
A mesoporous silica-supported CeO2/cellulose cathode catalyst for efficient bioelectrochemical reduction of inorganic carbon to biofuels†
Dayakar
Thatikayala
a,
Deepak
Pant
 b and
Booki
Min
*a
b and
Booki
Min
*a
aDepartment of Environmental Science and Engineering, Kyung Hee University, Yongin, Republic of Korea. E-mail: bkmin@khu.ac.kr
bSeparation and Conversion Technology, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400 Mol, Belgium
First published on 21st July 2021
Abstract
In this study, a novel efficient cathode electrode was fabricated to convert inorganic carbon to volatile fatty acids (VFAs) through microbial electrosynthesis (MES) in a single chamber reactor. The cathode catalyst was made up of mesoporous silica (mS) coated with cerium oxide (CeO2) and carbonized cellulose (C) in which mS acted as a core material and both CeO2 and C acted as a shell material. CeO2/C was loaded on the porous surface of mS, which acted as catalytic centers to enhance the biochemical reactions. The C/CeO2@mS composite catalyst coated on carbon cloth (Cc) was characterized by XRD and FESEM and showed high crystallinity and a porous core–shell morphology. The cyclic voltammetry analysis indicated that the cathode with C/CeO2@mS exhibited higher catalytic activity (−0.59 mA cm−2 (background current)) than the other controls (0.26 mA cm−2 for MES-C and −0.06 mA cm−2 for MES-mS). Three MES reactors with different cathodes were comparatively operated for the conversion of CO2 (8 g L−1 of HCO3−), and MES-C/CeO2@mS exhibited maximum acetate production (19.1 ± 0.95 mM) followed by MES-C (10.8 ± 0.51 mM) and MES-mS (9.5 ± 0.33 mM). The coulombic efficiency (CE%) in MES-C/CeO2@mS was 76%, and it was 42% and 34% for MES-mS and MES-C, respectively. The maximum current generation (0.48 ± 0.21 mA cm−2) was obtained with MES-C/CeO2@mS at a relatively higher cathode potential (−0.61 mV) as compared with the other cathodes. MES-C/CeO2@mS showed a lower Tafel slope of 220 mV dec−1, which was 2.71 times lower than that of abiotic MES-C/CeO2@mS (598 mV dec−1) suggesting enhanced electrokinetics with exoelectrogenic biofilm development on the cathode electrode. This study clearly demonstrates that the C/CeO2@mS catalyst can be successfully used for highly efficient bioelectrochemical conversion of CO2 to value added products via a MES route.
Introduction
The increase in atmospheric CO2 is mainly due to anthropogenic activities, causing an increase in the Earth's atmospheric temperature in the urbanized world.1 To control the excess amount of atmospheric CO2, researchers have been focusing on reducing the demand of fossil fuels and utilizing low carbon renewable resources by chemical treatment/electrocatalysis of CO2 and carbon capture, storage and utilization (CCS/CCU).2 Microbial electrosynthesis (MES) is an upcoming and innovative approach and is considered as a sustainable technology to diminish greenhouse gas emissions in the atmosphere or from point sources of CO2 generation.3,4 In MES, CO2 is directly captured and converted to valuable chemicals by microbial catalytic reactions on the cathode electrode with less energy input under environmentally friendly conditions.5 In recent studies, cathodes modified with ferrites, reduced graphene oxide, metal oxides, cobalt phosphide, and molybdenum alloys have been explored for the production of VFAs.6,7 Highly porous FexMnOy microspheres as a cathode catalyst produced 6.53 mM acetate with a CE of 58% with reduced charge transfer resistance and an enhanced exchange current density by 7.2 times compared to that of carbon cloth.8 Utilization of a MXene-coated biochar electrode for production of VFA resulted in a 2.3 times improved current density and a 1.7 times increase in VFA production as compared to those of an uncoated electrode.9 A copper ferrite/rGO cathode catalyst produced 11.4 ± 0.57 mM VFA with a current density of 2.9 A m−2 and CE of 77.7%.10 The novel cathode such as rGO enhanced the electron transfer rate with an acetate production rate of 168.5 ± 22.4 mmol m−2 d−1, current density of 2580 ± 540 mA m−2 and CE of 83.8%, and enhanced cell attachment over the cathode electrode was observed.11 These studies suggest that the innovative strategies of catalyst design can lead to a change in the conductivity and morphology, further enhancing the production rates of VFAs.12Cellulose has been widely used in a vast number of applications due to its great flexibility, high crystallinity, high mechanical strength and high electrical conductivity.13–15 Nano-cellulose-based electrodes in MFC mainly focused on the electrodes' electrochemical properties, bioelectricity generation, and bioremediation because nanocellulose is an excellent alternative to the expensive nanomaterial/membrane.16–20 Cerium oxide (CeO2) has several advantages such as superior resistance to chemical corrosion, antioxidant effect, non-toxicity, being a catalyst for fuel oxidation, enzyme-like catalytic activity, and outstanding electrochemical properties.21 Therefore, CeO2 has been attracting significant interest in fuel cell applications to synthesize valuable products.22 The addition of silica to CeO2 like metal oxides stabilized the nanostructure and enhanced the catalytic activity in several applications such as wastewater treatment, electrochemical sensors, batteries, supercapacitors, fuel cells, and solar cells.23,24 Recently, conducting macroporous/microporous electrodes have caused profound interest in biofuel generation because of addressing key issues like power density and the life span of devices.25,26 Robust nanoscale electrodes will be widely used as electrocatalysts in fuel cells and BES systems, and surface modification and morphology control will become the main strategies to improve the performance of electrodes.27,28
In this work, CeO2/mesoporous silica (mSiO2) was innovatively combined with carbonized cellulose nanoparticles as a cathode catalyst (C/CeO2@mS) to enhance the conductivity and surface area of the cathode catalyst and to enhance the microbial reduction of inorganic carbon to VFA in a single chamber MES reactor. The performance of MES-C/CeO2@mS was compared with that of carbonized cellulose (MES-C) and mesoporous silica (MES-mS) in terms of current generation, cathode potential, coulombic efficiency, and VFA generation. XRD studies investigated the crystallinity of the C/CeO2@mS cathode material which improves the electron transfer between the electrode and biofilm. FESEM studies revealed the formation of a core–shell structure and the porous nature of the material which provide the surface area for biofilm formation. The electrochemical techniques, CV and EIS, further investigated the efficiency of the C/CeO2@mS cathode in terms of reduction current and charge transfer resistance towards the microbial reduction of CO2 source.
Materials and methods
Synthesis of C/CeO2@mS
For the fabrication of the CeO2@mSiO2 core–shell composite (Fig. 1), first, mesoporous SiO2 was synthesized by an oil–water-based method using n-hexane. In the synthesis process, triethanolamine (TEA) (0.3 g) and hexadecyltrimethylammonium bromide (CTAB) (1.2 g) were added into deionized water (60 ml) and stirred at 60 °C for 1 h, and in this mixture, TEA acted as a catalyst and CTAB acted as the templating agent to achieve a mesoporous structure. Afterward, a solution mixture of TEOS (8.0 ml) and n-hexane (20 ml) was added to the TEA and CTAB solution and stirred for 24 h. After that, the formed oil and aqueous phases are separated by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm min−1). Further, the resulting product was washed with solvent (ethanol) and deionized water several times to remove the impurities and dried at 80 °C. Finally, the dried powder was calcined at 550 °C for 2 h.
000 rpm min−1). Further, the resulting product was washed with solvent (ethanol) and deionized water several times to remove the impurities and dried at 80 °C. Finally, the dried powder was calcined at 550 °C for 2 h.
 | ||
| Fig. 1 Schematic representation for the synthesis of CeO2 and carbonized cellulose-loaded mesoporous silica (C/CeO2@mS composite). | ||
Cerium nitrate hexahydrate (0.2 g) and 1 g of hexamethylenetetramine were dissolved in 40 ml of deionized water. The as-synthesized 0.1 g mSiO2 and 0.4 g cellulose were dispersed in 25 ml ethanol with ultrasonication. Two solutions were added together and continuously stirred for 2 h at 75 °C to form a uniform coating of ceria and cellulose precursors. The final product was collected by centrifugation, washed, and dried at 80 °C. To obtain final purity, the product was calcined at 550 °C for 2 h.
Electrode fabrication and MES setup
C/CeO2@mS NCs were coated on carbon cloth (W0S1002, CeTech Co., South Korea). Pretreated carbon cloth with dimensions of 4.5 × 4.5 cm was selected and weaved with a titanium wire (D: 0.25 mm). For the coating process, a catalytic ink was prepared by mixing 1 mg cm−2 of the catalyst, 5 ml of isopropyl alcohol, and 5% Nafion binder 117 in sequential steps, and ultrasonication was carried out for 3 h to achieve a perfect dispersion. The catalytic ink was coated on both sides of the carbon cloth using an airbrush technique (N2 gas: 99.9%).29 Finally, the catalyst-coated carbon cloth was dried in a vacuum oven at 50 °C and stored at room temperature for further use (Fig. S1a†).For the construction of MES reactors, three single chamber reactors (membraneless) with a total volume of 350 ml were designed. All the reactors were made up of acrylic cylinder (outer diameter: 8.5 cm and inner diameter: 7.5 cm); the bottom and top of the reactor were sealed with an acrylic disk with high strength glue. The top disk was provided with two sampling ports (inlets for the liquid sample and to connect the gas bag) and three electrode ports; the bottom end of the reactor provided an outlet port to discharge liquid, and a stone diffuser was placed through the reactor side wall port to purge gas. The electrodes were placed on the top lid in their respective position with a distance of 5 cm to each other (working (C/CeO2@mS), reference (Ag/AgCl, +196 mV, Basi Inc., USA), and counter electrodes (fibrous carbon brush, 2.5 × 4 cm, brush 21, South Korea)). After attaching all the components on the top lid, a silicone gasket was placed on the main reactor body and the reactor was closed using a nut-bolt joint (Fig. S1b†). All the connection points were sealed with silicon glue to avoid further diffusion of air and to maintain strict anaerobic conditions throughout the experiment.30
MES operation and analysis and calculation
The microbial sludge was collected from Suwon waste treatment plant (Suwon, South Korea) and was kept at 4 °C under dark conditions until further use. The characteristics of the collected sludge were analyzed using standard methods (APHA, 1998) (pH: 7.21 ± 0.01; SCOD: 761 ± 28 mg L−1; TCOD: 35463 ± 1182 mg L−1; TSS: 22250 ± 1150 mg L−1; VSS: 17083 ± 444 mg L−1). Before using it in the MES experiment, the microbial sludge was incubated at 35 °C for 6 h to enhance the activity of microbes in the sludge. To promote the growth of autotrophic microbes and to limit the heterotrophic microbial community, the reactors were operated for five cycles with each cycle for 10 days. In the first cycle, the MES reactors were added with 50 ml of activated microbial sludge along with 200 ml of media (8 g of NaHCO3; 1.5 g l−1 KH2PO4; 2.9 g l−1 K2HPO4; 0.5 g l−1 NH4Cl; 0.09 g l−1 CaCl2; 0.21 g l−1 MgCl2; with 12 ml of mineral solution, 5 ml of vitamin solution, 2 ml of sodium-2-bromoethane-sulphonate (SBS) solution-methane suppressor and 1 g l−1 glucose). The total working volume of the reactor was 250 ml sparged with N2:CO2 (70:30) for 10 to 15 min. The reactor was kept in an incubator (Vision, Korea) at 35 ± 2 °C temperature with a 270 rpm stirring rate using a magnetic stirrer (ATL-4200, Anytech Co., Korea). All the MES reactors were run with a fixed potential of 0.8 V through a DC power supply (Hwasung Electronics Co., Republic of Korea). The negative terminal of the voltage source was connected to the cathode (working electrode) and the positive terminal was connected to the anode as designed in the early studies.31 A 10 ohm external resistor was placed between the cathode electrode and the negative terminal of the power source (Fig. S1c†). The voltage and current of the MES reactor were continuously monitored using an automatic digital multimeter (National Instruments 9205, USA).31 In the subsequent 2nd, 3rd, 4th, and 5th cycles of the MES reactor, the fresh medium was replaced with glucose of varying concentrations (0.5, 0.3, 0.1 and 0 L) to grow autotrophic microbes. After 50 days of MES operation, the microbial community was able to fix CO2 efficiently to electro-reduce CO2 to VFA. Further, the reactors were operated without glucose for 40 days (4 cycles) by considering 8 g L−1 sodium bicarbonate as the carbon source.
X-ray diffraction (XRD) was performed to study the crystallinity and phase of the materials (Bruker diffractometer: scan rate: 10 to 80°; speed: 0.12° s−1). FESEM analysis was conducted to study the morphology of the samples (FESEM, Germany; Zeiss machine). A potentiostat (Versastat 3, USA) with a three-electrode cell was used to study the electrochemical properties of the material. The auto-digital multimeter (National Instrument 9205, USA) was used to record the cell potential and cathodic current of the MES reactors for every 30 min. The collected MES reactor samples were analyzed to estimate VFA concentrations on days 0, 2, 4, 6, 8 and 10 using ion chromatography (IC-861, Metrohm, USA chromatogram). The coulombic efficiency (CE%) was calculated using eqn (1):
 | (1) |
485 C mol−1).
Results and discussion
Characterization of C/CeO2@mSiO2
XRD was performed to analyze the phase purity and chemical composition of all the synthesized samples (Fig. 2a). For mSiO2, at a low angle of 2 theta, it has characteristic peaks at (100) and (110) corresponding to the highly ordered mesoporous nanostructure and a broad peak at 22.7° indicating its amorphous nature.32 In C/CeO2@mS, the diffraction peaks of CeO2 with respect to the (111), (200), (220), (311), and (222) planes were well matched with pure CeO2. From the diffraction planes, CeO2 was identified having a cubic fluorite structure using standard JCPDS card no: 34-0394.33 The presence of a weak broad peak at 30° theta indicates the carbonized form of cellulose. The loading amount of CeO2 and carbonized cellulose reduced the intensity of the mSiO2 peaks at (100), (110), and (101), confirming the formation of the C/CeO2@mS composite structure. The C/CeO2@mS composite contains amorphous SiO2, crystalline CeO2 and carbonized cellulose, which significantly enhance the crystallinity of the composite to improve the conductivity and electrocatalytic activity of the composite.34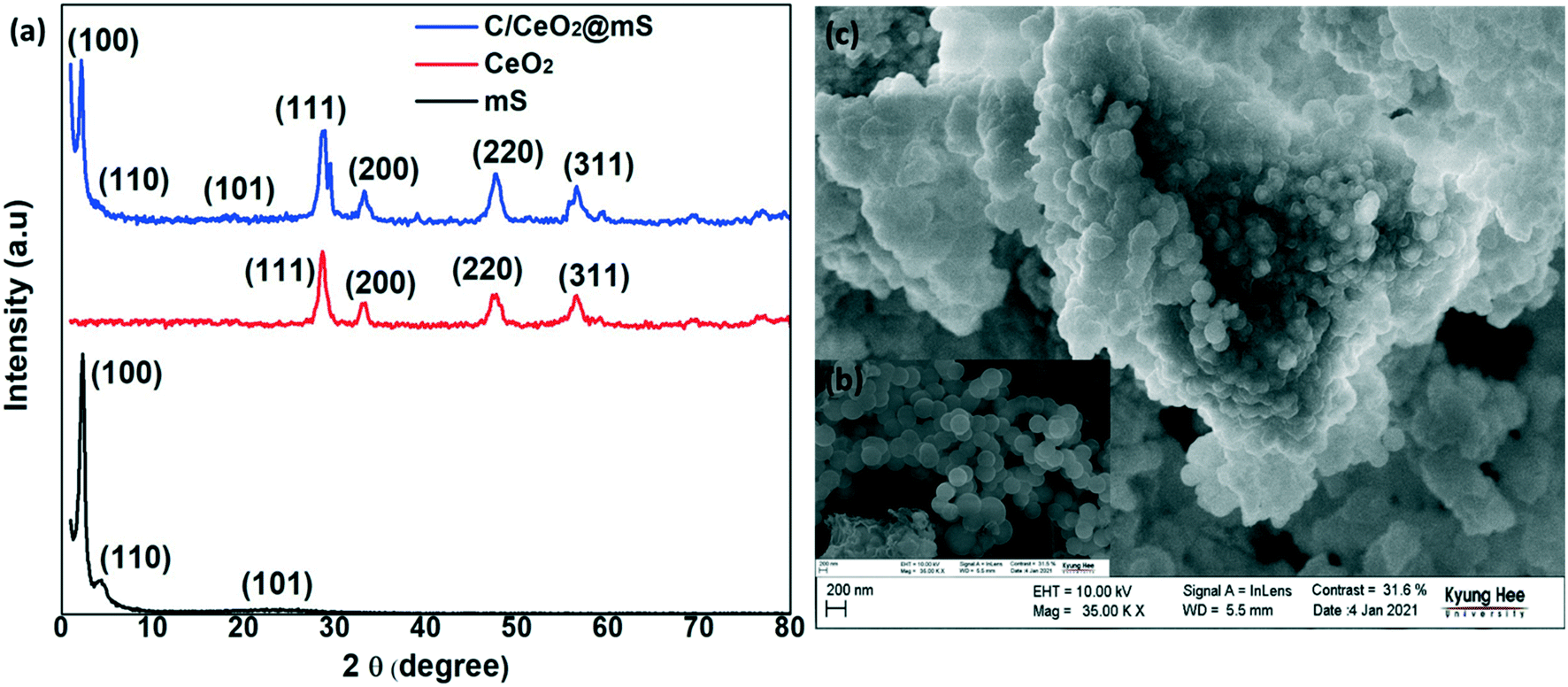 | ||
| Fig. 2 (a) XRD patterns of mesoporous silica (mS), CeO2, and C/CeO2@mS; (b and c) FESEM images of mS (inset) and C/CeO2@mS. | ||
Furthermore, the morphology of mSiO2 and the C/CeO2@mS composite was observed by FESEM measurements. From Fig. 2b (inset), spherical shape mesoporous SiO2 (mSiO2) nanoparticles were observed with a uniform size of 200 to 300 nm and acted as the core part in the catalyst. The shell (CeO2 and carbonized cellulose) was decorated over and in the pores of mSiO2 with a diameter of 10–12 nm. The contrast difference shown in Fig. 2c shows the core and shell regions in the composite structure, which was agglomerated because of the interactions with carbonized cellulose.
Volatile fatty acid (VFA) generation from inorganic carbon in MES
The VFA production of MES-mS, MES-C, and MES-C/CeO2@mS is shown in Fig. 3a. From 0 to 48 h of MES operation, a high quantity of VFA production was observed in all the MES reactors due to the utilization of the carbon substrate source and the higher metabolic rate of microbes on the working biocathodes.35 In MES-C/CeO2@mS, the maximum acetate production of 19.1 ± 0.95 mM at 48 h was observed, and the decline of VFA biosynthesis was noticed up to 10 days of operation, and the similar trend was observed for MES-C (10.8 ± 0.51 mM) and MES-mS (9.5 ± 0.33 mM). A slight decrease of VFA was observed during 48 h to 144 h and a sudden decrease of VFA was observed from 144 to 240 h due to the oxidation of synthesized VFAs at the anodic bioelectrode in the single-chamber MES reactor. In MES-C/CeO2@mS, the hybrid electrode catalyst favored bacterial biofilm formation because of the biocompatibility and enhanced surface area of the cathode electrode.36 In the single-chamber MES reactor, bioelectrochemical reactions on the biocathode favored the enhanced synthesis of VFA with increased movement of electrons and protons towards the cathode electrode.37,38 The total concentration of VFA production with MES-C/CeO2@mS was 23.8 ± 0.19 mM (MES-mS: 13.6 ± 0.68 mM and MES-C: 11.4 ± 0.53 mM), and this value was higher than previous studies.39,40 The total VFA production compared to acetate suggests that acetate-producing bacteria was higher than that of the other microbes. After 48 h of MES operation, the acetate is converted to other VFA, and most acetate is oxidized at the anodic electrode. | ||
| Fig. 3 MES operation vs. time: (a) VFA generation from CO2; (b) pH changes. | ||
In MES, pH plays an essential role in the bioconversion of CO2 to VFA. Fig. 3b shows the pH profiles in the three MES reactors for 10 days of MES operation. Generally, in dual-chamber MES systems, the pH increase in the cathode and a decrease in the anode were observed due to the accumulation of protons in the anode and consumption of hydrogen ions in the cathode.41 From a previous report, in single-chamber MES systems, pH was decreased due to the accumulation of VFA.42 In another case, the pH was increased due to the lower proton concentration and the immediate consumption of VFA by anodic microbes.43 In the present work, initially, the pH was around 7 for all three MES reactors, which was favorable to perform redox microbial metabolic activities to synthesize VFA. After 48 h, a drop in the pH was noticed, and the same phenomena were observed similar to those in previous work suggesting that the pH drop is due to reversal binding of HCO3− with H+ ions, which helps in buffering the solution to increase the VFA production. The pH drop for MES-C/mS/CeO2 was 6.81, which was low compared to that for MES-C (6.86) and MES-mS (6.99). The pH varied from 0th day to 10th day for MES-mS (6.98–7.44), MES-C (6.99–7.24), and MES-C/CeO2@mS (6.94–7.29). Overall, MES-C/CeO2@mS maintained a pH of around 7 to produce high concentrations of VFA compared to MES-mS and MES-C. To compare the performance of MES-mS, MES-C, and MES-C/CeO2@mS, the coulombic efficiency was considered as the average of the last 3 cycles (Fig. 4). At a +0.8 V applied potential, the coulombic efficiency (CE%) in MES-C was 34 ± 0.81%, and it was 42 ± 0.15% for MES-mS and 76 ± 0.69% for MES-C/CeO2@mS, respectively. The CE% of this work was higher compared to that of the other nanomaterial-based electrodes like Mo2C (64 ± 0.7%), 3D graphene/Ni (70%), and Perspex circular rings (49%).44–46 The obtained total Coulomb (Fig. 4) for MES-C/CeO2@mS was 4956.19 ± 0.80, and it was 2598.58 ± 0.92 for MES-C and 1487.17 ± 0.35 for MES-mS. The improvement in the CE% and total coulombs in MES-C/CeO2@mS indicates the activity of the cathodic biofilm, particularly the electroactive bacteria performing effectively; hence, the bicarbonate substrate was converted to current to produce VFAs.
 | ||
| Fig. 4 CE% and total Coulombs of MES-mS, MES-C, and MES-C/CeO2@mS. | ||
Current generation and cathode potential
The development of biofilm was carried out by applying the similar media and same applied potential conditions (+0.8 V) for all the MES reactors in each cycle (Fig. 5a). The current generation was increased with the biocathode and stabilized in the subsequent cycles, which indicates the successful and uniform formation of biofilm over the surface of the cathode electrode. In the 1st cycle, a current generation of 0.33 mA cm−2 for MES-C/CeO2@mS, 0.14 mA cm−2 (MES-C) and 0.09 mA cm−2 (MES-mS) was observed and increased in the further cycles. In the 2nd cycle, the current reached to 0.47 mA cm−2 for MES-C/CeO2@mS, and it was 0.21 mA cm−2 and 0.11 mA cm−2 for MES-C and MES-mS. During the last cycle, the current generation was 0.49 mA cm−2 (MES-C/CeO2@mS), 0.22 (MES-C) and 0.13 (MES-mS). The average current generation for the last 3 cycles for MES-C/CeO2@mS (0.48 ± 0.21 mA cm−2), MES-C (0.22 ± 0.67 mA cm−2), and MES-mS (0.12 ± 0.18 mA cm−2) indicates that the current generation was in the narrow range for the last 3 cycles, signifying the stable performance of the MES systems. The current densities of the present work were shown to be comparable with those from previous works: NiMoZn/stainless steel (0.5–1.1 mA cm−2), carbon cloth (0.07–0.09 mA cm−2), Si and TiO2 nanowire arrays (0.3 mA cm−2) and chitosan modified carbon cloth (0.6 mA cm−2).47–50 | ||
| Fig. 5 MES operation vs. time: (a) current generation; (b) cathode potential. | ||
The biocathode performance was further analyzed with the obtained cathode potentials of all the MES reactors (Fig. 5b). The cathode potential (average of the first 5 days) for MES-C/CeO2@mS was −0.65 mV in the 1st cycle and changed to −0.61 mV in the last cycle, and it was −0.97 for MES-C and changed to −0.81 mV (last cycle), whereas for MES-mS, it was −0.90 mV in the first cycle and changed to −0.82 mV (last cycle). The obtained cathode potential in this work is favorable for thermodynamic redox potential for CO2 to acetate (−0.48 V vs. Ag/AgCl, pH 7).51 In previous MES studies, a high positive potential (−0.6 V vs. Ag/AgCl) resulted in direct transfer of electrons52 was observed with the enhanced VFA production, whereas at low cathode potentials (−0.8 to −1 V Ag/AgCl), most of the previous studies suggested an indirect electron transfer mechanism.53 The direct electron transfer is dominant at high positive theoretical potential (−0.6 V vs. Ag/AgCl, hydrogen evolution) and the indirect electron transfer mechanism is dominant at low positive theoretical potentials. In few cases, the direct delivery of CO2 over a cathode electrode resulted in higher production of VFAs.54
Among all the three MES reactors, high acetate productivity was observed for MES-C/CeO2@mS (35.53 g m−2 d) on an average of 0 to 2 days, whereas it was 18.83 g m−2 d and 17.61 g m−2 d in MES-C and MES-mS, respectively, indicating that the hybrid combination of the catalyst in MES-C/CeO2@mS facilitated the enhanced electron transfer between the electrode and biocatalyst to synthesize higher production rates of acetate. The production rate of acetate in MES-C/CeO2@mS was 1.88 and 2.01 times higher than that of MES-C and MES-mS. A small amount of other VFAs was detected in the all MES reactors, and the acetate compound was reduced with more operation periods possibly due to the fast consumption of acetate by anodic bacteria.55 The total VFA production rate in MES-C/CeO2@mS was 47.70 g m−2 d, which was 1.74 and 1.95 times more than that in MES-C (27.84 g m−2 d) and MES-mS (24.35 g m−2 d). The production rates of the present work are higher compared to previous reports, FexMnOy (21.33 g m−2 d), CuFe2O4/rGO (35.37 g m−2 d), and 3D iron–carbon felt (25.4 g m−2 d),8,10,56 suggesting that the present catalysts effectively supported the microbial biofilm to enhance the synthesis of VFA. The overall performance of the present catalyst is compared with that of the other reported electrodes as shown in Table 1. The carbon conversion efficiency was analyzed based on the VFA production. An 8 g L−1 bicarbonate source in 200 ml had 0.27 g of carbon content. In MES-C/CeO2@mS, from the acetate (19.1 ± 0.95 mM), the converted carbon content was 0.19 g, and from the total VFA (23.8 ± 0.19 mM), it was 0.25 g, indicating that the present catalyst has a 93.5% carbon conversion efficiency. Similarly, the carbon conversion efficiency for MES-C and MES-mS was 53.56% and 46.84%. However, to estimate the exact mechanism for bio-electrokinetics (substrate and products) in the single-chamber MES reactor is quite challenging because of the presence of a three-electrode system in the single-chamber even though the interactions between the electrodes in the single-chamber may stimulate the VFA production.
| Cathode electrode | Microbe inoculum | Cathode potential (V) | Production rate of VFAs (g/m2/d) | Current densities (A m−2 or mA m−2) | CE (%) | Ref. |
|---|---|---|---|---|---|---|
| Graphene paper | S. ovata | −0.69 | 39.8 | −2.5 (A m−2) | 90.7 | 9 |
| Graphite granules | Enriched sludge | −0.59 | 1.03 (g L−1 per day) | −0.8 (kA m−3) | 69 | 58 |
| MXene Ti3C2TX-coated carbon felt | Mixed cultures | −0.6 | 12.24 (g L−1 per day) | −0.17 (A m−2) | 41 | 59 |
| CC coated with PEDOT:PSS | S. ovata | −0.69 | 59.5 | −3.2 (A m−2) | — | 45 |
| 3D graphene Ni-foam | Mixed cultures | −0.85 | 0.15 (g L−1 per day) | −10.2 (A m−2) | 70 | 60 |
| NiMo deposition on doped Si wafer | S. ovata | −0.7 | 6.7 (g L−1 per day) | −10 (A m−2) | 95–100 | 8 |
| Graphite stick Ni nanowire | S. ovata | −0.4 | 3.4 | −0.63 (A m−2) | 82 ± 14 | 54 |
| Porous Ni-hollow fiber with MWCNTs | S. ovata | −0.4 | 1.85 | −0.33 (A m−2) | 83 ± 8 | 61 |
| Gas diffusion cathode VITO-CoRE | Enriched anerobic sludge | −1 | 36.6 | −20 (A m−2) | 35 ± 8 | 62 |
| C/CeO2/mS | Enriched anerobic sludge | −0.61 | 47.70 | 0.48 ± 0.21 (mA cm−2) | 76 ± 0.69 | This work |
Electrochemical characterization
The electrochemical performance of abiotic and biotic MES reactors was analyzed using CV in the potential range of −1.0 V to 1.0 V at a scan rate of 20 mV s−1 (Fig. 6a and b). The electrocatalytic activity of biotic and abiotic was used to determine the role of microbes in bioelectrochemical reactions. The abiotic MES has shown a difference in the background current without any appearance of redox peaks compared to biotic MES, indicating that the microbes play a vital role in the current generation process. The background current of abiotic MES-C/CeO2@mS was −0.59 mA cm−2 which was 2.26 (−0.26 mA cm−2) and 9.83 (−0.06 mA cm−2) times higher than the that of MES-C and MES-mS.57 The biotic MES-C/mS/CeO2 has shown a prominent reduction peak at a potential of −0.22 V with a reduction current of −0.383 mA cm−2 and an increment in current compared to MES-C and MES-mS due to the formation of sufficient biofilm to convert CO2 to VFA. In the Tafel plot, the abiotic and biotic cathode catalysts were compared (Fig. 6c and d) and a lower slope of 220 mV dec−1 was obtained for biotic MES-C/CeO2@mS and for abiotic, it was 598 mV dec−1. The exchange current density for biotic MES was 0.12 mA cm−2, which was 1.33 times higher than that for abiotic MES (0.09 mA cm−2). The lower Tafel slope and enhanced exchange current density for biotic MES suggests the improved electrokinetics for the reduction of CO2 to VFA. | ||
| Fig. 6 (a) CV analysis of abiotic MES (cellulose, mesoporous silica (mS), and C/CeO2@mS); (b) CV analysis of biotic MES (MES-C, MES-mS, and MES-C/CeO2@mS); (c) Tafel graph of C/CeO2@mS and MES-C/CeO2@mS; (d) linear regression results (Tafel slope) of C/CeO2@mS and MES-C/CeO2@mS. | ||
Conclusion
A cathode catalyst C/CeO2@mS was successfully used to convert inorganic carbon to VFA in MES. The maximum VFA production of 23.8 ± 0.19 mM was obtained from MES with the C/CeO2@mS electrode catalyst containing CeO2/carbonized cellulose of high conductivity and mesoporous SiO2, whereas it was 13.6 ± 0.68 mM for MES-mS and 11.4 ± 0.53 mM for MES-C. The MES operation with C/CeO2@mS also showed the maximum VFA production rate of 47.70 g m−2 d with enhanced current generation and a relatively increased cathode potential. The maximum carbon conversion efficiency of MES-C/CeO2@mS was 93.50%, which was 1.74 and 1.99 times higher than that of MES-C (53.5%) and MES-mS (46.8%). The Tafel slope of biotic (220 mV dec−1) was very low compared to that of abiotic (598 mV dec−1) and the exchange current density for biotic MES (0.12 mA cm−2) was 1.33 times higher than that for abiotic MES (0.09 mA cm−2), indicating improved electrokinetics for the reduction of CO2 to VFA. These results suggested that the C/CeO2@mS cathode electrode with high biocompatibility and catalytic activity could enhance the CO2 conversion to valuable VFAs in MES operation for sustainable carbon recycling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported with a grant from the National Research Foundation of Korea (2018R1A2B6001507).References
- Z. Liu, D. Guan, W. Wei, S. J. Davis, P. Ciais, J. Bai, S. Peng, Q. Zhang, K. Hubacek, G. Marland and R. J. Andres, Nature, 2015, 524(7565), 335–338 CrossRef CAS.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett and J. P. Hallett, Energy Environ. Sci., 2018, 11(5), 1062–1176 RSC.
- S. Gildemyn, K. Verbeeck, R. Slabbinck, S. J. Andersen, A. Prévoteau and K. Rabaey, Environ. Sci. Technol. Lett., 2015, 2(11), 325–328 CrossRef CAS.
- R. Mateos, A. Sotres, R. M. Alonso, A. Morán and A. Escapa, Energies, 2019, 12(17), 3297 CrossRef CAS.
- Y. Jiang, H. D. May, L. Lu, P. Liang, X. Huang and J. Z. Ren, Water Res., 2019, 149, 42–55 CrossRef CAS PubMed.
- S. Bajracharya, S. Srikanth, G. Mohanakrishna, R. Zacharia, D. P. B. T. B. Strik and D. Pant, J. Power Sources, 2017, 356, 256–273 CrossRef CAS.
- S. Kalathil and D. Pant, RSC Adv., 2016, 6, 30582–30597 RSC.
- M. Tabish Noori and B. Min, ChemElectroChem, 2019, 6, 5973–5983 CrossRef CAS.
- K. Tahir, W. Miran, J. Jang, N. Maile, A. Shahzad, M. Moztahida, A. A. Ghani, B. Kim, H. Jeon and D. S. Lee, Sci. Total Environ., 2021, 773, 145677 CrossRef CAS.
- D. Thatikayala and B. Min, Sci. Total Environ., 2021, 768, 144477 CrossRef CAS PubMed.
- N. Aryal, A. Halder, M. Zhang, P. R. Whelan and P. Tremblay, Sci. Rep., 2017, 7, 9107 CrossRef PubMed.
- B. Bian, S. Bajracharya, J. Xu, D. Pant and P. E. Saikaly, Bioresour. Technol., 2020, 302, 122863 CrossRef CAS.
- J. Lamaming, R. Hashim, C. P. Leh, O. Sulaiman, T. Sugimoto and M. Nasir, Carbohydr. Polym., 2015, 134, 534–540 CrossRef CAS PubMed.
- C. H. Ye, S. T. Malak, K. Hu, W. Wu and V. V. Tsukruk, ACS Nano, 2015, 9, 1087–10895 Search PubMed.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- C. Vilela, A. J. D. Silvestre, F. M. L. Figueiredo and C. S. R. Freire, J. Mater. Chem. A, 2019, 35, 20045–20074 RSC.
- G. Jiang, J. Zhang, J. Qiao, Y. Jiang, H. Zarrin, Z. Chen and F. Hong, J. Power Sources, 2015, 273, 697–706 CrossRef CAS.
- H. Zhu, F. Shen, W. Luo, S. Zhu, M. Zhao, B. Natarajan, J. Dai, L. Zhou, X. Ji, R. S. Yassar, T. Li and L. Hu, Nano Energy, 2017, 33, 37–44 CrossRef CAS.
- M. Mashkour, M. Rahimnejad, M. Mashkour and F. Soavi, Appl. Energy, 2021, 282, 1–11 CrossRef.
- J. H. Kim, B. S. Shim and H. S. Kim, et al. , Int. J. Precis. Eng. Manuf., 2015, 2, 197–213 Search PubMed.
- Y. Zhang, S. Zhao, J. Feng, S. Song, W. Shi, D. Wang and H. Zhang, Chem, 2021 DOI:10.1016/j.chempr.2021.02.015 , in press.
- K. Tomishige, Y. Gu, T. Chang, M. Tamura and Y. Nakagawa, Mater. Today Sustainability, 2020, 9, 100035 CrossRef.
- L. Liao, H. X. Mai, Q. Yuan, H. B. Lu, J. C. Li, C. Liu, C. H. Yan, Z. X. Shen and T. Yu, J. Phys. Chem. C, 2008, 112, 9061–9065 CrossRef CAS.
- G. Aguila, S. Guerrero and P. Araya, Appl. Catal., A, 2013, 462, 56–63 CrossRef.
- S. Phanichphant, A. Nakaruk and D. Channei, Appl. Surf. Sci., 2016, 387, 214–220 CrossRef CAS.
- A. Serov, K. Artyushkova, E. Niangar, C. Wang, N. Dale, F. Jaouen, M. T. Sougrati, Q. Jia, S. Mukerjee and P. Atanassov, Nano Energy, 2015, 16, 293–300 CrossRef CAS.
- G. Iwona, G. John, S. Carlo, S. Alexey, M. Chris, A. Plamen and A. I. Ioannis, Energy, 2018, 144, 1073–1079 CrossRef PubMed.
- A. Sivasankaran, D. Sangeetha and Y. H. Ahn, Chem. Eng. J., 2016, 289, 442–451 CrossRef CAS.
- C. Flores-Rodriguez, C. Nagendranatha Reddy and B. Min, Biomass Bioenergy, 2019, 127, 105261 CrossRef CAS.
- M. Lee, C. N. Reddy and B. Min, Int. J. Hydrogen Energy, 2018, 2(11), 2380–2389 Search PubMed.
- M. T. Vu, Md TabishNoori and B. Min, Chem. Eng. J., 2020, 393, 124613 CrossRef CAS.
- A. Khare, R. J. Choudhary, K. Bapna, D. M. Phase and S. P. Sanyal, J. Appl. Phys., 2010, 108, 1–5 CrossRef.
- G. F. Cerofolini, C. Galati and L. Renna, Surf. Interface Anal., 2003, 35, 968–973 CrossRef CAS.
- S. A. Almuhtaseb and J. A. Ritter, Adv. Mater., 2010, 15, 101–114 CrossRef.
- C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman and H. D. May, Environ. Sci. Technol., 2013, 47(11), 6023–6029 CrossRef CAS.
- L. Chen, P.-L. Tremblay, S. Mohanty, K. Xu and T. Zhang, J. Mater. Chem. A, 2016, 4, 8395–8401 RSC.
- N. Aryal, A. Halder, P. L. Tremblay, Q. Chi and T. Zhang, Electrochim. Acta, 2016, 217, 117–122 CrossRef CAS.
- G. Mohanakrishna, J. S. Seelam, K. Vanbroekhoven and D. Pant, Faraday Discuss., 2015, 183, 445–462 RSC.
- A. H. Anwer, M. D. Khan, N. Khan, A. S. Nizami, M. Rehan and M. Z. Khan, J. Environ. Manage., 2019, 249, 109376 CrossRef CAS.
- G. Mohanakrishna, I. Abu-Reesh, K. Vanbroekhoven and D. Pant, Sci. Total Environ., 2020, 715, 137003 CrossRef CAS.
- G. Mohanakrishna, S. K. Butti, R. K. Goud and S. Venkata Mohan, Bioelectrochemistry, 2017, 115, 11–18 CrossRef CAS.
- J. A. Modestra, R. Katakojwala and S. Venkata Mohan, Chem. Eng. J., 2020, 394, 124759 CrossRef.
- J. R. Phillips, H. K. Atiyeh, R. S. Tanner, J. R. Torres, J. Saxena, M. R. Wilkins and R. L. Huhnke, Bioresour. Technol., 2015, 190, 114–121 CrossRef CAS PubMed.
- S. Tian, H. Wang, Z. Dong, Y. Yang, H. Yuan, Q. Huang, T. S. Song and J. Xie, Biotechnol. Biofuels, 2019, 71, 12 Search PubMed.
- I. Vassilev, P. A. Hernandez, P. Batlle-Vilanova, S. Freguia, J. O. Kromer, J. Keller, P. Ledezma and B. Virdis, ACS Sustainable Chem. Eng., 2018, 6(7), 8485–8493 CrossRef CAS.
- S. Srikanth, D. Singh, K. Vanbroekhoven, D. Pant, M. Kumar, S. K. Puri and S. S. V. Ramakumar, Bioresour. Technol., 2018, 265, 45–51 CrossRef CAS PubMed.
- J. P. Torella, C. J. Gagliardi, J. S. Chen, D. K. Bediako, B. Colón, J. C. Way, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2337–2342 CrossRef CAS.
- Q. Fu, S. Xiao, Z. Li, Y. Li, H. Kobayashi, J. Li, Y. Yang, Qu. Liao, X. Zhu, X. He, D. Ye, L. Zhang and M. Zhong, Nano Energy, 2018, 53, 232–239 CrossRef CAS.
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Y. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS PubMed.
- S. Xiao, Z. Li, Q. Fu, Y. Li, J. Li, L. Zhang, Q. Liao and X. Zhu, Chem. Eng. J., 2020, 390, 124530 CrossRef CAS.
- S. Bajracharya, K. Vanbroekhoven, C. J. Buisman, D. P. Strik and D. Pant, Faraday Discuss., 2017, 202, 433–449 RSC.
- H. Liu, T. Song, K. Fei, H. Wang and J. Xie, Bioresour. Bioprocess., 2018, 5(1), 1–10 CrossRef CAS.
- S. Bajracharya, A. ter Heijne, X. Dominguez Benetton, K. Vanbroekhoven, C. J. N. Buisman, D. P. B. T. B. Strik and D. Pant, Bioresour. Technol., 2015, 195, 14–24 CrossRef CAS PubMed.
- B. Bian, M. F. Alqahtani, K. P. Katuri, D. F. Liu, S. Bajracharya, Z. P. Lai, K. Rabaey and P. E. Saikaly, J. Mater. Chem. A, 2018, 6(35), 17201–17211 RSC.
- P. Batlle-Vilanova, R. Ganigué, S. Ramió-Pujol, L. Bañeras, G. Jiménez, M. Hidalgo, M. D. Balaguer, J. Colprim and S. Puig, Bioelectrochemistry, 2017, 117, 57–64 CrossRef CAS PubMed.
- Z. Dong, H. Wang, S. Tian, Y. Yang, H. Yuan, Q. Huang, T. Shun Song and J. Xie, Bioresour. Technol., 2018, 269, 203–209 CrossRef CAS.
- Y. E. Song, C. Kim, J. Baek, C. H. Im, E. Seol, J. Jae, Y. Nygård and J. R. Kim, Sustainable Energy Fuels, 2020, 4, 5952–5957 RSC.
- C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman and H. D. May, Environ. Sci. Technol., 2013, 47(11), 6023–6029 CrossRef CAS.
- K. Tahir, W. Miran, J. Jang, A. Shahzad, M. Moztahida, B. Kim and D. S. Lee, Chem. Eng. J., 2020, 381, 122687 CrossRef CAS.
- N. Aryal, P.-L. Tremblay, M. Xu, A. E. Daugaard and T. Zhang, Front. Energy Res., 2018, 6, 72 CrossRef.
- H. R. Nie, T. Zhang, M. M. Cui, H. Y. Lu, D. R. Lovley and T. P. Russel, Phys. Chem. Chem. Phys., 2013, 15(34), 14290–14294 RSC.
- S. Bajracharya, K. Vanbroekhoven, C. J. N. Buisman, D. Pant and D. P. B. T. B. Strik, Environ. Sci. Pollut. Res., 2016, 23(22), 22292–22308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00166c |
| This journal is © The Royal Society of Chemistry 2021 |