity at steady state. The other major products were 4 and 17, with 26% and 10% steady-state selectivity respectively. The selectivity distribution based on the number of carbons is shown in Fig. 7.
Following the approach of Goulas et al., who observed increased aldol condensation activity for Cu and Pd supported on MgO–Al2O3,35 we also evaluated Cu/MgO–Al2O3 and Pd/MgO–Al2O3 catalysts in an attempt to produce saturated, long-chain products. Cu and Pd were selected because of their high selectivity for reduction of CO and CC bonds, respectively. The reaction conditions were again kept the same, and the reactor was fed with 2, with the product distributions again shown in Fig. 7. Cu/MgO–Al2O3 achieved >99% conversion of 2 and favored production of 4 (68% selectivity at steady state), followed by 17 (10% selectivity at steady state) and 26 (8% selectivity at steady state). Notably, we also observed the reverse reaction to produce 1 from 2, with ca. 3% selectivity to 1. Pd/MgO–Al2O3 produced mostly 3 (60% selectivity at steady state), with some 17 (22% selectivity at steady state).
For comparison purposes we refer to the results of the Pd/CeZrOx-catalyzed reaction as a benchmark, and all four catalysts were evaluated at identical reaction conditions. The major product obtained over Pd/CeZrOx was 4, with 43% selectivity at steady state selectivity, followed by 20 with 16% steady-state selectivity, and 17 with 12% steady-state selectivity. Fig. 7 compares the overall product distributions, grouped by carbon-number, for all four catalysts. The largest fraction of long-chain carbon products from 2 was obtained using Pd/CeZrOx, while the smallest fraction of cracking products (i.e., C5–C9 products) was obtained using Cu/MgO–Al2O3. Accordingly, we suggest that the best approach from a processing standpoint for producing jet-fuel-range products is to use very low space velocities and a Pd/CeZrOx catalyst. The lighter cracking products could be recycled back into the reactor feed if needed, leading to similar oligomerization reactions as those described by Shylesh et al.36
Finally, to evaluate the effect of the presence of residual impurities in the fermentation broth on catalyst deactivation, we obtained results at the same reaction conditions (reaction temperature = 623 K, 135.8 kPa, and WHSV = 0.046 h−1) with esterified fermentation broth over Pd/CeZrOx. We also evaluated conversion of ethyl hexanoate esterified as described above as a control. The product distributions for the conversion of real fermentation broth is shown in Table 5. The conversion of 1 remained complete for both experiments. For the esterified hexanoic acid sample the main product was 6 with a steady-state selectivity of 37%. The other major products were 3 and 28 with 19% and 14% steady-state selectivities, respectively. The steady-state selectivity distribution based on carbon numbers of products was 1% for C5–C9, 82% for C10–C14, 14% for C15–C19, and 4% for C20+. The results for the control experiment were similar (see Table S1†). To assess whether impurities in the esterified broth led to catalyst deactivation, we evaluated deactivation rate constants based on the appearance of the primary product, 2. Because we are operating at low space velocities, the product distribution is dominated by conversion of 2 to downstream products. As the catalyst deactivates, the selectivity to downstream products decreases while the selectivity to 2 increases. For simplicity, we quantified the decrease in secondary product formation based on the increase in production of 2. Following this approach, we found a first-order deactivation rate constant of 1.4 ± 3 h−1 for the fermentation-derived esters, while the first-order deactivation rate constant was 0.002 ± 0.002 h−1 for the pure feed. That the catalyst undergoes substantial deactivation in the presence of real, biologically-derived feed is unsurprising, given the likely presence of biogenic impurities,28 a problem that has been previously addressed by the use of polymer-derived microenvironments.31
| Cmpd. | Product | #C | Initial Sel. (%) | S.S. Sel. (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.25 wt% Pd/CeZrOx at 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. | ||||
| — | Nonane | 9 | 1 | <1 |
| 20 | 4-Nonanone | 9 | <1 | <1 |
| — | Decane | 10 | 1 | 1 |
| 24 | 4-Methyl nonane | 10 | 5 | 4 |
| 28 | 4-Decanone | 10 | 5 | 14 |
| 6 | Undecane | 11 | 54 | 37 |
| — | 3-Methyl decane | 11 | <1 | <1 |
| 2 | 6-Undecanone | 11 | <1 | <1 |
| 3 | 6-Undecanol | 11 | <1 | 19 |
| — | 6-Methyl undecane | 12 | 8 | 4 |
| — | 1-Hexanone 1-phenyl | 12 | <1 | <1 |
| — | 6-Ethyl undecane | 13 | 1 | 1 |
| 18 | 6-Tridecanone | 13 | <1 | 1 |
| — | 2-Hexyl 1-octanol | 14 | <1 | <1 |
| — | Tetradecane | 14 | 1 | 1 |
| — | 6-Tetradecanone | 14 | <1 | 1 |
| — | 5-Methyl tetradecane | 15 | 2 | 1 |
| — | pentadecane | 15 | 2 | 1 |
| 26 | 8-pentadecanone | 15 | <1 | <1 |
| — | 6-Methyl pentadecane | 16 | 2 | <1 |
| — | 6-Propyl tridecane | 16 | <1 | 1 |
| 27 | Hexadecane | 16 | 1 | 2 |
| — | Heptadecane | 17 | 2 | 1 |
| — | 7-Methyl hexadecane | 17 | 1 | <1 |
| — | Octadecane | 18 | 9 | 4 |
| — | 2-Methyl octadecane | 19 | 1 | <1 |
| — | Nonadecane | 19 | 1 | 2 |
| — | Eicosane | 20 | 1 | <1 |
| — | 2-Methyl eicosane | 21 | <1 | 2 |
| — | 2,6,10,15-Tetramethyl heptadecane | 21 | 6 | 1 |
| — | Docosane | 22 | <1 | <1 |
| — | 9-Hexyl heptadecane | 23 | <1 | <1 |
| — | 6-Methyl docosane | 23 | 1 | <1 |
| — | Tricosane | 23 | <1 | <1 |
| — | 2-Methyl tricosane | 24 | 2 | <1 |
| — | Tetracosane | 24 | <1 | <1 |
| — | pentacosane | 25 | <1 | <1 |
| — | 7-Hexyl eicosane | 26 | <1 | <1 |
| — | 7-Butyl docosane | 26 | <1 | <1 |
| — | Heptacosane | 27 | <1 | <1 |
Reaction network
It has previously been shown that Pd/CeZrOx catalysts contain a distribution of acidic, basic, and metallic sites,37–43 which enable a wide range of organic chemical reactions.21,44 Judging from the broad distribution of products generated from our experiments, it seems a complex reaction network must occur. Scheme 2 shows a potential reaction network that would explain all of the major products observed here.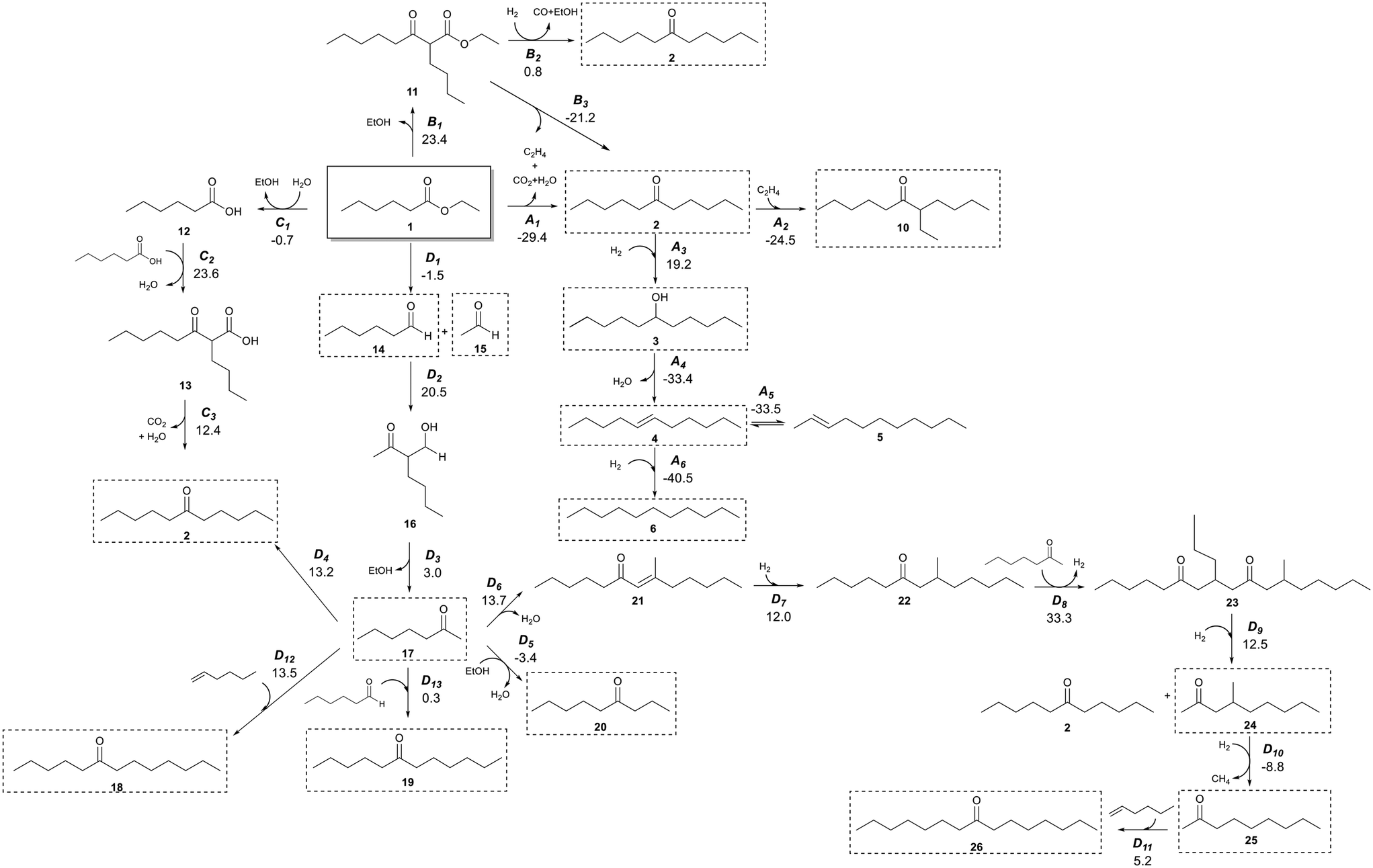 | ||
| Scheme 2 Reaction network of the conversion of ethyl hexanoate with H2 on Pd/CeZrOx. Compound numbers in bold correspond to Table 2. Pathway labels are given in italics, with the standard Gibbs free energy change for production of each species from 1 given below in kcal mol−1. For example, species 3 is produced by reaction A3, and the standard Gibbs free energy to produce 3 from 1 is 19.2 kcal mol−1. Compounds highlighted in dashed boxes were experimentally observed. | ||
The primary reaction pathway in Scheme 2 involves the self-ketonization of 1 to yield 2, which then undergoes further hydrogenation over metal sites to yield C11 alcohols and alkanes (3 and 6). As shown in pathway A of Scheme 2, a side product of this ketonization is ethylene, which itself can undergo oligomerization over acidic sites to yield longer-chain alkenes that can be rapidly hydrogenated to form alkanes.45,46 As illustrated in pathway B in Scheme 2, 1 can also undergo a Claisen–Dieckmann condensation to produce a C14 β-keto ester (11) which can convert to 2 by further hydrogenation and releasing CO and ethanol, or it can convert to 2 by releasing CO2 and ethylene. 1 can also undergo hydrolysis releasing the free acid and alcohol moieties, which react in pathway C of Scheme 2 by carboxylic acid condensation to produce a C12 β-keto acid (13) which undergoes subsequent decarboxylation (i.e., a Kagan reaction) to also yield a symmetric C11 ketone (2). The ethanol released in this pathway can also form ethylene by dehydration.47 Additionally, a C7 ketone (17) can form by C–C cleavage (i.e., the retro-Tishchenko reaction) 1, which can be followed by further aldol condensation as shown pathway D of Scheme 2.4417 can then undergo self-aldol condensation followed by a Michael addition of another C7 ketone to form a C21 dione (23). Subsequent retro-Michael C–C cleavage of this molecule leads to branched C10 and linear C11 ketones (24 and 2, respectively).2117 can also react with ethanol to form a C9 linear ketone (20)22 or 1-butene (itself produced from oligomerization of ethylene) to form a linear C11 ketone (2).48 Furthermore, the condensation of smaller molecules can ultimately lead to longer-chain hydrocarbons; for example, the coupling of a C11 ketone and ethylene can form a branched ketone (10).49 To evaluate whether the reaction network shown in Scheme 2 is thermodynamically feasible, we obtained standard Gibbs free energy changes for each reaction using vapor-phase density functional theory (DFT) calculations performed with Gaussian 16.50 The hybrid B3LYP functional was used51 with the 6-311G(d,p) basis set. The electronic energy changes were converted to enthalpies by inclusion of zero-point energies and thermal corrections. Entropies were calculated from statistical mechanics equations using partition functions obtained from frequency calculations. Scheme 2 and Fig. S2† show the Gibbs free energy changes for each reaction at 623 K and 101 kPa. These Gibbs free energies were then used to calculate equilibrium constants for each step in the network (Table S2†) at this temperature. The major product in all reactions of 1 was 2, which can be produced by three primary pathways, of which pathway A is the only one which is exergonic at these conditions.
Species 17 is observed even over catalysts that do not include Pd nanoparticles, which suggests that pathway D in Scheme 2 must catalyzed by basic sites on either the MgO–Al2O3 or CeZrOx supports. Indeed, pathway D leads to many of the minor products observed in our reactions, and while step D2 is quite endergonic, the subsequent steps are quite exergonic. The low selectivity to these products would thus be unsurprising, and if the remaining activation barriers are all low, then it is reasonable to assume this pathway is responsible for production of these minority species. Notably, for the reactions with particularly small equilibrium constants, the products generally are not observed experimentally. For example, the retro-Tischenko reaction of 1 (D1) is facile, with an equilibrium constant of 3.5, and 14 and 15 are observed as reaction products. However, the product of aldol condensation of 14 with 15 is not observed likely because of a combination between the small equilibrium constant for this reaction coupled with a low barrier for sequential decarboxylation to produce 17, which in turn is observed experimentally. Additionally, this pathway has been observed to occur commonly on ceria-based catalysts,44,52 and so despite the small equilibrium constants for a few steps, we suggest that the overall reaction is viable, and indeed the overall equilibrium constant for producing 17 from 1 is favorable. The presence of Pd nanoparticles facilitates CC and CO hydrogenation and allows for reactions according to pathway A in Scheme 2. This pathway is thermodynamically feasible (see Table S2†), and we accordingly observe production of 3, 4, and 6 starting from 2 for catalysts containing Pd and, to a lesser extent, Cu (see Table S2†). Interestingly, for unpromoted CeZrOx we also observe small amounts of 4 and 6, although we do not see substantial production of 3. These saturated species could be produced via transfer hydrogenation as described by Vivier et al.52
To identify the potential for base-catalyzed coupling of ketones, we studied the conversion of 2 over Pd/CeZrOx and Pd/MgO–Al2O3 (Table 6), which led to species 17 and 8, likely by the reverse of pathway D (Scheme 2), with an equilibrium constant of 10−5, which is thermodynamically feasible based on the argument presented above. Both 1 and 14 were detected in the product mixture, further suggesting that pathway D is reversible. Species 19 can also be obtained via pathway D, by condensation of 14 and 17. Notably, significant amounts of 19 are only observed for MgO–Al2O3-based catalysts, suggesting that stronger base sites are needed for the aldol condensation of 14 and 17.
| Compoundb | MgO–Al2O3 | Cu/MgO–Al2O3 | Pd/MgO-Al2O3 | Pd/CeZrOx |
|---|---|---|---|---|
| a Reaction conditions: 623 K, 134.9 kPa H2 pressure, 135.8 kPa total pressure, and WHSV = 0.046 h−1. b Not detected. | ||||
| 2-Heptanone | 9 | 10 | 22 | 12 |
| 4-Nonanone | N.D.b | N.D.b | N.D.b | 16 |
| 5-Undecanone | 25 | 68 | N.D.b | 43 |
| 6-Dodecanone | 58 | N.D.b | 60 | 1 |
| 8-pentadecanone | N.D.b | 8 | N.D.b | 1 |
Conclusions
In summary, we have applied a three-step process to convert lignocellulosic biomass to species containing between 7 and 19 carbons, which are in the correct carbon-chain range to be used as jet fuel blendstocks following mild HDO. The first step uses open culture fermentation (OCF) for digestion of lignocellulosic biomass to produce fatty acids with chains between 4 and 8 carbons long. In the second step of our process, the carboxylic-rich fermentation broth undergoes acid-catalyzed esterification, where reactive distillation is used to avoid equilibrium limitations. The mixture of esterification products is subsequently subjected to catalytic C–C coupling to produce longer chain molecules required for jet fuel applications. We have achieved >99% conversion of a model feedstock (ethyl hexanoate) into a wide range of C7–C19 molecules using bifunctional metal/mixed-oxide catalysts. In particular, we found that Pd/CeZrOx is highly selective and stable under reaction conditions. Of the products detected using this catalyst, more 90 mol% are C11 or greater compounds that contain little oxygen and would be appropriate for blending with jet fuel. These species are present as aliphatic alcohols, aldehydes, and ketones, all of which can be reduced to form hydrocarbons by simple treatment with an alumina-supported bimetallic catalyst system (e.g., Pd–Ni and Pd–Fe) at moderate temperatures, as described by Lee, et al.53The catalyst was largely stable with respect to time-on-stream for at least 30 days, with small decreases in the selectivity to deoxygenation products (i.e., alkanes), while the selectivity to C–C coupling products (i.e., ketones, alcohols, etc.) increases over the same time range. This trend suggests that the Pd sites on the catalyst can undergo deactivation during the first several days of time-on-stream, while the C–C bond-forming reactions occur mostly over sites on the CeZrOx support, which remains fully active.
In an effort to increase the carbon numbers of the products by favoring sequential reactions, we evaluated subsequent reaction of 6-undecannone (2) over Pd/CeZrO, MgO–Al2O3,Cu/MgO–Al2O3 and Pd/MgO–Al2O3, and the use of MgO–Al2O3 catalysts showed no advantages over CeZrOx catalysts in this study. Finally, we postulated a reaction network that accounts for the major products observed in this work and verified that the reaction network is thermodynamically feasible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Profs. Clayton Wheeler, Brian Frederick, and Carl Tripp for helpful discussions and Dr. George Bernhardt for experimental assistance. Funding for this work was provided by the United States Department of Agriculture (USDA) through the Northeast Sun Grant Center, Grant Number: 5702-UM-SDSU-G640. Computational resources were provided by the University of Maine High Performance Computing Group.References
- B. Sharafedin, T. Hepher and K. Samanta, Jet fuel demand to remain low as airlines buckle up for tough ride, https://www.reuters.com/article/us-global-oil-jet-fuel/jet-fuel-demand-to-remain-low-as-airlines-buckle-up-for-tough-ride-idUSKCN21X1DS, (accessed October 10, 2020) Search PubMed.
- U.S. Energy Information Administration (EIA) - Qb, https://www.eia.gov/opendata/qb.php?sdid=INTL.63-2-WORL-TBPD.A.
- E. Terrenoire, D. A. Hauglustaine, T. Gasser and O. Penanhoat, Environ. Res. Lett., 2019, 14(8), 084019 CrossRef CAS.
- T. J. Schwartz, A. R. P. van Heiningen and M. C. Wheeler, Green Chem., 2010, 12, 1353–1356 RSC.
- P. A. Case, A. R. P. van Heiningen and M. C. Wheeler, Green Chem., 2012, 14, 85–89 RSC.
- W. C. Wang and L. Tao, Renewable Sustainable Energy Rev., 2016, 53, 801–822 CrossRef CAS.
- M. Crocker, Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, Royal Society of Chemistry, 2010 Search PubMed.
- D. R. Shonnard, L. Williams and T. N. Kalnes, Environ. Prog. Sustainable Energy, 2010, 28, 382–392 CrossRef.
- D. Klein-Marcuschamer, P. Oleskowicz-Popiel, B. A. Simmons and H. W. Blanch, Biotechnol. Bioeng., 2012, 109, 1083–1087 CrossRef CAS.
- H. N. Chang, N. J. Kim, J. Kang and C. M. Jeong, Biotechnol. Bioprocess Eng., 2010, 15, 1–10 CrossRef CAS.
- J. Han, A. Elgowainy, H. Cai and M. Q. Wang, Bioresour. Technol., 2013, 150, 447–456 CrossRef CAS.
- P. Gegg, L. Budd and S. Ison, J. Air Transp. Manag., 2014, 39, 34–40 CrossRef.
- A. Milbrandt, C. Kinchin and R. McCormick, The Feasibility of Producing and Using Biomass-Based Diesel and Jet Fuel in the United States, 2013 Search PubMed.
- J. Janaun and N. Ellis, Renewable Sustainable Energy Rev., 2010, 14, 1312–1320 CrossRef CAS.
- H. Olcay, A. V. Subrahmanyam, R. Xing, J. Lajoie, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2013, 6, 205–216 RSC.
- J. Han, L. Tao and M. Wang, Biotechnol. Biofuels, 2017, 10, 1–15 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
- H. W. B. Daniel Klein-Marcuschamer, P. Oleskowicz-Popiel and B. A. Simmons, Biotechnol. Bioeng., 2011, 109, 1083–1087 CrossRef.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- C. A. Gaertner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027–6033 CrossRef CAS.
- E. L. Kunkes, E. I. Gürbüz and J. A. Dumesic, J. Catal., 2009, 266, 236–249 CrossRef CAS.
- K. A. Goulas, G. Gunbas, P. J. Dietrich, S. Sreekumar, A. Grippo, J. P. Chen, A. A. Gokhale and F. D. Toste, ChemCatChem, 2017, 9, 677–684 CrossRef CAS.
- R. A. Pollock, B. R. Walsh, J. Fry, I. T. Ghampson, Y. B. Melnichenko, H. Kaiser, R. Pynn, W. J. Desisto, M. C. Wheeler and B. G. Frederick, Chem. Mater., 2011, 23, 3828–3840 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. Kruk, M. Jaroniec and A. Sayari, Langmuir, 1997, 13, 6267–6273 CrossRef CAS.
- K. Kashkooli, MS Thesis, University of Maine, 2019 Search PubMed.
- T. J. Schwartz, B. J. O'Neill, B. H. Shanks and J. A. Dumesic, ACS Catal., 2014, 4, 2060–2069 CrossRef CAS.
- Z. Zhang, J. E. Jackson and D. J. Miller, Bioresour. Technol., 2008, 99, 5873–5880 CrossRef CAS.
- R. Luque, C. S. K. Lin, C. Du, D. J. Macquarrie, A. Koutinas, R. Wang, C. Webb and J. H. Clark, Green Chem., 2009, 11, 193–200 RSC.
- T. J. Schwartz, R. L. Johnson, J. Cardenas, A. Okerlund, N. A. Da Silva, K. Schmidt-Rohr and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 12718–12722 CrossRef CAS.
- A. Corma, V. Fornés, R. M. Martín-Aranda and F. Rey, J. Catal., 1992, 134, 58–65 CrossRef.
- D. Tichit, M. H. Lhouty, A. Guida, B. H. Chiche, F. Figueras, A. Auroux, D. Bartalini and E. Garrone, J. Catal., 1995, 151, 50–59 CrossRef CAS.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- K. A. Goulas, S. Sreekumar, Y. Song, P. Kharidehal, G. Gunbas, P. J. Dietrich, G. R. Johnson, Y. C. Wang, A. M. Grippo, L. C. Grabow, A. A. Gokhale and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 6805–6812 CrossRef CAS.
- S. Shylesh, D. Kim, A. A. Gokhale, C. G. Canlas, J. O. Struppe, C. R. Ho, D. Jadhav, A. Yeh and A. T. Bell, Ind. Eng. Chem. Res., 2016, 55, 10635–10644 CrossRef CAS.
- L. F. Liotta, G. Pantaleo, A. Macaluso, G. Marcì, S. Gialanella and G. Deganello, J. Sol-Gel Sci. Technol., 2003, 28, 119–132 CrossRef CAS.
- M. Glorius, M. A. C. Markovits and C. Breitkopf, Catalysts, 2018, 8, 1–25 CrossRef.
- K. Otsuka, Y. Wang and M. Nakamura, Appl. Catal., A, 1999, 183, 317–324 CrossRef CAS.
- J. C. Serrano-Ruiz, J. Luettich, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, J. Catal., 2006, 241, 45–55 CrossRef CAS.
- S. M. de Lima, A. M. Silva, U. M. Graham, G. Jacobs, B. H. Davis, L. V. Mattos and F. B. Noronha, Appl. Catal., A, 2009, 352, 95–113 CrossRef CAS.
- F. Al-Wadaani, E. F. Kozhevnikova and I. V. Kozhevnikov, J. Catal., 2008, 257, 199–205 CrossRef CAS.
- S. M. de Lima, I. O. da Cruz, G. Jacobs, B. H. Davis, L. V. Mattos and F. B. Noronha, J. Catal., 2008, 257, 356–368 CrossRef CAS.
- R. Kumar, N. Enjamuri, S. Shah, A. S. Al-Fatesh, J. J. Bravo-Suárez and B. Chowdhury, Catal. Today, 2018, 302, 16–49 CrossRef CAS.
- P. Cossee, J. Catal., 1964, 3, 80–88 CrossRef CAS.
- S. Moussa, P. Concepción, M. A. Arribas and A. Martínez, ACS Catal., 2018, 8, 3903–3912 CrossRef CAS.
- R. Klimkiewicz, H. Grabowska and L. Syper, Kinet. Catal., 2003, 44, 283–286 CrossRef CAS.
- C. Y. Ho, K. D. Schleicher, C. W. Chan and T. F. Jamison, Synlett, 2009, 2009, 2565–2582 CrossRef.
- F. Mo and G. Dong, Science, 2014, 345, 68–72 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision B.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- L. Vivier and D. Duprez, ChemSusChem, 2010, 3, 654–678 CrossRef CAS.
- J. Lee, Y. T. Kim and G. W. Huber, Green Chem., 2014, 16, 708–718 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated equilibrium constants, reaction coordinate diagram showing changes in Gibbs free energies, and catalyst characterization data. See DOI: 10.1039/d0re00401d |
| This journal is © The Royal Society of Chemistry 2021 |