
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scaffold-based [Fe]-hydrogenase model: H2 activation initiates Fe(0)-hydride extrusion and non-biomimetic hydride transfer†
Spencer A.
Kerns‡
 a,
Junhyeok
Seo‡
b,
Vincent M.
Lynch
a,
Jason
Shearer
c,
Sean T.
Goralski
a,
Eileen R.
Sullivan
a and
Michael J.
Rose
*a
a,
Junhyeok
Seo‡
b,
Vincent M.
Lynch
a,
Jason
Shearer
c,
Sean T.
Goralski
a,
Eileen R.
Sullivan
a and
Michael J.
Rose
*a
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mrose@cm.utexas.edu
bDepartment of Chemistry, Gwangju Institute of Science and Technology, Gwangju 61005, Republic of Korea
cDepartment of Chemistry, Trinity University, One Trinity Place, San Antonio, Texas 78212, USA
First published on 10th September 2021
Abstract
We report the synthesis and reactivity of a model of [Fe]-hydrogenase derived from an anthracene-based scaffold that includes the endogenous, organometallic acyl(methylene) donor. In comparison to other non-scaffolded acyl-containing complexes, the complex described herein retains molecularly well-defined chemistry upon addition of multiple equivalents of exogenous base. Clean deprotonation of the acyl(methylene) C–H bond with a phenolate base results in the formation of a dimeric motif that contains a new Fe–C(methine) bond resulting from coordination of the deprotonated methylene unit to an adjacent iron center. This effective second carbanion in the ligand framework was demonstrated to drive heterolytic H2 activation across the Fe(II) center. However, this process results in reductive elimination and liberation of the ligand to extrude a lower-valent Fe–carbonyl complex. Through a series of isotopic labelling experiments, structural characterization (XRD, XAS), and spectroscopic characterization (IR, NMR, EXAFS), a mechanistic pathway is presented for H2/hydride-induced loss of the organometallic acyl unit (i.e. pyCH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O → pyCH3+C
O → pyCH3+C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O). The known reduced hydride species [HFe(CO)4]− and [HFe3(CO)11]− have been observed as products by 1H/2H NMR and IR spectroscopies, as well as independent syntheses of PNP[HFe(CO)4]. The former species (i.e. [HFe(CO)4]−) is deduced to be the actual hydride transfer agent in the hydride transfer reaction (nominally catalyzed by the title compound) to a biomimetic substrate ([TolIm](BArF) = fluorinated imidazolium as hydride acceptor). This work provides mechanistic insight into the reasons for lack of functional biomimetic behavior (hydride transfer) in acyl(methylene)pyridine based mimics of [Fe]-hydrogenase.
O). The known reduced hydride species [HFe(CO)4]− and [HFe3(CO)11]− have been observed as products by 1H/2H NMR and IR spectroscopies, as well as independent syntheses of PNP[HFe(CO)4]. The former species (i.e. [HFe(CO)4]−) is deduced to be the actual hydride transfer agent in the hydride transfer reaction (nominally catalyzed by the title compound) to a biomimetic substrate ([TolIm](BArF) = fluorinated imidazolium as hydride acceptor). This work provides mechanistic insight into the reasons for lack of functional biomimetic behavior (hydride transfer) in acyl(methylene)pyridine based mimics of [Fe]-hydrogenase.
Introduction
The search for earth abundant substitutes for precious metal catalysts in energy-related chemical transformations has led researchers to investigate biological precedents that utilize first-row transition metals.1–6 Of these enzymes, the [FeFe] and [NiFe] H2ases have been studied in detail for their redox active sites for the generation and metabolism of dihydrogen (H2).7–9 Less studied is the ‘third hydrogenase’ — namely the redox inactive [Fe]-hydrogenase (Hmd). The single iron site in this enzyme heterolytically activates H2 and catalyzes hydride transfer to the C1 carrier substrate methenyl-tetrahydromethanopterin (H4MPT+, Scheme 1), thus generating methylene-tetrahydromethanopterin (H4MPT).10 The refined crystal structure reported by Shima in 2009 identified the active site environment,11,12 and a 2019 report13 described the crystallized enzyme in both the open (inactive) and closed (active, substrate-bound) conformations. The latter report precisely defined the proximity of the H4MPT+ hydride transfer substrate to the iron center, and proposed detailed a mechanism of H2 activation and hydride transfer using QM/MM calculations14 based on the new protein crystal structures.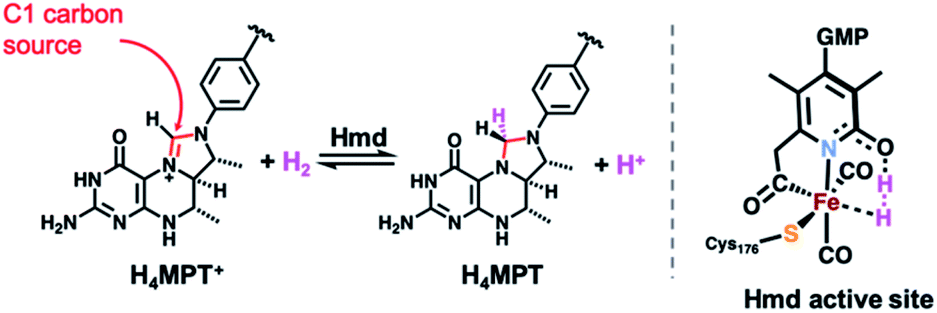 | ||
| Scheme 1 H2 activation and hydride transfer reaction catalyzed by Hmd (left) and active site's putative key intermediate in H2 activation and hydride transfer (right). | ||
Since 2009, researchers have significantly advanced structural models of Hmd. However, the scope of functional mimics of Hmd remains limited. Hu and coworkers developed functional systems derived from hybrid molecule|protein systems15 and a small molecule system that incorporates an abiotic diphosphine ligand with a pendant amine base.16 Our group has reported model systems capable of hydride abstraction17 (the enzymatic ‘reverse’ reaction) and hydride transfer18 (enzymatic ‘forward’ reaction) with biomimetic substrates. However, both of our reported systems replicated the strong trans influence of the Fe–Cacyl σ bond in the form of ‘carbamoyl’ ligation (i.e. –NHCO) as a synthetically more accessible proxy for the endogenous methylene-containing acyl unit (i.e. –CH2CO); synthesis of the former was originally demonstrated by Pickett.19,20 Indeed, the preparation of acyl-containing synthetic systems that rigorously replicate the primary coordination sphere of Hmd and exhibit biomimetic reactivity has proven to be a particular challenge due the inherent instability of such compounds and their apparent — and as yet unexplained — sensitivity to base.
In this report, we have more faithfully replicated the Hmd active site in comparison to our previous work by installing the biomimetic methylene linkage. Our synthetic approach uniquely uses an ‘anthracene scaffold’ that provides an accurate and stable means of emulating the biomimetic fac-CNS ligation motif. We first describe the synthesis of the model complex and its well-described reactivity in the presence of base. We then demonstrate functional H2 activation by a deprotonated iron-acyl model complex that results in liberation of ligand and reduction of the Fe center instead of hydride transfer to a model substrate. Additional base in solution did, in fact, result in successful hydride transfer to the model substrate. However, through a series of control experiments we identify the active hydride transfer agent as the tetracarbonylhydridoferrate species, [HFe(CO)4]−. Lastly, we describe a mechanistic pathway for reductive conversion of the Fe-acyl unit based on our observations from the structural (XRD, XAS, EXAFS) and spectroscopic (1H/2H NMR, IR) data collected. These observations provide clear benchmarks and ‘warning signs’ of false positives for other researchers working in the area of biomimetic [Fe]-hydrogenase systems.
Results and discussion
Ligand and metal complex syntheses
The desired methylpyridine/thioether ligand Anth·CH3NSMe (Scheme 2) was synthesized via selective mono-coupling of the 2-methylpyridine unit to 1,8-dichloroanthracene, followed by introduction of the aryl-thioether moiety. Briefly, 5-bromo-2-methylpyridine undergoes tandem borylation/Suzuki coupling using B2Pin2, Pd2(dba)3/SPhos (∼2 mol%), and weak base (KOAc). The 1,8-dichloroanthracene unit then coupled with the in situ prepared boronic acid, affording the asymmetric synthon Anth·CH3N·Cl (58% yield, 2.07 g). Subsequent coupling of Anth·CH3N·Cl to 3-(methylthio)phenylboronic acid catalyzed by Pd2(dba)3/XPhos (4 mol%) afforded the target ligand Anth·CH3NSMe (Fig. S1†) in good yield (70%, 1.58 g). Similar to reported procedures,21 the methylpyridine moiety of the Anth·CH3NSMe was lithiated with nBuLi in THF at 0 °C, followed by addition of Fe(CO)5 (−80 → −20 °C) and Br2 (−70 °C) to generate the target complex [(Anth·CH2NSMe)Fe(CO)2(Br)] (1) in 77% yield.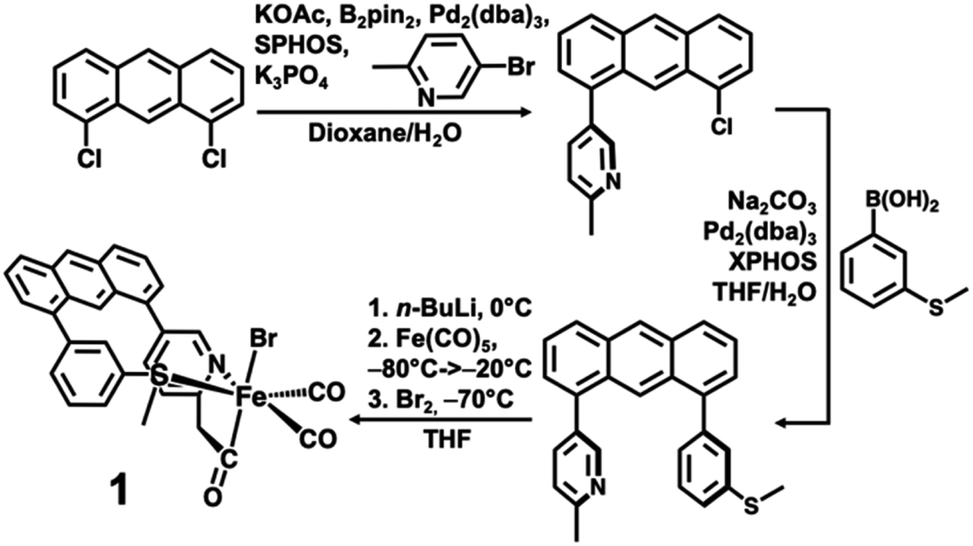 | ||
| Scheme 2 Ligand and metal complex syntheses. | ||
The 1H NMR spectrum of 1 in d8-THF solution (Fig. S2†) exhibits diamagnetic proton resonances with the characteristic methylene proton resonances observed as diastereotopic doublets at 3.97 and 4.52 ppm consistent with the ligation of the anionic acyl (–CH2CO) group to the iron center. The 13C NMR under 1 atm 13CO (Fig. S3†) revealed the iron-bound carbon of the acyl moiety (δ 254 ppm) to be exchangeable (t1/2 ≈ 3 d), while the 13CO ligands exchange slightly faster (t1/2 ≈ 2 d). Facile CO exchange of the acyl moiety was also reported in a complex reported by Hu.21
Attempts at isolation of single crystals of 1 were unsuccessful. Structural evidence supporting the core motif of 1 was obtained from the derivative complex bound with AsPh3. Addition of one equiv. of AsPh3 to 1 enabled the isolation of single crystals of the closely related complex [(Anth·CH2NSoff)Fe(CO)2(Br)(AsPh3)] (Fig. 1). The AsPh3 adduct exhibits fac-arrangement of the C, N, As donor atoms, with the AsPh3 ligand displacing the thioether-S ligand. The orthogonal face is occupied by cis carbonyl ligands and the bromide is located trans to the acyl–C ligand as proposed in the structure of 1. Upon coordination of AsPh3, a small red-shift is observed in the ν(CO) stretches to 2024 and 1971 cm−1 and a notable blue-shift (∼13 cm−1) to 1642 cm−1 is observed in ν(CO) stretch of the acyl unit (Fig. S22†). Notably, the bound state of the original thioether-S in 1 was supported by XPS analysis (Fig. S36†).
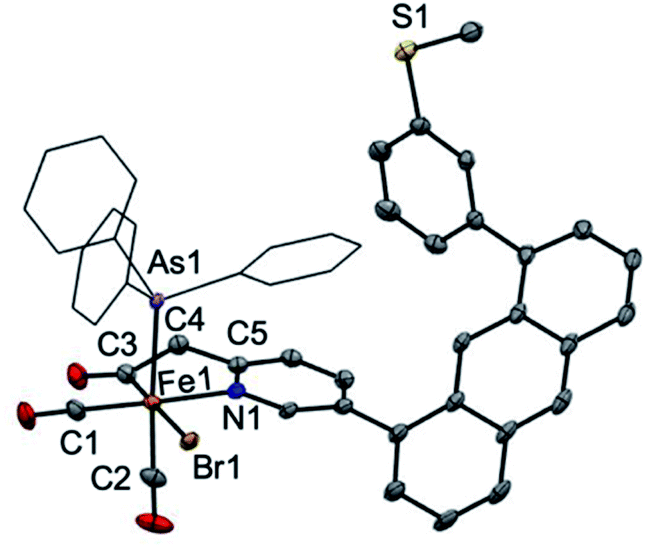 | ||
| Fig. 1 ORTEP representation (30% thermal ellipsoids) for [(Anth·CH2NSoff)Fe(CO)2(Br)(AsPh3)]. Hydrogen atoms are omitted for clarity and the phenyl groups of AsPh3 are depicted as wireframes. Selected bond distances (Å) and bond angles (°): Fe1–C3 = 1.942(3), Fe1–N1 = 2.036(2), Fe1–As1 = 2.4050(5), C3–Fe1–N1 = 83.55(11), C3–Fe1–As1 = 91.76(11), N1–Fe1–As1 = 88.97(6). | ||
Methylene-acyl deprotonation by exogenous base
It is proposed that Hmd utilizes the pendant pyridonate-O as a proton acceptor to facilitate heterolytic cleavage of H2. Due to the absence of this basic functionality in the present ligand design, we previously reported18 a system in which a bulky phenolate base, NEt4[MeOtBu2ArO], participated in H2 activation to ultimately drive hydride transfer. We thus attempted the analogous H2 activation in the presence of this base. However, in a synthetic scale reaction, treatment of 1 in THF with one equiv. NEt4[MeOtBu2ArO] immediately generated a red-orange solution, accompanied by a precipitate (NEt4Br). This contrasts carbamoyl-based systems (NH linkage, not CH2), wherein no direct reaction with the same bulky phenolate is observed. Concentration of the filtered solution and successive washes with pentane and Et2O removed the protonated phenol byproduct (MeOtBu2ArOH), which was identified by 1H NMR.Extraction of the resulting powder into MeCN produced X-ray quality crystals at −20 °C. The resulting structure (Fig. 2) revealed a remarkable result: a dimeric complex in which two iron centers bridge via the formation of a new Fe–C bond between the deprotonated methine-C (formerly the methylene unit) of adjacent, identical units. The new dimeric species is formulated as [(Anth·CHNSoff)Fe(CO)2(MeCN)]2 (2). The bond distances of the new bridging Fe–C bonds are quite long at 2.186(6) and 2.194(6) Å. These bond distances are significantly longer than the Fe–Cacyl bonds at 1.973(7) and 1.943(7) Å.23 Notably, the C–C and C–N bond lengths in the pyridine ring of 2 do not significantly deviate from those observed in [(Anth·CH2NSoff)Fe(CO)2(Br)(AsPh3)] and are thus inconsistent with de-aromatization observed in other methylene-bridged pincer systems upon deprotonation.22–25
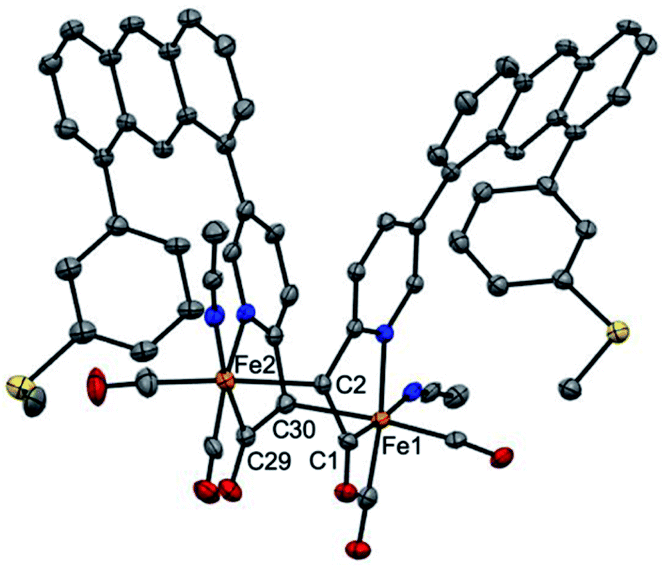 | ||
| Fig. 2 ORTEP diagram (30% thermal ellipsoids) of one of two molecules of 2 in the asymmetric part of the unit cell; hydrogen atoms and solvent molecules are omitted for clarity. Selected bond distances (Å): Fe1–C1 = 1.943(7), Fe1–C30 = 2.186(6), Fe2–C29 = 1.973(7), Fe2–C2 = 2.194(6). | ||
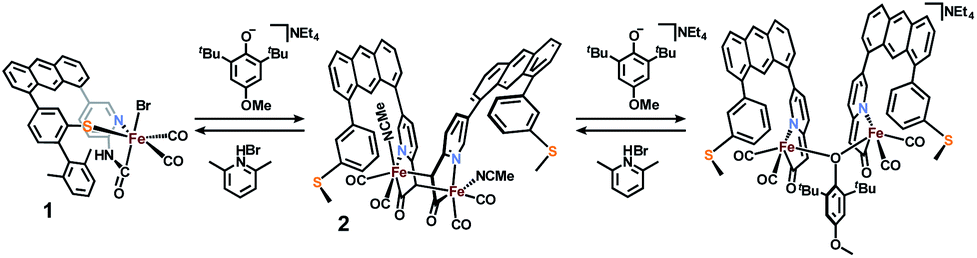
Deprotonation of a methylene proton was also evident through shifts in the IR spectrum and changes in the 1H NMR spectrum resulting from base addition. The solution ν(CO) features in the IR spectrum of 1 (2021, 1956 cm−1) red-shifted significantly to 2005, 1947 cm−1 upon addition of base. The expected four ν(CO) features for the C2-symmetric dimer 2 are only observable in the ground crystalline sample at 2021, 1998, 1962, and 1943 cm−1 (Fig. S23†). The deprotonation event (Scheme 3) resulting in generation of 2 was also achieved with weaker bases such as NEt4[p-BrtBu2ArO] or NEt4[p-CNtBu2ArO] but not NEt4[p-NO2tBu2ArO] — underscoring the surprising acidity of this C–H bond. The deprotonation was clearly reversible upon addition of one equiv. of the weak acid Lut·HBr (2021, 1955 cm−1) (Fig. 3). This conversion was also evidenced in the 1H NMR spectrum by disappearance of the characteristic diastereotopic methylene proton resonances of 1, and a new resonance at 4.45 ppm in 2.
 | ||
| Scheme 3 Reversible deprotonation of 1 to form 2, and proposed bridging coordination of base. Note that the sequence to isolate 2 was performed in MeCN, while the sequence to examine the base-bridged dimer (far right) by EXAFS was performed in THF. | ||
 | ||
| Fig. 3 IR spectra demonstrating reversible deprotonation of monomeric 1 (top, black line) to form dimeric 2 (middle, red line), and protonation of 2 to regenerate 1 (bottom, blue line). | ||
The structure of 2 unequivocally confirms deprotonation of the methylene proton as proposed (but not unambiguously proven) in another acyl-containing model compound (a mer-CNS dicarbonyl) recently published by our group,26 suggesting that this mechanism is broadly applicable. Furthermore, deprotonation of the methylene-acyl moiety has been observed in another model compound by Song and coworkers through a suggested keto–enol tautomerization and acylation mechanism, although the analogous intermediate was not identified in that case.27 These observations suggest that this acyl moiety is rather reactive, and must be fully understood in structural and functional synthetic mimics of this enzyme. Indeed, exogenous base has been noted to decompose previous non-scaffolded acyl-containing model compounds,16 perhaps related to this process. The scaffold-supported {Fe(CO)2}2+ motif of complex 2, however, is stable and even accommodates further addition of base.
Bridging coordination of base to the Fe centers (XAS)
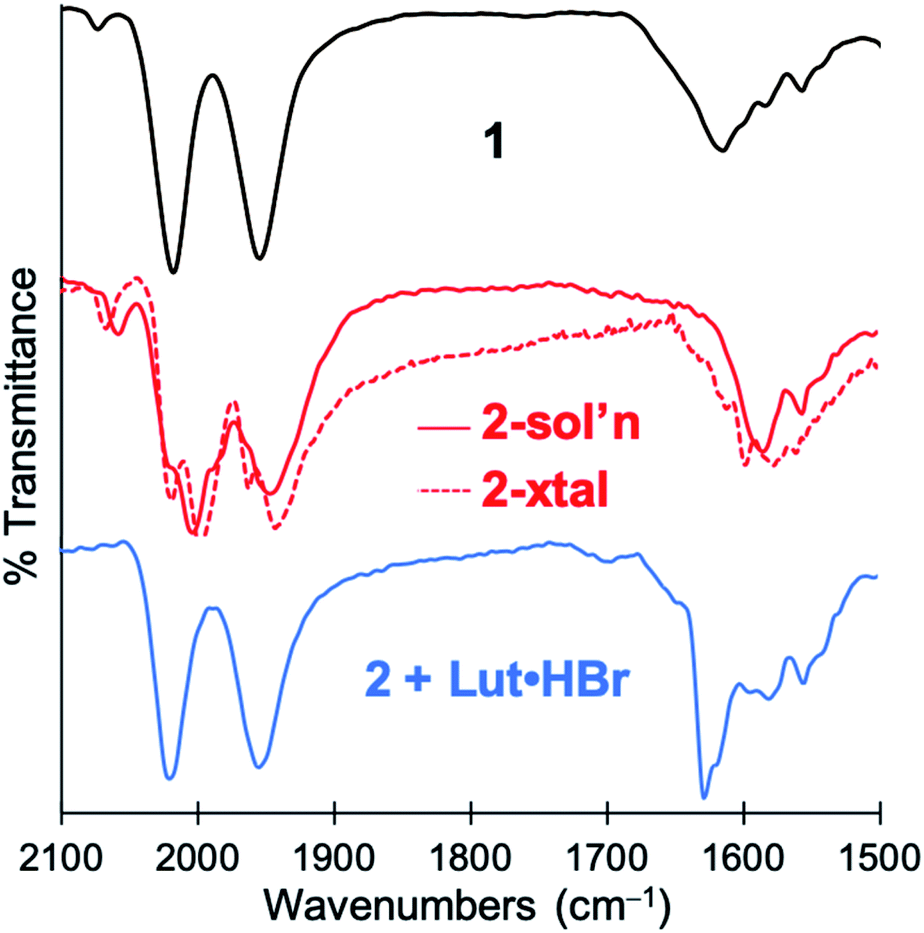
Treatment of 1 with two equiv. of NEt4[MeOtBu2ArO] in THF resulted in a more dramatic color change from orange to dark red. Additionally, the IR spectrum of the resulting solution exhibited further red-shifted carbonyl stretching frequencies observed at 1996 and 1923 cm−1 (Fig. S24†) in comparison to 1 or 2. The significant red-shift is consistent with binding of the anionic phenolate donor to displace the Fe–Cmethine bonds. Coordination of bridging or terminal 2,6-di-tert-butylphenolates is not unprecedented in the generation of low-coordinate iron centers.28,29 The fully reversible nature of this event was demonstrated by treatment of the dark red solution with two equiv. of 2,6-lutidine·HBr to re-generate a solution of 1 as followed by IR spectroscopy (Fig. S25†).Attempts to determine the molecular structure resulting from the treatment of 1 with two equiv. of NEt4[MeOtBu2ArO] (or, equally, treatment of 2 with one equiv. of base) by X-ray crystallography were unsuccessful. The resulting species was thus probed by iron K-edge X-ray absorption spectroscopy (Fig. 4). The XANES region of the iron K-edge X-ray absorption spectrum displays a pronounced pre-edge peak at 7113.5(1) eV corresponding to a nominal Fe(1s → 3d) transition (Fig. 4A); the intensity of this peak is consistent with iron contained in a non-centrosymmetric coordination environment (e.g. 5-coordinate distorted square pyramidal).30 The EXAFS data for 1 treated with two equiv. of base are best modeled as a dimer of five-coordinate Fe centers ligated by two short CO ligands at 1.77 Å and three additional light atom ligand donors, modeled as N-scatterers, at 2.03 Å, which is similar to the two short carbonyl ligands (1.79 Å) and 3–4 light atom donors, modeled as N-scatterers, at 2.05 Å obtained from the model to the EXAFS data for 2. It is therefore likely that the three light-atom ligand donors modeled at 2.03 Å in 1 treated with two equiv. of NEt4[MeOtBu2ArO] — are the acyl-C donor, a pyridine-N donor, and an additional coordinated phenolate-O donor. The Fe–CO bond length observed in 1 with two equiv. of NEt4[MeOtBu2ArO] is slightly shorter than the average Fe–CO distance observed in 2, and is consistent with the increased π-backbonding as corroborated by the red-shifted carbonyl stretching frequencies. In addition to the Fe–CO significant multiple scattering pathways found between R′ = 2.5–3.5 Å in the Fourier transform, which dominates the EXAFS of both 1 treated with two equiv. of NEt4[MeOtBu2ArO] and 2, an Fe⋯Fe vector could also be located. For 1 treated with two equiv. of NEt4[MeOtBu2ArO], the Fe⋯Fe vector is found at 3.44 Å; a wavelet transform of the EXAFS data of 2 clearly shows the Fe⋯Fe single scattering pathway is resolvable from the Fe–CO multiple scattering pathways, supporting this assignment (Fig. S42†). In contrast, the XAS data for 2 yields an Fe⋯Fe single scattering pathway at 3.80 Å, which is consistent with the crystallographic results. Taken together, these data are fully consistent with the formulation of 1 with two equiv. of NEt4[MeOtBu2ArO] as a phenoxyl-bridged Fe⋯Fe dimer (Fig. 4).
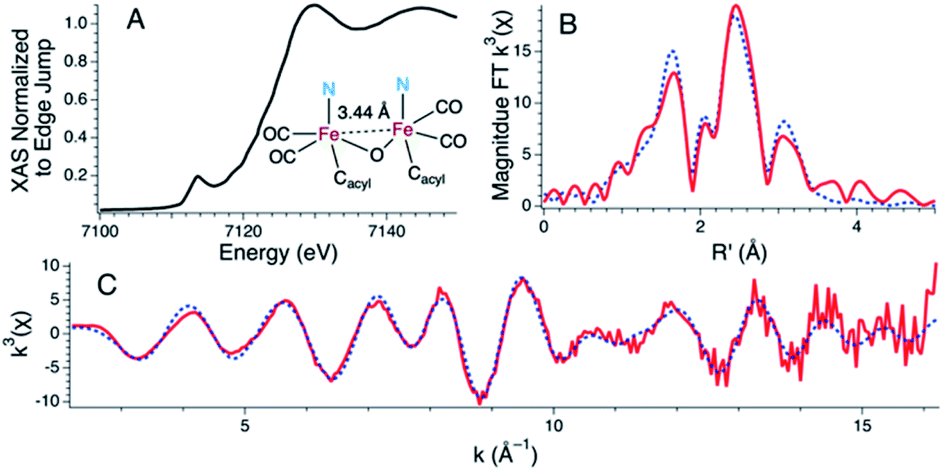 | ||
| Fig. 4 Fe K-edge X-ray absorption data for 1 following treatment with two equiv. of NEt4[MeOtBu2ArO] base with an inset ChemDraw representation of the proposed primary coordination sphere as modeled by the EXAFS data. (A) The XANES region of the Fe K-edge X-ray absorption spectrum (B) The magnitude Fourier transform of the k3-weighted EXAFS data depicted with experimental data (red line) overlaid on the best fit model (dotted blue line) (C) k3-weighted EXAFS data are depicted with experimental data (red line) overlaid on the best fit model (dotted blue line). Reported Model: Eo = 7131.9 eV; Shell #1 (Fe–CO): N = 2, R = 1.767(2) Å, σ2 = 0.002(1) Å2, Fe–C–O θ = 176.8(7)°, Fe⋯O R′ = 2.883(3) Å; Shell #2 (Fe–N) N = 3, R = 2.028(9) Å, σ2 = 0.004(2) Å2; Shell #3 (Fe–Fe) N = 1, R = 3.442(4) Å, σ2 = 0.002(1) Å2; Shell #4 (Fe–C): N = 3.2(4), R = 2.54(1) Å, σ2 = 0.005(1) Å2. ε2 = 1.51 (over the range of k = 2.2–16.2 Å and R = 1.00–3.75 Å). | ||
Biomimetic H2 activation by the first dimer (2)
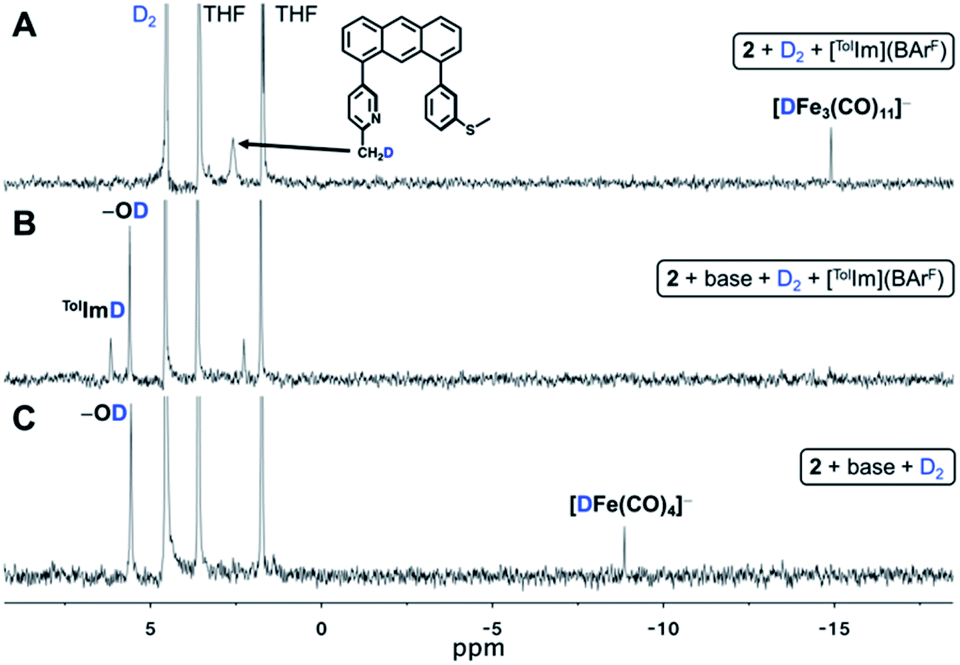 | ||
| Fig. 5 2H NMR spectra demonstrating (A) D2 activation by 2 in the presence of model substrate [TolIm](BArF). Deuterium labelling is observed at 2.59 and −14.90 ppm, corresponding to the formation of D-labelled free Anth·CH2DNSMe ligand and [DFe3(CO)11]−, respectively; (B) D2 activation by 2 in the presence of an additional equivalent of base and model substrate [TolIm](BArF). Deuterium labelling is observed at 5.57 and 6.11 ppm, corresponding to the formation of D-labelled MeOtBu2ArOD and TolImD, respectively; (C) D2 activation by 2 in the presence of an additional equivalent of base. Deuterium labelling is observed at 5.56 and −8.87 ppm, corresponding to the formation of D-labelled MeOtBu2ArOD and [DFe(CO)4]−, respectively. | ||
Competitive formation of reduced Fe-carbonyl species
We serendipitously obtained dark red crystals from the THF solution of both the H2/D2 and NaHBEt3 reactions in the NMR reaction tube which were — contrary to our optimistic expectation — identified as the known di-iron carbonyl dianion (NEt4)2[Fe2(CO)8] by X-ray diffraction, proving the reduction of the ferrous starting material to Fe(−1). Provided the overwhelming evidence of reductive chemistry and our previous observance of unbound ligand, we considered a conversion pathway to better explain the formation of (NEt4)2[Fe2(CO)8] (Scheme 4) in the context of the observed Fe–H or D resonance and extrusion of the metal center from the anthracene scaffold.
 | ||
| Scheme 4 Mechanistic pathway for the reactions of the iron-acyl unit with H2 and substrate, with the corroborating structural and spectroscopic evidence as indicated for both the observed and proposed intermediates. | ||
We first contemplated the retrosynthesis of the observed (NEt4)2[Fe2(CO)8] product, hypothesizing its derivation from bond formation between two simple {Fe(CO)4} building blocks. Upon inspection of known, simple iron tetracarbonyl compounds, we intuited that the product could be derived from initial protonation or deprotonation of one NEt4[HFe(CO)4] unit, thus providing the necessary 2e− for the reduction of 2Fe0 to 2Fe−1, concomitant with generation of H2 (i.e. Fe0–H + B → (Fe2− + BH) + Fe0–H → 2Fe1− + H2 + B). Furthermore, the 1H NMR resonance of the Fe–H of NEt4[HFe(CO)4] was previously reported at −8.8 ppm (d8-THF),32,33 which is obviously consistent with the Fe–H resonance (δ H/D ≈ −8.8) observed upon H2 activation in our studies. To confirm this hypothesis, we independently synthesized PPN[HFe(CO)4] (Fig. S11 and S26†) according to literature procedure32 and treated it with one equiv. of NEt4[MeOtBu2ArO] base to deprotonate the Fe–H species. Within minutes of base addition, we observed line broadening in the 1H NMR spectrum (Fig. S12†), consistent with reduction to form the intermediate paramagnetic Fe(−1) species concomitant with formation of a red precipitate, confirmed as (NEt4)2[Fe2(CO)8] by IR spectroscopy (Fig. S28†). Indeed, PPN[HFe(CO)4] is a known reductant33 and the control experiment reacting independently synthesized PPN[HFe(CO)4] and [TolIm](BArF) (Fig. S13†) proved successful hydride transfer, thus strongly indicating NEt4[HFe(CO)4] was the active hydride transfer agent in our previous experiments. Furthermore, at longer timepoints in this reaction (days), a new resonance at −14.79 ppm was observed — similar to the previously observed, unassigned Fe–H/D species in Fig. 5A. We now conclusively assign this Fe–H species as NEt4[HFe3(CO)11], a known side-product in hydride transfer reactions of NEt4[HFe(CO)4].33 Indeed, [TolIm](BArF) was separately treated with NEt4[HFe3(CO)11], but no hydride transfer reaction was observed over the course of several days (Fig. S14†), further supporting the role of NEt4[HFe(CO)4] as the exclusive active hydride transfer agent.
Identification of NEt4[HFe(CO)4] also confirms the loss of ligand which was observed by 1H NMR spectroscopy in both gas reactions utilizing H2 (Fig. S15†) and upon treatment with NaHBEt3 (Fig. S16†). Furthermore, we re-emphasize the observation of a feature at 2.51 ppm corresponding to deuteration of the methylpyridine moiety of the ligand in the 2H NMR spectrum upon generation NEt4[DFe(CO)4] (Fig. S8†).
The liberation of ligand is predicated upon de-insertion of the acyl unit, which is capable of serving as a CO source in the generation NEt4[HFe(CO)4]. Upon de-insertion (Scheme 4, right side), the methyl carbanion coordinates to the Fe center to generate an intermediate related to that proposed in the synthesis of the acyl unit by Song and coworkers.34 These observations are also consistent with a less electrophilic CO ligand bound to Fe(0) in comparison to Fe(II) and the demonstrated lability of the acyl unit from labeled 13CO exchange experiments.21
We investigated the reactivity of the proposed carbanion bound intermediate NEt4[(Anth·CH2NoffSoff)Fe0(CO)4] by independent synthesis of the lithium methyl-carbanion salt via lithiation of Anth·CH3NSMe and addition of Fe(CO)5 (i.e. omitting oxidation by Br2 from the synthesis of 1). The IR spectrum of Li[(Anth·CH2NoffSoff)Fe0(CO)4] exhibited CO stretching frequencies of similar energy to the related complex described by Song34 and to NEt4[HFe(CO)4] and did not exhibit an ν(CO) feature above 1600 cm−1, as would otherwise indicate acyl ligation (Fig. S29†). We hypothesized heterolysis of H2 across the Fe center and bound ligand could explain the generation of NEt4[HFe(CO)4] and protonation to liberate the free ligand; however, no reaction was observed upon treatment of Li[(Anth·CH2NoffSoff)Fe0(CO)4] with D2 by 2H NMR spectroscopy (Scheme 4, bottom). Instead, treatment of Li[(Anth·CH2NoffSoff)Fe0(CO)4] with two equiv. MeOtBu2ArOD indicated formation of D-labeled free ligand, Anth·CH2DNSMe, and NEt4[DFe(CO)4] by 2H NMR spectroscopy (Fig. S17†). Analogous control experiments performed with 2,6-lutidine·HCl provided similar results, supporting that the phenolic proton was the active agent — rather than H-atom or other radical chemistry. As indicated in Scheme 4, the extruded {Fe(CO)4} unit undergoes further chemistry to form NEt4[HFe(CO)4]; however, the nature or mechanism of this particular reaction remains elusive at this time.
Lastly, we considered the initial reduction event of the ferrous starting complex to Fe(0). Based on the activation of H2/D2 mediated by 2 and the control reaction treating 2 with NaHBEt3—and the spectroscopically detected reduced Fe carbonyl species—we postulate that reduction of the ferrous metal center occurs by loss of the unobserved, reactive hydride as a proton along with two electron reduction to form Fe(0). Consistent with our previous work,18 detection of the highly reactive (especially anionic) Fe–H species of [Fe]-hydrogenase synthetic models is difficult. Intriguingly, this reductive pathway contrasts the well-characterized intramolecular hydride transfer reaction resulting in methylthiol extrusion observed in another model system from our group (mer-CNS; no scaffold),35 likely due to the unbound state of the thioether-SMe unit downstream of 1 in this case.
Conclusions
In summary, we have prepared an acyl-containing anthracene-scaffolded [Fe]-hydrogenase model compound that exhibits a dynamic fac-CNS donor motif and performs H2 activation. The subtle structural replacement of the previously studied carbamoyl ligation for the methylene-acyl moiety provides a dramatically different reaction pathway to H2 activation, which first involves clean and structurally characterized deprotonation of the methylene linker. Notably, the anthracene-scaffolded model complex exhibits well-controlled reactivity upon base treatment in comparison to non-scaffolded systems, possibly due to the controlled hemi-lability of the thioether-S. The methine-ligated dimer 2 resulting from base addition was, itself, competent for H2 activation, but hydride transfer to a biomimetic substrate was not observed. Instead, isotopic D-labeled gas experiments revealed formation of free ligand and the reductively extruded hydridoferrate species [HFe(CO)4]− (which converts to [HFe3(CO)11]− over several days). The former species is unambiguously proven to be the active hydride transfer agent in the present study, while the latter species is more stable and thus ineffective for hydride transfer in this system.Attempts to utilize exogenous base for H2 activation in concert with 2 to prevent the loss of ligand and Fe reduction were unsuccessful, but importantly enabled us to structurally and spectroscopically characterize relevant intermediates in this process. Numerous control reactions delineate a mechanistic pathway describing these conversions. This enhanced understanding of this deleterious, competitive process may contribute to the design of a more robust biomimetic reactivity system for understanding the reactivity of acyl(methylene)-containing synthetic analogues of [Fe]-hydrogenase. The inclusion of the authentic and biomimetic pyridone and/or thiolate motifs may drastically alter the reactivity profile(s) described herein, thereby providing more enlightened insight into Nature's delicate choice of donor identity and location in the [Fe]-hydrogenase active site.
Experimental
General considerations
Commercially available reagents were used without further purification unless otherwise noted. Suppliers of relevant reagents are described in the ESI.† Solvents used for synthesis were procured from Fisher Scientific and dried over alumina columns using a Pure Process Technology solvent purification system, and stored over 3 Å molecular sieves until use; THF was stored over 3 Å molecular sieves and small pieces of sodium. High-pressure NMR tubes were purchased from Wilmad Labglass (Cat No. 524-PV-7). Infrared spectra were recorded on a Bruker Alpha spectrometer equipped with a diamond ATR crystal, all contained under inert atmosphere. UV/vis spectra were recorded on an Agilent Cary 6000i spectrometer. The 1H, 2H, and 13C were collected using Varian Direct Drive 400 MHz, 500 MHz or 600 MHz instruments. X-ray diffraction and X-ray absorption instrumentation and experimental techniques are described in the ESI.† All cross-coupling reactions and syntheses of metal complexes were performed under N2 atmosphere using Schlenk technique or glovebox conditions.Ligand syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 4:1 hexane/EA) to afford a yellow solid. Yield: 2.07 g (58%). 1H NMR (400 MHz, CDCl3): δ 2.72 (s, 3H), 7.36 (d, J = 7.5 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 5.8 Hz, 1H), 7.57 (m, 2H), 7.85 (dd, J = 7.9, 2.3 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 8.52 (s, 1H), 8.75 (d, J = 2.3 Hz, 1H), 8.86 (s, 1H). 13C NMR (100 MHz, CDCl3): 24.36, 121.73, 122.81, 125.26, 125.54, 125.68, 127.25, 127.35, 127.46, 128.30, 129.17, 130.59, 132.20, 132.28, 132.29, 133.14, 137.05, 137.67, 149.75, 157.67. IR (solid-state): 3036, 1614, 1533, 1307, 1028, 888, 735 cm−1. HR-MS (ESI): calcd for [C20H14ClN + H]+ 304.0888; found: 304.0899.
:H2O (7:1) under N2 atmosphere. The reaction solution was heated at 85 °C for 12 h under N2 atmosphere. After cooling the solution to room temperature, the mixture was quenched with a saturated NH4Cl(aq) solution (∼10 mL). The organic product was extracted with DCM and washed with saturated brine (2 × 100 mL). The product was dried over Na2SO4 and concentrated under vacuum, and further purified by silica gel column chromatography (4:1 to 1:1 hexane/EA) to afford a yellow solid. Yield: 1.58 g (70%). 1H NMR (400 MHz, d8-THF): δ 2.45 (s, 3H; thioether-CH3), 2.55 (s, 3H; pyridine-CH3), 7.26 (s, 1H), 7.28 (s, 2H), 7.34 (m, 1H), 7.41 (m, 3H), 7.53 (t, J = 7.6 Hz, 2H), 7.75 (dd, J = 8.0, 2.4 Hz, 1H), 8.07 (t, J = 7.5 Hz, 2H), 8.55 (s, 1H), 8.60 (s, 1H), 8.61 (s, 1H). 13C NMR (100 MHz, d8-THF): 15.66, 24.51, 122.95, 124.07, 126.23, 126.32, 126.40, 127.36, 127.50, 127.54, 128.11, 128.56, 128.87, 129.16, 129.64, 131.12, 131.20, 133.15, 133.25, 133.98, 138.05, 138.27, 140.19, 141.20, 142.20, 150.64, 158.40. HR-MS (ESI) calcd for [C27H21NS + H]+: 392.1467; found: 392.1479.
:1 to 4:1 hexane/EA) to afford a yellow solid. Yield: 2.07 g (58%). 1H NMR (400 MHz, CDCl3): δ 2.72 (s, 3H), 7.36 (d, J = 7.5 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.45 (d, J = 5.8 Hz, 1H), 7.57 (m, 2H), 7.85 (dd, J = 7.9, 2.3 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 8.52 (s, 1H), 8.75 (d, J = 2.3 Hz, 1H), 8.86 (s, 1H). 13C NMR (100 MHz, CDCl3): 24.36, 121.73, 122.81, 125.26, 125.54, 125.68, 127.25, 127.35, 127.46, 128.30, 129.17, 130.59, 132.20, 132.28, 132.29, 133.14, 137.05, 137.67, 149.75, 157.67. IR (solid-state): 3036, 1614, 1533, 1307, 1028, 888, 735 cm−1. HR-MS (ESI): calcd for [C20H14ClN + H]+ 304.0888; found: 304.0899.
:H2O (7:1) under N2 atmosphere. The reaction solution was heated at 85 °C for 12 h under N2 atmosphere. After cooling the solution to room temperature, the mixture was quenched with a saturated NH4Cl(aq) solution (∼10 mL). The organic product was extracted with DCM and washed with saturated brine (2 × 100 mL). The product was dried over Na2SO4 and concentrated under vacuum, and further purified by silica gel column chromatography (4:1 to 1:1 hexane/EA) to afford a yellow solid. Yield: 1.58 g (70%). 1H NMR (400 MHz, d8-THF): δ 2.45 (s, 3H; thioether-CH3), 2.55 (s, 3H; pyridine-CH3), 7.26 (s, 1H), 7.28 (s, 2H), 7.34 (m, 1H), 7.41 (m, 3H), 7.53 (t, J = 7.6 Hz, 2H), 7.75 (dd, J = 8.0, 2.4 Hz, 1H), 8.07 (t, J = 7.5 Hz, 2H), 8.55 (s, 1H), 8.60 (s, 1H), 8.61 (s, 1H). 13C NMR (100 MHz, d8-THF): 15.66, 24.51, 122.95, 124.07, 126.23, 126.32, 126.40, 127.36, 127.50, 127.54, 128.11, 128.56, 128.87, 129.16, 129.64, 131.12, 131.20, 133.15, 133.25, 133.98, 138.05, 138.27, 140.19, 141.20, 142.20, 150.64, 158.40. HR-MS (ESI) calcd for [C27H21NS + H]+: 392.1467; found: 392.1479.
Metal complex syntheses
O 2039 (s), 1978 (s), νCO 1629 (m), νCN 1584 (m). Anal. calcd for C30H20BrFeNO3S: C 59.04, H 3.30, N 2.30; found: C 58.97, H 3.44, N 2.54.
O 2024, 1971; νCO 1642. Anal. calcd for C48H36BrAsFeNO3S: C 62.83, H 3.95, N 1.53; found: C 58.24, H 4.08, N 1.08.
:1 CD2Cl2, d3-MeCN, 100 MHz): 212.98, 208.67, 172.33, 148.72, 140.92, 139.94, 138.91, 136.71, 136.06, 132.04, 131.85, 129.99, 129.15, 128.12, 127.99, 127.80, 127.27, 126.64, 126.38, 125.71, 125.46, 125.33, 122.92, 115.83, 67.13, 15.45. IR (crystalline solid, cm−1): νCO 2021 (s), 1998 (s), 1962 (s), 1943 (s) νCN 1599 (m). Anal. calcd for C64H44Fe2N4O6S2: C 67.38, H 3.89, N 4.91; found: C 67.21, H 4.04, N 4.76.
Data availability
Crystal structure data has been deposited in the Cambridge Crystal Structure Database, and additional spectra and experimental details are contained in the ESI.†Author contributions
Experiments were conceived and designed by J. Seo, SAK and MJR and executed by all co-authors. EXAFS data were acquired and analyzed by J. Shearer. X-ray diffraction data were analysed by VML. ERR and STG performed supporting syntheses and spectroscopic characterizations, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (NSF CHE-1808311 to MJR and CHE-1854854 to JS) and the Welch Foundation (F-1822, MJR). We thank Angela Spangenberg, Garrett Blake and Steve Sorey for critical assistance with NMR data collection. The authors also thank Chris Joseph for assistance in X-ray crystallography data collection.Notes and references
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- I. Čorić and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200–7211 CrossRef PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- P. Ghosh, S. Ding, R. B. Chupik, M. Quiroz, C.-H. Hsieh, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2017, 8, 8291–8300 RSC.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed.
- L.-C. Song, X.-Y. Yang, M. Cao, X.-Y. Gao, B.-B. Liu, L. Zhu and F. Jiang, Chem. Commun., 2017, 53, 3818–3821 RSC.
- H. Abul-Futouh, Y. Zagranyarski, C. Müller, M. Schulz, S. Kupfer, H. Görls, M. El-khateeb, S. Gräfe, B. Dietzek, K. Peneva and W. Weigand, Dalton Trans., 2017, 46, 11180–11191 RSC.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS PubMed.
- T. Hiromoto, K. Ataka, O. Pilak, S. Vogt, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer, S. Shima and U. Ermler, FEBS Lett., 2009, 583, 585–590 CrossRef CAS PubMed.
- G. Huang, T. Wagner, M. D. Wodrich, K. Ataka, E. Bill, U. Ermler, X. Hu and S. Shima, Nat. Catal., 2019, 2, 537–543 CrossRef CAS.
- A. R. Finkelmann, H. M. Senn and M. Reiher, Chem. Sci., 2014, 5, 4474–4482 RSC.
- S. Shima, T. Fujishiro, J. Kahnt, S. Shima, D. Chen, T. Xu, M. D. Wodrich, K. M. Schultz, X. Hu, M. D. Wodrich and K. Ataka, Nat. Chem., 2015, 7, 995–1002 CrossRef CAS PubMed.
- T. Xu, C.-J. M. Yin, M. D. Wodrich, S. Mazza, K. M. Schultz, R. Scopelliti and X. Hu, J. Am. Chem. Soc., 2016, 138, 3270–3273 CrossRef CAS PubMed.
- J. Seo, T. A. Manes and M. J. Rose, Nat. Chem., 2017, 9, 552–557 CrossRef CAS PubMed.
- S. A. Kerns, A.-C. Magtaan, P. R. Vong and M. J. Rose, Angew. Chem., Int. Ed., 2018, 57, 2855–2858 CrossRef CAS PubMed.
- P. J. Turrell, J. A. Wright, J. N. T. Peck, V. S. Oganesyan and C. J. Pickett, Angew. Chem., Int. Ed., 2010, 49, 7508–7511 CrossRef CAS PubMed.
- P. J. Turrell, A. D. Hill, S. K. Ibrahim, J. A. Wright and C. J. Pickett, Dalton Trans., 2013, 42, 8140–8146 RSC.
- D. Chen, R. Scopelliti and X. Hu, Angew. Chem., Int. Ed., 2010, 49, 7512–7515 CrossRef CAS PubMed.
- A. Murso and D. Stalke, Dalton Trans., 2004, 2563–2569 RSC.
- L. Schwartsburd, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2010, 29, 3817–3827 CrossRef CAS.
- T. Zell, R. Langer, M. A. Iron, L. Konstantinovski, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, E. Balaraman, Y. Ben-David and D. Milstein, Inorg. Chem., 2013, 52, 9636–9649 CrossRef CAS PubMed.
- S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed.
- Y. I. Cho, G. Durgaprasad and M. J. Rose, Inorg. Chem., 2019, 58, 12689–12699 CrossRef CAS PubMed.
- L.-C. Song, L. Zhu, F.-Q. Hu and Y.-X. Wang, Inorg. Chem., 2017, 56, 15216–15230 CrossRef CAS PubMed.
- R. A. Bartlett, J. J. Ellison, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2888–2894 CrossRef CAS.
- T. J. Boyle, L. A. M. Ottley, C. A. Apblett, C. A. Stewart, S. M. Hoppe, K. L. Hawthorne and M. A. Rodriguez, Inorg. Chem., 2011, 50, 6174–6182 CrossRef CAS PubMed.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- M. Y. Darensbourg, D. J. Darensbourg and H. L. C. Barros, Inorg. Chem., 1978, 17, 297–301 CrossRef CAS.
- J. J. Brunet, Chem. Rev., 1990, 90, 1041–1059 CrossRef CAS.
- L.-C. Song, Z.-J. Xie, M.-M. Wang, G.-Y. Zhao and H.-B. Song, Inorg. Chem., 2012, 51, 7466–7468 CrossRef CAS PubMed.
- G. Durgaprasad, Z.-L. Xie and M. J. Rose, Inorg. Chem., 2016, 55, 386–389 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1973989–1973991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03154b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |