
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected evolution of cyclic peptides for inhibition of autophagy†
Joshua P.
Gray
 a,
Md. Nasir
Uddin
a,
Rajan
Chaudhari
b,
Margie N.
Sutton
a,
Hailing
Yang
b,
Philip
Rask
b,
Hannah
Locke
c,
Brian J.
Engel
a,
Nefeli
Batistatou
d,
Jing
Wang
d,
Brian J.
Grindel
a,
Pratip
Bhattacharya
a,
Seth T.
Gammon
a,
Shuxing
Zhang
b,
David
Piwnica-Worms
a,
Joshua A.
Kritzer
d,
Zhen
Lu
b,
Robert C.
Bast
Jr.
b and
Steven W.
Millward
*a
a,
Md. Nasir
Uddin
a,
Rajan
Chaudhari
b,
Margie N.
Sutton
a,
Hailing
Yang
b,
Philip
Rask
b,
Hannah
Locke
c,
Brian J.
Engel
a,
Nefeli
Batistatou
d,
Jing
Wang
d,
Brian J.
Grindel
a,
Pratip
Bhattacharya
a,
Seth T.
Gammon
a,
Shuxing
Zhang
b,
David
Piwnica-Worms
a,
Joshua A.
Kritzer
d,
Zhen
Lu
b,
Robert C.
Bast
Jr.
b and
Steven W.
Millward
*a
aDepartment of Cancer Systems Imaging, University of Texas MD Anderson Cancer Center, USA. E-mail: smillward@mdanderson.org
bDepartment of Experimental Therapeutics, University of Texas MD Anderson Cancer Center, USA
cDepartment of Biology and Biochemistry, University of Houston, USA
dDepartment of Chemistry, Tufts University, USA
First published on 13th January 2021
Abstract
In recent decades it has become increasingly clear that induction of autophagy plays an important role in the development of treatment resistance and dormancy in many cancer types. Unfortunately, chloroquine (CQ) and hydroxychloroquine (HCQ), two autophagy inhibitors in clinical trials, suffer from poor pharmacokinetics and high toxicity at therapeutic dosages. This has prompted intense interest in the development of targeted autophagy inhibitors to re-sensitize disease to treatment with minimal impact on normal tissue. We utilized Scanning Unnatural Protease Resistant (SUPR) mRNA display to develop macrocyclic peptides targeting the autophagy protein LC3. The resulting peptides bound LC3A and LC3B—two essential components of the autophagosome maturation machinery—with mid-nanomolar affinities and disrupted protein–protein interactions (PPIs) between LC3 and its binding partners in vitro. The most promising LC3-binding SUPR peptide accessed the cytosol at low micromolar concentrations as measured by chloroalkane penetration assay (CAPA) and inhibited starvation-mediated GFP-LC3 puncta formation in a concentration-dependent manner. LC3-binding SUPR peptides re-sensitized platinum-resistant ovarian cancer cells to cisplatin treatment and triggered accumulation of the adapter protein p62 suggesting decreased autophagic flux through successful disruption of LC3 PPIs in cell culture. In mouse models of metastatic ovarian cancer, treatment with LC3-binding SUPR peptides and carboplatin resulted in almost complete inhibition of tumor growth after four weeks of treatment. These results indicate that SUPR peptide mRNA display can be used to develop cell-penetrating macrocyclic peptides that target and disrupt the autophagic machinery in vitro and in vivo.
Introduction
Macroautophagy (hereafter autophagy) is a conserved cellular catabolic process responsible for recycling damaged, misfolded, or superfluous cytosolic proteins and organelles.2 Upon initiation of autophagy, intracellular double membrane vesicles called autophagosomes are formed which engulf and sequester organelles, lipids, starches, and proteins.3 Cargo adapter proteins (e.g. p62) facilitate the transfer of organelles and protein aggregates to the interior of the autophagosome.4,5 Mature autophagosomes then fuse with lysosomes to degrade their contents to monomers for downstream anabolism and catabolism. Basal autophagy maintains cellular homeostasis through clearance of superfluous or damaged proteins and organelles, and autophagic flux is upregulated as an adaptive response to nutrient deprivation and exposure to cytotoxic agents. In recent years it has become increasingly apparent that autophagy upregulation plays a major role in the development of cancer and its response to treatment.6,7 Many tumors types—including ovarian,8 pancreatic,9 breast,10 and colon cancers11—depend on persistent activation of autophagy to sustain growth in the poorly vascularized, hypoxic, and nutrient-deprived conditions of the tumor microenvironment. Activation of autophagy in response to chemotherapy12 and radiation13 has been identified as a major contributing factor to acquisition of treatment resistance.14As a result of its central role in tumor development and treatment resistance, there has been intense clinical interest in autophagy inhibitors to augment frontline radiation and chemotherapy. As of 2019, over 50 Phase-I and Phase-II clinical trials focusing on autophagy inhibitors to treat cancer have been initiated, with the vast majority combining chloroquine (CQ) or hydroxychloroquine (HCQ) with current standard-of-care therapies.15 Unfortunately these non-specific lysosomotropic compounds inhibit the function of lysosomes in normal tissue leading to unwanted toxicities at therapeutically relevant doses.16 The narrow therapeutic window of chloroquine-based drugs argues for the development of targeted agents that selectively disrupt the fundamental protein–protein interactions (PPIs) of the autophagic machinery.
Microtubule-associate protein light chain 3 (LC3) is a 14 kDa ubiquitin-like protein that is essential for the maturation of the autophagosome, delivery of cargo to the autophagosome interior, and fusion with the lysosome.2 Following translation, pro-LC3 is cleaved by Atg4 to expose a C-terminal glycine residue, and the resulting product (LC3-I) is uniformly distributed throughout the cytosol. Upon induction of autophagy, LC3-I is modified by a series of autophagy-related proteins leading to conjugation of phosphatidylethanolamine (PE) to the exposed C-terminus (LC3-II). LC3-II is embedded in both inner and outer membranes of the nascent autophagosome where it facilitates its elongation and closure. Membrane-bound LC3-II also interacts with autophagy adapter proteins such as p62 (SQSTM1), NIX, NBR1, and NDP52 to transport damaged organelles and protein aggregates to the autophagosome interior. Disruption of LC3 processing and embedding—i.e. through LC3 knockout or knockout/knockdown of Atg5 or Atg7 which mediate LC3 conjugation to PE—leads to dysfunctional phagophore elongation, hampered autophagosome closure, and inhibition of fusion between the autophagosome and lysosome.2
Due to its essential role in the maturation of the autophagosome, blockade of LC3 PPIs represents a promising strategy for targeted autophagy inhibition. The vast majority of LC3 PPIs are thought to occur via a “hot spot” on the surface of LC3 consisting of two hydrophobic binding pockets (W site and L site) which recognize a core amino acid motif—(W/F/Y)–XX–(L/I/V) where X can be any amino acid.5,17 This sequence is referred to as the LC3-interacting motif (LIM) and is found in nearly all proteins that interact with LC3.
Inhibiting PPIs with small molecules remains challenging due to their small size and molecular radius.18 Small, linear peptides, which have shown great promise as PPI disruptors in vitro, cannot readily cross biological membranes and suffer from poor serum stability. LC3-binding peptides derived from naturally occurring LIMs possess relatively low affinities (mid to low micromolar range) and would likely suffer from the same stability and permeability issues that typically limit the usefulness of small, linear peptides in vivo.19
In natural product space, there are numerous examples of cyclic peptides that readily cross the cell membrane (e.g. microsytin LR, α-amanitin,20 and phalloidin21), exhibit nanomolar potencies against intracellular targets, and show sufficient biostability for in vivo applications. Consequently, macrocyclic peptide scaffolds have received a great deal of attention as a means to generate PPI-disrupting compounds with drug-like affinity, stability, and cell permeability.22 One strategy for designing cell-permeable cyclic peptides focuses heavily on N-methylation of amino acids within the context of macrocyclic scaffolds. The work of Lokey and co-workers, for example, has shown that extensive methylation of backbone amides within the peptide macrocycle effectively eliminates hydrogen bonds between backbone amide protons and water resulting in significant enhancement of passive cell permeability.23,24 Pei and co-workers have demonstrated that cyclic, amphipathic cell-penetrating peptides can drive the uptake of otherwise non-permeable peptide and protein cargo. These cyclic sequences can be incorporated within the context of bioactive macrocycles to greatly enhance cell uptake with minimal perturbation of biological function.25 The use of “stapled” peptides represents a third approach to enhancing cell uptake.26 In this approach, olefin metathesis is employed to cross-link the side chains of α-helical peptides, providing conformational constraint and stabilizing i + 4 and i, i + 7 hydrogen bonding patterns.26 This approach has been used to design proteolytically stable, cell permeable peptides that target MDM2/4 and inhibit proteasome-mediated degradation of p53.27
Scanning Unnatural Protease Resistant (SUPR) mRNA display provides a method to rapidly evolve macrocyclic, protease-resistant peptides for in vivo applications.28,29 SUPR peptide mRNA display, a recent advancement of mRNA display,30 integrates disuccinimidyl glutarate-based macrocyclization, incorporation of non-proteogenic N-methyl amino acids, and pre-screening for protease resistance.31 This technique leads to the selection of macrocyclic peptides with high target affinities and dramatically enhanced stability to hydrolytic degradation. The SUPR peptide scaffold also recapitulates many of the physiochemcial properties of cell permeable macrocyclic peptides (backbone amide N-methylation and a hydrophobic crosslinker) suggesting that SUPR peptides may possess the requisite cell permeability for disruption of intracellular PPIs.
We carried out SUPR mRNA display selections against recombinant LC3 to identify LC3-binding SUPR peptides with sufficient affinity, stability, and cell permeability for in vivo applications. The most promising candidate from the selection (SUPR4B1W) binds to LC3A and LC3B with mid-nanomolar affinity and was significantly more stable to proteolytic degradation than its linear and non-N-methylated counterparts. SUPR4B1W was found to penetrate the cytosol at low micromolar concentrations in cultured cells, and inhibited autophagosome formation in a concentration-dependent manner. SUPR4B1W sensitized multiple cancer cell lines to cisplatin treatment, resulted in accumulation of intracellular p62, and significantly modulated the LC3-I/LC3-II ratio in cell culture. Finally, SUPR4B1W, in combination with carboplatin, effectively prevented intraperitoneal tumor outgrowth in orthotopic mouse models of ovarian cancer suggesting that it possesses sufficient affinity, stability, and cell-permeability for further development as an in vivo autophagy inhibitor.
Experimental
Cell lines
OVCAR5, OVCAR8, and HEY cell lines were maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM Na-pyruvate, and 1X penicillin–streptomycin. SKOV3 cells were maintained in McCoy's 5A medium supplemented with 10% FBS, 2 mM L-glutamine, and 1X penicillin-streptomycin. MDA-MB-231, PANC-1, and MIA PaCa-2 cell lines were maintained in DMEM medium supplemented with 10% FBS and 1X penicillin–streptomycin.HeLa cells were maintained in RPMI 1640 supplemented with 5% FBS. HeLa cells stably expressing EGFP-LC3B were generated using pEGFP-LC3 (Addgene #24920). HeLa cells were directly transfected with pEGFP-LC3 using Mirus TransIT-LT1 transfection reagent and then maintained in RPMI 1640 supplemented with 5% fetal bovine serum and 400 μg mL−1 active G418 for 14 days. Single-cell sorting was performed on a FACSAria IIIu instrument. HeLA-GFP-LC3B cell lines derived from single cells were maintained in RPMI 1640 supplemented with 5% fetal bovine serum and 200 μg mL−1 active G418. All cell lines were obtained from and authenticated by the MD Anderson cytogenetics and cell authentication core.
SUPR mRNA display library preparation
To perform selections, we assembled a DNA library encoding eight random positions between an N-terminal methionine residue and a C-terminal lysine residue. The library was constructed by PCR amplification of the antisense template oligo (5′ TCC GCT GCC GGA TTT SNN SNN SNN SNN SNN SNN SNN SNN CAT TGT AAT TGT AAA TAT AAT T 3′ where N = A, C, G or T and S = C or G) with the primers MKForward (5′ TAA TAC GAC TCA CTA TAG GGA CAA TTA CTA TTT ACA ATT ACA 3′) and MKReverse (5′ GCC AGA TCC GCT GCC GGA TTT 3′). In addition to the open reading frame, the DNA library contains a T7 promoter sequence and a ribosome binding site.The mRNA display library was generated by in vitro transcription of the DNA library. Transcription was performed by incubating DNA libraries with transcription buffer (80 mM HEPES–KOH, pH 7.5, 2 mM spermidine, 40 mM DTT, and 25 mM MgCl2) and rNTP mixture (4 mM ATP, GTP, CTP, UTP) at 65 °C for 15 minutes before cooling briefly at room temp and adding T7 RNA polymerase. Transcription reactions were allowed to proceed overnight at 37 °C and were terminated by phenol chloroform extraction and ethanol precipitation. The mRNA library was then purified by Urea-PAGE followed by electroelution.
The mRNA library was then ligated at the 3′ end to a puromycin-DNA linker (pF30P – 5′ 8AA AAA AAA AAA AAA AAA AAA A77 7AC C6 3′) where 6 = puromycin CPG, 7 = spacer phosphoramidite 9, 8 = phosphate). Puromycin acts as an aminoacyl tRNA mimetic that can enter the active site of the ribosome to form a covalent link between the translated peptide and its encoding mRNA resulting in an mRNA–peptide fusion. Ligation reactions were performed by incubating 3000 pmol of mRNA library with 1.2 molar excess PF30P, 1.4 molar excess splint oligo (5′ TTT TTT TTT TTT TGC CAG ATC CGC TG 3′), and T4 DNA ligase at room temperature for 75 minutes. The ligated mRNA library was then purified by Urea-PAGE and electroelution as described above.
mRNA display selection
In vitro translations were performed using rabbit reticulocyte lysate kits (Ambion). To assemble the translation reaction, 10 pmol ligated mRNA library was added to rabbit reticulocyte lysate (17 μL), EasyTag [35S]-Methionine (3 μL, 8 μCi μL−1, PerkinElmer), amber suppressor tRNA charged with N-methyl alanine (2.25 μL, 1 μg μL−1), 20× high salt mix (1.25 μL, KOAc 3 M, MgOAc 10 mM, creatine phosphate 200 mM, and 1 mM of each amino acid except methionine). After a 70 minute incubation at 30 °C the translation mix was supplemented with 100 mM KCl and 0.5 mM MgCl2 to promote the formation of mRNA–peptide fusions. mRNA–peptide fusions were purified from the translation mix using oligo-dTcellulose (GE) which selectively binds to the poly dA linker between the mRNA template and puromycin. Oligo dT cellulose was washed with dT binding buffer (Tris–HCl pH 8.0 20 mM, NaCl 1 M, EDTA 1 mM, Triton-X 0.2% (v/v)) and translation mixes were rotated with 10 mg of oligo dT cellulose in 500 μL dT binding buffer at 4 °C for 60 minutes. The oligo-dT cellulose was then washed with dT wash buffer (Tris–HCl pH 8.0 20 mM, 300 mM NaCl) and mRNA–peptide fusions were eluted in 60 °C water.Following dT purification, mRNA–peptide fusions were ethanol precipitated and resuspended in 50 mM phosphate buffer pH 8.0 for cyclization between the primary amine of the lysine side chain and at the N-terminal amine using the crosslinker disuccinimidyl glutarate (DSG). The cyclization reaction was carried out by adding DSG to the mRNA–peptide fusions to a final concentration of 200 μg mL−1 and rotating at room temperature for 60 minutes. Cyclized mRNA–peptide fusions were then dT purified using the method described above. Following purification, mRNA–peptide fusions were reverse transcribed by M-MulV reverse transcriptase (NEB). Reverse transcribed fusions were subjected to protease screening by incubating with proteinase-K agarose (Sigma, 1 mg mL−1 resin) for 30 s before filtration with a Spin-X microcentrifuge column. To eliminate sequences with significant off-target binding to the resin, proteinase-treated fusions were incubated with streptavidin-agarose (Life Technologies) at 4 °C for 60 minutes before recovering unbound fusions using a Spin-X microcentrifuge filter.
Selection targets were prepared by incubating streptavidin-agarose with 1.5× molar excess biotinylated-LC3A at 4 °C with rotation for 30 minutes to saturate the resin with target. The immobilized target was then washed thoroughly with selection buffer (20 mM HEPES–KOH pH 7.5, 150 mM NaCl, 0.2% (v/v) Tween-20, 1 mg mL−1 BSA, 50 μg mL−1 tRNA, 1 mM β-mercaptoethanol). Selections were carried out by incubating fusions with immobilized target with rotation for 2 hours. Selection incubations were carried out at 4 °C for the first four rounds of selection and then split into parallel selections carried out at 25 °C or 4 °C thereafter. Following incubation, the selection resin was washed 3 times with selection buffer and filtered with a Spin-X column. The resin was then washed two additional times in selection buffer without BSA or tRNA and bound fusions were eluted in 0.15% sodium dodecyl sulfate (SDS) (Thermo-Fisher). SDS was subsequently precipitated using SDS-Out (Thermo-Fisher) and eluted fusions were filtered and ethanol precipitated. The precipitated pellet was resuspended in dH2O and one fifth of the total volume was subjected to PCR amplification using the MKForward and MKReverse primers. Small volumes of PCR product were removed every 3 PCR cycles and tested by 4% agarose gel to identify formation of the amplified product. The remaining eluted fusions were amplified using the minimum PCR cycles as defined in the previous step. Further selection rounds were conducted using previous round's enriched dsDNA library. The library was considered converged when the number of PCR cycles required to recover the enriched dsDNA library did not decrease in three consecutive rounds.
Radioligand binding assays
Plasmid minipreps for individual clones were PCR amplified using the MKForward and MKReverse primers to obtain dsDNA for individual sequences from the final pool. These dsDNA sequences were transcribed, ligated, and translated in the presence of [35S]-Methionine as described above. Following incubation for 70 minutes, RNAse A (1 μL, 10 mg mL−1, MilliporeSigma) was added to the translation mix to digest the fused mRNA. The peptides were purified from translation using dT cellulose and subjected to cyclization as described above. A small portion of the cyclized and purified translation product was reserved for scintillation counting and the remaining product was incubated with biotinylated-LC3A immobilized on neutravidin–acrylamide (Thermo-Fisher) at 25 °C with rotation for 1 hour. The incubation mix was pelleted, the supernatant reserved, and the pelleted resin washed three times with Spin-X microcentrifuge columns. The resin pellet and portions of the supernatant and each wash were subjected to scintillation counting and a percentage of input activity that remained bound to the immobilized target after washing was calculated for each clone.Atg4B activity assay
LC3-AMC (R&D Systems) was diluted in reaction buffer (25 mM Tris, 150 mM NaCl, 10 mM DTT) to a final concentration of 230 nM. 20 μL of LC3-AMC was added to 5 μL SUPR peptide (1 mM, 500 μM, 250 μM, 0 μM in reaction buffer) in a 96-well microtiter plate pre-blocked with 5 mg mL−1 BSA. After incubating for 5 minutes at room temperature, 25 μL 400 pM Atg4B (R&D Systems) was added to initiate the reaction. The final concentrations of each component in the hydrolysis reaction were LC3-AMC (92 nM), Atg4B (200 pM), and SUPR peptide (0–100 μM), and the final volume was 50 μL. The hydrolysis reaction was allowed to progress at room temperature for 40 minutes and the fluorescence measured each minute (λEx = 380 nm, λEm = 460 nm) on a BioTek Synergy H4 microplate reader. The resulting fluorescence at each time point (AU) was normalized to the fluorescence at t = 0 to compute the normalized product. Each reaction condition was carried out in triplicate and the values shown represent the mean value of three measurements.Proteinase-K stability assays
Protease stability was assessed by treating peptide solutions (1 mM in PBS, final volume 500 μL) with 0.3 mg proteinase-K agarose (Sigma). At time intervals of 0, 30, 60, 120, and 240 minutes, 50 μL samples were taken from the digestion mixture and separated from proteinase-K agarose by centrifugal filtration with Spin-X columns. Samples were then subjected to analytical HPLC (Agilent 1100 Series HPLC equipped with a Phenomenex Gemini 5μ C18 110A column) using gradient elution (10–30% buffer B over 30 minutes; buffer A dH2O + 0.1% (v/v) TFA, buffer B CH3CN + 0.1% (v/v) TFA). Peak area was calculated for the peak corresponding to the intact peptide for each sample and the fraction intact was calculated for each time point. Half-lives for each peptide were calculated in Graphpad Prism 8.Chloroalkane penetration assay (CAPA)
CAPA was performed as described.32,33 HeLa cells stably expressing the HaloTag-GFP-Mito construct34 were cultured in DMEM + 10% (v/v) FBS + 1X Pen/Strep + 1 μg mL−1 puromycin. Cells were seeded in a 96-well plate the day before the experiment at a density of 4 × 104 cells per well for the 4 hour incubation and 2 × 104 cells per well for the 24 hour incubation. The day of the experiment, the media was aspirated, and cells were treated with fresh media (as described above) and serial dilutions of ct-peptides [40.5 μΜ to 0.0021 μΜ] or ct-W control [2 μΜ to 0.0001 μΜ]. The plates were incubated for 4 h and 24 h at 37 °C with 5% CO2. The media was aspirated and cells were washed with additional media. Cells were then treated with 5 μM ct-TAMRA for 15 min, except for the no-ct-TAMRA control wells, which were incubated with media. Excess dye was washed away, and cells were trypsinized and resuspended in 180 μL phosphate-buffered saline. The cells were analyzed by flow cytometry (Guava easyCyte HT benchtop flow cytometer), gating for live, HaloTag expressing cells and measuring 5000 cells per well. Fluorescence was normalized to a no-molecule control (no ct-molecule during incubation, chased with ct-TAMRA, to indicate value for 100% fluorescence) and a no-dye control (no ct-molecule during incubation, chased with media without ct-TAMRA, to indicate value for 0% fluorescence).ATG8 homologue selectivity
SUPR-4B1W-biotin was conjugated to neutravidin acrylamide by adding 2 μL, 1 mM SUPR-4B1W-biotin to 50 μL neutravidin acrylamide slurry (Thermo-Fisher) with 200 μL binding buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7.8) and rotating for 1 h at room temperature. The immobilized peptide resin was then washed twice with 200 μL binding buffer using Spin-X centrifugal filter columns.pET151/D-TOPO bacterial expression vectors containing the genes for LC3A, LC3B, LC3C GABARAPL1, GABARAPL2, ATG8, and ubiquitin were designed and ordered from Thermo-Fisher. Forward and reverse primers were designed to allow PCR amplification of each gene from the bacterial vectors with the addition of 5′ T7 promotor and ribosome binding site sequences. Following PCR amplification, DNA templates were extracted with 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24:1 Phenol:chloroform:isoamyl alcohol (Sigma) and precipitated with >3 volumes cold ethanol at −80 °C. The centrifuged precipitate was dried by vacuum and resuspended in 100 μL dH2O. DNA templates were transcribed in vitro by T7 RNA polymerase and purified by UreaPAGE. 10 pmol of each RNA template were translated in vitro in the presence of [35S]-Methionine as described previously.
:24:1 Phenol:chloroform:isoamyl alcohol (Sigma) and precipitated with >3 volumes cold ethanol at −80 °C. The centrifuged precipitate was dried by vacuum and resuspended in 100 μL dH2O. DNA templates were transcribed in vitro by T7 RNA polymerase and purified by UreaPAGE. 10 pmol of each RNA template were translated in vitro in the presence of [35S]-Methionine as described previously.
To test selectivity of SUPR4B1W for the various ATG8 homologues, 8 μL of translation reaction was added to 10 μL of either peptide-saturated resin (125 pmol μL−1 resin) or naked resin in triplicate and brought to a final volume of 50 μL with binding buffer (20 mM HEPES–KOH pH 7.5, 150 mM NaCl). Reaction mixtures were rotated at room temperature for 60 minutes before transfer to a Spin-X column. After centrifugation, the resins were washed 3× with 200 μL binding buffer, and washed resin was subjected to scintillation counting. The resulting bound counts were normalized to the relative amounts of each radiolabeled protein in the translation mixture as determined by SDS-PAGE followed by autoradiography and densitometry (Fig. S1†).
Computational modeling of SUPR peptides and their interactions with LC3
The linear sequence of SUPR4B1W was built using protein builder panel of Molecular Operating Environment (MOE) software (v2019.0102). DSG linker was added to the N-terminal of Met residue and Ala8 was modified to NMA. To generate cyclic peptide conformations, a 1–1.5 angstrom distance restrains was applied between the carboxyl carbon of the DSG linker and the side chain ε-amino group of lysine. Conformational sampling was carried out using LowModeMD method with default settings except that the rejection limit was relaxed to 1000. We selected 10 conformers generated from conformational sampling for further optimization. The distance restraint between carboxyl carbon of DSG linker and ε-amino group of lysine was replaced with a covalent bond. The resulting structure was then subjected to all atom MD simulations with Amber10:EHT forcefield using Node–Poincare–Anderson equations derived from NPT Hamiltonian. The peptide structure was inserted into a cubic box of water molecules. To simulate the biological conditions, 0.1 M NaCl ions were added and the system was then equilibrated for 10 ps. Production runs were carried out for 50 ps, 500 ps and 1 ns and the trajectories were analyzed in MOE database viewer. A stable, low energy snapshot conformation was selected from the trajectories based on the intra-molecular hydrogen bonding pattern for further investigation.To model the interactions between LC3 and SUPR4B1W, we imported an LC3 crystal structure (PDB: 5CX3) into MOE. The selected conformation of cyclic SUPR peptide was aligned and superimposed with the LIM peptide from the crystal structure followed by tethered minimization to resolve atomic clashes with LC3. For linear peptide docking, initial orientation and docking poses were generated using CABS-dock server.35 Briefly, the LC3 structure and peptide sequence was provided to the CABS-dock server with contact information defining the interaction between Trp2 of the peptide and Phe108 of the protein (108:A 2:PEP 5.0 1.0). The CABS-dock experiments were carried out in triplicate. CABS-dock generates random structures of the peptide on the protein surface and employs replica exchange monte carlo simulations to generate peptide orientation and docking poses. The top ten poses from each triplicate run were collected and Ala8 was mutated to NMA8 and energy minimized to remove any steric clashes.
To identify per residue binding contribution of the cyclic peptide, computational alanine scan was carried out. Briefly, rotamers of the NMA residues were calculated and added to the system. Alanine scan calculations were carried out by mutating each residue with alanine except Ala7 which was mutated to Gly as per convention. The N-terminal Met and Lys9 residues were ignored as they were involved in cyclization. A LowMode ensemble technique was used to generate 25 conformations for each wild type and mutant peptide. The binding affinities were calculated using the GBVI/WSA scoring function.
Quantification of GFP-LC3 puncta
HeLa cells stably expressing eGFP-LC3 were seeded at a density of 30000 cells per well in an 8-well chambered coverslide (Corning) 24 hours before beginning treatment. Cells were treated with complete media supplemented with SUPR4B1W or SUPR4B1W scramble at final concentrations of 500 μM, 50 μM, 5 μM, or 0.5 μM for 24 hours. After 24 hours cells were starved in Hank's Balanced Salt Solution (Corning) for 2 hours and then fixed in 10% paraformaldehyde for 10 min, washed twice with PBS, mounted with fluoroshield containing DAPI (Sigma-Aldrich) and sealed with coverslips. Slides were imaged by epifluorescence microscopy at 60× magnification.
Average number of GFP-LC3 puncta per cell were quantified in ImageJ using established methods36 for each of the treatment conditions. Independent and interactive effects of SUPR peptide treatment on average puncta count were determined by two-way ANOVA with Type III sum of squares. Log10 transformations were used on average puncta count data to achieve satisfactory normality. Following analysis, Tukey's Honestly Significant Difference test (HSD) via the package lsmeans was used to determine which treatment means were significantly different from one another.
Cellular cisplatin sensitization assays
OVCAR8, OVCAR5, HEY, SKOv3, MDA-MB-231, PANC-1, and MIA PaCa-2 cells were seeded in black-walled 96-well plates at a density of 5,000 cells per well 24 hours prior to treatment. Cells were treated with SUPR peptides (50 μM) or chloroquine (5 μM, Sigma) both in the presence and absence of cisplatin (10 μM, Sigma) in order to determine the effect of autophagy inhibition on cellular viability in treatment resistant cell lines. Cells were treated for a total of 48 hours (n = 6 per treatment group) prior to viability testing by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Differences in cellular viability measurements between treatment groups were evaluated in R by applying two-way ANOVA after applying a log10 or square root transformation to satisfy normality.
Cisplatin sensitization in animal models
Athymic nude mice were injected with 2 million OVCAR8 cells IP. Mice were treated with SUPR4B1W (10 mg kg−1) combined with carboplatin (25 mg kg−1) or saline 3× per week by IP injection. Each treatment group had 8 mice, with the exception of the group treated with carboplatin alone which had 4 mice. Treatments were administered for 3 weeks before mice were sacrificed and tumors were dissected and weighed. Differences in tumor mass between treatment groups were evaluated in R by a nonparametric Mann–Whitney–Wilcoxon rank-sum test.The assay was repeated with the additional treatment groups for combinations of scrambled SUPR4B1W (10 mg kg−1) with carboplatin (25 mg kg−1) or saline. Treatment group size was expanded to 10 mice and the duration of the experiment was extended from 3 weeks to 4 weeks.
Statistical analysis
To determine the independent and interactive effects of cisplatin and SUPR4B1W on cell viability, we used a two-way ANOVA with Type I sum of squares on A570 absorbance. Log10 and square root transformations were used on OVCAR8 and HEY cell line absorbance data, respectively, to achieve satisfactory normality. To determine the independent and interactive effects of carboplatin and SUPR4B1W structure on tumor weight in mice, we used a two-way ANOVA with Type III sum of squares. A square root transformation was necessary to handle right skewedness in week 4 data. Following both analyses, Tukey's Honestly Significant Difference test (HSD) via the package lsmeans was used to determine which treatment means were significantly different from one another.
Results & discussion
Our goal was to generate small, protease-stable peptides with high affinity and selectivity for LC3. In order to generate target for selection, His6-tagged LC3A was expressed in E. coli and purified by Ni-NTA affinity chromatography (8.1 mg L−1 culture). Biotinylation of recombinant LC3A was carried out using biotin–NHS and confirmed by HABA–avidin assay. LC3A–biotin was then immobilized on streptavidin agarose for selection.Selection against LC3A using MK8 library
Selections against LC3A were carried out using SUPR peptide mRNA display.28 The DNA template library was designed with eight random codons and a fixed lysine codon to allow cyclization between the side chain amine of lysine and the N-terminal amino group (Fig. 1A). The resulting library was translated in the presence of amber suppressor tRNA acylated with N-methyl-L-alanine. After seven rounds of selection against LC3A, binding of the library to the immobilized target increased dramatically as evidenced by a reduction in the number of PCR cycles required to amplify the enriched library (Fig. 1B). Sequencing revealed three dominant sequence families in the final pool, two of which share a consensus motif (Y/F/N)–XXX–(L/V) analogous to canonical LIM albeit with an additional amino acid in the spacer region (Fig. 1C). It is possible that this additional amino acid arose from the more constrained conformation of the cyclic peptide compared to the linear LIMs found in LC3 adaptor proteins which typically occur as elongated β-strands.37 Sequencing data also revealed that >80% of the clones analyzed from the round 7 pool incorporated N-methyl-L-alanine (NMA) at position 8 within the random region indicating that the selection provides a significant selection pressure for N-methyl-L-alanine incorporation at this position. In order to identify the highest affinity clones from the round 7 pool, we performed radioligand binding assays. Individual clones were translated as mRNA–peptide fusions in the presence of [35S]-methionine, cyclized, and treated with RNAse A to remove the RNA portion yielding peptides conjugated only to the poly-dA linker. Radiolabeled peptide–DNA conjugates were then panned against LC3A immobilized on neutravidin–acrylamide or resin alone. After an hour incubation at room temperature, the resins were washed thoroughly and analyzed by scintillation counting. While several clones showed a moderate level of target-specific binding, clone 4B (cycMFPHRVTAZK), hereafter SUPR4B, showed the highest target-specific binding with 86% of the input activity remaining bound to the immobilized target and minimal nonspecific binding to the resin alone (Fig. 1D).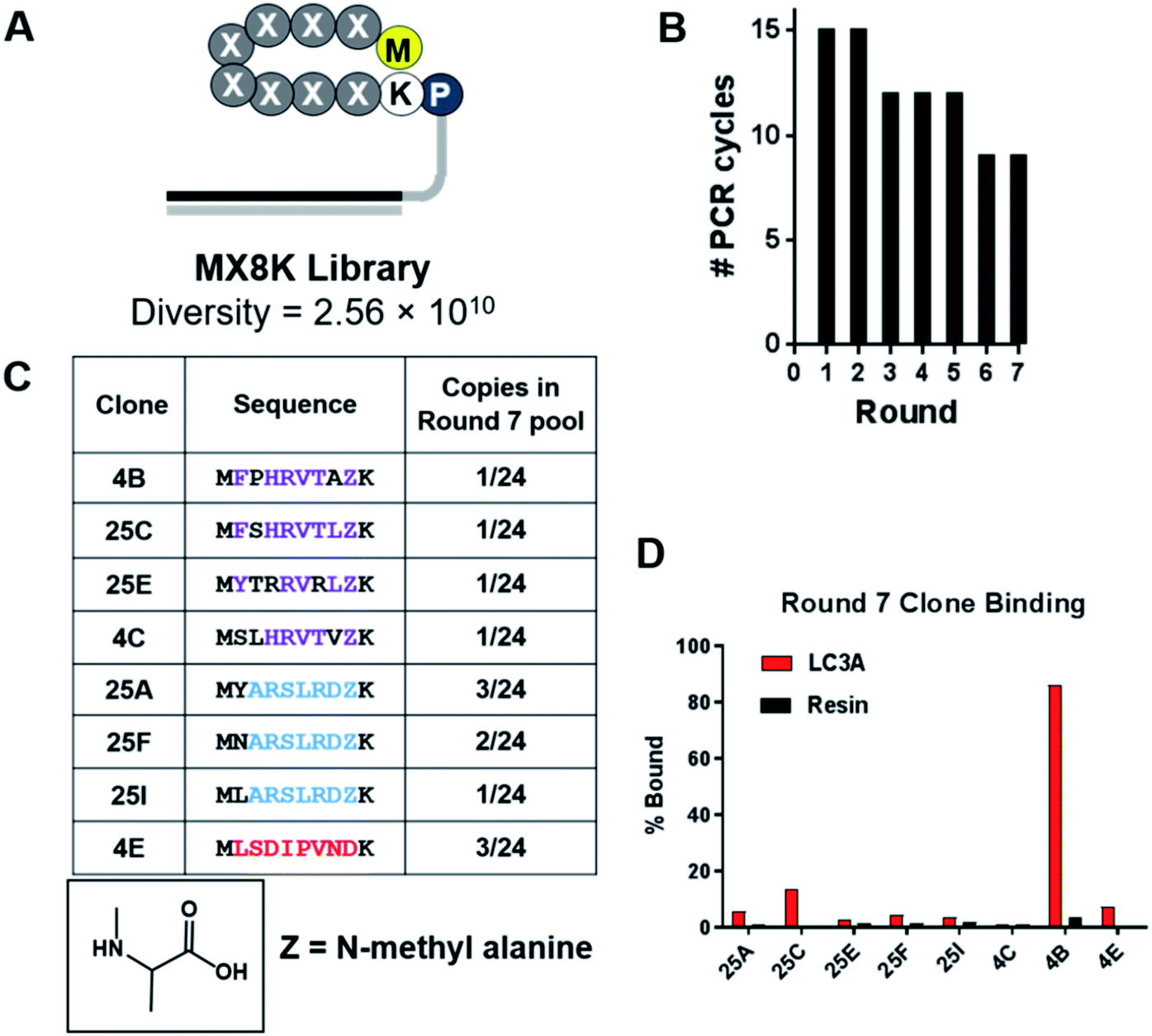 | ||
| Fig. 1 Selection of LC3-binding SUPR peptides. (A) Schematic of the MX8K library. Eight randomized codons were installed between the N-terminal methionine and a locked lysine residue. The library was cyclized by disuccinimidyl glutarate (DSG)-mediated crosslinking of the N-terminal amino group and the side-chain amino group of lysine. (B) After seven rounds of selection the number of PCR cycles required to amplify the enriched dsDNA library declined and plateaued indicating convergence of the library. (C) Sequencing of the final pool revealed three dominant peptide families (purple, blue and red). >80% of sequences identified had incorporated an N-methyl-L-alanine residue (Z) in the final random position of the macrocycle. (D) Each sequence was synthesized as an mRNA–peptide fusion and panned against LC3A immobilized on streptavidin acrylamide or streptavidin acrylamide alone (resin). Clone 4B showed 86% binding to immobilized LC3 and minimal binding to the resin alone. | ||
Binding affinities of SUPR-4B and variants towards LC3A and LC3B
While SUPR4B appears to serve as a non-canonical LIM, we noted that phenylalanine occurred at the first randomized position within the macrocycle. Based on known LIM sequences, this residue is likely to interact with the W-site of the LC3 hot spot. Although some LIMs have phenylalanine in this position, it is the least commonly occurring amino acid residue.31 Binding to the W-site is achieved by burying an aromatic residue into the hydrophobic pocket, and the sidechain of tryptophan is well-suited to perform this interaction. While phenylalanine and tyrosine appeared in the first position of the macrocycle in several clones, tryptophan was not incorporated in any of the peptides sequenced. This may have arisen from the low frequency of tryptophan-encoding codons in the library (1/32) or poor incorporation efficiency at the N-terminus. Our sequencing may also have been too sparse to detect clones with tryptophan at the first position and next-generation high throughput sequencing may reveal such clones. We hypothesized that substitution of tryptophan for phenylalanine at position 1 in SUPR4B (SUPR4B1W) would improve affinity for LC3.To evaluate the impact of tryptophan substitution on binding affinity for LC3, we measured the binding affinity of both SUPR4B (Fig. 2A) and SUPR4B1W (Fig. 2B) using fluorescence polarization anisotropy. Fluorescein-labeled SUPR peptides were incubated with increasing concentrations of GST-LC3A and GST-LC3B. Fluorescence polarization was measured (λEx = 485 nm, λEm = 528 nm) and converted to fluorescence polarization anisotropy which was plotted against concentration of protein and fit to a one-site saturation binding model. SUPR4B1W was found to bind to GST-LC3A with a KD of 121 ± 19 nM while SUPR4B bound to GST-LC3A with a KD of 355 ± 84 nM (Fig. 2C and D). This data supported our prediction that the larger aromatic side-chain of tryptophan would engage more strongly with the hydrophobic W-site binding pocket and increase the affinity of SUPR4B1W for LC3A relative to SUPR4B. SUPR4B1W was also found to bind to GST-LC3B with a KD of 192 ± 39 nM while SUPR4B bound to GST-LC3B with a KD of 387 ± 83 nM (Table 1). This indicates that selection against LC3A resulted in ligands with similar affinities for the LC3B homolog which is unsurprising given the 83% residue identity and 90% residue similarity between the two proteins (Fig. S2†). A scrambled variant of SUPR4B1W showed no detectable binding to either LC3A or LC3B, confirming the sequence-dependence of this interaction (Fig. S3†).
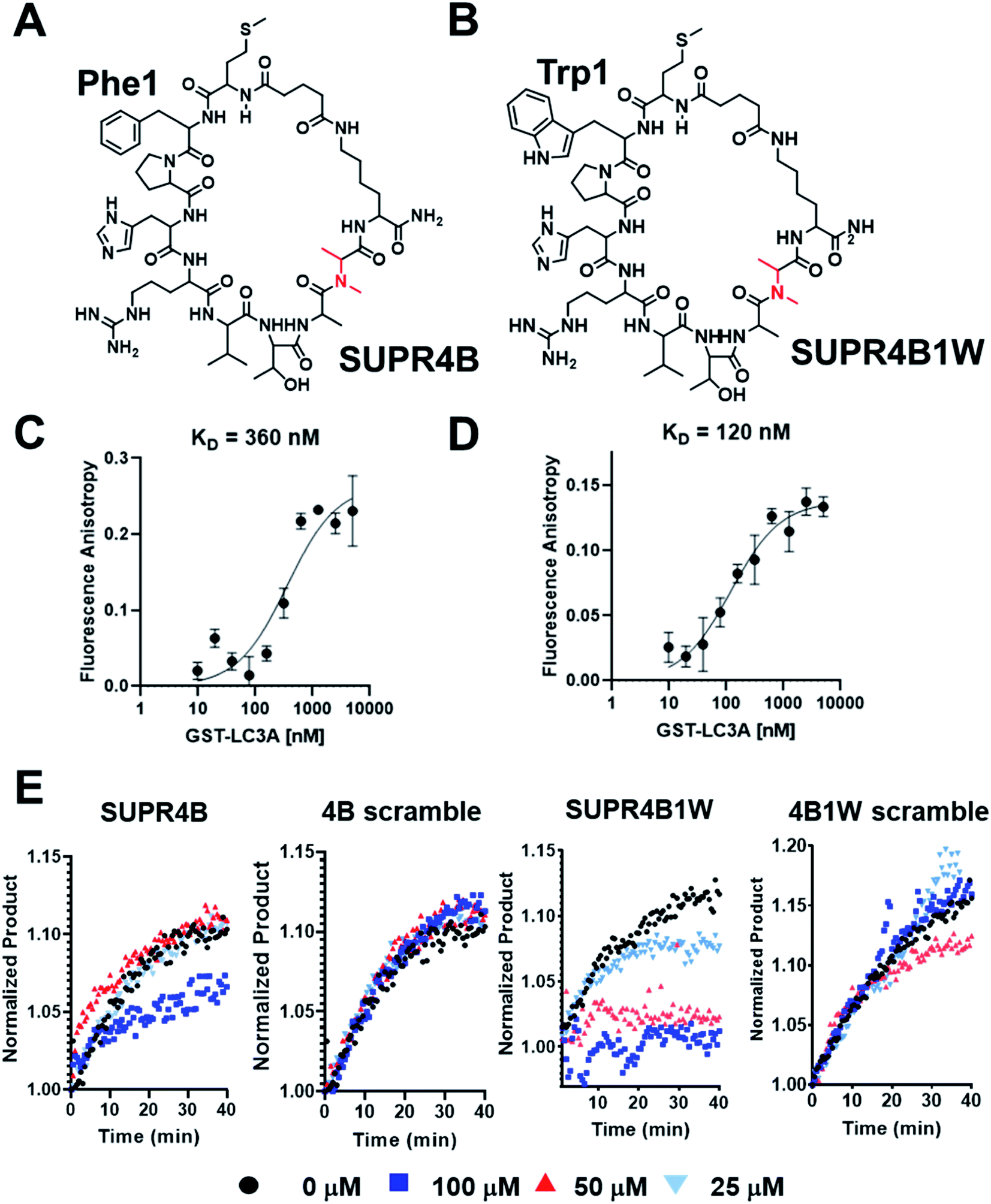 | ||
| Fig. 2 Binding affinities of SUPR4B peptides. (A) Structure of SUPR4B. (B) Structure of SUPR4B1W. FAM labeled SUPR peptides were incubated with increasing concentrations of GST-LC3A and fluorescence polarization measurements were made in triplicate. Each measurement was converted to anisotropy and fit to a one-site saturation binding model in Graphpad Prism to obtain the equilibrium binding constant. (C) SUPR4B binds LC3A with a KD = 360 nM (D) SUPR4B1W binds LC3A with a KD = 120 nM. (E) Atg4B enzyme inhibition assay. Increasing concentrations of both SUPR4B and SUPR4B1W impair the ability of Atg4B to cleave the C-terminus of LC3-AMC leading to diminished AMC fluorescence. Neither scrambled peptide inhibited Atg4B's ability to cleave LC3-AMC. | ||
| LC3A KD (nM) | LC3B KD (nM) | |
|---|---|---|
| SUPR4B1W linear | 375 ± 157 | 418 ± 154 |
| SUPR4B1W cyclic | 121 ± 19 | 192 ± 39 |
| SUPR4B linear | 691 ± 252 | 312 ± 143 |
| SUPR4B cyclic | 355 ± 84 | 387 ± 83 |
| SUPR4B1W8A linear | 1123 ± 356 | 1250 ± 453 |
| SUPR4B1W8A cyclic | 101 ± 22 | 194 ± 52 |
Binding affinity of both SUPR4B and SUPR4B1W for LC3A is significantly enhanced by cyclization. Fluorescence polarization assays were carried out using fluorescein-labeled linear SUPR4B and SUPR4B1W. Linear SUPR4B1W was found to bind to GST-LC3A with a KD = 375 ± 157 nM while linear SUPR4B bound to GST-LC3A with a KD = 691 ± 252 nM. Linear SUPR4B1W was found to bind to GST-LC3B with a KD = 418 ± 154 nM while linear SUPR4B bound to GST-LC3B with a KD = 312 ± 143 nM (Table 1). These data indicate that cyclization provides approximately 0.7 kcal mol−1 of binding energy to stabilize the SUPR4B1W:LC3A interaction, likely through reduction of conformational entropy.
We also synthesized a version of SUPR4B1W in which the N-methyl-L-alanine at position 9 was replaced with L-alanine (SUPR4B1W8A) to determine the effect of N-methylation on binding affinity. Fluorescein-labeled SUPR4B1W8A bound to GST-LC3A with a KD of 101 ± 22 nM and GST-LC3B with a KD of 194 ± 52 nM. These affinities are very similar to those observed for SUPR4B1W and indicate that incorporation of N-methylated amino acids does not positively contribute to binding energy of cyclic SUPR4B1W. In contrast, the affinity of linear SUPR4B1W8A for LC3A and LC3B is 3-fold lower relative to its N-methylated counterpart (linear SUPR4B1W). This may indicate that NMA imposes structural contstraints that positively influence affinity in the context of a linear peptide but not in the context of the macrocycle. A summary of binding affinities obtained by fluorescence anisotropy is presented in Table 1.
SUPR peptides attenuate Atg4b-mediated cleavage of LC3
Atg4 is a cysteine protease that plays a critical role in the regulation of autophagy through post-translational modification of LC3 homologs. Following translation, Atg4 binds to LC3 – an interaction mediated in part by a C-terminal LIM – and cleaves the protein at the C-terminus to expose a glycine residue which is lipidated with phosphatidylethanolamine (PE) prior to membrane insertion. We hypothesized that if our SUPR peptides bound to the Atg4 LIM-recognizing site on LC3, Atg4 binding and proteolytic cleavage would be inhibited. LC3A conjugated at the C-terminal with 7-amino-3-methylcoumarin (LC3-AMC) was pre-incubated with increasing concentrations of SUPR4B, SUPR4B1W, or a scrambled version of either SUPR peptide. Since AMC is only fluorescent when free in solution (λEx = 380 nm, λEm = 460 nm), cleavage of LC3 by Atg4 can be monitored by increase in fluorescence at 460 nm. Atg4B was added to each combination of LC3-AMC and SUPR peptide (0–100 μM) and fluorescence emission was monitored for 40 minutes (Fig. 2E). SUPR4B inhibits Atg4B cleavage of LC3-AMC with an apparent Ki > 100 μM while SUPR-4B1W inhibits cleavage with apparent Ki ∼ 35 μM. Neither scrambled peptide had any inhibitory effect on Atg4B cleavage of LC3-AMC indicating that inhibition is sequence-dependent. We note that the apparent Ki for SUPR4B1W is significantly higher than its dissociation constant as measured by fluorescence polarization anisotopy. While the reason for this mismatch is unclear, we speculate that it arises from substrate-level inhibition of LC3B rather than direct inhibition of the Atg4B protease in the enzymatic reaction. We also note that, LC3B can still be bound and processed by Atg4B lacking a functional C-terminal LIM (FEIL, residues 388–391), albeit at approximately 50% efficiency in vitro.38 This partial dependence of enzymatic activity on LIM binding may lead to significantly lower SUPR peptide inhibitory potencies than would be expected based on affinity alone. These results suggest that the SUPR peptides occupy the PPI hot spot of LC3 and inhibit formation of the Atg4:LC3 complex leading to inhibition of C-terminal proteolysis. Additional site-directed mutagenesis studies of the LC3 hot spot are required to confirm the SUPR4B1W binding site and to define the relative contributions of the W and L subsites to the energetics of this interaction.Protease stability and cytosolic access are dependent on cyclization and N-methyl incorporation
Peptides are highly susceptible to proteolysis in human serum and this feature has compromised the translation of many peptides into therapeutic compounds. SUPR mRNA display attempts to mitigate this drawback through macrocyclization and incorporation of N-methyl amino acids which have been shown to increase biological half-lives of peptides by orders of magnitude.28 We evaluated the protease stability of both cyclic and linear SUPR4B1W and as well as a variant where the N-methyl alanine residues is replaced with alanine (SUPR4B1W8A). SUPR peptides were incubated for increasing time with agarose-immobilized proteinase-K and subjected to analysis by HPLC. SUPR4B1W was found to have a half-life in proteinase-K of 110 minutes, while the linearized version of the peptide was found to have a half-life of 57 minutes (Fig. 3A). Removing the N-methyl-L-alanine at position 8 dramatically increased susceptibility to proteolysis; SUPR4B1W8A was completely degraded within 60 minutes of incubation with proteinase-K (half-life ∼ 6 min). Based on these data, it appears that N-methyl-L-alanine is a major driver of protease stability, as substitution with L-alanine completely abrogates resistance to proteinase-K degradation. Previous SUPR mRNA display selections suggest that while incorporation of a single N-methyl amino acid moderately improves protease resistance, dramatic improvements in protease stability (i.e. order of magnitude) are driven by multiple tertiary amide linkages.28 Indeed, SUPR4B1W incorporates two residues with tertiary amide linkages: the N-methyl-L-alanine incorporated at position 8 and the proline at position 2. When considered alongside the fluorescence polarization binding data, the stability data suggests that incorporation of N-methyl-L-alanine was driven primarily by selection for protease resistance, not selection for binding affinity in the context of the macrocycle.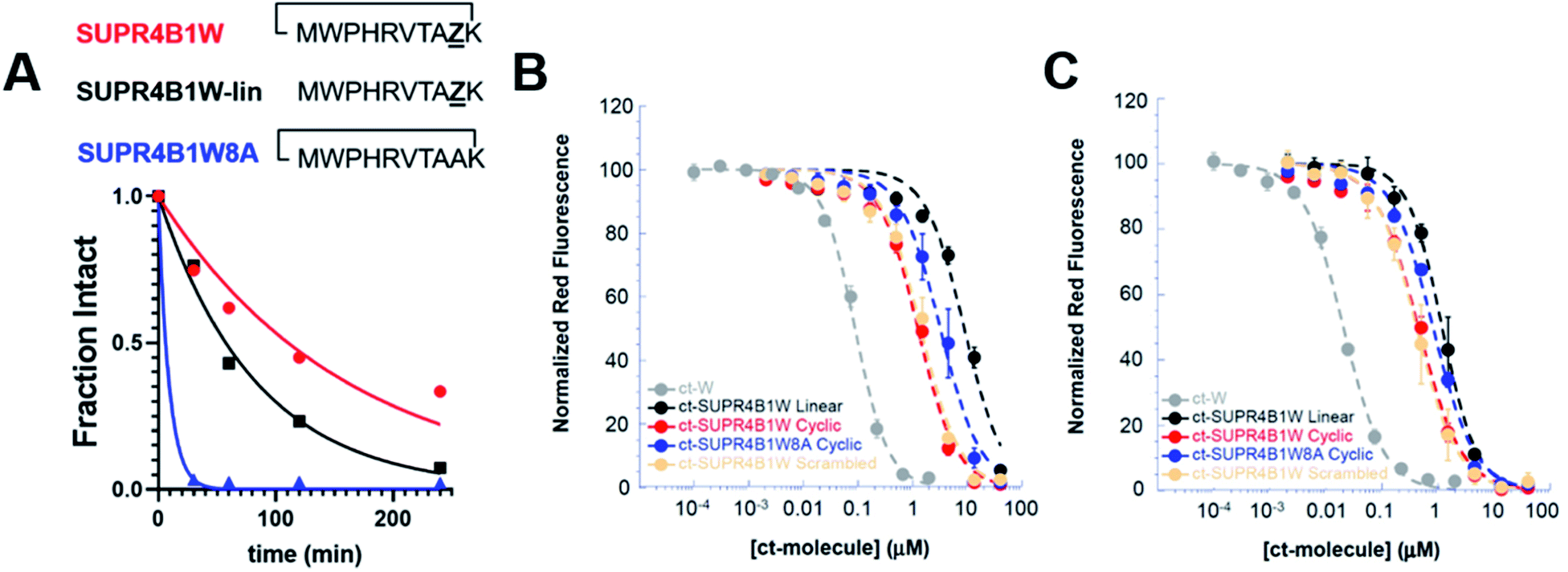 | ||
| Fig. 3 Cyclization and N-methyl-L-alanine improve the protease stability and cytosolic access of SUPR4B1W. (A) SUPR4B1W has a half-life in proteinase-K of ∼110 min (red line). Without cyclization, linear SUPR4B1W has a half-life of 57 min, a nearly 2-fold reduction (black line). Substitution of N-methylalanine with alanine reduces the protease half-life of the SUPR peptide to 6 min (blue line). (B) CAPA data showed that at 4 h, cyclic SUPR4B1W (red line) showed approximately 7-fold and 3-fold higher cytosolic access than the linear (black line) and non-N-methylated (blue line) variants, repectively. (C) CAPA data at 24 h showed increased cytosolic access relative to 4 h incubation, but the rank-order among derivatives was maintained. Both cyclic SUPR4B1W and scrambled cyclic SUPR4B1W (salmon line) showed the same extent of cytosolic access at both time points. Chloroalkane-tagged tryptophan (grey line) is included as a positive control. | ||
In addition to conferring protease resistance, cyclization and backbone N-methylation have also been shown to impart cell permeability.22 To measure the cell permeability and cytosolic access of SUPR4B1W as well as the effect of cyclization and N-methylation on these properties, we employed the chloroalkane penetration assay (CAPA).33 In CAPA, a mutant bacterial haloalkane dehydrogenase is expressed in the cytosol.34 This enzyme catalyzes the formation of a covalent bond with chloroalkane-tagged molecules irreversibly blocking the enzyme active site. CAPA involves pulsing the cells with a chloroalkane-tagged molecule of interest, then chasing with a tagged fluorescent dye. Thus, the extent to which the chloroalkane-tagged dye is retained by the cells is inversely proportional to the extent to which the chloroalkane-tagged molecule of interest penetrated the cytosol. Dose-dependent CAPA experiments produce a sigmoidal curve, which is used to derive a CP50 value which corresponds to the concentration of chloroalkane-tagged peptide at which 50% cytosolic penetration is observed.32
We synthesized chloroalkane-tagged versions of cyclic SUPR4B1W, linear SUPR4B1W, cyclic SUPR4B1W8A, and scrambled cyclic SUPR4B1W and measured their cell penetration at 4 h and 24 h in HeLa cells by CAPA (Fig. 3B and C). At 4 h, the CP50 of cyclic SUPR4B1W was found to be 1.26 ± 0.13 μM while linear SUPR4B1W and cyclic SUPR4B1W8A showed CP50 values of 8.82 ± 0.81 μM and 3.58 ± 0.79 μM respectively. At 24 h, the CP50 values of cyclic SUPR4B1W, linear SUPR4B1W, and cyclic SUPR4B1W8A decreased to 0.46 ± 0.06 μM, 1.19 ± 0.16 μM, and 0.81 ± 0.04 μM respectively. A sequence scrambled version of cyclic SUPR4B1W showed essentially the same cell penetration as cyclic SUPR4B1W at both 4 h and 24 h time points. These results indicate that cyclic SUPR4B1W readily gains access to the cytosolic compartment at low micromolar concentrations. It is also clear that both cyclization and backbone N-methylation contribute positively to cytosolic access. Finally, these experiments suggest that SUPR4B1W cell uptake is dependent on composition and/or structure rather than primary sequence.
SUPR4B1W selectivity for LC3 homologs
Fluorescence polarization assays showed that SUPR4B1W binds to LC3B with an affinity similar to that of the selection target, LC3A. This is not entirely unsurprising, as LC3A and LC3B share a high degree of sequence similarity, particularly in the regions that define the protein-binding hot spot of each protein (Fig. 4A). We sought to determine whether SUPR4B1W, with its non-canonical LIM, exhibits binding for other Atg8 mammalian orthologues or if binding was specific for LC3A/LC3B.1 Open reading frames (ORFs) for LC3A, LC3B, LC3C, GABARAP1, GABARAP2, Atg8, and Ubiquitin were transcribed into mRNA in vitro and translated in vitro in the presence of [35S]-Methionine. SUPR4B1W was biotinylated and immobilized on neutravidin–acrylamide resin and the radiolabeled translation products were incubated with the resin. SUPR4B1W showed the highest binding to LC3A followed closely by LC3B (Fig. 4B), an observation in line with our fluorescence anisotropy binding data. Moderate binding was also observed for both isoforms of GABARAP as well as Atg8 (the yeast ortholog of LC3). We observed virtually no binding by LC3C, which can be explained by LC3C's strong preference for the LVV motif which is not found in SUPR4B1W.39 SUPR4B1W showed no affinity for ubiquitin suggesting that the peptide binds to the LC3 hot spot (defined by the W and L sub-sites) rather than the core ubiquitin fold.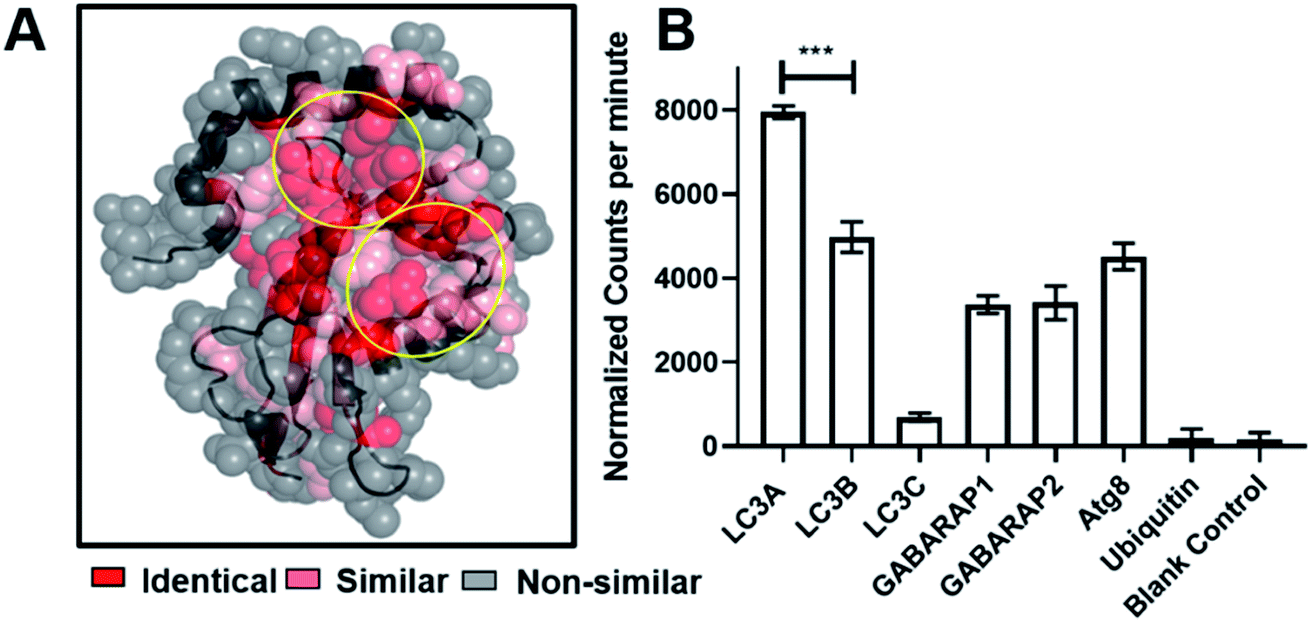 | ||
| Fig. 4 SUPR4B1W selectivity for LC3 homologs. (A) Crystal structure of LC3 (5CX3)1 showing the extent of residue conservation between LC3A, LC3B, LC3C, GABARAP1, GABARA2, and Atg8. The W and L sub-sites are highly conserved (circled in yellow). (B) SUPR4B1W was immobilized on neutravidin–acrylamide an incubated with [35S]-labeled LC3 homologs (LC3A, B, and C along with GABARAP L1 and L2, Atg8 (the yeast ortholog of LC3)) and ubiquitin. SUPR4B1W showed modest selectivity for LC3A and LC3B compared with all other homologs and orthologs. No binding to ubiquitin was observed. | ||
Autophagy-related proteins that contain canonical LIMs often show affinity for multiple isoforms of LC3 as well as isoforms of GABARAP, though affinity for each isoform varies.39 There also exists a GABARAP interacting motif (GIM)—(W/F)–(V/I)–X–V—which confers high selectivity for GABARAP proteins.19 Given its small size and non-canonical LIM, the isoform selectivity of SUPR4B1W is very much in line with previously described linear LIM peptides.
Modeling the SUPR4B1W:LC3 interaction
To gain insight into the mechanism of LC3 binding, we computationally modeled the SUPR4B1W peptide using the Molecular Operating Environment (MOE) software package. We began with the linear version of SUPR4B1W with the DSG crosslinker conjugated to the N-terminal amine. Conformational sampling was then carried out with a 1–1.5 angstrom distance restraint between the carboxyl carbon of the DSG linker and the side chain ε-amino group of lysine. Ten conformations with pairwise RMSD of >1.5 angstroms were generated and the linker between Met and Lys was constructed. The resulting structures were solvated with water, energy minimized, and subjected to 50 ps, 500 ps and 1 ns MD simulations to ensure that the dynamics reaches equilibria. We did not see significant difference among all models, and one model is illustrated in Fig. 5A. Notably, this structure shows the pre-positioning of the Trp1 and Val5 side chains for interaction with LC3, suggesting that cyclization enforces a binding-competent conformation. In addition, the guanidinium side chain of Arg4 mediates an extensive hydrogen bond network with the backbone carbonyl oxygens of Trp1, Val5, and the DSG crosslinker. Although preliminary, these results suggest that Arg4 may facilitate cell permeability and conformational constraint by supporting an intramolecular hydrogen bond network.22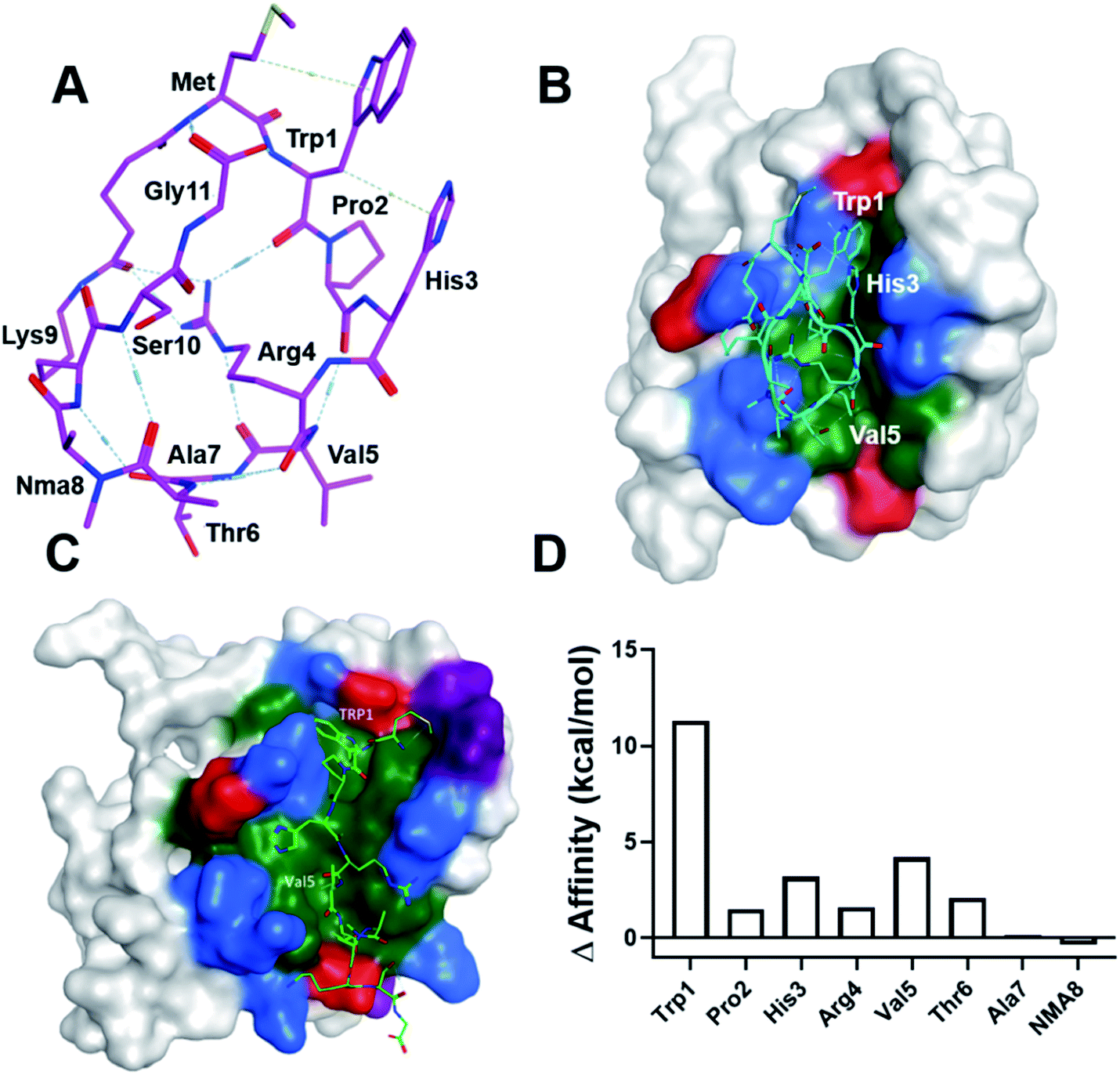 | ||
| Fig. 5 Computational modeling of the SUPR4B1W:LC3 interaction. (A) One of the conformational ensemble structures of SUPR4B1W reveals that the Arg4 side chain forms multiple internal hydrogen bonds with the backbone carbonyls of several other residues including Trp1 and Val5. (B) SUPR4B1W:LC3 structural model indicating that Pro2 facilitates formation of a beta turn involving residues His3, Arg4, and Val5 allowing Trp1 and Val5 to interact with the W- and L-sites. Red: negatively charged amino acids, blue: positively charged amino acids, green: hydrophobic amino acids. Only binding site residues are colored. (C) Linear SUPR4B1W:LC3 structural model showing the Trp1 and Val5 side chains occupying the W-site and L-site of LC3, respectively. (D) Results of computational alanine scanning showing the predicted energetic contributions of each SUPR4B1W residue. In this analysis, a positive change in affinity indicates that alanine substitution destabilizes the binding interaction while a negative change in affinity indicates a strengthening of the interaction. Trp1, His3, and Val 5 are predicted to make the strongest energetic contributions to binding. | ||
To model the interactions between LC3 and SUPR4B1W, we imported an LC3 crystal structure (PDB: 5CX3)1 into MOE. The modeled structure of the cyclic SUPR peptide was aligned and superimposed with the LIM peptide in the crystal structure followed by tethered minimization to resolve atomic clashes with LC3. A docked structure is shown in Fig. 5B. Notably, this pose is similar to the energy minimized structure with RMSD = 1.58 Å and RMSD = 2.26 Å for main chain and all atoms, respectively. The docked structure shows Pro2 driving the formation of a β turn allowing Trp1 and Val5 side chains to interact with the W-site and L-site, respectively. Indeed, our initial SUPR peptide screening experiments (Fig. 1D) indicate that loss of the proline at position 2 leads to significant degradation of binding affinity (compare clones 4B and 25C). Based on this data, we provisionally conclude that the conformation enforced by the combination of Pro2 and cyclization may allow the extra residue between W- and L-site interacting residues in SUPR4B1W relative to naturally occurring, linear LIM peptides. As mentioned, the side chain guanidinium of Arg4 mediates an extensive network of intramolecular hydrogen bonds with backbone carbonyl oxygens.
Modeling of the linear version of SUPR4B1W (Fig. 5C) shows Trp1 in the W-site and Val5 in the L-site. This suggests that the linear peptide has a similar, lower affinity binding mode which is in agreement with our experimental observations (Table 1). Computational alanine scanning (Fig. 5D) was also carried out to assess the energetic contribution of each residue in the SUPR4B1W macrocycle. The results of this analysis indicate that Trp1, His3, and Val5 each make a significant contribution to the total binding energy. It is worth noting that Pro2 was not identified as making critical energetic contributions to the binding affinity in this analysis. Proline is an unusual residue with a rigid conformation which cannot be readily replaced with any of the other 19 natural amino acid residues without significantly changing the backbone conformation of the peptide. A large-scale mutagenesis study confirms that proline is the least tolerated residue40 and it is not surprising that computational alanine scanning methods either do not consider proline in their method validation or suffer from poor accuracy when proline is considered. In this study, we employed the alanine scan method in MOE, which assumes no large conformational change in the backbone following alanine substitution which may explain why computational alanine scanning did not reveal a significant contribution of Pro2. Conversely, residues outside the non-canonical LIM motif of SUPR4B1W (e.g. Ala7 and NMA8) were not predicted to significantly affect binding. These observations are in broad agreement with our empirical data (Fig. 1D and Table 1).
SUPR4B1W is internalized into cells and co-localizes with punctate LC3 after starvation
Before assessing its potential as a therapeutic inhibitor of autophagy, we sought to validate the uptake and target engagement of SUPR4B1W in human cell lines. We utilized copper-catalyzed click chemistry to conjugate the rhodamine derivative ROX-azide to SUPR4B1W via the side chain of a C-terminal propargylglycine residue. HeLa cells stably expressing eGFP-LC3B were grown in media supplemented with SUPR4B1W-ROX (500 nM) for 24 hours. After 24 hours, cells were starved for 2 hours to induce autophagy prior to confocal microscopy imaging. Induction of autophagy in this cell line results in a diffuse to punctate GFP staining pattern reflecting transition of diffuse cytosolic LC3-GFP to autophagosomes. SUPR4B1W-ROX is taken up by both unstarved (Fig. 6A) and starved (Fig. 6B) HeLa-eGFP-LC3 cells, and in both cases displayed a punctate pattern, possibly suggesting an endocytotic/pinocytotic mechanism of uptake. To quantify the extent of colocalization between SUPR4B1W-ROX and GFP-LC3 puncta we calculated Manders' M1 and M2 colocalization coefficients for three fields of both starved and unstarved cells (Fig. 6C). The M1 coefficient gives the percentage of pixels in the red channel (SUPR4B1W-ROX) that overlap with pixels in the green channel (eGFP-LC3). The M2 coefficient gives the percentage of pixels in the green channel that overlap pixels in the red channel. Starvation increases the M2 coefficient suggesting an increased co-localization of LC3 puncta with SUPR4B1W-ROX after starvation-induced autophagy. These data support cell uptake of SUPR4B1W and engagement with intracellular LC3.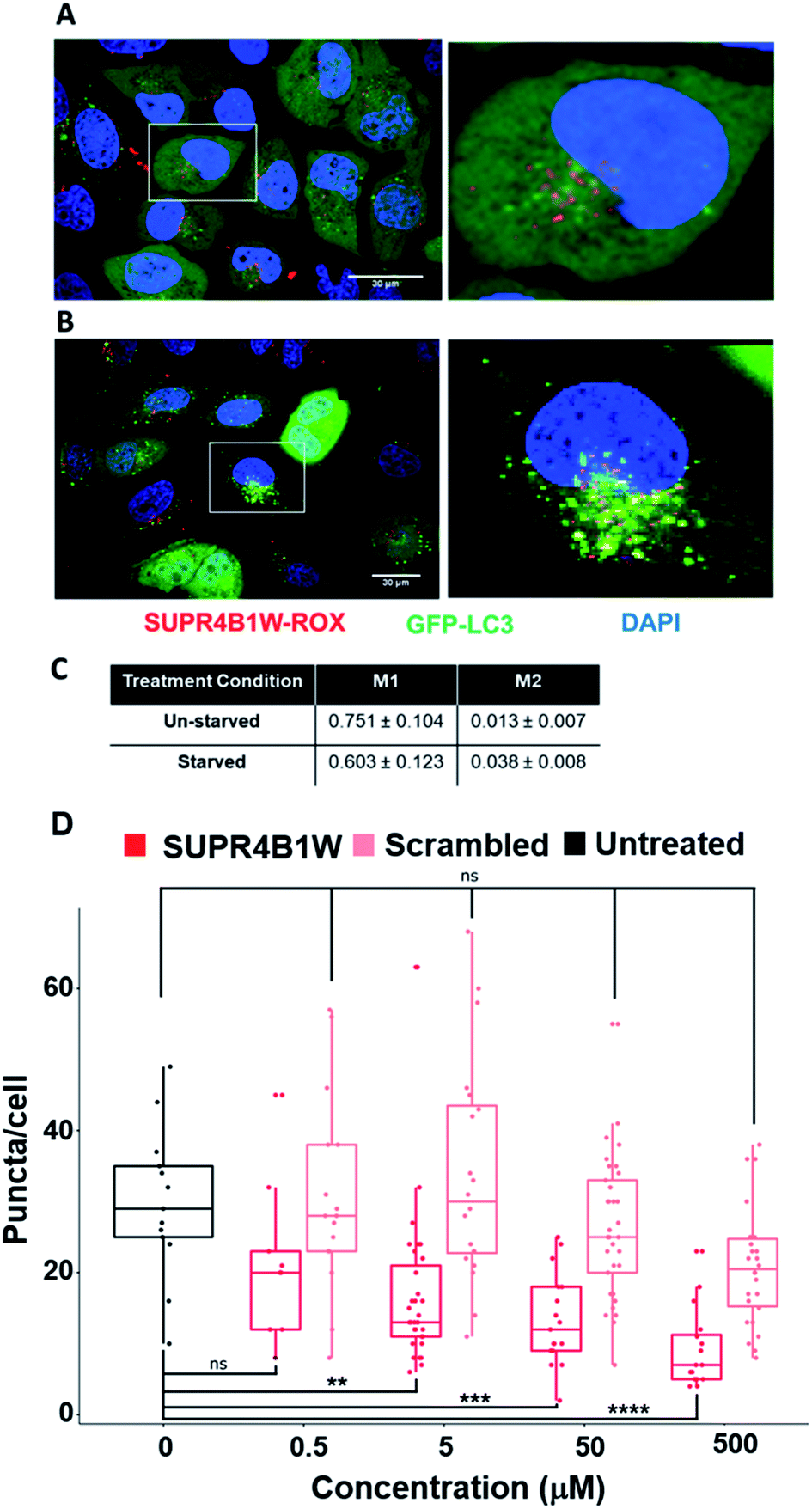 | ||
| Fig. 6 SUPR4B1W co-localizes with LC3 and inhibits LC3-GFP puncta formation during starvation-induced autophagy. SUPR4B1W-ROX was incubated with HeLa cells expressing GFP-LC3 (green) in normal media (A) or starvation media (B) and visualized by confocal fluorescence microscopy. After starvation, the EGFP-LC3 transitions from diffuse to punctate staining indicating induction of autophagy. Significant overlap (yellow) of red and green puncta in starved cells indicates co-localization of LC3 and SUPR4B1W inside the cell. (C) M1 and M2 co-localization coefficients were calculated for three fields of starved and unstarved cells. The M2 coefficient (co-localization green signal with red signal) increases after starvation. Scale bars represent 30 μm. (D) HeLa cells were starved to induce autophagy and GFP-LC3 puncta formation. Treatment with SUPR4B1W (red) resulted in a dose-dependent reduction in the average number of puncta per cell while treatment with the scrambled peptide (pink) had no statistically significant effect. | ||
SUPR4B1W inhibits GFP-LC3 puncta formation in starved HeLa cells
To determine if LC3 engagement by SUPR4B1W results in autophagosome inhibition, we carried out a titration of SUPR4B1W and its scrambled control in HeLa cells stably expressing eGFP-LC3. As seen in Fig. 6D, starvation-mediated autophagy induces robust GFP-LC3 puncta count which is inhibited in a concentration-dependent fashion by SUPR4B1W (Fig. S4†). In contrast, scrambled SUPR4B1W has no statistically significant effect on GFP-LC3 puncta formation even at concentrations as high as 500 μM. This data further supports the conclusion that SUPR4B1W inhibits autophagsome maturation through LC3 blockade and that this inhibition is sequence-dependent.SUPR peptides sensitize resistant cell lines to cisplatin in vitro
One of the overarching goals of this project was to produce cyclic peptide autophagy inhibitors that re-sensitized tumors to frontline chemotherapeutics. Previous reports have shown that induction of autophagy is correlated with cisplatin resistance in many human ovarian cancer cell lines including SKOv3, OVCAR8, Hey, CAOv3, and OVCA420. Cisplatin activates extracellular signal-regulated kinase (ERK) and promotes autophagy induction.41,42 Inhibition of MAPK/ERK signaling blocks autophagy induction and enhances cisplatin-induced apoptosis. This suggests that targeting cisplatin-induced autophagy may be a viable strategy for overcoming chemotherapy resistance in ovarian cancer.To determine the utility of SUPR4B1W in this context, two platinum-resistant epithelial ovarian cancer cell lines—OVCAR8 and HEY—were subjected to treatment with combinations of cisplatin and SUPR peptide for 48 hours before cellular viability was evaluated by MTT assay (Fig. 7A and B). In both OVCAR8 and HEY cell lines, the presence of cisplatin and SUPR4B1W affected cell viability, although interactive effects were only detected in the OVCAR8 cell line (ESI Table S1†). In both OVCAR8 and Hey, chloroquine and cyclic SUPR4B1W significantly reduced cell viability in the absence of cisplatin whereas linear SUPR4B1W did not (Tukey's HSD, p < 0.05). Combining chloroquine or SUPR4B1W treatment with cisplatin reduced cell viability to a greater degree than chloroquine or SUPR4B1W treatment alone in both OVCAR8 and Hey cells (p < 0.0001) In OVCAR8 cells, cisplatin and the cyclic SUPR peptide interacted synergistically to decrease cell viability more than either cisplatin alone (p < 0.001) or chloroquine combined with cisplatin (p = 0.0002). Treatment with scrambled SUPR4B1W resulted in no significant change in viability compared to untreated cells and combination of cisplatin and scrambled SUPR4B1W had no effect on viability compared to treatment with cisplatin alone (Fig. S5†). A linearized version of SUPR4B1W showed no significant effects on viability alone or in conjunction with cisplatin (Tukey's HSD, p > 0.05). These data suggest that viability changes are not the result of bulk uptake of non-specific peptide into cells, but rather a specific interaction mediated by cyclic SUPR4B1W. Enhancement of cisplatin efficacy was also seen in other ovarian cell lines (SKOV3, OVCAR5), triple-negative breast cancer cell lines (MDA-MB-231), and pancreatic cancer cell lines (Mia-PaCa-2, Panc1) (Fig. S6†). These data suggest that cyclic SUPR4B1W, rather than a linearized or scrambled version, inhibits autophagy in cancer cell lines and sensitizes them to cisplatin-mediated cytotoxicity. This observation is very much in line with the results in Table 1 and Fig. 3 demonstrating the enhanced affinity, stability, and cell permeability of SUPR4B1W relative to the linearized variant.
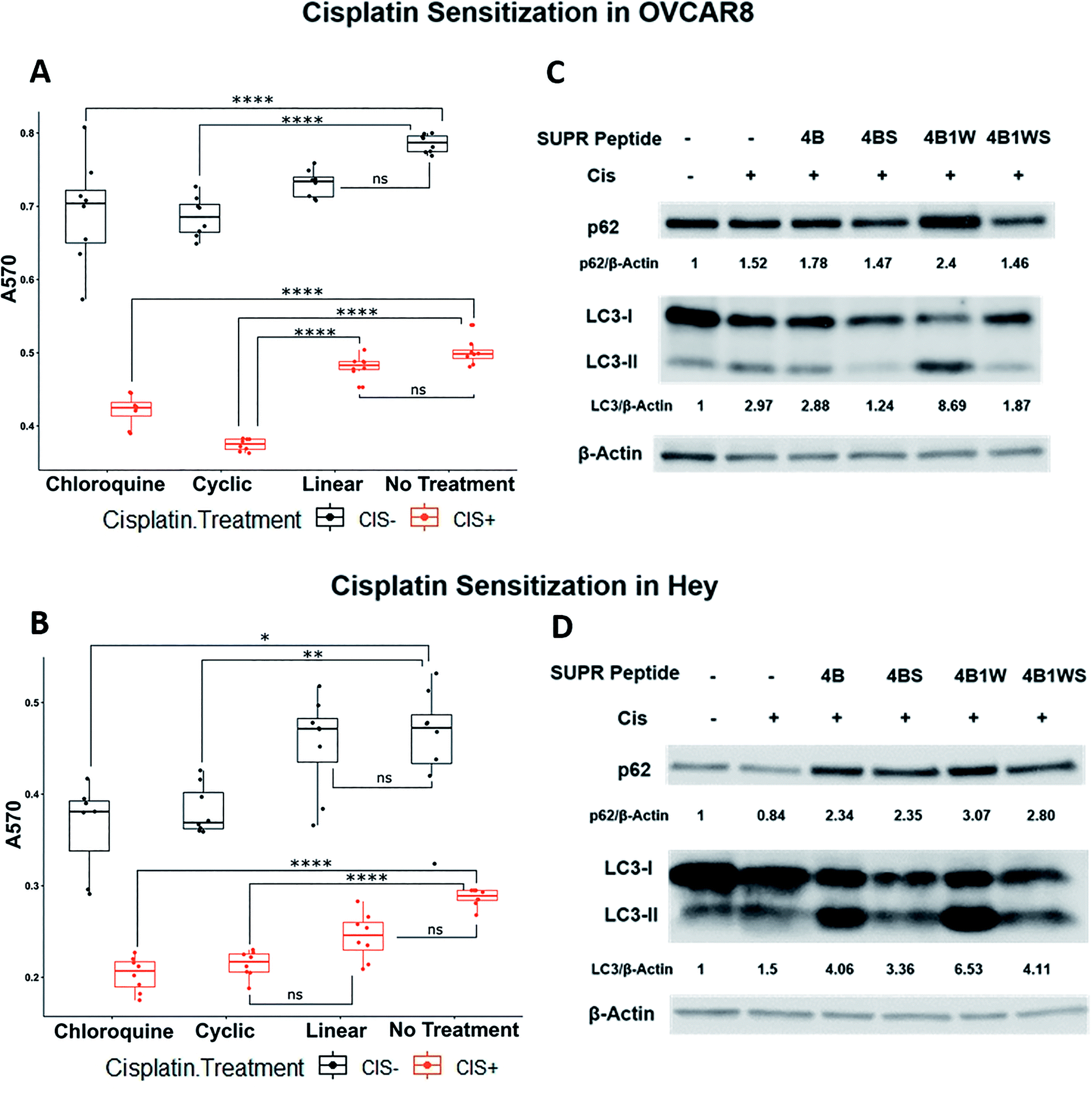 | ||
| Fig. 7 SUPR4B1W sensitizes ovarian cancer cells to cisplatin and results in p62 accumulation and LC3-II/LC3-I perturbation. Treatment of OVCAR8 (A) and HEY (B) ovarian cancer cells with SUPR4B1W and cisplatin significantly reduces cell viability after 48 h relative to cisplatin alone. A linearized version of SUPR4B1W showed no significant effect on viability in the presence or absence of cisplatin. Incubation of cisplatin-treated OVCAR8 cells (C) and HEY cells (D) with SUPR4B1W resulted in accumulation of p62 and an increase in the LC3II/LC3I ratio indicating inhibition of autophagy while a scrambled version of SUPR4B1W showed no effect. LC3/β-actin indicates the LC3II/LC3I ratio for each condition normalized to β-actin followed by normalization to the untreated condition (no SUPR peptide, no cisplatin). | ||
Western blots of treated cells show that combining cisplatin with SUPR4B1W leads to a dramatic accumulation of lipidated LC3-II and p62 in both OVCAR8 and HEY cell lines (Fig. 7C and D). Comparatively, the combination of CQ and cisplatin induces an accumulation of p62 and a dramatic reduction of cytosolic LC3-I and LC3-II (Fig. S7†). Cells undergoing autophagy typically show decreased levels of p62 as the protein is degraded in the lysosome following autolysosome fusion, and accumulation of p62 indicates disruption of autophagic flux.5 Taken together, increased p62 and LC3 levels provides a strong indication that SUPR4B1W inhibits cisplatin-induced autophagy in cell culture. Treatment with a scrambled version of SUPR4B1W showed no significant effects on p62 accumulation or LC3 lipidation indicating that this effect is sequence-dependent. Notably, SUPR4B showed some effect on the LC3II/LC3I ratio in Hey cells but not in OVCAR8. SUPR4B showed a modest effect on p62 accumulation in Hey and no effect in OVCAR8, which is consistent with its reduced LC3 affinity relative to SUPR4B1W.
SUPR4B1W sensitizes OVCAR8 tumors to carboplatin in nude mice
Given the encouraging cell-based data, we next sought to determine if SUPR4B1W could sensitize tumors to carboplatin treatment in an orthotopic mouse model. Cisplatin resistance in ovarian cancer is frequently observed in recurrent, metastatic nodules that appear throughout the peritoneum.43 When injected i.p., OVCAR8 cells form tumor nodules throughout the abdominal peritoneum and gastrointestinal tract thus, intraperitoneal injection of OVCAR8 cells provides a suitable mouse model for recurrent, resistant metastatic disease. To determine the effect of SUPR4B1W on tumor nodule formation, nude mice were IP-injected with 2 million OVCAR8 cells and subsequently treated 3 times weekly with combinations of carboplatin and SUPR4B1W. At 3 weeks, the mice were sacrificed, the tumors were dissected, and total tumor mass for each treatment condition was evaluated (Fig. 8A). Treatment with SUPR4B1W alone had no impact on tumor development (Tukey's HSD, p = 0.9827), but combining SUPR4B1W with carboplatin resulted in a significant reduction in total tumor mass compared to treatment with carboplatin alone (Tukey's HSD, p = 0.0032). This experiment was repeated, extending the treatment period to 4 weeks and adding additional treatment groups receiving a scrambled version of SUPR4B1W (Fig. 8B). At 4 weeks, combined SUPR4B1W and carboplatin treatment again had a strong effect on tumor growth with 70% of mice in the SUPR4B1W + carboplatin group showing no observable tumor development. Combining scrambled SUPR4B1W with carboplatin resulted in no difference in total tumor mass compared to treatment with carboplatin alone (Tukey's HSD, p = 0.9911). These results provide a strong indication that SUPR4B1W selectively inhibits autophagy in tumors and may provide a potent lead compound for future therapeutic development.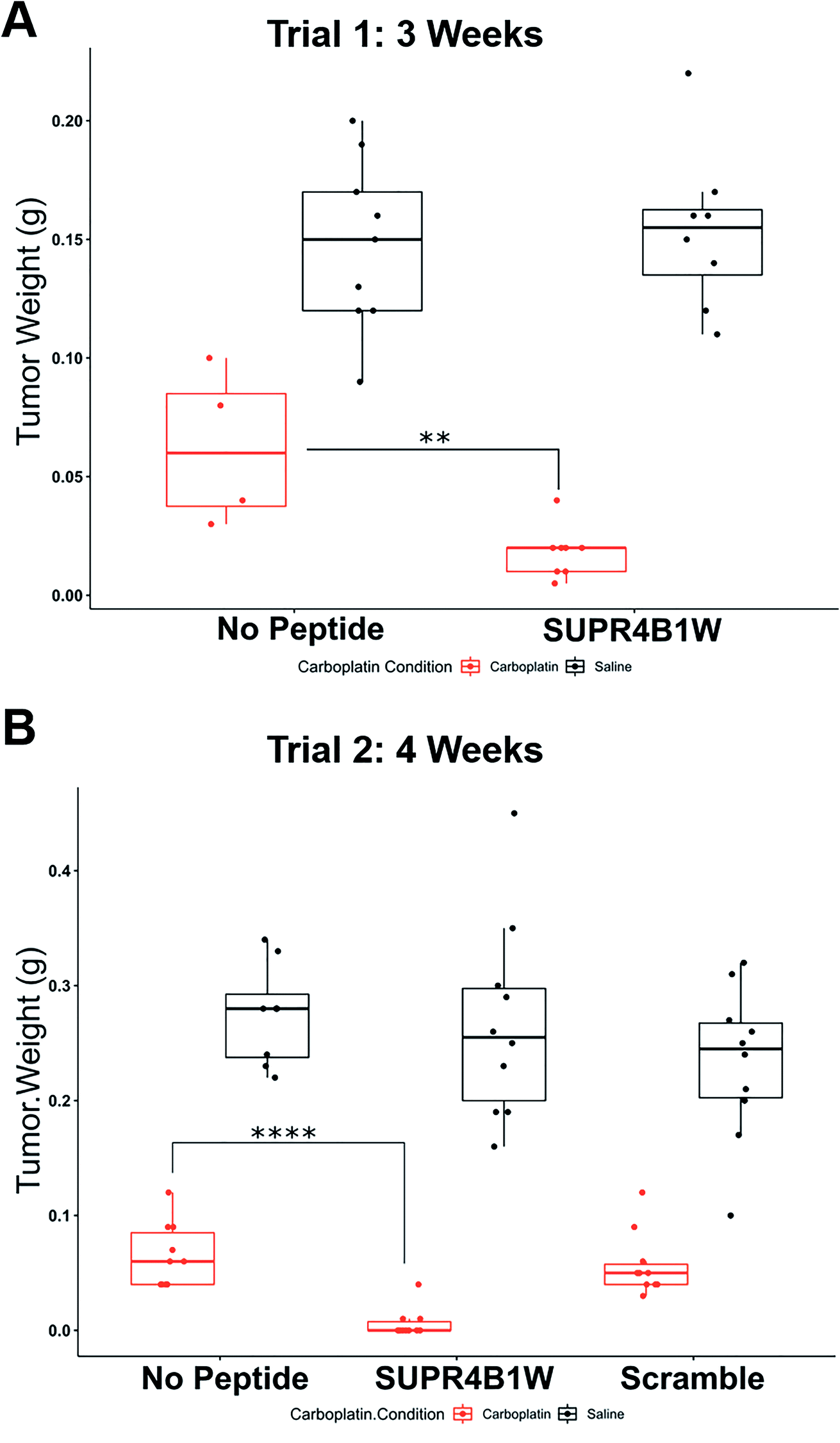 | ||
| Fig. 8 SUPR4B1W inhibits tumor growth in vivo. (A) OVCAR8 cells were injected into the peritoneum of nude mice (n = 8 per group) followed by i.p. treatment with carboplatin (25 mg kg−1, 3 times per week), SUPR4B1W (10 mg kg−1, 3 times per week), or a combination of the two for 3 weeks. SUPR4B1W + carboplatin dramatically reduced tumor mass relative to carboplatin alone while SUPR4B1W alone had no effect. (B) The same therapeutic trial was carried out for 4 weeks with the introduction of a scrambled version of SUPR4B1W. The combination of SUPR4B1W and carboplatin almost completely eliminated intraperitoneal growth in 4 weeks while the combination of carboplatin and a scrambled peptide showed no improvement relative to carboplatin alone. | ||
Conclusions
We have employed SUPR peptide mRNA display to design SUPR4B1W, a potent, cell-permeable inhibitor of the LC3 protein. This macrocyclic peptide shows mid-nanomolar affinity for LC3A and LC3B and selectivity for LC3A/B over other Atg8 orthologs. Binding studies revealed the cyclization contributes modestly, but significantly, to the binding energetics of SUPR4B1W while the non-proteogenic N-methylalanine is dispensable for affinity in the context of the macrocycle. In contrast, the N-methyl alanine residue is absolutely essential for protease resistance while the contribution of cyclization to stability is relatively modest.SUPR4B1W is taken up into HeLa cells at low micromolar concentrations and appears to readily access the cytosolic compartment. Here, cyclization and backbone N-methylation also appear to play critical roles in driving cell uptake. Cyclic SUPR4B1W (but not its scrambled control) was found to potently inhibit GFP-LC3 puncta formation in starved HeLa cells which strongly suggests inhibition of autophagosome formation/maturation by LC3 blockade. Fluorophore-tagged SUPR4B1W is also readily taken up in HeLa cells and displays a punctate staining pattern consistent with endocytosis or pinocytosis. While heavily N-methylated cyclic peptides have previously been shown to cross the cell membrane by diffusion,23 this mechanism of uptake would presumably have resulted in a diffuse staining pattern, particularly in unstarved cells where LC3-GFP is distributed evenly throughout the cytosol. Given the relatively low fraction of N-methyl amino acids (∼12% of the randomized region) and the presence of an aliphatic macrocyclic bridge, we propose that the mechanism of uptake may be similar to that of stapled peptides which are also believed to transit the cell membrane by endocytosis and/or pinocytosis.44 While it has also been proposed that stapled peptides enter the cell following membrane disruption,45,46 we found no evidence of this mechanism by microscopy or by the appearance of significant toxicity in cell culture and animal models. Future studies will address the effect of N-methylation, positive charge,47 and hydrophobicity on the mechanism of SUPR peptide uptake and/or endosomal escape.
SUPR4B1W sensitizes multiple cell lines to cisplatin treatment and results in accumulation of p62 and significant perturbation of the LC3II/LC3I ratio. These results suggest that SUPR4B1W disrupts the PPIs of LC3 inside the cell and inhibits autophagosome maturation. Accumulation of LC3II, the lipidated form of LC3, may indicate that SUPR4B1W inhibits Atg4 binding and subsequent proteolytic recycling of LC3II – a conclusion supported by in vitro data showing inhibition of Atg4B proteolysis by SUPR4B1W (Fig. 2E). Inhibition of LC3II recycling could arrest autophagic flux by attenuating the rate of formation of new autophagosomes, although this mechanism of action remains to be proven experimentally.
The combination of SUPR4B1W and carboplatin results in almost complete inhibition of tumor outgrowth in vivo suggesting that this compound could be administered in an adjuvant setting to enhance the efficacy of platinum-based therapies or to prevent the outgrowth of platinum-resistant disease. Although the combined effect of SUPR4B1W and cisplatin was also observed in cell culture, the effect size was significantly lower relative to the in vivo models. This divergence may be the result of context. It has previously been shown that induction of autophagy in ovarian cancer cell lines results in a strikingly different survival phenotype than autophagy induction in the corresponding animal models.8 This was attributed to the effects of a relatively nutrient-poor tumor microenvironment compared to the nutrient-rich conditions found in cell culture. Alternatively the striking effect of SUPR peptide/carboplatin combinations in vivo may be the result of both chemotherapeutic effects and enhancement of the innate immune system. Although athymic nude mice were used to generate the orthotopic models, these mice still retain a functional innate immune system.48,49 Previous work in breast cancer50 and renal cell carcinoma (RCC)51 mouse xenograft models has shown that autophagy inhibition sensitizes tumors to natural killer (NK)-mediated cell killing by preventing degradation of secreted granzyme. Inhibition of autophagy in macrophages has also been shown to alter polarization from M2 to M1 phenotype which could facilitate a pro-inflammatory, anti-tumoral response.52 Future studies will be carried out to deconvolute the effect of SUPR peptide-mediated autophagy inhibition on tumor viability, drug resistance, and the immune response.
Conflicts of interest
J. P. G. and S. W. M. are co-inventors on a patent application related to this work (US patent application no. 62/799,388).Acknowledgements
All animal studies were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals and were approved by the UT MD Anderson Institutional Animal Care and Use Committee (IACUC). This work was supported by UT MDACC Startup Funds (SWM), a UT MDACC Moonshot Knowledge Gap Pilot Project, a Cancer Prevention and Research Institute of Texas (CPRIT) Individual Investigator Research Award (RP200166-IIRA, SWM), and the University of Texas MD Anderson Cancer Center Institutional Research Grant (IRG) Program. Additional support came from the MD Anderson SPORE in Ovarian Cancer NCI P50 CA217685, R01 CA135354, R21 CA181994, R01 GM127585, and the shared resources of the MD Anderson CCSG grant NCI P30 CA 16672. We also acknowledge support from T32CA196561 (BJG), F32EB024379 (BJE), and a CPRIT Early Translational Research Award RP170333 (SZ). We also gratefully acknowledge support from The National Foundation for Cancer Research, philanthropic support from the Anne and Henry Zarrow Foundation, and generous donations from Stuart and Gaye-Lynn Zarrow, the Mossy Foundation and the Roberson endowment.References
- X. Cheng, Y. Wang, Y. Gong, F. Li, Y. Guo, S. Hu, J. Liu and L. Pan, Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins, Autophagy, 2016, 12(8), 1330–1339 CrossRef CAS
.
- Y. Feng, D. He, Z. Yao and D. J. Klionsky, The machinery of macroautophagy, Cell Res., 2014, 24(1), 24–41 CrossRef CAS
- Z. Xie and D. J. Klionsky, Autophagosome formation: core machinery and adaptations, Nat. Cell Biol., 2007, 9(10), 1102–1109 CrossRef CAS
- T. Johansen and T. Lamark, Selective autophagy mediated by autophagic adapter proteins, Autophagy, 2011, 7(3), 279–296 CrossRef CAS
- S. Pankiv, T. H. Clausen, T. Lamark, A. Brech, J. A. Bruun, H. Outzen, A. Overvatn, G. Bjorkoy and T. Johansen, p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy, J. Biol. Chem., 2007, 282(33), 24131–24145 CrossRef CAS
- E. White, Deconvoluting the context-dependent role for autophagy in cancer, Nat. Rev. Cancer, 2012, 12(6), 401–410 CrossRef CAS
- E. White, The role for autophagy in cancer, J. Clin. Invest., 2015, 125(1), 42–46 CrossRef
- Z. Lu, R. Z. Luo, Y. Lu, X. Zhang, Q. Yu, S. Khare, S. Kondo, Y. Kondo, Y. Yu, G. B. Mills, W. S. Liao and R. C. Bast Jr, The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells, J. Clin. Invest., 2008, 118(12), 3917–3929 CAS
- D. Hashimoto, M. Blauer, M. Hirota, N. H. Ikonen, J. Sand and J. Laukkarinen, Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs, Eur. J. Cancer, 2014, 50(7), 1382–1390 CrossRef CAS
- Y. Han, S. Fan, T. Qin, J. Yang, Y. Sun, Y. Lu, J. Mao and L. Li, Role of autophagy in breast cancer and breast cancer stem cells (Review), Int. J. Oncol., 2018, 52(4), 1057–1070 CAS
- Y. A. Wen, X. Xing, J. W. Harris, Y. Y. Zaytseva, M. I. Mitov, D. L. Napier, H. L. Weiss, B. Mark Evers and T. Gao, Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer, Cell Death Dis., 2017, 8(2), e2593 CrossRef CAS
- X. Sui, R. Chen, Z. Wang, Z. Huang, N. Kong, M. Zhang, W. Han, F. Lou, J. Yang, Q. Zhang, X. Wang, C. He and H. Pan, Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment, Cell Death Dis., 2013, 4, e838 CrossRef CAS
- S. Paglin, T. Hollister, T. Delohery, N. Hackett, M. McMahill, E. Sphicas, D. Domingo and J. Yahalom, A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles, Cancer Res., 2001, 61(2), 439–444 CAS
- A. G. Smith and K. F. Macleod, Autophagy, cancer stem cells and drug resistance, J. Pathol.: Clin. Res., 2019, 247(5), 708–718 CrossRef
- R. K. Amaravadi, A. C. Kimmelman and J. Debnath, Targeting Autophagy in Cancer: Recent Advances and Future Directions, Cancer Discovery, 2019, 9(9), 1167–1181 CrossRef CAS
- M. Marinkovic, M. Sprung, M. Buljubasic and I. Novak, Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy, Oxid. Med. Cell. Longevity, 2018, 2018, 8023821 Search PubMed
- N. N. Noda, H. Kumeta, H. Nakatogawa, K. Satoo, W. Adachi, J. Ishii, Y. Fujioka, Y. Ohsumi and F. Inagaki, Structural basis of target recognition by Atg8/LC3 during selective autophagy, Genes Cells, 2008, 13(12), 1211–1218 CrossRef CAS
- J. C. Fuller, N. J. Burgoyne and R. M. Jackson, Predicting druggable binding sites at the protein-protein interface, Drug Discovery Today, 2009, 14(3–4), 155–161 CrossRef CAS
- M. Wirth, W. Zhang, M. Razi, L. Nyoni, D. Joshi, N. O'Reilly, T. Johansen, S. A. Tooze and S. Mouilleron, Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins, Nat. Commun., 2019, 10(1), 2055 CrossRef
- K. Letschert, H. Faulstich, D. Keller and D. Keppler, Molecular characterization and inhibition of amanitin uptake into human hepatocytes, Toxicol. Sci., 2006, 91(1), 140–149 CrossRef CAS
- H. Faulstich, H. Trischmann and D. Mayer, Preparation of tetramethylrhodaminyl-phalloidin and uptake of the toxin into short-term cultured hepatocytes by endocytosis, Exp. Cell Res., 1983, 144(1), 73–82 CrossRef CAS
- P. G. Dougherty, A. Sahni and D. Pei, Understanding Cell Penetration of Cyclic Peptides, Chem. Rev., 2019, 119(17), 10241–10287 CrossRef CAS
- T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobson and R. S. Lokey, Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers, J. Am. Chem. Soc., 2006, 128(8), 2510–2511 CrossRef CAS
- T. R. White, C. M. Renzelman, A. C. Rand, T. Rezai, C. M. McEwen, V. M. Gelev, R. A. Turner, R. G. Linington, S. S. Leung, A. S. Kalgutkar, J. N. Bauman, Y. Zhang, S. Liras, D. A. Price, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds, Nat. Chem. Biol., 2011, 7(11), 810–817 CrossRef CAS
- C. A. Rhodes, P. G. Dougherty, J. K. Cooper, Z. Qian, S. Lindert, Q. E. Wang and D. Pei, Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IkappaB Kinase Interaction Directly from a Combinatorial Library, J. Am. Chem. Soc., 2018, 140(38), 12102–12110 CrossRef CAS
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y.-W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, Towards understanding cell penetration by stapled peptides, MedChemComm, 2015, 6(1), 111–119 RSC
- J. A. Kritzer, J. D. Lear, M. E. Hodsdon and A. Schepartz, Helical beta-peptide inhibitors of the p53-hDM2 interaction, J. Am. Chem. Soc., 2004, 126(31), 9468–9469 CrossRef CAS
- S. V. Fiacco, L. E. Kelderhouse, A. Hardy, Y. Peleg, B. Hu, A. Ornelas, P. Yang, S. T. Gammon, S. M. Howell, P. Wang, T. T. Takahashi, S. W. Millward and R. W. Roberts, Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in Vivo Applications, Chembiochem, 2016, 17(17), 1643–1651 CrossRef CAS
- F. Pisaneschi, L. E. Kelderhouse, A. Hardy, B. J. Engel, U. Mukhopadhyay, C. Gonzalez-Lepera, J. P. Gray, A. Ornelas, T. T. Takahashi, R. W. Roberts, S. V. Fiacco, D. Piwnica-Worms and S. W. Millward, Automated, Resin-Based Method to Enhance the Specific Activity of Fluorine-18 Clicked PET Radiotracers, Bioconjugate Chem., 2017, 28(2), 583–589 CrossRef CAS
- R. W. Roberts and J. W. Szostak, RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(23), 12297–12302 CrossRef CAS
- S. W. Millward, T. T. Takahashi and R. W. Roberts, A general route for post-translational cyclization of mRNA display libraries, J. Am. Chem. Soc., 2005, 127(41), 14142–14143 CrossRef CAS
- K. Deprey and J. A. Kritzer, Quantitative measurement of cytosolic penetration using the chloroalkane penetration assay, Methods Enzymol., 2020, 641, 277–309 Search PubMed
- L. Peraro, K. L. Deprey, M. K. Moser, Z. Zou, H. L. Ball, B. Levine and J. A. Kritzer, Cell Penetration Profiling Using the Chloroalkane Penetration Assay, J. Am. Chem. Soc., 2018, 140(36), 11360–11369 CrossRef CAS
- E. R. Ballister, C. Aonbangkhen, A. M. Mayo, M. A. Lampson and D. M. Chenoweth, Localized light-induced protein dimerization in living cells using a photocaged dimerizer, Nat. Commun., 2014, 5, 5475 CrossRef
- M. Kurcinski, M. Jamroz, M. Blaszczyk, A. Kolinski and S. Kmiecik, CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site, Nucleic Acids Res., 2015, 43(W1), W419–W424 CrossRef CAS
- C. T. Chu, E. D. Plowey, R. K. Dagda, R. W. Hickey, S. J. Cherra 3rd and R. S. Clark, Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models, Methods Enzymol., 2009, 453, 217–249 CAS
- A. B. Birgisdottir, T. Lamark and T. Johansen, The LIR motif - crucial for selective autophagy, J. Cell Sci., 2013, 126(pt 15), 3237–3247 CAS
- M. Skytte Rasmussen, S. Mouilleron, B. Kumar Shrestha, M. Wirth, R. Lee, K. Bowitz Larsen, Y. Abudu Princely, N. O'Reilly, E. Sjottem, S. A. Tooze, T. Lamark and T. Johansen, ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8, Autophagy, 2017, 13(5), 834–853 CrossRef CAS
- T. Johansen and T. Lamark, Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors, J. Mol. Biol., 2020, 432(1), 80–103 CrossRef CAS
- V. E. Gray, R. J. Hause and D. M. Fowler, Analysis of Large-Scale Mutagenesis Data To Assess the Impact of Single Amino Acid Substitutions, Genetics, 2017, 207(1), 53–61 CrossRef CAS
- Y. Jiang, F. Ji, Y. Liu, M. He, Z. Zhang, J. Yang, N. Wang, C. Zhong, Q. Jin, X. Ye and T. Chen, Cisplatin-induced autophagy protects breast cancer cells from apoptosis by regulating yes-associated protein, Oncol. Rep., 2017, 38(6), 3668–3676 CAS
- J. Wang and G. S. Wu, Role of autophagy in cisplatin resistance in ovarian cancer cells, J. Biol. Chem., 2014, 289(24), 17163–17173 CrossRef CAS
- A. K. Mitra, D. A. Davis, S. Tomar, L. Roy, H. Gurler, J. Xie, D. D. Lantvit, H. Cardenas, F. Fang, Y. Liu, E. Loughran, J. Yang, M. Sharon Stack, R. E. Emerson, K. D. Cowden Dahl, M. V. Barbolina, K. P. Nephew, D. Matei and J. E. Burdette,
In vivo tumor growth of high-grade serous ovarian cancer cell lines, Gynecol. Oncol., 2015, 138(2), 372–377 CrossRef CAS
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix, Science, 2004, 305(5689), 1466–1470 CrossRef CAS
- Y. C. Li, L. W. Rodewald, C. Hoppmann, E. T. Wong, S. Lebreton, P. Safar, M. Patek, L. Wang, K. F. Wertman and G. M. Wahl, A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time, Cell Rep., 2014, 9(5), 1946–1958 CrossRef CAS
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity, ACS Chem. Biol., 2013, 8(2), 297–302 CrossRef CAS
- A. Komin, L. M. Russell, K. A. Hristova and P. C. Searson, Peptide-based strategies for enhanced cell uptake, transcellular transport, and circulation: Mechanisms and challenges, Adv. Drug Delivery Rev., 2017, 110–111, 52–64 CrossRef CAS
- L. R. Kelland, Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development, Eur. J. Cancer, 2004, 40(6), 827–836 CrossRef CAS
- I. Szadvari, O. Krizanova and P. Babula, Athymic nude mice as an experimental model for cancer treatment, Physiol. Res., 2016, 65(suppl. 4), S441–S453 CAS
- J. Baginska, E. Viry, G. Berchem, A. Poli, M. Z. Noman, K. van Moer, S. Medves, J. Zimmer, A. Oudin, S. P. Niclou, R. C. Bleackley, I. S. Goping, S. Chouaib and B. Janji, Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(43), 17450–17455 CrossRef CAS
- Y. Messai, M. Z. Noman, M. Hasmim, B. Janji, A. Tittarelli, M. Boutet, V. Baud, E. Viry, K. Billot, A. Nanbakhsh, T. Ben Safta, C. Richon, S. Ferlicot, E. Donnadieu, S. Couve, B. Gardie, F. Orlanducci, L. Albiges, J. Thiery, D. Olive, B. Escudier and S. Chouaib, ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy, Cancer Res., 2014, 74(23), 6820–6832 CrossRef CAS
- K. Liu, E. Zhao, G. Ilyas, G. Lalazar, Y. Lin, M. Haseeb, K. E. Tanaka and M. J. Czaja, Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization, Autophagy, 2015, 11(2), 271–284 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details are available and include all synthetic methods for small molecules and SUPR peptides, supplemental figures (Table S1 and Fig. S1–S7), and compound characterization data (Fig. S8–S28). See DOI: 10.1039/d0sc03603j |
| This journal is © The Royal Society of Chemistry 2021 |