
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinct electronic structures and bonding interactions in inverse-sandwich samarium and ytterbium biphenyl complexes†
Yuyuan
Xiao‡
 a,
Xiao-Kun
Zhao‡
b,
Tianpin
Wu§
c,
Jeffrey T.
Miller¶
c,
Han-Shi
Hu
*b,
Jun
Li
b,
Wenliang
Huang
*a and
Paula L.
Diaconescu
*d
a,
Xiao-Kun
Zhao‡
b,
Tianpin
Wu§
c,
Jeffrey T.
Miller¶
c,
Han-Shi
Hu
*b,
Jun
Li
b,
Wenliang
Huang
*a and
Paula L.
Diaconescu
*d
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Material Chemistry and Application, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: wlhuang@pku.edu.cn
bDepartment of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of the Ministry of Education, Tsinghua University, Beijing 100084, P. R. China. E-mail: hshu@mail.tsinghua.edu.cn
cChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, Illinois 60439, USA
dDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: pld@chem.ucla.edu
First published on 29th October 2020
Abstract
Inverse-sandwich samarium and ytterbium biphenyl complexes were synthesized by the reduction of their trivalent halide precursors with potassium graphite in the presence of biphenyl. While the samarium complex had a similar structure as previously reported rare earth metal biphenyl complexes, with the two samarium ions bound to the same phenyl ring, the ytterbium counterpart adopted a different structure, with the two ytterbium ions bound to different phenyl rings. Upon the addition of crown ether to encapsulate the potassium ions, the inverse-sandwich samarium biphenyl structure remained intact; however, the ytterbium biphenyl structure fell apart with the concomitant formation of a divalent ytterbium crown ether complex and potassium biphenylide. Spectroscopic and computational studies were performed to gain insight into the electronic structures and bonding interactions of these samarium and ytterbium biphenyl complexes. While the ytterbium ions were found to be divalent with a 4f14 electron configuration and form a primarily ionic bonding interaction with biphenyl dianion, the samarium ions were in the trivalent state with a 4f5 electron configuration and mainly utilized the 5d orbitals to form a δ-type bonding interaction with the π* orbitals of the biphenyl tetraanion, showing covalent character.
Introduction
The chemistry of rare earth elements (Sc, Y, and lanthanides) is still dominated by their trivalent ions, with the few exceptions of Ce(IV), Eu(II), Yb(II), and Sm(II), while the field of unusual redox chemistry is expanding.1–3 The overwhelming stability of the trivalent state results from the high electropositivity and the core-like 4f orbitals of the rare earth ions.4 Unlike the d orbitals of transition metals, the 4f orbitals of lanthanides are strongly shielded by the 5s2 and 5p6 electrons so that they barely interact with the ligand-based orbitals.4–9 As a result, the bonding interactions are primarily ionic with a negligible covalent character.10 This is in contrast to actinides, especially light actinides, which have been shown to exhibit distinctive redox chemistry and significant covalent interactions.10–14 However, a breakthrough in both high and low valent rare earth chemistry has emerged in recent years. For example, Mazzanti and La Pierre independently reported the synthesis and characterization of Tb(IV)15–17 and Pr(IV) complexes,18 which were the first examples of tetravalent molecular rare earth metal complexes beyond Ce(IV). In addition, Li and Zhou showed that pentavalent praseodymium oxides or nitrides could be trapped and characterized in a noble gas matrix.19,20 On the other hand, low valent rare earth chemistry had gradually progressed at the turn of the century from Sm(II)21 to Tm(II),22 Dy(II)23 and Nd(II).24 Further advance to afford divalent molecular complexes for other rare earth metals was once considered impractical due to the extreme negative M3+/2+ reduction potentials.25–28 This is in accordance with the solid state rare earth metal diiodides: Eu, Yb, Sm, Tm, Dy, and Nd form a genuine M(II)I2 salt, while other rare earth metal diiodides exist as metallic or semi-conducting M(III)I2(e−) with extra electrons delocalized in the band composed of the metal d orbitals (Chart 1).29–31 The seminal example of [Cp′′3La]− (Cp′′ = 1,3-C5H3(SiMe3)2) reported by Lappert et al. demonstrated that divalent molecular rare earth metal complexes beyond Nd(II) were accessible.32 Later, Evans et al. expanded this chemistry and completed the series of divalent complexes for all rare earth metals, except the radioactive Pm.33–37 Recently, the syntheses of the neutral sandwich complexes Ln(II) (CpiPr52) (Ln = Tb and Dy, CpiPr5 = C5(iPr)5) were reported by Long et al.38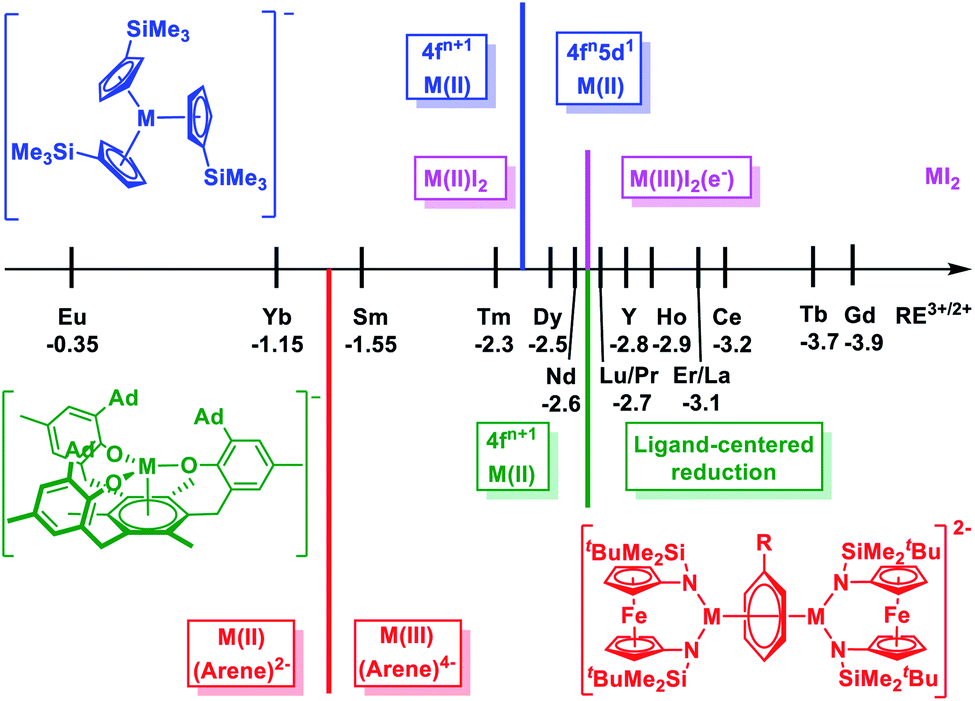 | ||
| Chart 1 Scale of M3+/2+ (M = rare earth metal) reduction potentials28 and electron configurations of divalent rare earth metal complexes in different coordination environment: pink, M(II)I2;29 blue, [K(crypt)](Cp′3M);39 green, [K(crypt)][((Ad,MeArO)3mes)M];42 red, [(NNTBS)M]2(μ-biphenyl)[K(solv)]2 (this work). | ||
Notably, in these compounds, rare earth(II) ions adopt a non-traditional 4fn5d1 configuration with the additional electron occupying on the 5dz2 orbital.28,39–41 Different from the core-like 4f orbitals, the 5d orbitals are diffused and greatly influenced by the ligand field.10 Systematic studies of the [K(crypt)][Cp′3M] (Cp′ = C5H4(SiMe3), M = Y and lanthanides)39,40 and [K(crypt)][((Ad,MeArO)3mes)M] (M = Y and lanthanides) ((Ad,MeArO)3mes = tris(aryloxide)arene)42,43 series reveal that the electron configuration of rare earth(II) ions depend on the coordination environment: in the former series, the switch from a 4fn+1 to 4fn5d1 ground state takes place at Dy(II) (−2.5 V for Dy3+/2+vs. standard hydrogen electrode, SHE),39,40 while in the latter series, this switch does not happen even for Nd(II) (−2.6 V for Nd3+/2+vs. SHE),42 and a ligand-centered instead of a metal-centered reduction occurred for rare earth ions that are more difficult to reduce than Nd (Chart 1).43 This dependence of ground state electronic configuration for divalent rare earth ions suggests that it is possible to facilitate the transition from 4fn+1 to 4fn5d1 ground states through ligand manipulation.31,44,45 Therefore, we were wondering how far this approach can push the limit for 4f → 5d transitions, especially for traditional divalent rare earth metals with a 4fn+1 electron configuration, such as Sm(II) and Yb(II).
Previously, we reported the synthesis and characterization of a series of inverse-sandwich rare earth metal biphenyl complexes [(NNTBS)M]2(μ-biphenyl)[K(solv)]2 (M2-biph-K2, NNTBS = fc(NSitBuMe2)2, fc = 1,1′-ferrocenediyl; M = Sc, Y, La, Lu, Gd, Dy, and Er) with an unprecedented tetranionic phenyl ring bridging two rare earth(III) ions.46,47 Structural, spectroscopic, and computational studies of the representative yttrium complex supported the assignment that the tetranionic phenyl ring is a 10π-electron aromatic system stabilized by a δ-type bonding interaction between the 4d orbitals of Y(III) ions and the antibonding π* orbitals of the phenyl ring. This δ bonding interaction has a moderate covalent character since the contribution of yttrium orbitals is over 20% in HOMO and HOMO−1 (HOMO = highest occupied molecular orbital).46 In the case of Dy2-biph-K2, instead of a reduction to 4f10 Dy(II), structural and magnetic data suggested the presence of 4f9 Dy(III) ions.47 We have then become interested in the cases of other traditional divalent rare earth metals, in particular, Sm and Yb, because their M3+/2+ reduction potentials are different from those of other rare earth metals and also apart from each other (Chart 1).
Herein, we report the synthesis of inverse-sandwich samarium and ytterbium biphenyl complexes, which are characterized by X-ray crystallography, NMR spectroscopy, elemental analysis, and UV/Vis/NIR spectroscopy. The structural and spectroscopic characterization supports the formulation of Sm(III) ions in the samarium biphenyl complexes and Yb(II) ions in the ytterbium biphenyl complexes. Upon encapsulation of the potassium ions, the coordination between rare earth ions with biphenyl remained intact for Sm but collapsed for Yb. Density functional theory (DFT) calculations show that the samarium and ytterbium biphenyl complexes have distinct electronic structures and bonding interactions: in the samarium biphenyl complex, Sm(III) ions have a 4f5 electron configuration and the 5d orbitals form a δ-type bonding interaction with the π* orbitals of the biphenyl tetraanion, showing a covalent character, while in the ytterbium biphenyl complex, Yb(II) ions have a 4f14 electron configuration and are bound to the biphenyl dianion via a primary ionic interaction.
Results and discussion
It has been shown that the ferrocene diamide ligand NNTBS is well suited to stabilize rare earth metal complexes with a variety of reduced arenes, including biphenyl,46,47 naphthalene,48–50 and (E)-stilbene.51 Therefore, we prepared (NNTBS)SmI(THF)2 (Sm-I) and (NNTBS)YbCl(THF)2 (Yb-Cl) according to literature procedures,52 and employed them as trivalent metal precursors. Analogous to the synthesis of yttrium biphenyl complexes,46 we attempted the reduction of Sm-I or Yb-Cl with potassium graphite (KC8) in the presence of biphenyl.Samarium biphenyl reduction
Upon the addition of 2.5 equivalents of KC8 into a pre-cooled THF solution of an equivalent of Sm-I and 0.5 equivalents of biphenyl at −78 °C, the solution color turned immediately to dark (Scheme 1a). The reaction mixture was warmed up to room temperature and stirred for 10 min. After quickly passing it through a Celite column to remove insoluble solids, the filtrate was dried under a reduced pressure to yield a crude product. The NMR spectrum of the crude product showed only paramagnetic signals with the absence of starting materials. X-ray crystallography confirmed the product was [(NNTBS)Sm]2(μ-biphenyl)[K(toluene)]2 (Sm2-biph-K2). An analytically pure material was obtained in a moderate yield of 55% after washing the crude product with Et2O.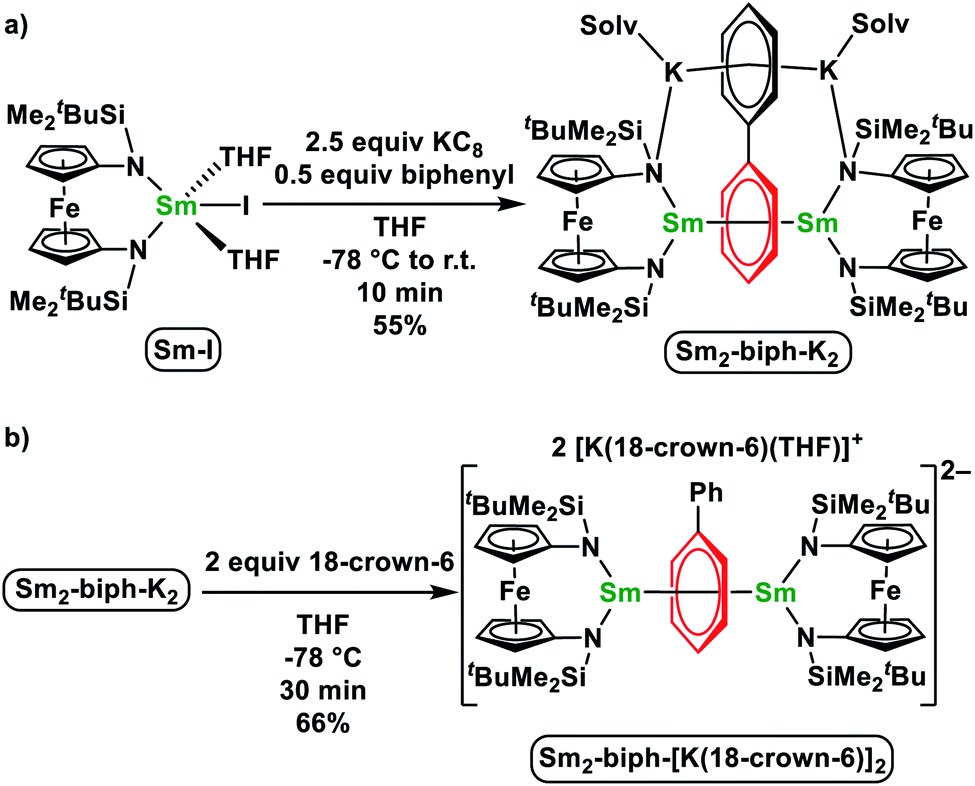 | ||
| Scheme 1 Synthesis of Sm2-biph-K2 (a) and Sm2-biph-[K(18-crown-6)]2 (b). | ||
The structure of Sm2-biph-K2 is similar to that of [(NNTBS)Y]2(μ-biphenyl)[K(toluene)]2 (Y2-biph-K2), with two Sm ions coordinated to the same phenyl ring and two potassium ions coordinated to the other phenyl ring (Fig. 1a). The phenyl ring bridging the two Sm ions adopted a μ-η6:η6-coordination mode with Sm–C distances ranging from 2.549(4) to 2.639(4) Å (average Sm–C distance of 2.60 Å) and a Sm–Sm distance of 4.336(1) Å, comparable to the corresponding values in Gd2-biph-K2, when the difference between their ionic radii (average Gd–C distance of 2.58 Å, Gd–Gd distance of 4.27 Å, R(Sm) − R(Gd) = 0.03 Å)53 is taken into account.47 The C–C distances of the Sm-bound phenyl ring range from 1.421(5) to 1.476(1) Å, with an average value of 1.45 Å and a Cipso–Cipso distance of 1.413(4) Å, while the average C–C distance of the other phenyl ring is 1.41 Å. These values are close to those of Y2-biph-K2 (the average C–C distance of the Y-bound phenyl ring: 1.46 Å; Cipso–Cipso: 1.414(4) Å; the average C–C distance of the other phenyl ring: 1.41 Å).46 The Sm1–N1 distance of 2.341(6) Å is 0.08 Å longer than the Sm–N distance of 2.263(3) Å in Sm-I.52 However, this elongation is likely due to the weakening of the Sm–N bond by the strong bonding between Sm and the phenyl ring, since a similar elongation of 0.07 Å was observed in the case of Y2-biph-K2 and (NNTBS)YI(THF)2 (2.292(2) Å vs. 2.222(3) Å).46,49 The Sm–Ccentroid distance is 2.196(7) Å, much shorter than the Sm–Ccentroid distances found in Sm(III) neutral arene complexes 2.521(5) Å in (Sm(C6Me6) (AlCl4)3 (ref. 54) and 2.638(4) Å in (trans-calix[2]benzene[2]pyrrole)SmCl).55 The angle Sm1–Ccentroid–Sm2 is 164.3(9)°, indicating a nearly linear arrangement of the three atoms. The Sm-bound phenyl ring is slightly distorted from planarity with a torsion angle of 6.3(9)°, while the two phenyl rings have a dihedral angle of 5.9(2)°. Overall, the structural parameters of Sm2-biph-K2 are consistent with the assignment of two Sm(III) ions and a charge-localized biphenyl tetraanion, as in the analogous Y2-biph-K2.
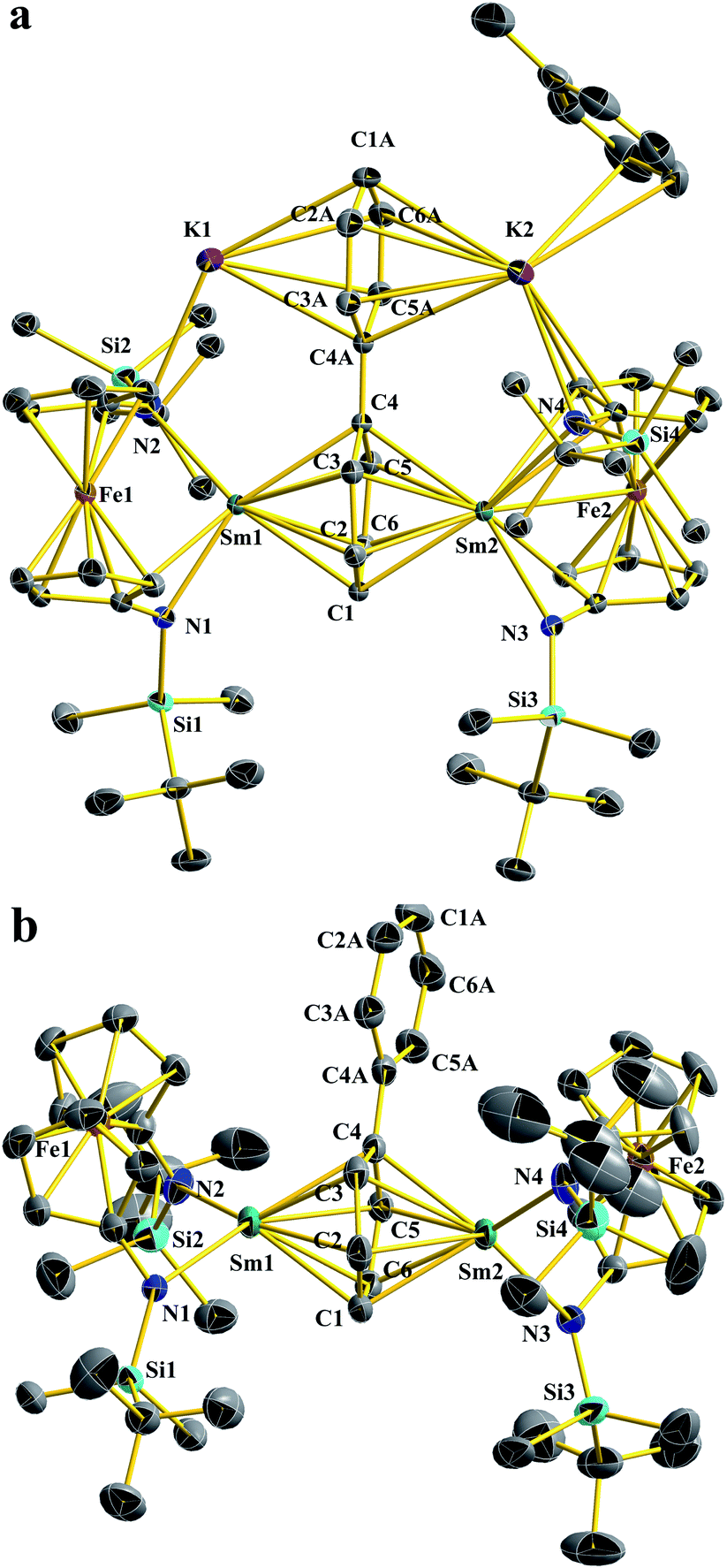 | ||
| Fig. 1 Thermal-ellipsoid (50% probability) representations of Sm2-biph-K2 (a) and Sm2-biph-[K(18-crown-6)]2 (b). Hydrogen and solvent atoms, disordered counterparts, and the counter cations for Sm2-biph-[K(18-crown-6)]2 were omitted for clarity. Selected distances [Å] and angles [°]: for Sm2-biph-K2: Sm1–N1 2.341(6), Sm1–N2 2.452(7), Sm1–C1 2.614(2), Sm1–C2 2.639(3), Sm1–C3 2.569(4), Sm1–C4 2.623(1), Sm1–C5 2.639(4), Sm1–C6 2.574(3), Sm1–Ccentroid 2.196(7), Sm1–Fe1 3.225(2), C1–C2 1.421(5), C2–C3 1.464(6), C3–C4 1.476(1), C4–C5 1.476(1), C5–C6 1.464(6), C6–C1 1.421(5), C4–C4A 1.412(7), C4A–C3A 1.442(2), C3A–C2A 1.389(7), C2A–C1A 1.389(1), C1A–C6A 1.389(1), C6A–C5A 1.389(7), C5A–C4A 1.442(2), K1–N2 2.817(7), K1–Ccentroid 2.813(6), Sm1–Sm2 4.336(3); N1–Sm1–N2 103.5(7), Sm1–Ccentroid–Sm2 164.3(9), torsion angle defined by (C2–C3) vs. (C5–C6) 6.3(9), dihedral angle between the phenyl rings 5.9(2). For Sm2-biph-[K(18-crown-6)]2: Sm1–N1 2.380(5), Sm1–N2 2.409(2), Sm1–C1 2.603(2), Sm1–C2 2.652(1), Sm1–C3 2.512(3), Sm1–C4 2.615(6), Sm1–C5 2.625(6), Sm1–C6 2.549(4), Sm1–Ccentroid 2.146(8), Sm1–Fe1 3.352(1), C1–C2 1.447(3), C2–C3 1.458(2), C3–C4 1.479(1), C4–C5 1.477(4), C5–C6 1.451(3), C6–C1 1.432(2), C4–C4A 1.439(8), C4A–C3A 1.407(9), C3A–C2A 1.392(1), C2A–C1A 1.374(3), C1A–C6A 1.379(9), C6A–C5A 1.384(2), C5A–C4A 1.428(4), Sm1–Sm2 4.300(8); N1–Sm1–N2 98.1(3), Sm1–Ccentroid–Sm2 178.8(5), torsion angles defined by (C2–C3) and (C5–C6) 10.7(9), dihedral angle between the two phenyl rings 9.0(7). | ||
It was previously found that the K ions are not required to maintain the inverse-sandwich structure in Y2-biph-K2.46 Therefore, we probed whether this is also the case for Sm2-biph-K2. The addition of two equivalents of 18-crown-6 to a THF solution of Sm2-biph-K2 at −78 °C (Scheme 1b) led to no obvious change. Crystallization of the reaction mixture resulted in the isolation of an ion pair complex, Sm2-biph-[K(18-crown-6)]2, in a 66% yield. X-ray crystallography established the molecular structure of Sm2-biph-[K(18-crown-6)]2 (Fig. 1b), which is analogous to Y2-biph-[K(18-crown-6)]2.46 The charge localization is more prominent in Sm2-biph-[K(18-crown-6)]2 than in Sm2-biph-K2 after the encapsulation of the potassium ions: average C–C distance of the Sm-bound phenyl ring increased to 1.46 Å and Cipso–Cipso distance increased to 1.440(6) Å, while the average C–C distance of the other phenyl ring decreased to 1.39 Å. The bonding between Sm and phenyl ring also strengthened as evidenced by the slightly shorter average Sm–C distance of 2.59 Å and shorter Sm–Sm distance of 4.301(1) Å compared to 2.60 Å and 4.336(1) Å in Sm2-biph-K2, respectively. The Sm–Ccentroid distance also decreased from 2.196(7) Å to 2.146(8) Å with an essentially linear arrangement of Sm1–Ccentroid–Sm2 (178.8(5)°). The torsion angle of the Sm-bound phenyl ring also increased slightly to 10.7(9)°, as did the dihedral angle between the two phenyl rings, to 9.0(7)°. These structural changes observed upon the removal of the potassium ions was also observed in the case of Y2-biph-K2 and Y2-biph-[K(18-crown-6)]2, interpreted as the strengthening of the bonding interaction between yttrium and the bound phenyl ring.46
Ytterbium biphenyl reduction
Initially, we attempted the synthesis of Yb biphenyl complexes through the one-pot reduction of Yb-Cl and biphenyl by KC8, similarly to the synthesis of Sm2-biph-K2 (Scheme 2a). The reaction proceeded with the formation of multiple diamagnetic products corresponding to various Yb(II) products. Crystallization of the crude products from Et2O yielded some dark single crystals that were determined by X-ray crystallography to be an ytterbium biphenyl complex [(NNTBS)Yb]2(μ-biphenyl)[K(Et2O)]2 (Yb2-biph-K2). However, the corresponding 1H NMR spectrum showed a persistent contamination from other Yb(II) side products, even after a number of purification attempts by multiple fractional crystallization. In addition, we found that the relative amount of Yb(II) side products was sensitive to the reaction conditions. Since the Yb3+/2+ reduction potential is −1.15 V vs. SHE,26 which is far less negative than the reduction potential of −2.45 V vs. SHE for (biphenyl)0/1−,56 we rationalized that a simultaneous reduction of Yb-Cl and biphenyl resulted in the formation of multiple products. Therefore, we tried pre-mixing an excess biphenyl and KC8 in order to generate a reduced biphenyl species (to simplify, we use [K2(biphenyl)] for representation) and then added Yb-Cl (Scheme 2b). Through this protocol, we were able to obtain Yb2-biph-K2 reproducibly in high purity and a moderate yield of 52%.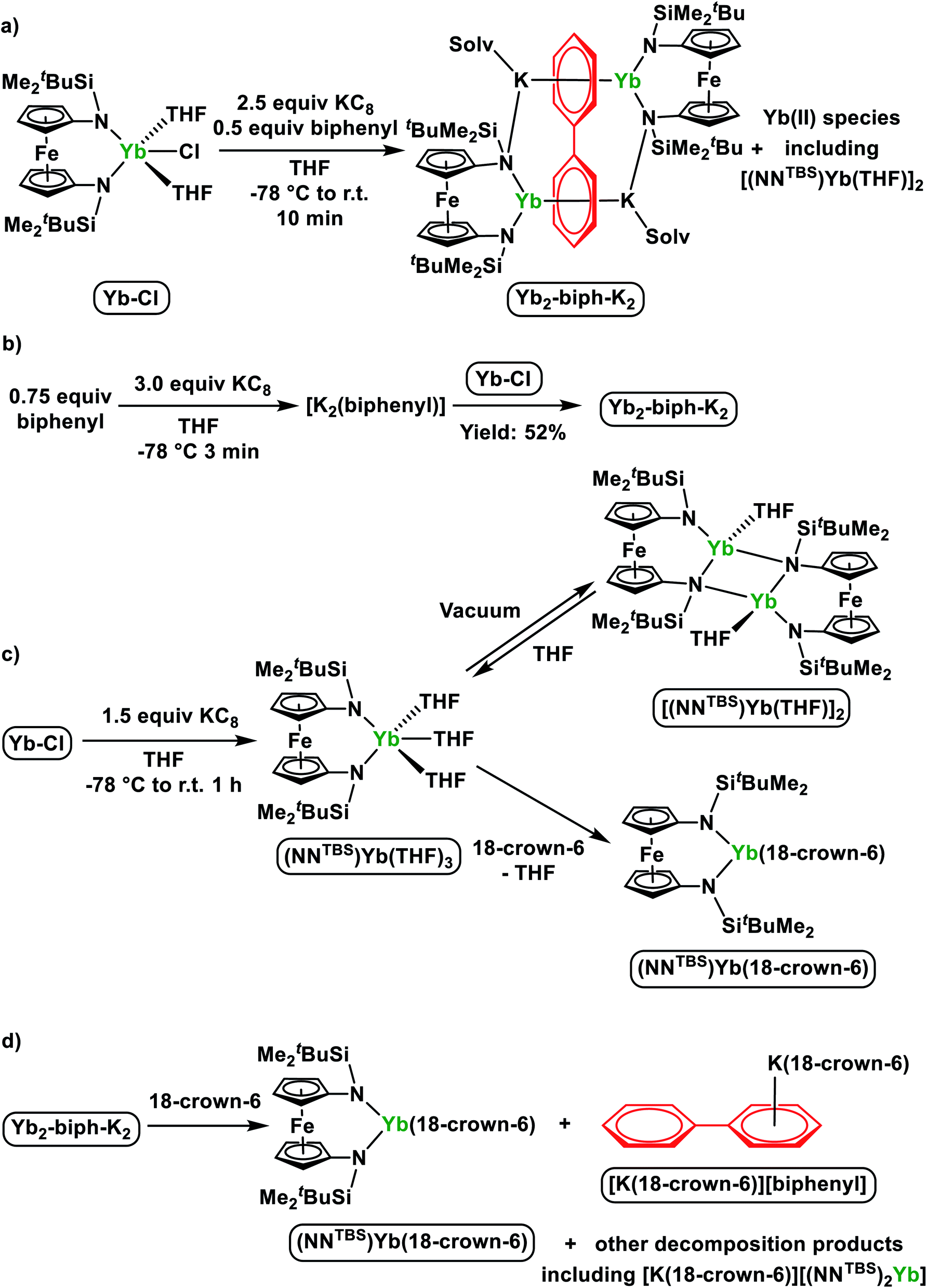 | ||
| Scheme 2 (a) One-pot reduction of Yb-Cl and biphenyl by KC8; (b) optimized synthesis of Yb2-biph-K2 by pre-mixing an excess of biphenyl and KC8; (c) direct reduction of Yb-Cl and transformations of Yb(II) products; (d) reaction of Yb2-biph-K2 with an excess of 18-crown-6. | ||
The 1H NMR spectrum of Yb2-biph-K2 in THF-d8 at room temperature showed broad peaks indicating the structure of compound is fluxional in solution. Signals of SiCH3 and SiC(CH3)3 of the NNTBS ligands are in the typical diamagnetic region, consistent with the presence of 4f14 Yb(II) ions. A broad peak at 5.41 ppm likely belongs to the meta proton of the biphenyl anion, based on a comparison with a previously reported rare earth metal stabilized biphenyl dianion.57 Four peaks at 127.9, 102.2, 101.8, and 87.2 ppm in the 13C NMR spectrum are assigned to the reduced biphenyl ligand, indicating that the two phenyl rings are equivalent. These 13C chemical shifts are distinct from those of Y2-biph-K2 (four peaks for the Y-bound phenyl ring at 86.8, 79.0, 73.1, and 58.6 ppm; four peaks for the K-bound phenyl ring at 138.5, 128.5, 114.8, and 103.5 ppm)46 but are similar to those of the alkali metal biphenyl dianion (four peaks at 128.5, 104.6, 102.3 and 74.2 ppm).58 Overall, the 1H and 13C NMR spectra support the assignment of two 4f14 Yb(II) ions and a charge-delocalized biphenyl dianion.
Despite of the similar molecular formula, the molecular structure of Yb2-biph-K2 is distinct from Sm2-biph-K2 and other M2-biph-K2 compounds (Fig. 2a). The two Yb and K ions coordinate to different phenyl rings. The Yb1–N2 distance of 2.312(2) Å is about 0.13 Å longer than the average Yb–N distance of 2.182(5) Å in Yb-Cl. This elongation is likely due to the change in ionic radii upon going from Yb(III) to Yb(II) (R(Yb(II)) − R(Yb(III)) = 0.15 Å).53 The two phenyl rings are equivalent, consistent with the symmetric solution structure determined by 1H and 13C NMR spectra. The alternating C–C distances (1.41, 1.38, and 1.47 Å) within the phenyl ring and the shortening of the Cipso–Cipso distance to 1.396(4) Å imply that the negative charges are equally delocalized over the two phenyl rings and the biphenyl ligand is best described as a dianion.57 The average Yb–C distance of 2.80 Å and the Yb1–Ccentroid distance of 2.415(6) Å are much longer than those of 2.60 Å and 2.196(7) Å in Sm2-biph-K2, respectively, even after adjusting for the difference in ionic radii (R(Sm(III)) = 1.08 Å, R(Yb(II)) = 1.14 Å)53, implying a much weaker interaction between the Yb ions and the phenyl rings. In addition, the two phenyl rings are not twisted but essentially coplanar, differing from other M2-biph-K2 but similar to the yttrium biphenyl dianion complex previously reported by Fryzuk et al.57
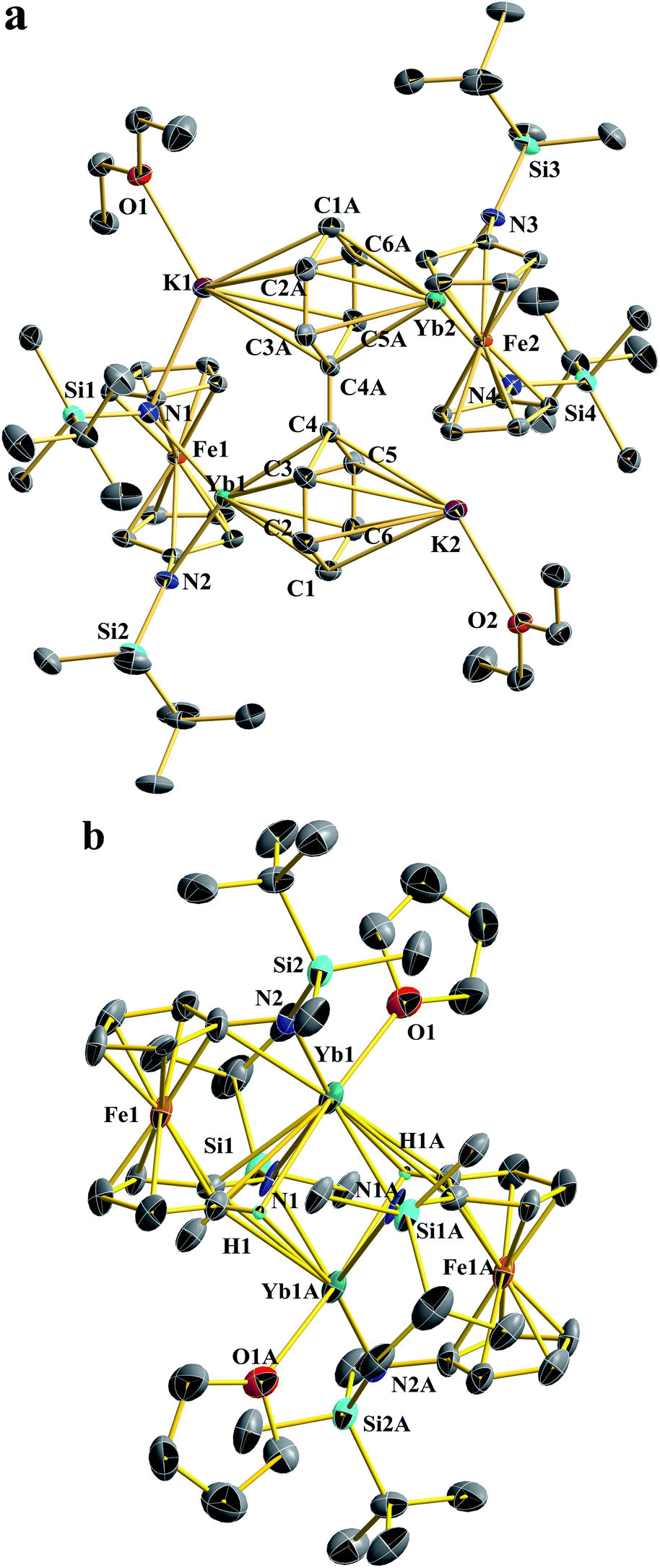 | ||
| Fig. 2 Thermal-ellipsoid (50% probability) representations of Yb2-biph-K2 (a) and [(NNTBS)Yb(THF)]2 (b). Hydrogen (except H involved in agostic interaction), disordered counterparts, and solvent atoms were omitted for clarity. Selected distances [Å] and angles [°]: For Yb2-biph-K2: Yb1–N1 2.395 (9), Yb1–N2 2.312(2), Yb1–C1 2.773(3), Yb1–C2 2.775(9), Yb1–C3 2.797(5), Yb1–C4 2.860(3), Yb1–C5 2.812(5), Yb1–C6 2.791(7), Yb1–Ccentroid 2.415(6), Yb1–Fe1 3.097(7), C1–C2 1.412(1), C2–C3 1.378(8), C3–C4 1.471(9), C4–C5 1.469(8), C5–C6 1.376(7), C6–C1 1.413(7), C4–C4A 1.396(0), K1–N1 2.881(4), K1–Ccentroid 2.829(8); N1–Yb1–N2 104.2(1), torsion angle defined by (C2–C3) vs. (C5–C6) 0.0(8), dihedral angle between the phenyl rings 0.0(1). For [(NNTBS)Yb(THF)]2: Yb1–N2 2.360(5), Yb1–N1 2.558(7), Yb1–O1 2.443(1), Yb1–Fe1 3.251(4), Yb1–C1 3.019(6), Yb1–H1 2.747(8); N1–Yb1–N2 133.8(5), Yb1–N1–Yb1A 92.7(1). | ||
Since our initial attempt to synthesize a ytterbium biphenyl complex by the one-pot reduction procedure resulted in the concomitant formation of unknown divalent ytterbium side product(s) together with Yb2-biph-K2, we were interested in finding out their identity. The corresponding 1H NMR spectra showed multiple peaks other than those belonging to Yb2-biph-K2 in the SiCH3 region of the NNTBS ligand. This implies that there are either multiple Yb(II) side products or one Yb(II) species of low symmetry. The direct reduction of Yb-Cl by 1.5 equivalents of KC8 in THF yielded a single Yb(II) product with high symmetry as determined by 1H NMR spectroscopy (Scheme 2c). However, upon removing volatiles and washing with hexanes, a poorly soluble brownish solid was obtained. The solid is insoluble in aliphatic solvents and barely soluble in aromatic solvents. The corresponding 1H NMR spectrum in C6D6 showed weak signals but matched those of the side product from the one-pot reduction.
X-ray crystallography determined the molecular structure of this Yb(II) product to be a dimer, [(NNTBS)Yb(THF)]2 (Fig. 2b). Each Yb(II) ion is coordinated to one terminal amide, two bridging amides and a THF molecule. The Yb–N distance of 2.558(7) Å for the bridging amide is much longer than the terminal one of 2.360(5) Å, which is close to 2.312(2) Å in Yb2-biph-K2. An agostic interaction of the Yb ion and a C–H bond from the ferrocene backbone (Yb1–H1: 2.747(8) Å and Yb1–C1: 3.019(6) Å) may play a role in stabilizing this dimeric structure. This rigid structure may be responsible for its poor solubility and very low symmetry in solution. When dissolved in THF-d8, the 1H NMR spectrum showed a highly symmetric pattern for the NNTBS ligand. We anticipated that the dimeric structure could be intercepted by the coordination of additional THF molecules. Crystallization from THF/hexanes yielded single crystals determined by X-ray crystallography to be the monomeric (NNTBS)Yb(THF)3 (Fig.S14†). The structural parameters are consistent with an Yb(II) ion. However, attempts to obtain an analytically pure sample of (NNTBS)Yb(THF)3 were not successful due to the loss of the coordinating THF molecules upon drying. This observation contrasts what we know about trivalent rare earth metal complexes supported by the NNTBS ligand and indicates a weak interaction between Yb(II) and THF. In an effort to obtain an Yb(II) complex that is stable in both the solid and solution state, a chelating ligand, 18-crown-6, was employed. The chelation effect allowed a complete replacement of the coordinating THF molecules (Scheme 2c). The 1H NMR spectrum of the resulting product, (NNTBS)Yb(18-crown-6), showed a single peak at 3.43 ppm for 18-crown-6 protons, indicating a fluxtional behavior in solution. In the solid state, we obtained two coordination isomers from different crystals of the same crystallization batch, (NNTBS)Yb(κ3-18-crown-6) (Fig. S16†) and (NNTBS)Yb(κ4-18-crown-6) (Fig. S17†), which differed by the number of coordinated O-donors from 18-crown-6.
In previously reported M2-biph-K2 (ref. 46 and 47) and Sm2-biph-K2, it was found that upon the addition of 18-crown-6, the inverse-sandwich structure remained intact and the bonding interaction between rare earth ions and biphenyl strengthened in the ion pair complexes M2-biph-[K(18-crown-6)]2. However, the reaction of 18-crown-6 with Yb2-biph-K2 led to the collapse of the whole structure (Scheme 2d). Upon the addition of excess 18-crown-6, the dark solution of Yb2-biph-K2 became clouded. After crystallization, the dark crystals that precipitated out were determined by X-ray crystallography to be the previously reported [K(18-crown-6)][biphenyl],59 while the orange supernatant was identified to be (NNTBS)Yb(18-crown-6) by 1H NMR spectroscopy (Fig. S10†). [K(18-crown-6)][biphenyl] is likely to be the decomposition product of the initially generated bare biphenyl dianion, which was not stable under the reaction conditions. In addition, we also identified [K(18-crown-6)][(NNTBS)2Yb] by 1H NMR spectroscopy. We rationalized that the decomposition of Yb2-biph-K2 upon the addition of 18-crown-6 is due to the much weaker interaction between the Yb(II) ions and the biphenyl dianion.
Spectroscopic and magnetic characterization
To probe the electronic structures of samarium and ytterbium biphenyl complexes further, we collected the UV/Vis/NIR spectra of Sm2-biph-K2, Sm2-biph-[K(18-crown-6)]2, and Yb2-biph-K2 in THF. The spectra of Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2 were almost identical, with an intense band around 403 nm (ε > 104 M−1 cm−1) and a broad intense band centered at 638 nm (ε > 104 M−1 cm−1, Fig. 3). The broad intense band in the visible region is responsible for the extremely dark color of their crystals and solutions; it may be attributed to ligand to metal charge transfer (LMCT) or an excitation of the ligand-based orbitals. However, the absorption spectrum of Yb2-biph-K2 lacked this broad intense band in the visible region (Fig. S18†). The near-infrared (NIR) region of Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2 has a higher intensity than that of Yb2-biph-K2, in agreement with the latter having a 4f14 electronic configuration, and, thus, no f–f transitions. Moreover, there was no Yb(III) characteristic f–f transition observed around 980 nm (ref. 60) for Yb2-biph-K2, further supporting the presence of Yb(II) instead of Yb(III).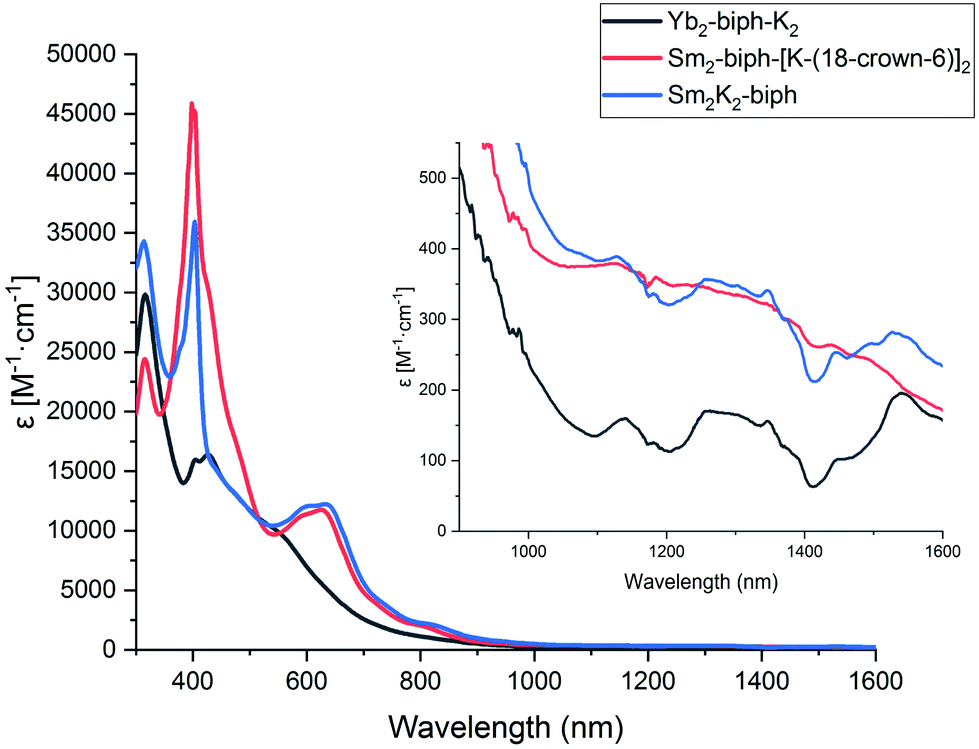 | ||
| Fig. 3 UV/Vis/NIR spectra of Yb2-biph-K2 (black), Sm2-biph-[K(18-crown-6)]2 (red), and Sm2-K2-biph (blue) in THF at room temperature. The NIR region from 1000–1600 nm is enlarged in the upper right corner. | ||
Magnetic susceptibility has been used to probe the ground state electronic configuration of divalent rare earth ions.61 In particular, Sm(II) and Sm(III) have distinct magnetic susceptibilities, with the normal range for Sm(II) being 3.4–3.8 μB, while the normal range for Sm(III) is 1.3–1.9 μB.62–67 The room temperature magnetic susceptibilities of Sm-I, Sm2-biph-K2, and Sm2-biph-[K(18-crown-6)]2 were determined by the Evans method to be 1.53, 2.47, and 2.39 μB, respectively (per formula unit). These results are consistent with the assignment of Sm(III) having a 4f5 electron configuration in Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2.
Preliminary X-ray absorption near edge structure (XANES) spectroscopy studies were conducted on Sm2-biph-K2 and Yb2-biph-K2 (see ESI for details†). Although the collected data agreed with the findings discussed above, the quality of the data was not high enough to allow decisive conclusions about the oxidation state of samarium and ytterbium in these biphenyl complexes.
Quantum chemical calculations
Theoretical calculations were carried out at the level of scalar relativistic density functional theory (DFT) to probe the electronic structures and bonding interactions of (Sm2-biph)2− (the structure is based on Sm2-biph-[K(18-crown-6)]2 with the potassium counter cations omitted for simplification) and Yb2-biph-K2 as well as two hypothetical isomeric structures of (Sm2-biph)iso2− and [Yb2-biph-K2]iso for comparison (in (Sm2-biph)iso2−, the two Sm ions are bound to different phenyl rings; in [Yb2-biph-K2]iso, the two Yb ions are bound to the same phenyl ring while the two potassium ions are bound to the other phenyl ring, see also Fig. S19†). Herein, the alkyl substituents on the silicon atoms were replaced by hydrogen atoms for simplification. In order to determine the ground state of the (Sm2-biph)2− complex, two electronic configurations 11A and 13A with different multiplicities were performed. The calculated energy of (Sm2-biph)2− (13A) is 26.6 kcal mol−1 higher than that of (Sm2-biph)2− (11A). Furthermore, there is also a larger discrepancy in the optimized structure parameters for (Sm2-biph)2− (13A) and those of the experimental compound (Table S3†). For example, the calculated average Sm–C distance of 2.68 Å and Sm-Ccentroid distance of 2.33 Å are significantly longer than the experimental ones of 2.59 Å and 2.15 Å. The calculated torsion angle of the Sm-bound phenyl ring is only 0.9°, while the experimental one is 10.7(9)°. In contrast, the optimized structure parameters for (Sm2-biph)2− (11A) are in good agreement with those of the experimental structure (Table S3†). The calculated average Sm–C distance of 2.56 Å and Sm–Ccentroid distance of 2.10 Å are comparable to the experimental ones of 2.59 Å and 2.15 Å, respectively. The torsion angle of the Sm-bound phenyl ring is also reproduced well (12.4° vs. 10.7(9)°). Moreover, the calculated average C–C distance of the Sm-bound phenyl ring is 1.47 Å, which is close to the experimental average value of 1.46 Å. For the unbound phenyl ring, the calculated average C–C distance is 1.40 Å, similar to the experimental one of 1.39 Å. Overall, according to the DFT results, the (Sm2-biph)2− complex has a 11A ground state, in which the oxidation state of the samarium ions is 3+ with a 4f5 electronic configuration.As depicted in Fig. 4a, the highest occupied molecular orbital (HOMO, 40b) and HOMO−1 (41a) confirm the δ bonding interaction between the samarium ions and the bound phenyl ring in (Sm2-biph)2−. Moreover, the contribution from the C 2p orbitals of the bound phenyl ring is around 60% and the contribution from the 5d orbitals of the samarium ions is over 20% on average. Furthermore, the natural population analysis (NPA) charges for the bound ring (−1.56) and the unbound ring (−0.25) are significantly different (Table S5†). Indeed, the δ bonding molecular orbital (MO) is formed by the interaction between the 5d atomic orbitals of samarium and the π* MOs of the Sm-bound phenyl ring. Based on the MO component percentage of the δ bonding interaction and NPA results, the four δ bonding electrons are mainly localized on the bound phenyl ring, resulting in a 10π-electron tetraanionic system in (Sm2-biph)2−, similar to the previously reported Y2-biph-K2.46 Meanwhile, the proposed isomer (Sm2-biph)iso2− can be formulated as two 8π-electron dianionic systems (Fig. S21†). Apart from that, the energy of the hypothetical structure (Sm2-biph)iso2− is calculated to be 30.3 kcal mol−1 higher than the experimental structure (Sm2-biph)2− (Fig. S24†). For Yb2-biph-K2, the biphenyl ring contributes around 70% to the HOMO with a small amount (ca. 10%) of 4f ytterbium atomic orbitals, indicating that there is a weak interaction between the ytterbium ions and the biphenyl ring (Fig. 4b). Furthermore, HOMO−1 to HOMO−14 are mostly composed of the 4f electrons of ytterbium with almost no biphenyl contribution. A similar bonding pattern is found for the Kohn–Sham frontier MOs in the proposed isomer [Yb2-biph-K2]iso (Fig. S22†).
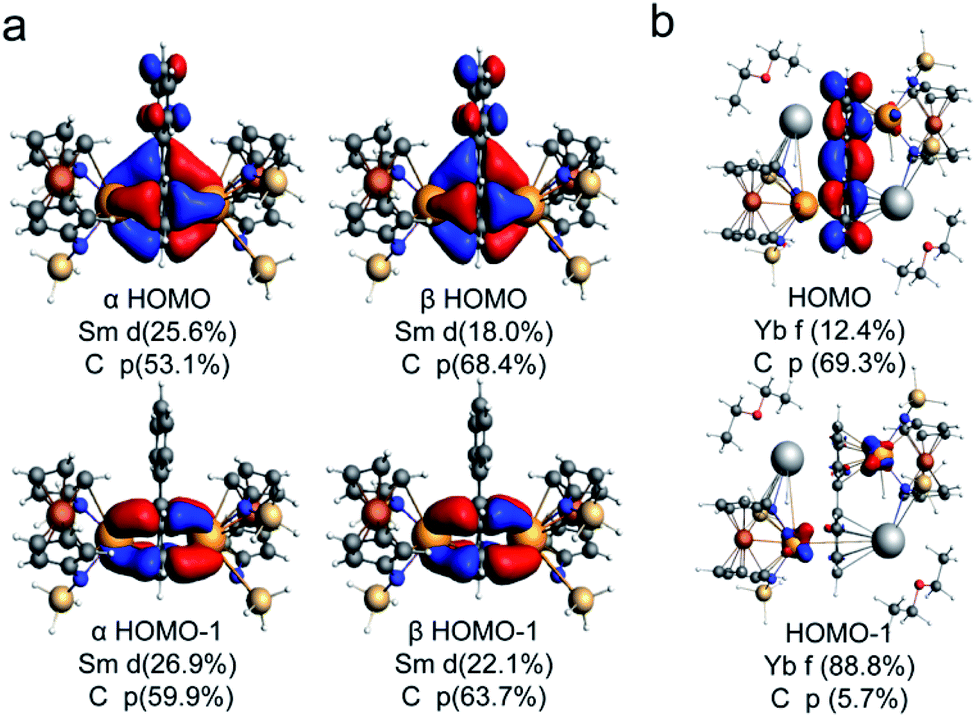 | ||
| Fig. 4 Kohn–Sham representations of HOMO and HOMO−1 and main atomic orbital contributions for (Sm2-biph)2− (a) and Yb2-biph-K2 (b). The alkyl substituents on the silicon atoms were replaced by hydrogen atoms for simplification (iso = 0.03). | ||
The δ bonding assignment in the simplified (Sm2-biph)2−, represented as (Sm2-biph)Cl2− hereafter, in which the NNTBS ligands are replaced by chlorine atoms, is also supported by adaptive natural density partitioning (AdNDP)68 analysis as shown in Fig. S20.† The semi-localized results indicate that the bonding interaction between the samarium ions and the bound phenyl ring involve two 8c–2e δ bonds, which resemble the Kohn–Sham frontier molecular orbitals. Fig. 4 illustrates the bonding interaction scheme of (Sm2-biph)Cl2− between the fragments of (Sm2Cl4)2+, with two Sm(III) ions each in a 4f5 electron configuration, and (biphenyl)4−.
The 4f orbitals are known to be radially too contracted in lanthanides to participate in chemical bonding, and form a nonbonding f-band shown as a red shadow in Fig. 5. On the other hand, the 5d orbitals are more radially extended than the 4f orbitals. Thus, the two unoccupied δ-type 5d orbitals (30a, 30b) in the [Sm2Cl4]2+ fragment and the two occupied π* orbitals (15b, 18a) of the biphenyl ring form two δ bonding MOs (41a, 40b). Furthermore, the stability of Sm(III) with a 4f5 electronic configuration is evident by the energy gap of 2.19 eV between the HOMO−1 and the 4f band. The stability of the whole compound is also revealed by the large HOMO–LUMO gap of 3.14 eV, indicating that the ground state is dominated by a single configuration and the description of structure and bonding based on single-configurational DFT methods is sensible.
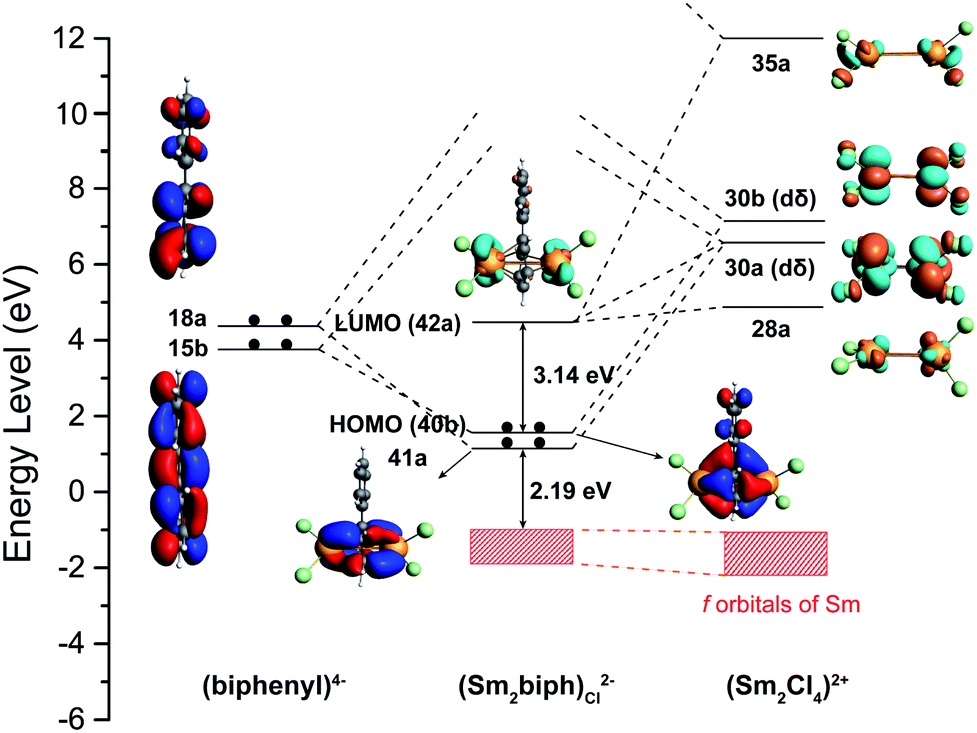 | ||
| Fig. 5 Bonding scheme of (Sm2-biph)Cl2− illustrating the MO interactions between [Sm2Cl4]2+ and the biphenyl tetraanion (iso = 0.03). | ||
The nature of the above-mentioned pairwise orbital interactions can be further analyzed by the energy decomposition analysis with natural orbitals for chemical valence (EDA-NOCV).69,70 Table S4† shows the numerical results for the interaction between fragments of [Sm2Cl4]2+ and (biphenyl)4− in (Sm2-biph)Cl2−. It shows that 60% of the attractive interaction ΔEint is from the electrostatic attraction ΔEelstat, 39% from the covalent orbital interaction ΔEorb and the rest 1% is from the dispersion interaction ΔEdisp. The breakdown of the ΔEorb into individual orbital contributions reveals that there are two major terms ΔEorb(δ1) and ΔEorb(δ2) due to the δ-type bonding with the energies of −278.75 and −192.77 kcal mol−1, respectively. The total amount of −471.52 kcal mol−1 for these two terms ΔEorb(δ1) and ΔEorb(δ2) is about the same as the total ΔEorb term for Sm(C5Me5)2 and Sm(C4Me4P)2, with values of −494.4 and −484.8 kcal mol−1, respectively.71 The other two terms ΔEorb(π1) and ΔEorb(π2) also show a charge flow from the bound phenyl ring to Sm centers, accounting for about 10% in the ΔEorb. The δ bonding interaction in (Sm2-biph)2− is further characterized using the principal interacting orbital (PIO) approach.72 The calculated PIOs and principal interacting molecular orbitals (PIMOs) shown in Fig. S23† clearly reveal the δ-type bonding interaction between samarium ions and the biphenyl fragment in (Sm2-biph)2−, which further supports the above electronic structure assignment. The δ bonding interaction in these inverse sandwich samarium biphenyl complexes is reminiscent of recently identified inverse sandwich lanthanide-boron binary clusters73,74 and actinide metallacycles.75
Discussion
Sm2-biph-K2 and Yb2-biph-K2 extend the series of M2-biph-K2 (M = Sc, Y, La, Lu, Gd, Dy, and Er) to traditional divalent lanthanide ions and provide a unique case study of the electronic structures of low valent rare earth ions. In the previously reported (Cp′3M)− series, despite the fact that Nd(II) and Dy(II) adopted a non-traditional 4fn5d1 electronic configuration, rare earth metals with less negative M3+/2+ reduction potentials, including Tm, Sm, and Yb, maintained a traditional 4fn+1 electronic configuration for their divalent ions.39,40 However, in the series of [((Ad,MeArO)3mes)M]−, all traditional divalent rare earth ions kept a 4fn+1 electron configuration.42,43 In the M2-biph-K2 series, not only was dysprosium found unambiguously in the 3+ oxidation state,47 but also samarium was confirmed to be Sm(III) with a 4f5 electronic configuration. The latter is remarkable since the (biphenyl)0/1− reduction potential of −2.45 V vs. SHE56 is ca. 0.90 V more negative than the Sm3+/2+ reduction potential of −1.55 V vs. SHE.25 Instead of a simultaneous reduction to the 4f6 Sm(II) ions and a biphenyl dianion or even a neutral biphenyl, Sm2-biph-K2 contained two Sm(III) ions and a biphenyl tetraanion with most negative charges localized in the Sm-bound phenyl ring.To the best of our knowledge, Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2 are the first structurally characterized and spectroscopically confirmed Sm(III) complexes with anionic arene ligands that are stable in the presence of coordinating solvents like THF.76 The syntheses of (C10H8)Sm(THF)3,77 [(CpV(C10H8)SmCp(THF)n] and [CpV(C10H8)]2Sm(THF) (DME),78 and (CpSm)2(C10H8) (THF)4 (ref. 79) were reported but their structures were not determined by X-ray crystallography. Magnetic and spectroscopic data suggested the formation of Sm(II) and a naphthalene dianion in these compounds.76 The only other structurally characterized Sm(III) complexes with anionic arene ligands are (Cp*2Sm)2(C14H10), (Cp*2Sm)2(C16H10), and (Cp*2Sm)2(C18H12), which were synthesized by the reduction of the corresponding arene by Cp*2Sm.80 The structures of these compounds were characterized by X-ray crystallography and showed an unusual η3-allylic type of bonding between Sm ions and the arene ligand.80 The structural and spectroscopic data supported the formulation of two Sm(III) ions and dianionic arene ligands. However, upon the addition of THF to their toluene solutions, the regeneration of neutral arenes and Cp*2Sm(THF)2 immediately occurred, implying that these Sm(III) arene complexes are intrinsically unstable and readily decompose to Sm(II) and neutral arenes.80 Two structurally relevant samarium inverse-sandwich toluene complexes (Sm2L3)2(μ-η6:η6-C7H8) and (KSmL3)2(μ-η6:η6-C7H8) (L = OSi(OtBu)3) were recently reported by Mazzanti et al.81 The former tetranuclear complex was formulated with four Sm(II) ions and a toluene dianion bridging between two Sm(II) ions, while the latter was assigned to two Sm(II) ions bridged by a neutral toluene ligand. It is also worth noting that in the series of formal zero-valent rare earth metal bis(arene) complexes reported by Cloke et al., Sm(1,3,5-tBu3C6H3)2 is the least stable one among all M(1,3,5-tBu3C6H3)2 (except for Ce, Eu, Tm, and Yb, which did not form isolable products).82,83 The difference in the thermal stability of rare earth metal bis(arene) complexes is rationalized by the difference in the promotion energy from fns2 to fn−1d1s2 for different rare earth metals since the bonding interaction is mainly the back-donation from the d and s orbitals of the rare earth metals to the π* orbitals of the arenes.82–85 These literature precedents suggest that samarium should have a low tendency to form a strong bonding interaction with arenes due to the relative stability of the 4f6 electronic configuration of the Sm(II) ion.
The above observations lead to the question why in the series of M2-biph-K2, most rare earth metals, including Sm, are in the trivalent state and utilize primarily the dxy and dx2−y2 orbitals to form a strong δ bonding interaction with the π* orbitals of the bridging arene ligand. We propose two main reasons. First, the resulting arene tetraanion is a 10π-electron aromatic system, giving an extra aromatic stability to these inverse-sandwich compounds. In the series of (Cp*2Sm)2(arene), neither are the Sm ions bound to the π surface of the arene nor do the arenes fulfill the (4n + 2) Hückel rule.80 In the case of (KSmL3)2(μ-η6:η6-C7H8), a formulation of two Sm(III) ions would result in a dianionic toluene ligand, which is anti-aromatic.81 Similarly, in the series of [((Ad,MeArO)3mes)M]–, a single electron reduction could take place at either the metal center or the arene, but the latter would result in a mesitylene radical anion.43 However, this aromatic stabilization could not explain why in other inverse-sandwich samarium arene complexes, such as (Sm2L3)2(μ-η6:η6-C7H8), the toluene is a dianion and all Sm ions were divalent.81 It is also worth noting that a relevant trinuclear cerium toluene complex, [K(2.2.2-crypt)]2[((KL3Ce) (μ-η6:η6-C7H8))2Ce], supported by the same siloxide ligand reported by Mazzanti et al. was formulated as three Ce(II) ions and two toluene dianions,86 despite the much more negative Ce3+/2+ reduction potential of −3.2 V vs. SHE, compared to Sm3+/2+ reduction potential of −1.55 V vs. SHE.28 Therefore, other than aromaticity, we considered that the ferrocene diamide ligand NNTBS may also play a key role in stabilizing the unique electronic structure of Sm2-biph-K2. The δ bonding interaction in the inverse-sandwich complex Sm2-biph-K2 could be viewed by analogy to the sandwich compounds M(1,3,5-tBu3C6H3)2. In the latter, the strength of the M–arene bond is correlated to the promotion energy from 4fn6s2 to 4fn−15d16s2: the lower the promotion energy, the stronger the M–arene bond.82 While the energy of the 4f orbitals is barely changed by the ligand field, the 5d orbitals interact strongly with the ligand orbitals and their energy could be readily tuned by the ligand field. Compared to O-donors in (Sm2L3)2(μ-η6:η6-C7H8), N-donors in Sm2-biph-K2 provide a stronger ligand field, which is also responsible for lowering the energy level of the 5d orbitals. Moreover, the flexibility of the NNTBS ligand may play a role here, since it allows a much smaller bite angle (N–Sm–N) of 103.5(7)° and 98.1(3)° in Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2, respectively, which are very close to the bite angle (Cl–Sm–Cl) of 101.9° in optimized (Sm2Cl4)4+ structure, but much smaller than that of 133.6(1)° in Sm-I. Combined, the strong ligand field of N-donors and the flexibility in coordination geometry of the NNTBS ligand may explain why Sm2-biph-K2 has a unique electronic structure among low valent samarium arene complexes.
Unlike Sm2-biph-K2, Yb2-biph-K2 contains two Yb(II) ions and a biphenyl dianion with the negative charges equally delocalized over the two phenyl rings. The Yb(II) ions and the biphenyl dianion were only weakly bound through a primarily ionic interaction, which could be readily disrupted by the addition of 18-crown-6. The divalent 4f14 electron configuration of ytterbium in Yb2-biph-K2 was supported by structural parameters, reactivity studies, absorption spectra, and diamagnetic nature of the compound. It is worth noting that ytterbium was reported to have a multiconfigurational character when complexed with redox active ligands, such as 2,2′-bipyiridine.87–90 However, we did not observe any evidence for such a multiconfigurational character in Yb2-biph-K2. This is likely due to the already highly reducing nature of the biphenyl dianion.
The distinct electronic structures of samarium and ytterbium biphenyl complexes can be rationalized by the difference in their M3+/2+ reduction potentials,25 as well as their fn to fn−1d1 promotion energies.82 The preference to stay in the completely filled 4f14 electronic configuration led to a switch in the electronic structure of Yb2-biph-K2 compared to other M2-biph-K2 with more negative M3+/2+ reduction potentials (Chart 1).
Conclusions
We successfully synthesized the inverse-sandwich samarium and ytterbium biphenyl complexes Sm2-biph-K2 and Yb2-biph-K2 and characterized them by X-ray crystallography, elemental analysis, NMR and UV/Vis/NIR spectroscopy, and room temperature magnetic susceptibility measured by the Evans method. The structural and spectroscopic data are consistent with the assignment of 4f5 Sm(III) ions and a biphenyl tetraanion in Sm2-biph-K2 and 4f14 Yb(II) ions and a biphenyl dianion in Yb2-biph-K2. The reaction of Sm2-biph-K2 or Yb2-biph-K2 with 18-crown-6 proceeded with distinct results: while the Sm–arene interaction remained intact, the Yb–arene interaction was readily disrupted by the coordination of 18-crown-6. DFT calculations were carried out on model complexes for Sm2-biph-K2 and Yb2-biph-K2 to elucidate the electronic structures and bonding interactions. Sm2-biph-K2 was confirmed to bear two Sm(III) ions and a charge-localized biphenyl tetraanion. The bonding interaction between the Sm(III) ions and the biphenyl tetraanion involves the δ-type 5d orbitals of Sm and the π* orbitals of the bound phenyl ring and features two δ bonds with a covalent character accounting for a 39% attractive interaction by computational analysis. On the contrary, Yb2-biph-K2 was found to contain two Yb(II) ions and a charge-delocalized biphenyl dianion, which are only weakly bound through a primarily ionic interaction. Sm2-biph-K2 and Yb2-biph-K2 extended the series of M2-biph-K2. The electronic structure of rare earth metals in this series was compared with the previously reported (Cp′3M)− and [((Ad,MeArO)3mes)M]− series as well as the solid state rare earth metal diiodides. It was found that the switch point for rare earth ions to adopt a divalent 4fn electronic configuration changed from Nd in MI2 and [((Ad,MeArO)3mes)M]− to Tm in (Cp′3M)− and further to Yb in M2-biph-K2, showing that the appropriate choice of ligands could compensate the positive shifts in M3+/2+ reduction potentials and the increase in the promotion energy for the fn+1 to fnd1 transition. The stability of Sm2-biph-K2 and Sm2-biph-[K(18-crown-6)]2 is remarkable since they are the only structurally characterized and stable Sm(III) complexes with a highly reducing anionic arene ligand. In comparison with other samarium arene complexes, the unique electronic structure of Sm2-biph-K2 is attributed to the features of the ferrocene diamide NNTBS ligand. Overall, this study extends the series of the inverse-sandwich complexes M2-biph-K2 to traditional divalent rare earth metals Sm and Yb, and sheds light on the relationship between the electronic structures of rare earth ions and their coordination environment, as well as the bonding interaction between rare earth ions and arene ligands in low valent metal chemistry. Future studies will focus on detailed XANES spectroscopic and magnetometry studies to gain further insight into the electronic structure of these highly reducing systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
YX, X-KZ, H-SH, and WH thank Peking University and Beijing National Laboratory for Molecular Sciences for funding. JTM was supported by the National Science Foundation under Cooperative Agreement no. EEC1647722. YX thanks Dr Jie Su for help with X-ray crystallography and Dr Hui Fu and Dr Xiu Zhang for help with NMR spectroscopy. PLD acknowledges support from NSF Grant CHE-1809116. The use of the Advance Photon Source was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under contract no. DE-AC02-06CH11357. MRCAT operations were supported by the Department of Energy and MRCAT member institutions. The Tsinghua Xuetang Talents Program is acknowledged for providing computational resources. We are grateful to Ms Rong Sun and Prof. Bing-wu Wang at Peking University for helpful discussion about magnetic data.References
- N. Kaltsoyannis and P. Scott, The f Elements, Oxford University Press, New York, 1999 Search PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, Wiley, Hoboken, NJ, 2006 Search PubMed.
- D. A. Atwood, The Rare Earth Elements: Fundamentals and Applications, Wiley, Hoboken, NJ, 2012 Search PubMed.
- A. J. Freeman and R. E. Watson, Phys. Rev., 1962, 127, 2058–2075 CrossRef CAS.
- G. H. Dieke, Spectra and Energy Levels of Rare Earth Ions in Crystals, Wiley, New York, 1968 Search PubMed.
- K. A. Gschneidner, J. Less-Common Met., 1971, 25, 405–422 CrossRef CAS.
- H. J. Emeléus and A. G. Sharpe, Modern Aspects of Inorganic Chemistry, Wiley, New York, 4th edn, 1973 Search PubMed.
- J. C. G. Bünzli and G. R. Choppin, Lanthanide probes in life, chemical, and earth sciences: theory and practice, Elsevier, Amsterdam, 1989 Search PubMed.
- K. A. Gschneidner, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2016, vol. 50, pp. 1–18 Search PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- G. R. Choppin, J. Alloys Compd., 2002, 344, 55–59 CrossRef CAS.
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS.
- S. M. Butorin, K. O. Kvashnina, J. R. Vegelius, D. Meyer and D. K. Shuh, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8093–8097 CrossRef CAS.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS.
- C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS.
- N. T. Rice, I. A. Popov, D. R. Russo, J. Bacsa, E. R. Batista, P. Yang, J. Telser and H. S. La Pierre, J. Am. Chem. Soc., 2019, 141, 13222–13233 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, R. Scopelliti, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2020, 59, 3549–3553 CrossRef CAS.
- A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS.
- Q. Zhang, S.-X. Hu, H. Qu, J. Su, G. Wang, J.-B. Lu, M. Chen, M. Zhou and J. Li, Angew. Chem., Int. Ed., 2016, 55, 6896–6900 CrossRef CAS.
- S.-X. Hu, J. Jian, J. Su, X. Wu, J. Li and M. Zhou, Chem. Sci., 2017, 8, 4035–4043 RSC.
- W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 941–946 CrossRef CAS.
- M. N. Bochkarev, I. L. Fedushkin, A. A. Fagin, T. V. Petrovskaya, J. W. Ziller, R. N. R. Broomhall-Dillard and W. J. Evans, Angew. Chem., Int. Ed. Engl., 1997, 36, 133–135 CrossRef CAS.
- W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2000, 122, 11749–11750 CrossRef CAS.
- M. N. Bochkarev, I. L. Fedushkin, S. Dechert, A. A. Fagin and H. Schumann, Angew. Chem., Int. Ed., 2001, 40, 3176–3178 CrossRef CAS.
- L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- L. J. Nugent, R. D. Baybarz, J. L. Burnett and J. L. Ryan, J. Phys. Chem., 1973, 77, 1528–1539 CrossRef CAS.
- N. B. Mikheev and A. N. Kamenskaya, Coord. Chem. Rev., 1991, 109, 1–59 CrossRef CAS.
- W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef CAS.
- G. Meyer, Chem. Rev., 1988, 88, 93–107 CrossRef CAS.
- G. Meyer, Z. Anorg. Allg. Chem., 2007, 633, 2537–2552 CrossRef CAS.
- G. Meyer, J. Solid State Chem., 2019, 270, 324–334 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488–1491 CrossRef CAS.
- M. R. MacDonald, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 15914–15917 CrossRef CAS.
- M. R. MacDonald, J. E. Bates, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 8420–8423 CrossRef CAS.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS.
- D. H. Woen, G. P. Chen, J. W. Ziller, T. J. Boyle, F. Furche and W. J. Evans, Angew. Chem., Int. Ed., 2017, 56, 2050–2053 CrossRef CAS.
- A. J. Ryan, L. E. Darago, S. G. Balasubramani, G. P. Chen, J. W. Ziller, F. Furche, J. R. Long and W. J. Evans, Chem.–Eur. J., 2018, 24, 7702–7709 CrossRef CAS.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS.
- M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2015, 137, 369–382 CrossRef CAS.
- M. E. Fieser, M. G. Ferrier, J. Su, E. Batista, S. K. Cary, J. W. Engle, W. J. Evans, J. S. Lezama Pacheco, S. A. Kozimor, A. C. Olson, A. J. Ryan, B. W. Stein, G. L. Wagner, D. H. Woen, T. Vitova and P. Yang, Chem. Sci., 2017, 8, 6076–6091 RSC.
- V. E. Fleischauer, G. Ganguly, D. H. Woen, N. J. Wolford, W. J. Evans, J. Autschbach and M. L. Neidig, Organometallics, 2019, 38, 3124–3131 CrossRef CAS.
- M. E. Fieser, C. T. Palumbo, H. S. La Pierre, D. P. Halter, V. K. Voora, J. W. Ziller, F. Furche, K. Meyer and W. J. Evans, Chem. Sci., 2017, 8, 7424–7433 RSC.
- C. T. Palumbo, D. P. Halter, V. K. Voora, G. P. Chen, A. K. Chan, M. E. Fieser, J. W. Ziller, W. Hieringer, F. Furche, K. Meyer and W. J. Evans, Inorg. Chem., 2018, 57, 2823–2833 CrossRef CAS.
- K. E. Johnson and J. N. Sandoe, J. Chem. Soc., Abstr., 1969, 1694–1697 RSC.
- S. A. Kulyukhin, Radiochemistry, 2018, 60, 451–469 CrossRef CAS.
- W. Huang, F. Dulong, T. Wu, S. I. Khan, J. T. Miller, T. Cantat and P. L. Diaconescu, Nat. Commun., 2013, 4, 1448 CrossRef.
- W. Huang, J. J. Le Roy, S. I. Khan, L. Ungur, M. Murugesu and P. L. Diaconescu, Inorg. Chem., 2015, 54, 2374–2382 CrossRef CAS.
- W. Huang, S. I. Khan and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 10410–10413 CrossRef CAS.
- W. Huang and P. L. Diaconescu, Chem. Commun., 2012, 48, 2216–2218 RSC.
- W. Huang and P. L. Diaconescu, Eur. J. Inorg. Chem., 2013, 2013, 4090–4096 CrossRef CAS.
- W. Huang, P. M. Abukhalil, S. I. Khan and P. L. Diaconescu, Chem. Commun., 2014, 50, 5221–5223 RSC.
- W. Huang, J. L. Brosmer and P. L. Diaconescu, New J. Chem., 2015, 39, 7696–7702 RSC.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- F. A. Cotton and W. Schwotzer, J. Am. Chem. Soc., 1986, 108, 4657–4658 CrossRef CAS.
- S. Ilango, B. Vidjayacoumar and S. Gambarotta, Dalton Trans., 2010, 39, 6853–6857 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, J. Am. Chem. Soc., 1997, 119, 9071–9072 CrossRef CAS.
- W. Huber, A. M. Und and K. Müllen, Chem. Ber., 1981, 114, 1318–1336 CrossRef CAS.
- M. Castillo, A. J. Metta-Magaña and S. Fortier, New J. Chem., 2016, 40, 1923–1926 RSC.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- K. R. Meihaus, M. E. Fieser, J. F. Corbey, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2015, 137, 9855–9860 CrossRef CAS.
- W. J. Evans, J. W. Grate, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 405–409 CrossRef CAS.
- W. J. Evans, J. W. Grate and R. J. Doedens, J. Am. Chem. Soc., 1985, 107, 1671–1679 CrossRef CAS.
- W. J. Evans, D. K. Drummond, S. G. Bott and J. L. Atwood, Organometallics, 1986, 5, 2389–2391 CrossRef CAS.
- W. J. Evans and T. A. Ulibarri, J. Am. Chem. Soc., 1987, 109, 4292–4297 CrossRef CAS.
- J. Jubb and S. Gambarotta, J. Am. Chem. Soc., 1994, 116, 4477–4478 CrossRef CAS.
- A. N. Selikhov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. del Rosal and L. Maron, Organometallics, 2015, 34, 555–562 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- S. Labouille, C. Clavaguéra and F. Nief, Organometallics, 2013, 32, 1265–1271 CrossRef CAS.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Chem.–Eur. J., 2018, 24, 9639–9650 CrossRef CAS.
- W.-L. Li, T.-T. Chen, D.-H. Xing, X. Chen, J. Li and L.-S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6972–E6977 CrossRef CAS.
- T.-T. Chen, W.-L. Li, J. Li and L.-S. Wang, Chem. Sci., 2019, 10, 2534–2542 RSC.
- M. P. Kelley, I. A. Popov, J. Jung, E. R. Batista and P. Yang, Nat. Commun., 2020, 11, 1558 CrossRef CAS.
- M. N. Bochkarev, Chem. Rev., 2002, 102, 2089–2118 CrossRef CAS.
- M. N. Bochkarev, A. A. Trifonov, E. A. Fedorova, N. S. Emelyanova, T. A. Basalgina, G. S. Kalinina and G. A. Razuvaev, J. Organomet. Chem., 1989, 372, 217–224 CrossRef CAS.
- I. L. Fedushkin, V. K. Nevodchikov, V. K. Cherkasov, M. N. Bochkarev, H. Schumann, F. Girgsdies, F. H. Görlitz, G. Kociok-Köhn and J. Pickardt, J. Organomet. Chem., 1996, 511, 157–162 CrossRef CAS.
- A. V. Protchenko, L. N. Zakharov, G. K. Fukin, Y. T. Struchkov and M. N. Bochkarev, Russ. Chem. Bull., 1996, 45, 950 CrossRef.
- W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1994, 116, 2600–2608 CrossRef CAS.
- R. P. Kelly, D. Toniolo, F. F. Tirani, L. Maron and M. Mazzanti, Chem. Commun., 2018, 54, 10268–10271 RSC.
- D. M. Anderson, F. G. N. Cloke, P. A. Cox, N. Edelstein, J. C. Green, T. Pang, A. A. Sameh and G. Shalimoff, J. Chem. Soc., Chem. Commun., 1989, 53–55 RSC.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–24 RSC.
- W. A. King, T. J. Marks, D. M. Anderson, D. J. Duncalf and F. G. N. Cloke, J. Am. Chem. Soc., 1992, 114, 9221–9223 CrossRef CAS.
- W. A. King, S. Di Bella, G. Lanza, K. Khan, D. J. Duncalf, F. G. N. Cloke, I. L. Fragala and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 627–635 CrossRef CAS.
- R. P. Kelly, L. Maron, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed., 2017, 56, 15663–15666 CrossRef CAS.
- M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- R. E. Da Re, C. J. Kuehl, M. G. Brown, R. C. Rocha, E. D. Bauer, K. D. John, D. E. Morris, A. P. Shreve and J. L. Sarrao, Inorg. Chem., 2003, 42, 5551–5559 CrossRef CAS.
- C. H. Booth, M. D. Walter, D. Kazhdan, Y.-J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, X-ray crystallography, XANES and UV/Vis/NIR spectroscopic data, and DFT calculation details. CCDC 2012143–2012149. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03555f |
| ‡ Y. X. and X.-K. Z. contributed equally to this work. |
| § Present address: X-ray Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA. |
| ¶ Present address: Davidson School of Chemical Engineering, Purdue University, West Lafayette, Indiana 47907, USA. |
| This journal is © The Royal Society of Chemistry 2021 |