
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green, scalable, low cost and reproducible flow synthesis of biocompatible PEG-functionalized iron oxide nanoparticles†
Julien
Mahin
 ,
Christoph O.
Franck
,
Luise
Fanslau
,
Hirak K.
Patra‡
,
Michael D.
Mantle
,
Ljiljana
Fruk
and
Laura
Torrente-Murciano
*
,
Christoph O.
Franck
,
Luise
Fanslau
,
Hirak K.
Patra‡
,
Michael D.
Mantle
,
Ljiljana
Fruk
and
Laura
Torrente-Murciano
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, CB3 0AS, UK. E-mail: lt416@cam.ac.uk
First published on 29th July 2021
Abstract
Functionalized iron oxide nanoparticles are of great interest for multiple biomedical applications. However, it remains a considerable challenge to manufacture these nanoparticles reproducibly on a large scale with the appropriate surface coating to render them completely stable and biocompatible. To overcome this problem, a novel combination of synthesis and functionalization using modular microreactor systems is presented here, avoiding the need of intermediate ligand exchange steps. Continuous flow technology enables reproducible synthesis of bare iron oxide nanoparticles (7 ± 2 nm) in water under mild conditions, in tandem with extremely fast and efficient functionalization with a custom heterobifunctional PEG stabilizer. The nanoparticles can be easily derivatized with any molecule of interest through simple amide coupling, demonstrating their capacity to act as a versatile platform for biomedical applications. The produced iron oxide nanoparticles are fully biocompatible based on a LDH cytotoxicity assay, highly stable in various biologically relevant media and suitable for T2 MRI contrast applications (r1 = 1.44 mM−1 s−1, r2 = 272 mM−1 s−1). A full cost analysis reveals the commercial viability of the process, with a total cost as low as £ 506 g−1, demonstrating the potential of this modular approach to enable the large-scale deployment of functionalized nanomaterials in real world applications.
1. Introduction
Iron oxide nanoparticles (Fe3O4NP) are widely investigated for biomedical applications, such as MRI contrast agents,1–4 drug delivery,5–7 magnetic hyperthermia cancer treatment,8–10 theranostics,11,12 cryopreservation,13,14etc. iron oxide is particularly suited for biomedical applications because of its magnetic properties and low toxicity compared to other ferromagnetic materials such as cobalt and nickel.15 Furthermore, an increasing amount of iron oxide-based nanomedicines have begun to be approved by the FDA for human use such as Feridex® for an image contrast agent for the detection of liver lesions16 and Feraheme® for the treatment of anaemia in adult patients with chronic kidney disease.17In order to use such particles in biomedical applications, stringent requirements have to be met. The nanoparticles must be stabilized against aggregation and opsonization by an irreversibly bound hydrophilic ligand and ideally their size and size distribution should be well-defined.18,19 Typical stabilizing agents consist of long polyethylene glycol (PEG) chains, which impart the particles with water dispersibility and stabilize the particles by steric repulsions.20 Additionally, PEG coatings have been shown to confer a “stealth” property which greatly reduces the likelihood of the nanoparticles being recognised as a foreign body by the immune system.21 PEG molecules must be strongly bound to the iron oxide surface by a high affinity anchor, to avoid desorption in biological media. Catechols, and in particular nitrocatechols, have been identified as excellent choices to anchor substrates to iron oxide surfaces.22–24
Despite keen interest to apply iron nanoparticle systems to a wide range of biomedical applications, the potential is currently limited to lab scale because the scale up of the synthesis processes and its reproducibility remain a considerable technological challenge. Until now, the general strategy to achieve the requirements for well-defined and highly stable iron oxide nanoparticles with a stabilizer coating consists of a two-step procedure.25–31 Firstly, the iron oxide cores are synthesized in batch typically by a thermal decomposition method. Secondly, the iron oxide particles are functionalized through a batch ligand exchange with a suitable stabilizing agent. Each of these steps requires further purification steps to remove excess reactants.32 In addition, thermal decomposition methods, unfortunately, produce particles coated by a layer of hydrophobic surfactants, requiring an additional ligand exchange step to disperse the nanoparticles in an aqueous phase,33 representing a significant barrier to large scale production. Furthermore, batch synthesis methods tend to have reproducibility issues and are difficult to scale as small differences in mixing or heat transfer can have large effects on the final nanoparticles.34
Therefore, it is of great interest to develop new synthetic methods which can reproducibly manufacture iron oxide nanoparticles under milder conditions on a large scale, but most importantly, directly produce nanoparticles with the necessary functional coating for biomedical applications. Recently, continuous flow synthetic strategies have been demonstrated to enable high throughput production of nanoparticles without sacrificing reproducibility.35–41 In particular, continuous synthesis has been demonstrated to greatly improve the reproducibility of precipitations methods, which have traditionally been known to produce polydisperse samples.42,43 The classical co-precipitation synthesis of iron oxide represents a cheaper, more energy efficient and green alternative to thermal decomposition methods, as it can be carried out in water at room temperature. Because of the bare nature of nanoparticles produced, such continuous precipitation methods can also be coupled with functionalization with small molecules44,45 or low affinity polymers.46 However, to the best of our knowledge continuous functionalization of highly stable nanoparticles has not been achieved so far. In this work, we demonstrate the continuous, green, scalable and reproducible synthesis of highly stable PEG-functionalized iron nanoparticles using a modular flow system. Synthesis of bare iron oxide cores is achieved in a first reactor by simple co-precipitation followed by functionalization in a downstream reactor, enabling synthesis and functionalization to be integrated into a single set-up without intermediate ligand exchange steps. Furthermore, designing a heterobifunctional nitrodopamine–PEG–COOH stabilizing agent results in free carboxylic acid moieties which can be used to further couple the iron oxide nanoparticles with any molecule of interest such as fluorophores or antibodies for bio-imaging and targeted drug delivery. This unique flexibility was illustrated here by functionalization of the nanoparticles with fluorescein. The resulting nanoparticles were found to be completely biocompatible through cytotoxicity assays and showed T1 and T2 relaxivity comparable to other commercial iron oxide-based MRI contrast agents. Finally, a full cost analysis was carried out, demonstrating the commercial viability of the synthetic process opening the door to the deployment of iron oxide nanoparticles in a wide range of biomedical applications.
2. Results and discussion
2.1. Synthetic strategy and reaction setup
A general strategy combining the synthesis and functionalization of iron oxide nanoparticles in continuous flow conditions is demonstrated in this work (Fig. 1). Colloidally stable bare iron oxide nanoparticles (Fe3O4NP) were prepared through a co-precipitation synthetic method (Scheme 1b). The co-precipitation method was selected because it is a cheap, efficient, and green synthesis approach that produces bare iron oxide particles. While co-precipitation techniques typically result in poor nanoparticle size control and wide particle size distributions, this is mostly true for batch processes because of mixing inconsistencies. Continuous flow co-precipitation has been shown to lead to better particle size control thanks to the high degree of control on mixing that it enabled by its high mass transfer rates.47 These bare iron oxide nanoparticles were subsequently functionalized with a custom nitrodopamine–polyethyleneglycol–carboxylic acid (NDA–PEG–COOH) stabilizer molecule (Scheme 1a). This stabilizer was designed with a nitrodopamine (NDA) moiety which possess high affinity to iron oxide acting as a strong anchoring group. The polyethylene glycol polymer (PEG) imparts water dispersibility and biocompatibility whilst providing a steric colloidal stabilization effect to the nanoparticles, preventing aggregation. Finally, the free carboxylic acid moiety enables further derivatization by amide coupling with any amine containing compound. The capacity for further derivatization was illustrated in this work by coupling the nanoparticles with fluorescein as a proof-of-concept molecule (Scheme 1c). This approach could be easily extended to any compounds containing an amine functionality, such as antibodies or antitumoral compounds to enable targeted drug delivery.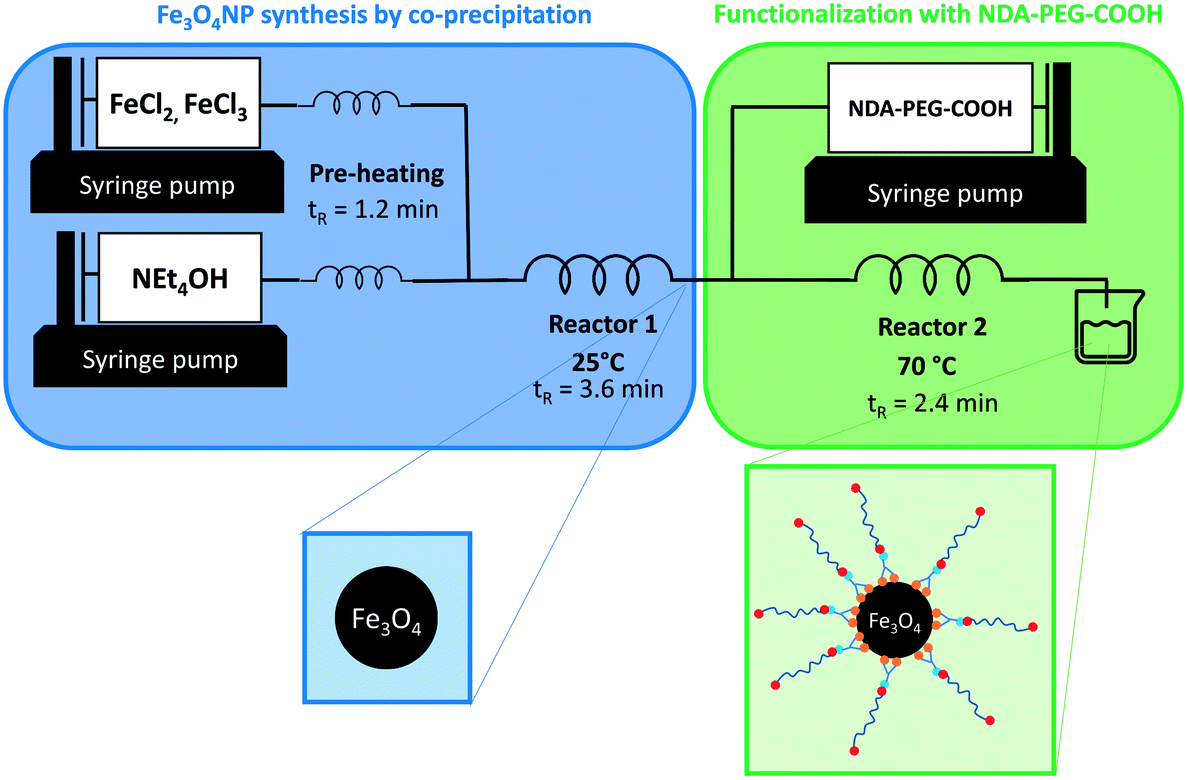 | ||
| Fig. 1 Reaction setup for the continuous synthesis of Fe3O4NP and functionalization with NDA–PEG–COOH in a single set-up. tR indicates the residence time in each reactor. | ||
 | ||
| Scheme 1 Synthetic scheme for a) NDA–PEG–COOH stabilizer b) Fe3O4NP and functionalization with NDA–PEG–COOH c) further batch derivatization of Fe3O4NP–NDA–PEG–COOH with aminofluorescein. | ||
The NDA–PEG–COOH stabilizing agent was prepared in a simple two-step scheme, starting with nitration of dopamine followed by amide coupling with a dicarboxylic acid PEG (Scheme 1a). The synthesis of NDA–PEG–COOH was carried out using standard batch chemistry. The bare iron oxide nanoparticles (Fe3O4NP) were synthesized in continuous flow in a fluoropolymer tubular microreactor, by the co-precipitation method (Fig. 1). In the same setup, the nanoparticles were immediately functionalized by mixing with the NDA–PEG–COOH stabilizing agent in a downstream reactor. This functionalization strategy is extremely efficient as it only requires a minimal amount of stabilizing agent (0.5 equiv.) and bypasses the need for a ligand exchange step entirely.
2.2. Iron oxide nanoparticles synthesis, functionalization and characterization
As mentioned above, the continuous synthesis of bare iron oxide nanoparticles stabilized electrostatically was achieved by the co-precipitation method, mixing a solution of Fe(II) and Fe(III) (0.02 M, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) with an alkaline tetraethylammonium hydroxide (NEt4OH, 0.1 M) solution (eqn (1)).
:2 molar ratio) with an alkaline tetraethylammonium hydroxide (NEt4OH, 0.1 M) solution (eqn (1)).| Fe2+ + 2Fe3+ + 8OH− → Fe3O4 + 4H2O | (1) |
In the synthesis reactor, the initially formed particles visibly aggregated immediately but this aggregation was found to be reversible depending on the base counter cation employed in the synthesis. The particles de-aggregate in the presence of NEt4+ at high pH (12–13) over the course of 1–2 minutes. This observation suggests that two competing processes are at play: aggregation and electrostatic stabilization. The aggregation is kinetically fast compared to the electrostatic stabilization, but the latter represents a more thermodynamically stable state. For this reason, NEt4OH was selected as the base for the co-precipitation synthesis. Conversely, when using sodium hydroxide as the base, aggregation could not be reversed. In this case, the aggregated state is not only kinetically fast but also thermodynamically stable. This observation highlights the importance of the counter cation in the system. A similar observation was already made by Mascolo48 and by Massart with NMe4OH.49 While reactor fouling and clogging can be an issue for material synthesis in microreactors, in this case negligible fouling was observed during operation. This co-precipitation method produced nanoparticles of 7.2 ± 1.6 nm in size (Fig. 3a–c). These nanoparticles were crystalline and exhibited characteristic magnetite/maghemite reflexions (Fig. 3d, S1†). Most likely, the magnetite (Fe3O4) phase is formed initially and then slowly oxidises to maghemite (γ-Fe2O3) over the course of a few weeks, as indicated by the colour change of the suspensions from an opaque black colour to lighter orange/brown (Fig. S3†). The magnetite and maghemite phases of iron oxide have very similar properties and are hard to distinguish using diffraction techniques as they both present the inverse spinel structure and similar diffraction signatures. For simplicity, the iron oxide nanoparticles obtained in this work are referred to as Fe3O4 NP, but the particles likely consist of a mixture of magnetite and maghemite phases, as is generally obtained with co-precipitation methods.49 The electrostatic stabilization by the NEt4+ cation enables the synthesis of essentially bare iron oxide nanoparticles with excellent colloidal stability. Zeta-potential measurements confirmed the electrostatic nature of the stabilization of the bare nanoparticles at alkaline pH (Fig. 4a). The zeta-potential of bare Fe3O4NP was measured at −40 mV at pH = 13.
The bare nature of the particles combined with their high colloidal stability enables the fast binding of the NDA–PEG–COOH stabilizer to the nanoparticles in the functionalization reactor. NDA–PEG–COOH stabilizer was synthesised according to the scheme in Scheme 1a. Nitrodopamine (NDA) was synthesised according to a method from the literature.50 PEG dicarboxylic acid (600 g mol−1) dissolved in dichloromethane was activated at room temperature for 20 min with 1 equiv. of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 1 equiv. of N-hydroxysuccinimide (NHS), followed by addition of 1 equiv. of nitrodopamine dissolved in dimethylformamide and stirred for 16 h. The final product was purified by addition of an acidic 1 M LiCl solution followed by extraction with dichloromethane. The formation of the desired NDA–PEG–COOH product was confirmed by Fourier transform infrared spectroscopy (FTIR) (Fig. S2†) and 1H-NMR spectroscopy (SI). The conversion of PEG(COOH)2 into NDA–PEG–COOH was estimated at 52% based on the ratio of acid/amide Hα peak area. Batch tests with quenched time aliquots were carried out to evaluate the kinetics of the functionalization step to evaluate the amount of NDA–PEG–COOH stabilizer required to achieve successful nanoparticle stabilization. The amount of Fe stabilized was calculated by quenching the nanoparticle suspensions in 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) pH buffer to quickly lower the reaction pH to 7. Because the bare iron oxide nanoparticles are only electrostatically stabilized and have a point of zero charge around 7, any unfunctionalized nanoparticles quickly aggregate at pH = 7. On the other hand, functionalized Fe3O4–NDA–PEG–COOH nanoparticles remain stable at pH = 7. Any unfunctionalized and aggregated nanoparticles were separated by filtration with a 200 nm syringe filter and an excess of hydrochloric acid was added to the filtrate to dissolve the stabilized nanoparticles. The iron concentration in the digested filtrate was determined through a UV-vis spectrophotometric assay, allowing a mass balance calculation of the amount of stabilized Fe. The minimal amount of NDA–PEG–COOH to achieve nanoparticle stabilization was determined to be 0.5 molar equivalents (equiv.) compared to Fe (Fig. 2), with around 85% of Fe stabilized under 1.5 min. Slightly more nanoparticles were stabilized by further increasing the reaction time although this increase is relatively small. A clear cut off was observed at 0.5 equiv. of NDA–PEG–COOH, using less resulted in almost no stabilized particles, indicating that the NDA–PEG–COOH binds to all the nanoparticles uniformly. Above this 0.5 equiv. threshold, essentially all the particles are stabilized. 0.5 equiv. corresponds to a theoretical grafting density of 24 NDA–PEG–COOH molecules per nm2, assuming 7 nm spherical particles. Note that the reaction time to achieve the functionalization could be even shorter, as the first aliquot was collected after 1.5 minutes.
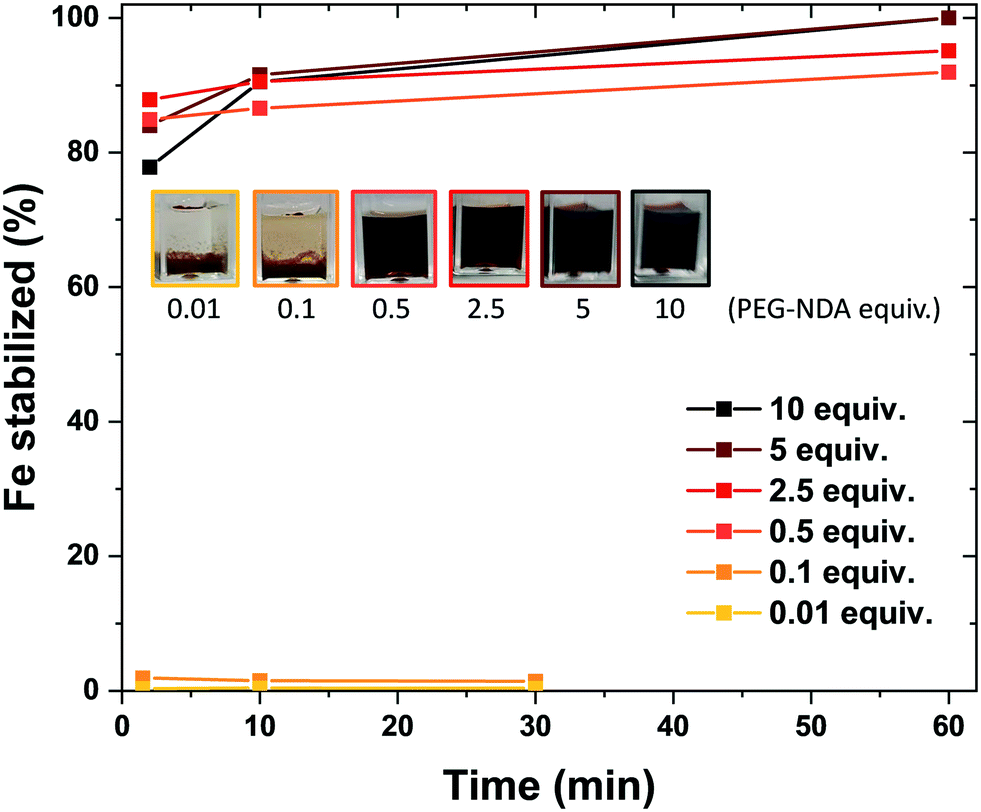 | ||
| Fig. 2 Evolution of the amount of iron oxide nanoparticle stabilized as a function of reaction time for various amounts of NDA–PEG–COOH stabilizer added, after the pH was adjusted to 7. Inset shows pictures of the respective suspensions, illustrating that good stabilization is achieved for 0.5 equivalents or more NDA–PEG–COOH. | ||
Once the optimal amount of NDA–PEG–COOH was determined, the functionalization of continuously synthesised Fe3O4NP was achieved by introducing a solution of NDA–PEG–COOH (adjusted to pH = 13 with NEt4OH) in a second reactor, with a residence time of 2.4 minutes. While the nanoparticle synthesis step was carried out at 25 °C, the functionalization step temperature was set at 70 °C as it was found to lead to improve the stabilization (Fig. S5†). The high heat transfer rates in microdevices enables this fast change of conditions. To ensure complete reproducibility, it is possible to quench the reaction by adding a large excess of a buffer such as HEPES in a third downstream reactor to rapidly lower the pH to 7. However, this step is optional as similar nanoparticles properties were obtained with or without a quenching step.
In order to purify the produced nanoparticles and remove excess NDA–PEG–COOH, a simple and scalable protocol was established. The water solvent was evaporated by rotary evaporation and the particles were subsequently dispersed in ethanol followed by reprecipitation by adding hexane as anti-solvent. This procedure was carried out thrice. Finally, the particles were dried in air and resuspended in deionized water and filtered through a 200 nm syringe filter. Using this method, large amounts of nanoparticles can be purified at once, with high yields of around 70–90% of the total iron content. Rotary evaporation is what enables this purification procedure to be carried out on a large scale, compared to the more commonly used freeze-drying approach. Although rotary evaporation represents comparatively much harsher conditions which can lead to particle aggregation, the fact that minimal aggregation was observed illustrates the excellent stability of the synthesized functionalized nanoparticles. Although we carried out this step in batch, it is important to highlight that continuous rotary evaporation is commercially available should this step needs to be carried out continuously.
The Fe3O4NP–NDA–PEG–COOH nanoparticles were characterized by transmission electron microscopy (TEM), zeta-potential, FTIR and dynamic light scattering (DLS). The functionalized nanoparticles appeared significantly better dispersed on TEM micrographs compared to bare iron oxide nanoparticles, indicating successful steric stabilization (Fig. 3). The average size of the iron oxide cores after functionalization (7.0 ± 1.9 nm) was not significantly different from the bare iron oxide nanoparticles, indicating that no surface etching process occurs during functionalization. Interestingly, the functionalized iron oxide nanoparticles remained black in colour even after 3 months of storage which suggest that the functionalization coating protects the magnetite nanoparticles from oxidation to maghemite (Fig. S3†).
 | ||
| Fig. 3 Representative transmission electron micrographs and particle size distributions of a–c) bare iron oxide nanoparticles and e–h) Fe3O4NP–NDA–PEG–COOH. d) Fourier transform image of the selected area in the white square on micrograph c, exhibiting the characteristic lattice spacings for magnetite/maghemite phase. The functionalized nanoparticles appear much better dispersed on the grid, as a result of their steric stabilization, whereas bare nanoparticles tend to clump together. | ||
Zeta-potential data indicated a shift in the point of zero charge (PZC) of the particles from pH = 7 to pH = 5 after functionalization, caused by acidic carboxylate surface groups (Fig. 4a). This observation suggests that the NDA–PEG–COOH preferentially binds with the NDA moiety, leaving free carboxylic acid functions available for downstream derivatization. FTIR of functionalized particles showed characteristic vibration bands attributed to NDA–PEG–COOH on their surface, confirming successful functionalization (Fig. 4b). Note that the difference in intensity between the pure NDA–PEG–COOH ligand and the functionalized Fe3O4NP–NDA–PEG–COOH is due to their state (liquid and solid respectively) and thus different signal transmission through the ATR detector. No aggregation was detected visually or by DLS even after 1 month of storage in 18 mM phosphate buffer (Fig. 4c). Similarly, the functionalized nanoparticles remained mostly stable in harsh 1 M NaCl dispersant. Stability in phosphate buffer indicates strong binding of PEG to the particles, as phosphate ions have strong affinity for the iron oxide surface and cause aggregation and precipitation of poorly stabilized iron oxide nanoparticles. Stability in 1 M NaCl confirms that the stabilization obtained is of steric nature, as a high salt concentration collapses any electric double layer responsible for electrostatic stabilization. The functionalized nanoparticles were stable in a wide range of pH (4–13), unlike bare nanoparticles which precipitated around their PZC (Fig. 4d). The hydrodynamic size of the particles is around 44 nm, which indicates that the 7 nm iron oxide nanoparticles likely exist in solution in the form of clusters of a few cores bundled together.
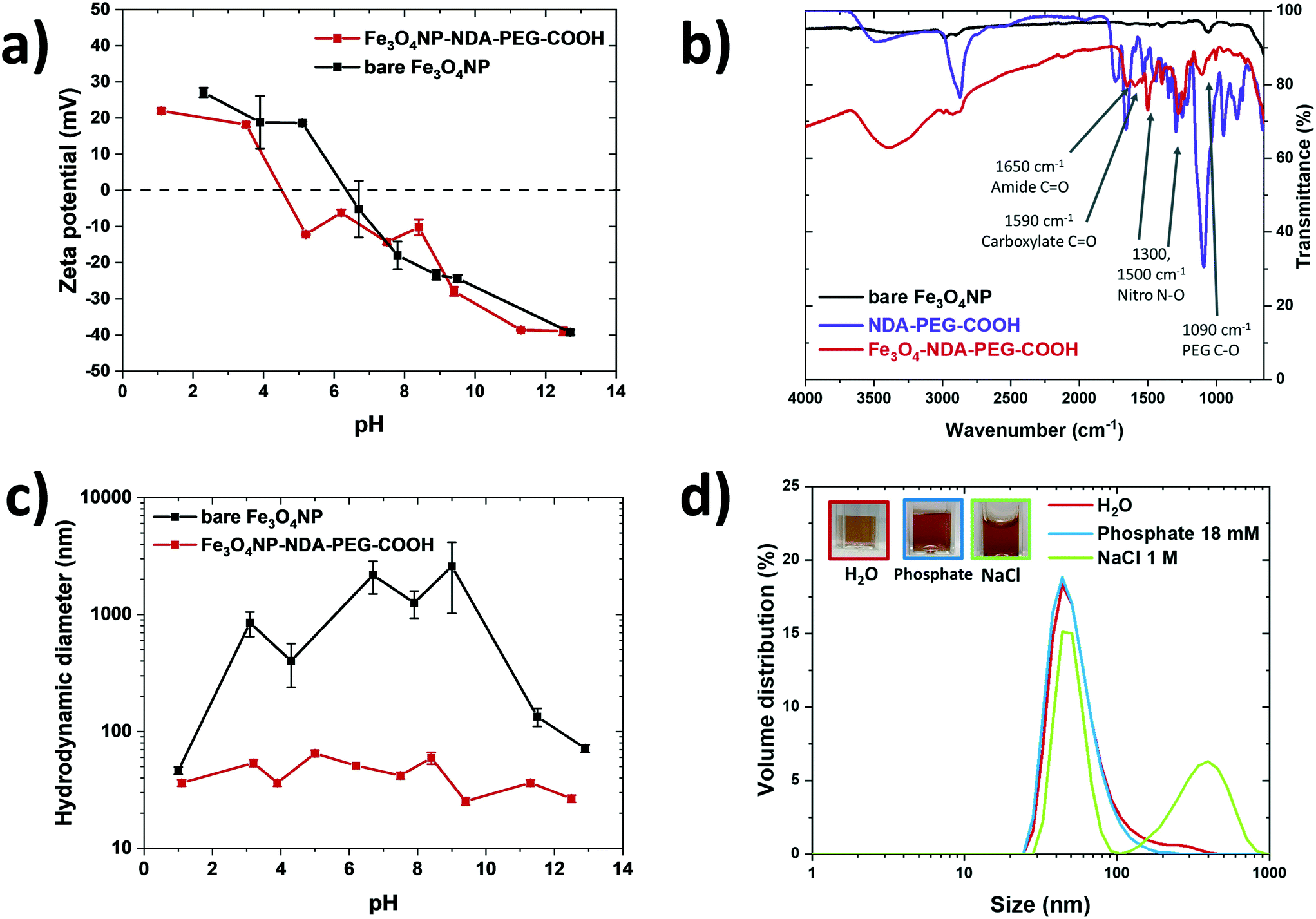 | ||
| Fig. 4 a) Zeta-potential measurement of the bare nanoparticles (black) compared to the functionalized nanoparticles (red). The functionalization shifts the point of zero charge from pH = 7 (for bare nanoparticles) to pH = 5. b) FTIR of bare nanoparticles (black), functionalized nanoparticles red) and pure NDA–PEG–COOH (purple). Characteristic vibration bands of NDA–PEG–COOH appear on the spectrum of the functionalized sample, indicating that NDA–PEG–COOH is present on the surface of the functionalized nanoparticles. c) Particle size distribution of functionalized iron oxide nanoparticles in deionised water (red), phosphate buffer 18 mM (blue) and NaCl 1 M (green) measured by dynamic light scattering. No significant aggregation of the particles was detected after 1 month in these harsh dispersion environments. Inset pictures show the stable suspensions. d) Hydrodynamic diameter of the bare nanoparticles (black) and functionalized nanoparticles (red) as a function of pH, measured by dynamic light scattering. | ||
2.3. Further derivatization
The NDA–PEG–COOH stabilizer molecule was designed to include a carboxylic acid group to enable derivatization of the nanoparticles with any compound of interest through simple coupling strategies. This property of the functional Fe3O4NP–NDA–PEG–COOH nanoparticles is illustrated here by grafting fluorescein on the nanoparticles as a model molecule. Functionalization of the nanoparticles with aminofluorescein was achieved by standard EDC/NHS carbodiimide amide coupling in water. Briefly, a solution of Fe3O4NP–NDA–PEG–COOH in HEPES buffer at pH = 8 was activated at room temperature by addition of 5 mol% of EDC/NHS for 5 min, followed by addition of 5 mol% fluorescein under stirring at pH = 8 for 16 h. The resulting Fe3O4NP–NDA–PEG–fluorescein nanoparticles were purified by a slightly modified purification procedure compared to the Fe3O4NP–NDA–PEG–COOH nanoparticles, consisting of rotary evaporation followed by one wash with ethanol and two washes with acetone to remove any unreacted fluorescein and EDC/NHS by-products. After purification, 36% of the total iron was recovered and the particles showed negligible aggregation (Fig. 5b).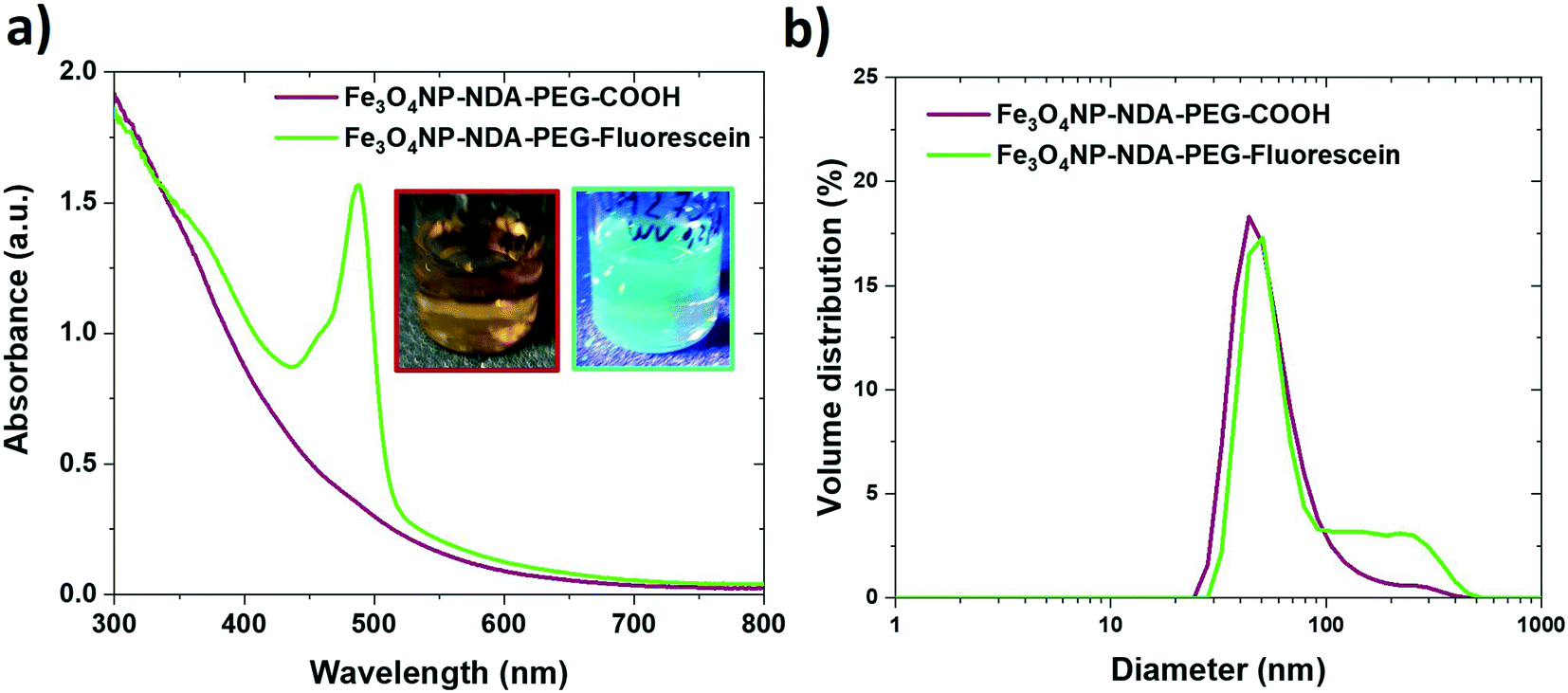 | ||
| Fig. 5 a) UV-visible absorption spectra of functionalized iron oxide nanoparticles before (red) and after coupling with aminofluorescein (green). The appearance of the characteristic absorption peak of fluorescein at 494 nm confirms the successful functionalization. Inset picture show the fluorescein-functionalized particles under visible light illumination (left) and UV illumination (right), exhibiting bright fluorescence. b) Particle size distribution of the functionalized iron oxide nanoparticles before (red) and after coupling with aminofluorescein (green), indicating that negligible aggregation occurs during the coupling process. | ||
The appearance of the characteristic fluorescein peak in the UV-vis spectrum of the nanoparticles confirmed successful fluorescein attachment to the particles (Fig. 5a). Based on the fluorescein absorbance peak height, the drug loading efficiency was estimated at 21% and the drug loading capacity was calculated at 15 wt% which is considered a high drug loading (>10%)51 suitable for drug delivery applications. Fluorescein serves here as a proof-of-concept, illustrating that the Fe3O4NP–NDA–PEG–COOH can act as a flexible nanoparticle platform to which any molecule of interest with an amine group can be coupled to. This strategy can easily be extended to functionalize the nanoparticles with antibodies and aptamers for targeting specific epitopes or with drug molecules such as doxorubicin.
A lactate dehydrogenase (LDH) cytotoxicity assay was performed to assess the biocompatibility of the Fe3O4NP–NDA–PEG–COOH and Fe3O4NP–NDA–PEG–fluorescein nanoparticle samples (Fig. 6a). No significant decrease in cell viability was detected after 24 h and 48 h, even at the highest nanoparticle concentration of 100 μg mL−1. This results supports the fact that the prepared nanoparticles are completely biocompatible and can be used in biomedical applications.
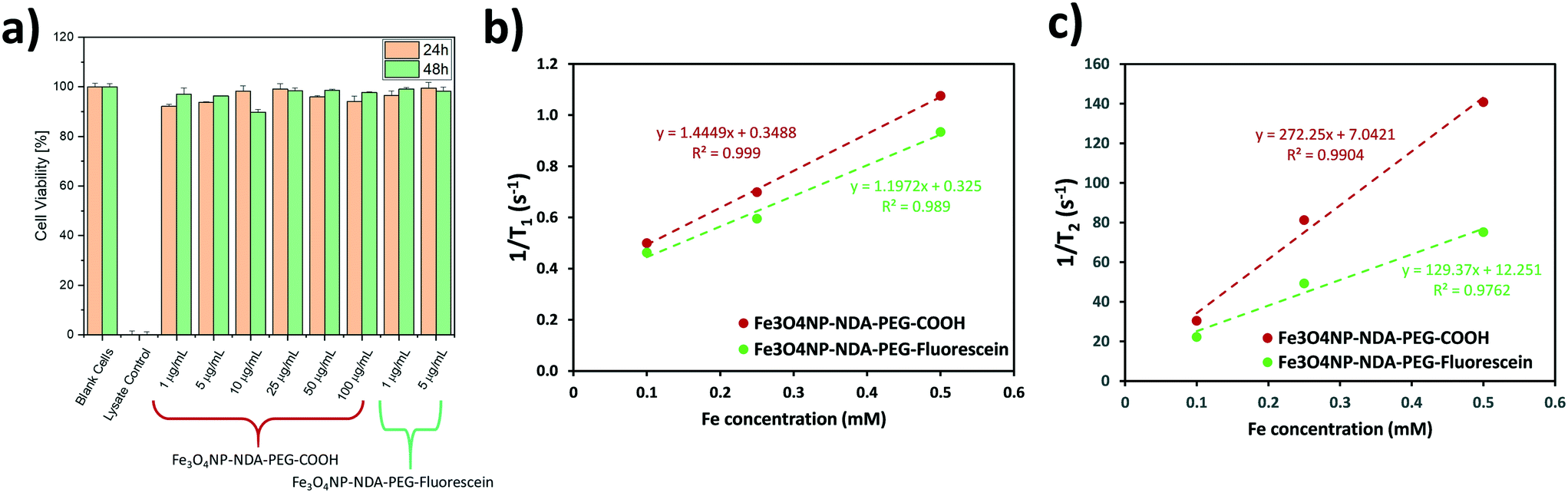 | ||
| Fig. 6 a) Cell viability measured by LDH assay at 24 h and 48 h in the presence of various concentrations of Fe3O4NP–NDA–PEG–COOH and Fe3O4NP–NDA–PEG–fluorescein nanoparticles. b) Inverse T1 and c) T2 relaxation times measured at 7 T as a function of Fe concentration with the corresponding linear regression fitting for Fe3O4NP–NDA–PEG–COOH and Fe3O4NP–NDA–PEG–fluorescein nanoparticles. | ||
T 1 and T2 nuclear magnetic resonance (NMR) relaxation experiments were carried out to determine the longitudinal and transverse relaxation rates respectively of the different functionalized nanoparticle samples to evaluate their possible application as MRI contrast agents. The T1 and T2 relaxation times were measured on a Bruker Biospin DMX 300 spectrometer, using standard inversion-recovery, and Carr–Purcell–Meiboom–Gill pulse sequences. The relaxation rates (1/T1 and 1/T2) were plotted as a function of iron concentration to determine the molar relaxivities (r1 and r2) of each sample by linear regression as shown in Fig. 6b and c. The resulting values are summarized in Table 1. The r2 values are relatively large compared to single core nanoparticle systems which suggests that the Fe3O4NP–NDA–PEG–COOH exist in clusters of multiple iron oxide particles, perhaps because of crosslinking of nanoparticles with free carboxylate moieties.52 Functionalization with fluorescein reduces the r2/r1 ratio, which aligns with the partial crosslinking hypothesis since less carboxylate groups are available in the fluorescein-functionalized particles. All samples demonstrated a high r2/r1 ratio which indicates that this nanoparticle system can act as a good T2-based contrast agent. These values are comparable to commercial iron oxide based contrast agent Resovist®, which demonstrated r1 = 1.67, r2 = 375, r2/r1 = 224 at 9.4 T.53
| Sample | r 1 [mM−1 s−1] | r 2 [mM−1 s−1] | r 2/r1 ratio |
|---|---|---|---|
| Fe3O4NP–NDA–PEG–COOH | 1.44 ± 0.05 | 272 ± 27 | 188 |
| Fe3O4NP–NDA–PEG–fluorescein | 1.20 ± 0.13 | 129 ± 20 | 108 |
2.4. Cost analysis
A full cost analysis of the process was carried out to demonstrate the commercial viability of this approach. Currently, most commercially available iron oxide nanoparticles are stabilized with citric acid, oleic acid or polyvinylpyrrolidone. Only one supplier, OceanNanotech (which also supplies Sigma-Aldrich), was found to offer PEG stabilized iron oxide nanoparticles.54 Note that these nanoparticles are oleic acid capped iron oxide nanoparticles, stabilized with an amphiphilic polymer and they do not present a reactive function for further derivatization. These nanoparticles are currently priced at £ 23000 g−1, which illustrates the need to develop cheaper and scalable alternatives to the traditional batch synthesis processes.
The setup presented in Fig. 1 was considered for the preparation of Fe3O4NP–NDA–PEG–COOH nanoparticles in continuous flow. A single operation period of 8 h was considered, split into 1 h of solution preparation, 5 h of reactor operation and 2 h of work up. Within this operation period, two operating flow rates were considered: 0.2 mL min−1 (which was used in this work) and 10 mL min−1. Taking the preparation and workup times into account, these flow rates correspond to mass production rates of 9.6 mg h−1 and 0.48 g h−1 respectively. It should be noted that in terms of mass, the nanoparticles were considered to be pure magnetite Fe3O4 and the contribution of NDA–PEG–COOH to the final nanoparticle mass was ignored. Therefore, the production rates are likely to be underestimated. An analysis of the chemical cost of the continuous flow reaction was carried out. First of all, the chemical costs for the batch syntheses of NDA (Table S1†) and NDA–PEG–COOH (Table S2†) were calculated, as these are the only non commercially available precursor compounds. Note that both these chemicals can be prepared on a relatively large scale compared to the amount of NDA–PEG–COOH required for a standard operation period of 5 h. Indeed, 3 mmol of NDA–PEG–COOH can be synthesised per batch which represents enough compound for 463 mg of Fe3O4 (i.e. three 5 h-runs in flow at 0.2 mL min−1). Once the cost of the NDA–PEG–COOH stabilizer was determined, the chemical costs for the continuous flow synthesis and functionalization process could be calculated (Table S3†). Interestingly, from a purely chemical perspective, it is clear that the main costs are associated with the solvents used during the purification procedure, as relatively large amounts of solvent are used to purify small quantities of nanoparticles. The chemical costs for the synthesis of Fe3O4NP–NDA–PEG–COOH were evaluated at £ 430 g−1 (compared to £ 61 g−1 without considering the solvents in the work up procedure). Thus, the chemical costs are 86% due to solvent use and optimising the work up procedure could lead to significant cost savings as well as making the process even greener. Similarly, this observation illustrates how using water as the main solvent for the reaction greatly reduces overall costs.
Beyond simply accounting for the cost of the chemicals, a fully accurate cost analysis requires careful consideration of all the associated capital, operational and labour costs of the process. The equipment costs are detailed on Table S4† and were evaluated by assuming a 2-year lifetime averaged over 500 working days. For the energy costs, the nominal power of the hot plate was considered as the power required to drive the pumps is negligible in comparison (Table S5†). Finally for the labour costs, the typical salary of a senior laboratory technician is estimated at £ 30000 per year.55 This value was multiplied by 2.5 to account for National Insurance contributions and other employer costs, resulting in an estimated salary cost of £ 75000 per year or £ 37.5 per h (Table S5†). The overall results of the cost analysis are compiled on Table 2. Note that there will not be any extra labour costs associated with NDA and NDA–PEG–COOH production as both syntheses for these compounds are not time intensive (1 to 2 h each including work up) therefore they can be carried out in parallel with the flow system. When factoring all these parameters, the production cost of the Fe3O4NP–NDA–PEG–COOH was evaluated at £ 3775 g−1 for 0.2 mL min−1 operating flow rate. This production cost could be reduced to £ 506 g−1 (7-fold reduction) by operating at a higher flow rate of 10 mL min−1. At lower operating flow rates, labour costs tend to dominate (86% of total costs at 0.2 mL min−1), while at higher flow rates the chemical costs constitute the bulk of the costs (85% of total costs at 10 mL min−1) (Fig. 7). As anticipated, increasing the production rate leads to an overall decrease in the production cost, as another example of economy of scale. This increase in operating flow rate could easily be achieved and would only require longer reactors to maintain the same residence times and different pump types such as microannular gear pumps to handle larger precursor solution volumes (both changes were factored into an increase in the equipment costs in Table S4†). Note that the production rate could also easily be further increased by numbering up reactors in parallel.
| Flow rate [mL min−1] | 0.2 | 10 | ||||
|---|---|---|---|---|---|---|
| Production rate [g run−1] | 0.077 | 3.8 | ||||
| Costs | Cost per run [£] | Mass cost [£ g−1] | Cost ratio [%] | Cost per run [£] | Mass cost [£ g−1] | Cost ratio [%] |
| Operating flow rates of 0.2 and 10 mL min−1 and a yield of 50% were considered. A run consists of an 8 h work shift with 1 h of set up, 5 h of reactor operation and 2 h of work up. | ||||||
| Chemicals | 40 | 430 | 11 | 1987 | 430 | 85 |
| Reactor | 8 | 87 | 2 | 52 | 11 | 2.2 |
| Energy | 1.0 | 11 | 0.3 | 1.0 | 0.2 | 0.04 |
| Labour | 300 | 3247 | 86 | 300 | 65 | 13 |
| Total | 349 | 3775 | 100 | 2339 | 506 | 100 |
 | ||
| Fig. 7 Pie charts of the distribution of the process operating costs at different flow rates. | ||
Therefore, this approach enables production of iron oxide nanoparticles stabilized by a functional PEG coating at a 1/6 the price of similar commercially available samples, and this cost can be further driven to 1/45 of the commercial samples at higher flow rates (10 mL min−1).
3. Conclusions
In this work, we pioneer the synthesis and functionalization of highly stable PEG-functionalized iron oxide nanoparticles for biomedical application in a single modular and flexible setup. The synthesis of the iron oxide nanoparticles by co-precipitation allows the process to be carried out in water under mild conditions while the continuous flow strategy ensures narrow size distribution, high reproducibility and easy scale up. A custom stabilizing heterobifunctional PEG-based molecule was designed with a nitrodopamine anchor on one side for strong binding to iron oxide and a carboxylic acid moiety on the other side to enable further derivatization by simple amide conjugation. The combination of electrostatically stabilized bare iron oxide nanoparticles with a high affinity nitrodopamine anchor leads to extremely fast and efficient functionalization, requiring only a minimal amount of the PEG-based stabilizer. The derivatization property was illustrated by coupling with fluorescein, conferring fluorescent properties to the iron oxide nanoparticles. The nanoparticles synthesized in this work can act as a platform that can be functionalized with any molecule of interest such as fluorophores, radioactive labels, or antibodies. The functionalized iron oxide nanoparticles were found to be completely biocompatible through a standard cytotoxicity assay and demonstrated good relaxivity values of r1 = 1.44 and r2 = 272 mM−1 s−1 at 7 T, making them suitable for MRI contrast agent applications. To highlight the commercial viability of the processes, a full cost analysis of the process was carried out, showing that the functionalized nanoparticles cost of manufacturing is as low as £ 430 g−1 only accounting for chemicals and £ 3775 g−1 if the capital, operating, and labour costs are included, which is significantly lower (>80% cheaper) than any commercially available similar products. This cost can easily be driven down further to £ 506 g−1 simply by increasing the setup flow rate and throughput. This work demonstrates that continuous flow synthetic methods have the capability to overcome the scale up challenges of complex nanomaterial synthesis and contribute to their deployment in exciting applications beyond lab scale. Furthermore, this novel approach can easily be extended to other materials, particularly metal oxides as nitrocatechols are known to bind strongly to other oxides.4. Experimental section
Materials
FeCl2·4H2O (97%, Alfa Aesar), FeCl3·6H2O (98% Alfa Aesar), 20 wt% aqueous NEt4OH (Sigma-Aldrich), dopamine hydrochloride (Sigma-Aldrich), 5-aminofluorescein (Sigma-Aldrich), sodium nitrite (98%, Alfa Aesar), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (TCI), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid sodium salt (TCI), hydroxylamine hydrochloride (96 + %, Alfa Aesar), 1,10-phenanthroline (TCI), hydrochloric acid 1 M (Fisher scientific), sulfuric acid (98%, Fisher scientific), PEG(COOH)2 (600 g mol−1, Sigma-Aldrich), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (TCI), N-hydrosuccinimide (TCI), LiCl (98+%, Alfa Aesar), ethanol (absolute, Fisher scientific), dimethylformamide (99.8% anhydrous, Sigma-Aldrich), dichloromethane (99.8 + %, stabilized with amylene, Acros organics), acetone (95 + %, Acros organics), hexane (95%, Acros organics) were used as received without any further purification.MilliQ deionized water (18 MΩ) was used for all the experiments.
Characterization
Absorbance spectra of Fe3O4NP–NDA–PEG–COOH and Fe3O4NP–NDA–PEG–fluorescein were recorded using an Agilent Cary 60 ultraviolet-visible (UV-vis) spectrophotometer. Dynamic light scattering (DLS) measurements were obtained using a Malvern Zetasizer Nano S90 instrument with a 633 nm laser.Zeta-potential data was recorded on a Malvern Zetasizer Nano ZS instrument with a 633 nm laser.
Fourier transform infraRed (FTIR) spectra were measured with a SpectrumOne instrument (Perkin Elmer). Transmission electron microscopy grid samples of iron oxide nanoparticle suspensions in water were prepared by drop-coating onto carbon-coated Cu grids (Agar Scientific, 400 mesh). A FEI Tecnai F20 G2 200 kV FEGTEM with a Gatan image filter (GIF) 200 followed by a 4 × 4 k CCD detector was used to acquire transmission electron microscopy images of the samples. Particle size distribution histograms were obtained from measuring at least 100 particles using ImageJ open source software.
Batch synthesis of nitrodopamine
Nitrodopamine was synthesised according to a method from the literature.50 Briefly, 5 mL of 20 wt% sulfuric acid were added dropwise to a solution of dopamine hydrochloride (1 g) and sodium nitrite (1.26 g) in 30 mL of water cooled in an ice bath. The solution was stirred overnight while being allowed to warm to room temperature. The resulting yellow precipitate was filtered and washed with 10 mL of ice-cold water and 10 mL of ice-cold EtOH and dried. 803 mg of nitrodopamine hydrogen sulfate were obtained (71% yield).Batch synthesis of NDA–PEG–COOH
1.8 g (3 mmol) of PEG(COOH)2 (600 g mol−1) were dissolved in 30 mL of dichloromethane. To this solution, 700 μL (3 mmol) of EDC and 460 mg (3 mmol) of NHS were added and stirred for 20 min. A solution of 564 mg (3 mmol) of nitrodopamine hydrogenosulfate in 15 mL of dimethylformamide was added and the final solution was stirred for 16 h. 50 mL of solution of 1 M LiCl adjusted to pH = 1 with HCl was prepared and added to the reaction solution, resulting in the separation of two phases. The bottom dichloromethane layer was collected and evaporated through rotary evaporation at 70 °C and 30 mbar, with a high boiling point solvent trap, in order to remove any leftover dimethylformamide. The final product, a dark brown oily substrate (2.05 g, 89% yield), was dissolved in a minimal amount of deionised water for later use.Continuous iron oxide nanoparticle synthesis and functionalization
Fe3O4NP–NDA–PEG–COOH were prepared through a continuous flow synthetic strategy. All solutions were freshly prepared right before carrying out a synthesis. 26 mg of FeCl2·4H2O and 72 mg FeCl3·6H2O (0.02 M Fe total) were dissolved in 20 mL of 0.01 M HCl. 1.458 mL of 20 wt% NEt4OH were diluted to 20 mL with deionised water to obtain a 0.1 M solution. A 20 mL solution of NDA–PEG–COOH of the desired concentration (typically 0.01 M, 0.5 equiv. compared to Fe) and NEt4OH (2 equiv. compared to NDA–PEG–COOH) was prepared in deionized water. The iron solution and the NEt4OH solutions were then sparged with nitrogen for 1 h to remove dissolved oxygen. The solutions were transferred to a polypropylene syringe and connected to the microreactor setup via Luer-lock connection and injected in the microreactor at a flow rate of 0.2 mL min−1 with a Chemyx 6000 syringe pump. The microreactors consisted of a polytetrafluoroethylene tubing (1/16 inch outer diameter, 1/32 inch inner diameter). The tubing was coiled over 3D printed supports (1 cm diameter). Each precursor solution passed first through a pre-heating region (50 cm), before mixing at a T-mixer (0.02 inch inner diameter) and flowing through the first reactor (2.9 m). For the case of functionalized nanoparticle synthesis, the NDA–PEG–COOH solution was added with a second T-mixer and connected to a second reactor (2.9 m). A solution volume equivalent to two reactor volumes was discarded before collecting any product to ensure steady state was achieved in the microreactor. In between reactions, the microreactor was washed with 1 M HCl and deionized water.Nanoparticle purification procedure
To purify the nanoparticles and remove any excess reactants, the water solvent was evaporated through rotary evaporation at 70 °C. The obtained solid nanoparticle residue was then dispersed in EtOH (2 mL per 10 mL of starting Fe3O4NP–NDA–PEG–COOH solution) followed by precipitation with hexane as an antisolvent (1:1 volume ratio with EtOH). The nanoparticles separation was facilitated with a strong permanent magnet and the supernatant was discarded. This procedure was repeated a total of 3 times. The particles were then dried in a vacuum at room temperature, re-dispersed in a minimal amount of deionised water (typically 4 mL of H2O) and filtered through 200 nm syringe filter.
Further derivatization of the nanoparticles with fluorescein
To functionalize the Fe3O4NP–NDA–PEG–COOH nanoparticles with aminofluorescein, 3 solutions denotated A–C were prepared. All solutions were freshly prepared before being used. Solution A was prepared by diluting 500 μL of stock Fe3O4NP–NDA–PEG–COOH (16.7 mM in [Fe] based on a mass balance, 13.3 mM measured [Fe]), 50 μL of 1 M HEPESNa and 25 μL of HEPES acid in 5 mL of deionised water (final pH = 8). Solution B consisted of EDC (4.2 × 10−5 M, 5 mol% compared to Fe) and NHS (8.4 × 10−5 M, 10 mol% compared to Fe) in 10 mL of deionised water. Solution C consisted of an aminofluorescein solution (9.3 × 10−6 M, 5 mol% compared to Fe) in 4.5 mL of deionised water.Solution B was added to solution A under vigorous stirring (COOH activation). After 5 minutes, solution C was added, and the mixture was stirred at room temperature for 16 h. The HEPES buffer enabled the pH to be maintained at 8 throughout the reaction. The Fe3O4NP–NDA–PEG–fluorescein particles were then purified and recovered by rotary evaporation, followed by washing and sonication (10 min) in EtOH (1 mL per 10 mL of starting solution), magnetic decantation and two more cycles of washing/magnetic decantation with acetone (1 mL per 10 mL of starting solution). Finally, the particles were dried in vacuum at room temperature, re-dispersed in a minimal amount of deionised water (typically 4 mL of H2O) and filtered through 200 nm syringe filter.
Semi-batch reaction procedure to determine functionalization kinetics
The kinetics of the functionalization step were determined through a semi-batch method. Bare iron oxide nanoparticles were synthesised under continuous flow conditions as described above. The resulting suspension (5 mL, 0.01 M in [Fe]) was transferred to a vial with a magnetic stir bar and heated to the desired reaction temperature. A solution of NDA–PEG–COOH and NEt4OH (2 equiv. compared to NDA–PEG–COOH) of the desired concentration was then quickly added and a timer was started. 750 μL aliquots were then extracted and quenched by mixing with 250 μL of MES buffer (0.2 M). The quenched aliquots were then filtered through a 200 nm syringe filter to remove any aggregated nanoparticles.Fe concentration determination
The concentration of Fe in the samples was determined by UV-vis spectrophotometric assay. The samples were dissolved in HCl, then an excess hydroxylamine was added to reduce the Fe3+ to Fe2+ and an excess 1,10-phenanthroline was added to form the red Fe(phen)32+ complex. The absorbance at 511 nm was measured and compared to a calibration curve to determine the Fe concentration through the Lambert–Beer law.Cytotoxicity tests
HEK293 cell line was used in this study to determine the cell toxicity of Fe3O4NP–NDA–PEG–COOH and Fe3O4NP–NDA–PEG–fluorescein nanoparticles. The cells were maintained in T75 flasks with Dulbecco's modified Eagle's medium (DMEM), containing 0.5% penicillin–streptomycin and 10% fetal bovine serum (FBS) at 37 °C and 5% CO2 in an incubator. The cell line was passaged every three days. For passaging, the culture was washed with Dulbecco's phosphate buffered saline (DPBS) and detached from the flask by incubating with 0.7 mL of Trypsin–EDTA at 37 °C and 5% CO2 for 5 min. After adding 8 mL of DMEM, cells were centrifuged at 500 rpm for 5 min and the supernatant removed. The cell pellet was re-suspended with 10 mL of fresh DMEM. To sustain cells, 2.5 mL of cell stock was transferred into a new T75 flask and diluted with DMEM to a final volume of 15 mL.For LDH assay analysis, cells were counted using a disposable haemocytometer (Two Channel C-Chip, NanoEntek) and the stock was diluted with DMEM to a final concentration of 105 cells per mL. 100 μL of 105 cells per mL cell stock solution were seeded into 96 Well plates (ThermoFisher Nunclon, TC treated) and allowed to grow to 80% confluency. Prior to the administration of nanoparticle samples and controls, DMEM was replaced. Next, 20 μL of NP stock solutions were added per well in triplicates and incubated at 37 °C and 5% CO2. 10 μL aliquots were taken of each well at each time point of the study (24 h and 48 h after initial administration) and centrifuged for 5 min on a SciSpin mini centrifuge to remove cell debris. Following the instructions given in the LDH assay manual (LDH Assay Kit ab65393, abcam), 5 μL aliquots were taken of each sample and were treated with 95 μL of freshly prepared LDH assay reaction mix. All samples were transferred in a 96 well plate (ThermoFisher Nunclon, black round bottom) and incubated on a shaker for 10 min. Fluorescence of samples was measured on a microplater reader (Tecan Infinite 200 Pro) at Ex./Em. = 535/587 nm with a bandwidth of 20 nm.
T 1/T2 NMR relaxation measurements
All measurements were performed on a Bruker Biospin Ltd, Avance DMX 300 spectrometer operating at a proton frequency of 300.13 MHz. A 5 mm radio-frequency coil was used and samples were prepared in 5 mm NMR tubes. The 90 degree pulse length was 8.75 μs. A standard Bruker T1 inversion-recovery pulse sequence was used (‘t1ir’) for T1 relaxation time measurements with 12 variable delays, spaced between 10 s and 5 ms. 2 averages were used and the data were processed offline using Microsoft Excel software. T2 relaxation time measurements were performed using a standard Carr–Purcell–Meiboom–Gill (‘cpmg’) spin-echo train, with 1024 echoes; echo times of either 1 ms or 10 ms were used depending upon the particular sample being investigated. 8 averages were acquired. All the data were processed offline using Microsoft Excel software.Author contributions
J. M. and L. T. M. conceived and designed the experiments. J. M. carried out the synthesis experiments and material characterization. C. O. F. and L. F. conducted the cytotoxicity test. M. D. M. conducted the NMR relaxation measurements. J. M. and L. T. M. analyzed the data and wrote the manuscript. M. D. M., L. F. and H. K. P. revised the manuscript. L. F. and L. T. M. provided the funding support. All authors have reviewed the manuscript.Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.Acknowledgements
Julien Mahin and Laura Torrente-Murciano are very thankful for the financial support from the UK Engineering and Physical Science and Research Council (grant number EP/L020443/2). Christoph Otto Franck acknowledges funding by the Engineering and Physical Sciences Research Council Centre for Doctoral Training in Sensor Technologies and Applications (EP/L015889/1) and the support by AstraZeneca in the framework of Project Beacon.References
- Y. Bao, J. A. Sherwood and Z. Sun, J. Mater. Chem. C, 2018, 6, 1280–1290 RSC.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS.
- A. Szpak, S. Fiejdasz, W. Prendota, T. Strączek, C. Kapusta, J. Szmyd, M. Nowakowska and S. Zapotoczny, J. Nanopart. Res., 2014, 16, 2678 CrossRef PubMed.
- H. Wei, O. T. Bruns, M. G. Kaul, E. C. Hansen, M. Barch, A. Wiśniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- L. Yang, Z. Cao, H. K. Sajja, H. Mao, L. Wang, H. Geng, H. Xu, T. Jiang, W. C. Wood, S. Nie and Y. A. Wang, J. Biomed. Nanotechnol., 2008, 4, 439–449 CrossRef CAS PubMed.
- M. M. Lin, D. K. Kim, A. J. El Haj and J. Dobson, IEEE Trans. Nanobioscience, 2008, 7, 298–305 Search PubMed.
- A. Hervault and N. T. K. Thanh, Nanoscale, 2014, 6, 11553–11573 RSC.
- B. Kozissnik, A. C. Bohorquez, J. Dobson and C. Rinaldi, Int. J. Hyperthermia, 2013, 29, 706–714 CrossRef PubMed.
- C. S. S. R. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789–808 CrossRef CAS.
- H. K. Patra, N. U. Khaliq, T. Romu, E. Wiechec, M. Borga, A. P. F. Turner and A. Tiwari, Adv. Healthcare Mater., 2014, 3, 526–535 CrossRef CAS PubMed.
- H. K. Patra, M. Azharuddin, M. M. Islam, G. Papapavlou, S. Deb, J. Osterrieth, G. H. Zhu, T. Romu, A. K. Dhara, M. J. Jafari, A. Ghaderi, J. Hinkula, M. S. Rajan and N. K. H. Slater, Adv. Funct. Mater., 2019, 29, 1903760 CrossRef.
- N. Manuchehrabadi, Z. Gao, J. Zhang, H. L. Ring, Q. Shao, F. Liu, M. McDermott, A. Fok, Y. Rabin, K. G. M. Brockbank, M. Garwood, C. L. Haynes and J. C. Bischof, Sci. Transl. Med., 2017, 9(379) DOI:10.1126/scitranslmed.aah4586.
- A. Chiu-Lam, E. Staples, C. J. Pepine and C. Rinaldi, Sci. Adv., 2021, 7, eabe3005 CrossRef.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS.
- J. W. M. Bulte, Am. J. Roentgenol., 2009, 193, 314–325 CrossRef PubMed.
- M. H. Schwenk, Pharmacotherapy, 2010, 30, 70–79 CrossRef CAS PubMed.
- R. M. Patil, N. D. Thorat, P. B. Shete, P. A. Bedge, S. Gavde, M. G. Joshi, S. A. M. Tofail and R. A. Bohara, Biochem. Biophys. Rep., 2018, 13, 63–72 CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- S. Zürcher, D. Wäckerlin, Y. Bethuel, B. Malisova, M. Textor, S. Tosatti and K. Gademann, J. Am. Chem. Soc., 2006, 128, 1064–1065 CrossRef.
- E. Amstad, S. Zurcher, A. Mashaghi, J. Y. Wong, M. Textor and E. Reimhult, Small, 2009, 5, 1334–1342 CrossRef CAS PubMed.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819 RSC.
- S. D. Anderson, V. V. Gwenin and C. D. Gwenin, Nanoscale Res. Lett., 2019, 14, 188 CrossRef PubMed.
- A. Walter, A. Garofalo, A. Parat, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Nanotechnol. Rev., 2015, 4, 581–593 CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- J. Zeng, L. Jing, Y. Hou, M. Jiao, R. Qiao, Q. Jia, C. Liu, F. Fang, H. Lei and M. Gao, Adv. Mater., 2014, 26, 2694–2698 CrossRef CAS PubMed.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van Den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2013, 7, 500–512 CrossRef CAS PubMed.
- A. Lassenberger, O. Bixner, T. Gruenewald, H. Lichtenegger, R. Zirbs and E. Reimhult, Langmuir, 2016, 32, 4259–4269 CrossRef CAS.
- S. Peng, C. Wang, J. Xie and S. Sun, J. Am. Chem. Soc., 2006, 128, 10676–10677 CrossRef CAS.
- P. R. Makgwane and S. S. Ray, J. Nanosci. Nanotechnol., 2014, 14, 1338–1363 CrossRef CAS PubMed.
- J. Mahin and L. Torrente-Murciano, Chem. Eng. J., 2020, 396, 125299 CrossRef CAS.
- M. O. Besenhard, A. P. LaGrow, S. Famiani, M. Pucciarelli, P. Lettieri, N. T. K. Thanh and A. Gavriilidis, React. Chem. Eng., 2020, 5, 1474–1483 RSC.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC.
- K.-J. Wu, G. M. De Varine Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116–128 RSC.
- K.-J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267–276 RSC.
- Y. Gao, B. Pinho and L. Torrente-Murciano, Curr. Opin. Chem. Eng., 2020, 29, 26–33 CrossRef.
- B. Pinho and L. Torrente-Murciano, React. Chem. Eng., 2020, 5, 342–355 RSC.
- L. Frenz, A. El Harrak, M. Pauly, S. Bégin-Colin, A. D. Griffiths and J. C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS.
- W. Bin Lee, C. H. Weng, F. Y. Cheng, C. S. Yeh, H. Y. Lei and G. Bin Lee, Biomed. Microdevices, 2009, 11, 161–171 CrossRef.
- M. O. Besenhard, A. P. LaGrow, A. Hodzic, M. Kriechbaum, L. Panariello, G. Bais, K. Loizou, S. Damilos, M. Margarida Cruz, N. T. K. Thanh and A. Gavriilidis, Chem. Eng. J., 2020, 399, 125740 CrossRef CAS.
- G. Thomas, F. Demoisson, J. Boudon and N. Millot, Dalton Trans., 2016, 45, 10821–10829 RSC.
- K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. Demello and J. C. Demello, J. Mater. Chem., 2012, 22, 4704–4708 RSC.
- A. Abou Hassan, O. Sandre, V. Cabuil and P. Tabeling, Chem. Commun., 2008, 1783–1785 RSC.
- M. Mascolo, Y. Pei and T. Ring, Materials, 2013, 6, 5549–5567 CrossRef CAS.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- A. Napolitano, M. d'Ischia, C. Costantini and G. Prota, Tetrahedron, 1992, 48, 8515–8522 CrossRef CAS.
- Y. Liu, G. Yang, S. Jin, L. Xu and C. X. Zhao, ChemPlusChem, 2020, 85, 2143–2157 CrossRef CAS PubMed.
- C. Blanco-Andujar, A. Walter, G. Cotin, C. Bordeianu, D. Mertz, D. Felder-Flesch and S. Begin-Colin, Nanomedicine, 2016, 11, 1889–1910 CrossRef CAS.
- A. Jedlovszky-Hajdú, E. Tombácz, I. Bányai, M. Babos and A. Palkó, J. Magn. Magn. Mater., 2012, 324, 3173–3180 CrossRef.
- https://oceannanotech.alwayly.com/products-type/iron-oxide-nanoparticles-5-30nm/functionalized-iron-oxide-nanoparticles/peg-iron-oxide-nanoparticles/peg-iron-oxide-nanoparticles-636.html, (accessed 5 January 2021).
- https://nationalcareers.service.gov.uk/job-profiles/laboratory-technician, (accessed 5 January 2021).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00239b |
| ‡ Present address: Department of Surgical Biotechnology, UCL Division of Surgery and Interventional Science, Royal Free Campus, Rowland Hill Street, London, NW3 2PF, UK |
| This journal is © The Royal Society of Chemistry 2021 |