
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular and supported Ti(III)-alkyls: efficient ethylene polymerization driven by the π-character of metal–carbon bonds and back donation from a singly occupied molecular orbital†‡
Anton
Ashuiev§
,
Florian
Allouche§
,
Nino
Wili
 ,
Keith
Searles
,
Daniel
Klose
,
Christophe
Copéret
* and
Gunnar
Jeschke
*
,
Keith
Searles
,
Daniel
Klose
,
Christophe
Copéret
* and
Gunnar
Jeschke
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1-5, CH-8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch; gunnar.jeschke@phys.chem.ethz.ch
First published on 11th November 2020
Abstract
While Ti(III) alkyl species are the proposed active sites in Ziegler–Natta ethylene polymerization catalysts, the corresponding well-defined homogeneous catalysts are not known. We report that well-defined neutral β-diiminato Ti(III) alkyl species, namely [Ti(nacnac)(CH2tBu)2] and its alumina-grafted derivative [(AlsO)Ti(nacnac)(CH2tBu)], are active towards ethylene polymerization at moderate pressures and temperatures and possess an electron configuration well-adapted to insertion of ethylene. Advanced EPR spectroscopy showed that ethylene insertion into a Ti(III)–C bond takes place during polymerization from Ti(nacnac)(CH2tBu)2. A combination of pulsed EPR spectroscopy and DFT calculations, based on a crystal structure of [Ti(nacnac)(CH2tBu)2], enabled us to reveal details about the structure and electronic configurations of both molecular and surface-grafted species. For both compounds, the α-agostic C–H interaction, which involves the singly occupied molecular orbital, indicates a π character of the metal–carbon bond; this π character is enhanced upon ethylene coordination, leading to a nearly barrier-less C2H4 insertion into Ti(III)–C bonds after this first step. During coordination, back donation from the SOMO to the π*(C2H4) occurs, leading to stabilization of π-ethylene complexes and to a significant lowering of the overall energy of the C2H4 insertion transition state. In d1 alkyl complexes, ethylene insertion follows an original “augmented” Cossee–Arlman mechanism that involves the delocalization of unpaired electrons between the SOMO, π*(C2H4) and σ*(Ti–C) in the transition state, which further favors ethylene insertion. All these factors facilitate ethylene polymerization on Ti(III) neutral alkyl species and make d1 alkyl complexes potentially more effective polymerization catalysts than their d0 analogues.
Introduction
Since the discovery of Ziegler–Natta ethylene polymerization catalysts in the early 1950s,1,2 the nature of the active site(s) has been a matter of debate. Later, group IV transition-metal metallocenes were developed as efficient homogeneous,3–7 as well as supported,8,9 olefin polymerization catalysts for which cationic M(IV) alkyl species have been proposed as the active sites.10 Such species have been isolated in the form of Lewis base adducts and have been shown to be competent in olefin polymerization; one noteworthy example is Ti(IV) amidinate species [(Cp*)Ti{NC(Ph)NiPr2}(OPPh3)Me][BArF4] (Scheme 1a).11 Taking into account the strong ionic character of MgCl2, the key support of Ziegler–Natta catalysts, surface Ti(IV) cationic alkyl species are sometimes proposed as the active sites in these systems in analogy to their metallocene equivalents.12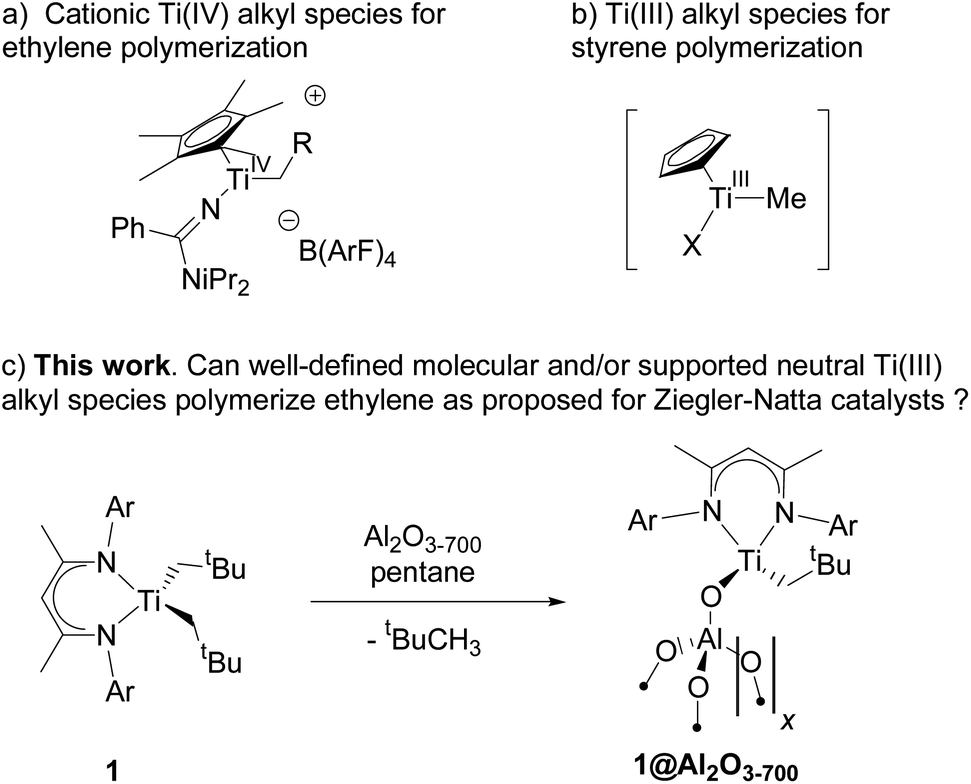 | ||
| Scheme 1 Known Ti(IV) (a) and Ti(III) (b) alkyl species, competent in olefin polymerization, together with synthesis of well-defined neutral supported Ti(III) alkyl species 1@Al2O3-700 (c) from molecular complex 1via surface organometallic chemistry. | ||
However, the evidence for Ti(IV) cationic species in Ziegler–Natta heterogeneous catalysts has remained elusive. In fact, the reaction with triethyl aluminum, a common cocatalyst used in the Ziegler–Natta process, undoubtedly leads to reduction and/or alkylation of certain sites, and Ti(III) centers have previously been observed by XPS and EPR.13–18 Since their role as active sites in the polymerization of ethylene has not been evidenced so far, it remains unclear whether or not titanium d1 complexes can be efficient in ethylene polymerization.19,20 In parallel, cationic monocyclopentadienyl Ti(III) compounds show high activities towards styrene polymerization and are catalytically comparable to or better than the corresponding Ti(IV) derivatives (Scheme 1b).21 Furthermore, it has been recently shown by pulsed EPR spectroscopy, combined with DFT calculations, that Ti(III)-alkyl surface species are formed when silica-supported titanium(III) hydride comes into contact with ethylene.22 This finding is consistent with previous calculations that show Ti(III) hydrides as competent for initiating ethylene polymerization;23 it further implies that Ti(III) d1 alkyl complexes could polymerize olefins such as ethylene.
Among well-defined Ti(III) alkyl species, the neutral β-diiminato Ti(III) dialkyl species (Scheme 1c, 1) are noteworthy for their stability.24,25 While reported to be unreactive towards ethylene polymerization without a cocatalyst at room temperature under 6 bar of ethylene,24 the presence of two alkyl ligands in this compound makes it particularly attractive to generate a supported Ti(III) alkyl catalyst through surface organometallic chemistry (SOMC)26 and to evaluate its polymerization activity. Indeed, SOMC has been shown to provide access to many active and stable catalysts through isolation of metal sites at the surface of oxide supports.26–29 In addition, the presence of strong Lewis acid surface sites, such as in alumina, can also help promote the formation of more active species.30–32 Alternatively, cationic surface sites can be stabilized using sulfated metal oxides as a support.32–34
Herein, we show that the molecular β-diiminato Ti(III) alkyl complex (Scheme 1c, 1) efficiently promotes ethylene polymerization without the need for co-catalysts at 80 °C and pressures higher than 4 bar and that its alumina-supported analogue, prepared via the SOMC26 approach (Scheme 1c, 1@Al2O3-700), shows significantly improved productivity. Using pulsed EPR spectroscopy, combined with DFT calculations and polymerization tests, we demonstrate that neutral Ti(III) alkyl species are indeed able to initiate ethylene polymerization to produce ultra-high molecular weight polyethylene via ethylene insertion into a Ti(III)–C bond. Detailed DFT calculations show that ethylene insertion into the Ti(III)–C bond, a key step of ethylene polymerization, is favored by a partial alkylidenic character of the metal–carbon bond, as in its d0 analogues,35,36 and the added possibility of a partial electron transfer to the coordinated olefin in Ti(III) compounds, which can be viewed as the π-back donation from the SOMO into the coordinated ethylene. This “augmented” Cossee–Arlman mechanism of olefin polymerization, possible for d1 metal-alkyl complexes, involves the delocalization of unpaired electrons in the transition state of olefin insertion into the metal–carbon bond with a strong alkylidenic character.
Results and discussion
Synthesis and characterization of neutral Ti(III) species and examination of their polymerization activity
Elemental analysis of 1@Al2O3-700 gives 0.24 wt% Ti, 2.06 wt% C, 0.20 wt% N and 0.24 wt% H, corresponding to 41.0 ± 1 C/Ti (34 expected), 3.4 ± 1 N/Ti (2 expected) and 57.0 H/Ti (52 expected). The grafting of 0.54 Ti per surface OH group is thus consistent with the formation of a monografted species, [(AlsO)Ti(nacnac)(CH2tBu)] (Als = surface aluminium). Further characterization of 1@Al2O3-700 by Fourier transform infrared spectroscopy (FTIR) shows the disappearance of the initial isolated hydroxyl groups and the appearance of a broad band from 3420 to 3760 cm−1, associated with hydroxyl groups interacting with the ligands of the grafted Ti centres (e.g. nacnac ligand).37 These fragments are also revealed via ν(C–H) vibrations at 3081–2874 cm−1 (Fig. S3‡).
Both materials 1 and 1@Al2O3-700 were then characterized with CW EPR spectroscopy. The X-band CW EPR spectrum of complex 1 recorded at 10 K (Fig. 1a, blue) originates from an S = 1/2 electron spin system with a nearly axial g tensor, having its principal values gx = 1.898 ± 0.023, gy = 1.981 ± 0.016, and gz = 1.996 ± 0.012 (the given intervals indicate Gaussian distributions of g principal values; the simulation of the EPR spectrum is shown in Fig. 1a, red).
 | ||
| Fig. 1 (a) CW EPR spectrum of 1 in a frozen toluene solution (blue) and simulation (red); (b) CW EPR spectrum of 1@Al2O3-700 (blue) and simulation (red). See text for simulation parameters. | ||
For the surface-grafted material 1@Al2O3-700, the X-band CW EPR spectrum (Fig. 1b blue) was measured at 10 K; the spectrum is consistent with the presence of an S = 1/2 system, with gx = 1.880 ± 0.087, gy = 1.970 ± 0.049 and gz = 1.984 ± 0.016 (the simulation is shown in Fig. 1b, red) associated with a paramagnetic Ti(III) surface species. Significant line broadening is observed in the grafted material 1@Al2O3-700 compared to molecular complex 1 which may result from the presence of different Ti3+ surface species, possibly due to small differences in local surface environments and thereby coordination geometry. In spite of replacing one Ti-coordinated carbon atom by a more electronegative oxygen atom, the g principal values for both 1 and 1@Al2O3-700 agree with each other within the given line widths, thus indicating a similar electronic structure and symmetry of molecular complex 1 and alumina-supported species 1@Al2O3-700.
 | ||
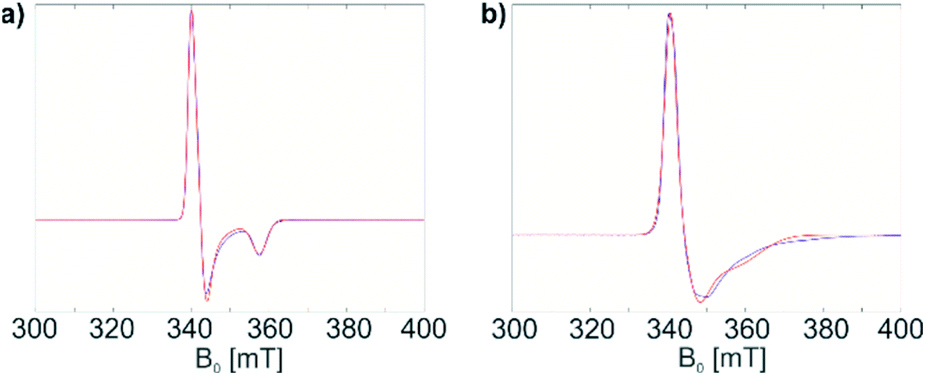
| Fig. 2 (a) Ethylene consumption of a toluene solution of 1 with time under different pressure and temperature conditions. (b) Molecular weight distribution of polyethylene produced by 1 under 7 bar of ethylene pressure and at 100 °C (blue), together with molecular weight distribution of polyethylene produced by 1@Al2O3-700 under 6 bar of ethylene pressure and at 50 °C (red). (c) SEM image of the 1@Al2O3-700 material before polymerization. (d) SEM image of the 1@Al2O3-700 material after polymerization under 1 bar of ethylene pressure and at room temperature. The changes in ethylene consumption curves up to minute 12 in (a) are due to the pressure changes in the system while reaching the reaction temperature. | ||
The material 1@Al2O3-700 is significantly more active towards ethylene polymerization than its molecular analogue 1, and the polymerization stops within minutes due to the formation of a dense PE layer that can be directly observed with scanning electron microscopy (SEM) (Fig. 2c and d). Overall, this material displays a productivity of ca. 36 kgPE (molTi h)−1 under 6 bar of ethylene at 50 °C. Note that, in contrast to 1, 1@Al2O3-700 initiates ethylene polymerization even under very mild conditions, i.e. room temperature, 80 mbar of ethylene.
The molecular weight distribution for PE, produced by 1 and 1@Al2O3-700, is obtained with size-exclusion chromatography (SEC, Fig. 2b). For both catalysts, the distribution is asymmetric with a main heavy fraction and a broad distribution of molecular weights of lighter PE fractions, possibly being the products of chain termination reactions. The main fraction of PE has a molecular weight of ca. 1130 kg mol−1 for 1 and ca. 5660 kg mol−1 for 1@Al2O3-700, which is typical for ultra-high molecular weight polyethylene (UHMWPE).38 As the PE molecular weight distribution has similar character for both 1 and 1@Al2O3-700, and as UHMWPE is produced in both cases, we propose that the polymerization on both catalysts takes place via a similar mechanism. The higher molecular weight of the main fraction of PE, produced with 1@Al2O3-700, can be explained by the absence and/or the slower rate of possible chain termination reactions, one example being the α-H abstraction reaction.
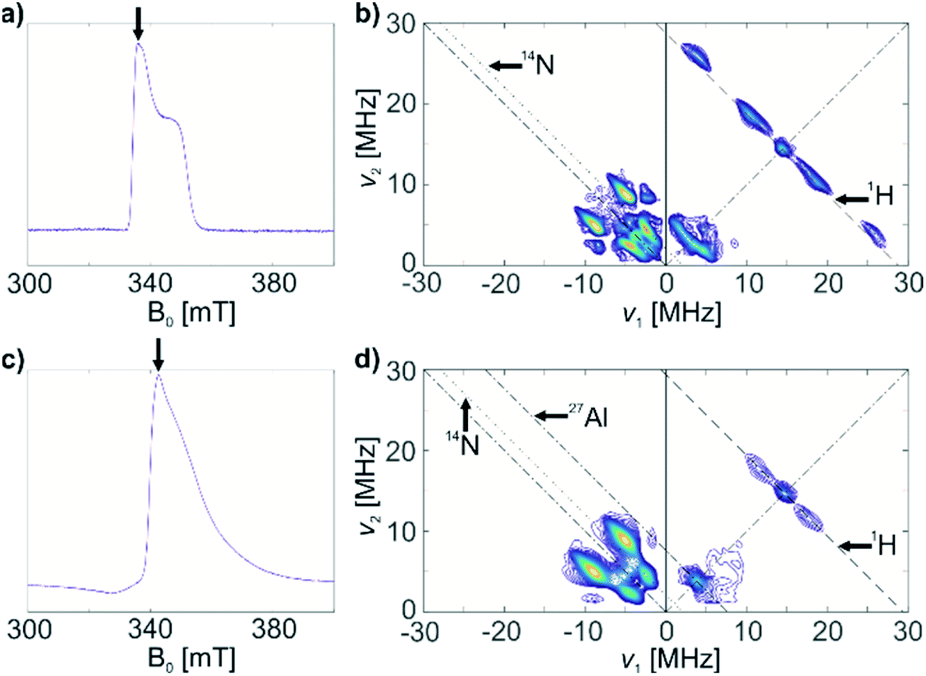 | ||
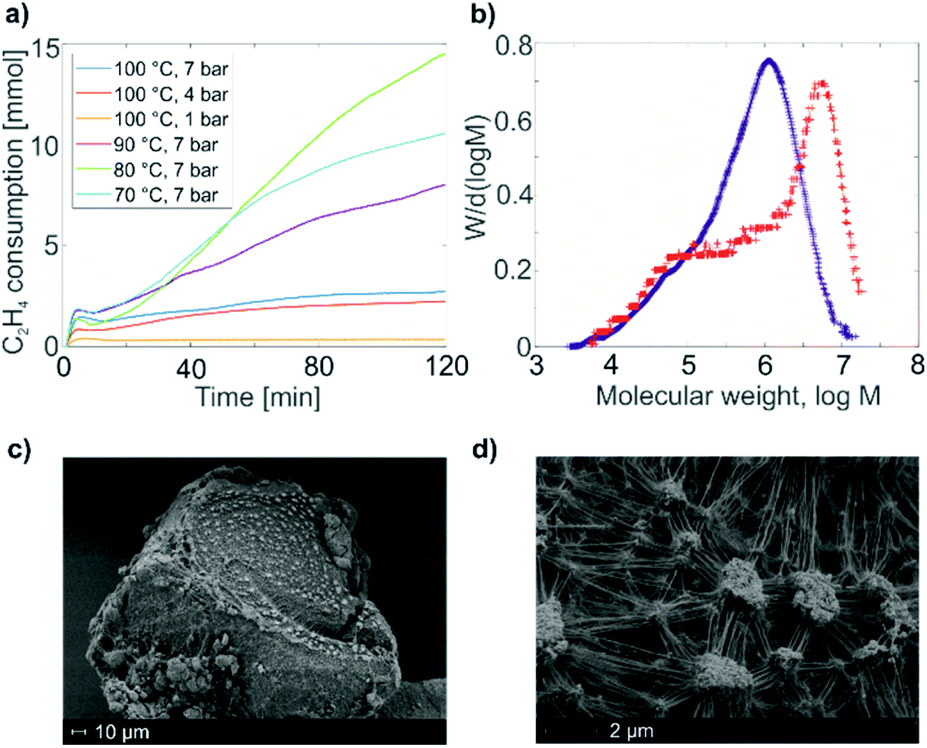
| Fig. 3 (a) Echo-detected EPR spectrum of a frozen toluene solution of 1; (b) HYSCORE spectrum of 1, τ = 128 ns; (c) echo-detected EPR spectrum of 1@Al2O3-700; (d) HYSCORE spectrum of solid 1@Al2O3-700, τ = 128 ns. Arrows on the echo-detected EPR spectra (a and c) indicate the magnetic field positions for HYSCORE measurements. Antidiagonal lines on the HYSCORE spectra (b and d) correspond to nuclear frequencies indicated on the spectra (black arrows). | ||
The HYSCORE spectrum of complex 1 (Fig. 3b) shows the presence of 1H and 14N nuclei in the Ti(III) coordination sphere, revealed by cross peaks along the 1H antidiagonal in the weak coupling (+, +) quadrant, corresponding to 1H hyperfine couplings, and by peaks in the low-frequency region both in the weak coupling (+, +) and strong coupling (−, +) quadrants, corresponding to 14N hyperfine and quadrupole couplings. For the material 1@Al2O3-700, the HYSCORE spectrum (Fig. 3d) reveals the presence of 14N hyperfine and quadrupole couplings as well, which are close to the ones observed for molecular complex 1 before grafting. This indicates that the (nacnac) ligand remains coordinated to the Ti(III) center after grafting onto the Al2O3-700 surface. For 1@Al2O3-700, a different set of 1H hyperfine couplings is observed, with a loss of strongly coupled 1H being the most prominent change after grafting. Most probably, this is due to the loss of one of the CH2tBu ligands upon grafting, as shown in Scheme 1. Furthermore, 27Al couplings are observed for 1@Al2O3-700 as a matrix peak on the 27Al antidiagonal line (Fig. 3d) and are well-resolved in Q-band HYSCORE spectra (Fig. 5c). This is consistent with the presence of nearby surface Al atoms in the surroundings of Ti(III), as expected for a grafted species.
Using the experimental HYSCORE spectra, we could further estimate the conformation of molecular complex 1 in toluene solution and provide detailed structural information regarding the surface species in 1@Al2O3-700 by comparing experimental and calculated hyperfine and quadrupole tensors. In order to find the explicit structure of complexes 1 and 1@Al2O3-700, the X-ray crystal structures of 1 (ref. 25) (see Fig. S6‡) and the derived model for the species in 1@Al2O3-700 were optimized with unrestricted Kohn–Sham density functional theory (DFT), using the functional PBE0 (ref. 40) in ORCA 3.41,42 For these geometry optimizations, a polarized triple-ζ def2-TZVPP basis set43 was used for all atoms, together with Becke's three-center dispersion correction.44 The COSMO continuum solvation model45 was applied for complex 1. Furthermore, the hyperfine and quadrupole tensor parameters were calculated with the def2-TZVPP basis set for Ti and Al atoms and the EPR-II basis set46 for all other atoms. Based on the calculated parameters, the simulations of HYSCORE spectra were carried out in EasySpin.47 Hyperfine and nuclear quadrupole couplings were previously found to be highly sensitive to small structural changes.22 Here we find that rotation of one of the CH2tBu ligands of 1 has a tremendous effect on the calculated isotropic part of the 1H hyperfine tensors of α-H atoms of the CH2tBu ligands (see Fig. S7‡). Together with their dipolar part, which is sensitive to the Ti–H distances, the calculated hyperfine tensors act as fingerprints of α-H positions in the calculated structures of 1 and 1@Al2O3-700. The calculated multiple-quantum (e.g. double-quantum) 14N transitions are affected by such a rotation as well (see Fig. S7‡), and hence are an effective probe for the molecular structure and conformation through both hyperfine and quadrupole tensors. This allows us to use DFT-based simulations of the experimental HYSCORE spectra as a tool for evaluation of the molecular structures of 1 and 1@Al2O3-700. After such an evaluation, the explicit structures and the Kohn–Sham molecular orbital sets for 1 and 1@Al2O3-700 were obtained simultaneously, both verified by a comparison of the experimental and simulated HYSCORE spectra.
The crystal structure of 1, however, does not yield the correct set of 1H hyperfine coupling parameters, since it fails to fully predict the experimental HYSCORE spectrum (Fig. S6‡). This indicates that complex 1 possesses a different conformation in frozen toluene solution than in the solid state. However, after the geometry optimization using the parameters indicated above, the obtained conformation (Fig. 4a) generates hyperfine and quadrupole tensors for 14N and 1H nuclei (Table 1), which simulate the entire X-band HYSCORE spectrum rather nicely (Fig. 4b). Both the isotropic and dipolar parts of 1H hyperfine tensors fit well to the experimental spectrum in Fig. 4b, thus indicating the correct positions of the α-H atoms of CH2tBu ligands in the optimized structure (Fig. 4a). Furthermore, the calculated 14N hyperfine and quadrupole tensor parameters are found to simulate both the X-band (Fig. 4b) and Q-band (see Fig. S8‡) HYSCORE spectra. We therefore propose that in a frozen toluene solution, molecular compound 1 is present in the form of the conformer shown in Fig. 4a, which differs from the solid-state structure by a slight rotation of the CH2tBu ligands around the Ti–C axis (see ESI Part 2.3‡).
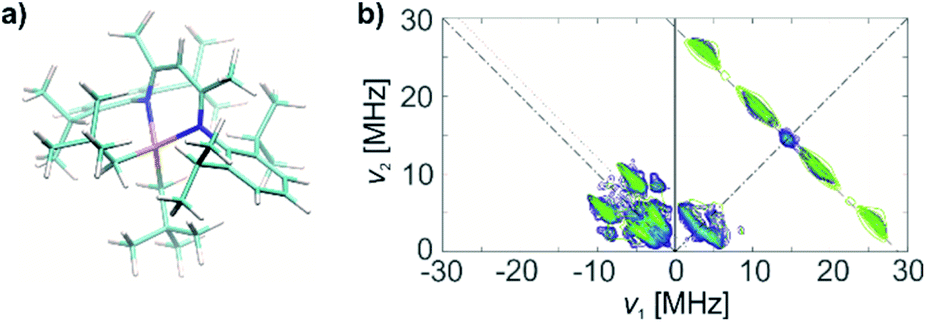 | ||
| Fig. 4 (a) Molecular structure of 1, based on DFT optimizations and on the agreement with HYSCORE spectra. (b) X band HYSCORE spectrum of 1 (blue) and simulation (green), based on DFT calculations on the structure in (a). | ||
| 1 | 1@Al2O3-700 | |
|---|---|---|
| 1H | a iso = 7.77 | a iso = 5.80 |
| a dip = [−4.94; −3.08; 8.02] | a dip = [−3.87; −2.57; 6.44] | |
| 1H | a iso = 13.37 | a iso = −3.34 |
| a dip = [−3.85; −2.57; 6.42] | a dip = [7.65; −3.34; −4.30] | |
| 1H | a iso = 18.76 | — |
| a dip = [−3.93; −2.82; 6.76] | ||
| 1H | a iso = 1.27 | — |
| a dip = [−3.40; −5.71; 9.11] | ||
| 14N | a iso = −7.98 | a iso = −6.37 |
| a dip = [2.05; 1.74; −3.79] | a dip = [0.61; −0.17; −0.43] | |
| P = 2.72 (η = 0.316) | P = 2.50 (η = 0.470) | |
| 14N | a iso = −7.07 | a iso = −7.03 |
| a dip = [1.08; 0.74; −1.82] | a dip = [0.80; 0.50; −1.30] | |
| P = 2.69 (η = 0.276) | P = 2.60 (η = 0.354) | |
| 27Al (least squares fit) | — | a iso = 2.68 |
| a dip = [−2.23; 3.56; −1.34] | ||
| P = 7.66 (η = 0.01) |
A similar approach of evaluating the molecular structure was used for the grafted species 1@Al2O3-700. We considered two possible types of 1@Al2O3-700 species, either neutral [(AlsO)Ti(nacnac)(CH2tBu)] surface Ti complexes obtained via grafting of 1 through surface OH groups (Scheme 1c) or cationic [Ti(nacnac)(OAls)]+⋯[(CH2tBu)Als]− where the alkyl group is transferred onto alumina (see Fig. S9‡) as previously observed for other complexes.31,48,49 Geometry optimizations with subsequent calculations of EPR parameters were performed for both cationic and neutral models. It appeared that the cationic model, however, does not result in 1H hyperfine couplings that are strong enough to simulate the elongated experimental HYSCORE ridges (see Fig. S9‡). The α-H atoms of the CH2tBu fragment in this model are far from the Ti center such that both dipolar and isotropic parts of the 1H hyperfine tensors appear too small (see ESI Part 2.3‡). Only the neutral models exhibit 1H hyperfine couplings that are strong enough to simulate the experimental HYSCORE spectrum.
The geometry optimization of the neutral [(AlsO)Ti(nacnac)(CH2tBu)] model was performed as described above for complex 1, followed by a DFT-based calculation of EPR parameters. The complex nature of the Al2O3 surface results in a number of different types of OH groups that can participate in grafting.31 Here, we represent the (OH)Als surface atom by the simplest possible tetracoordinated neutral Al model, namely (HO)Als = (HO)Al(OH)2(H2O). Within this approximation, the symmetry of the {AlO4} cluster may be decreased compared to the structure of an alumina surface center due to different Al–OH2 and Al–OH bond lengths; this may result in overestimation of quadrupole coupling for the 27Al nucleus. Furthermore, it is worth noting that the spin density on Al nuclei tends to be underestimated even with basis sets that contain diffuse functions, which are expected to better describe spin density near the nucleus.50 Taking this into account, it appeared to be better to rely on experimentally determined 27Al hyperfine and quadrupole couplings rather than to evaluate these from DFT calculations.
The 1H and 14N couplings (Table 1) computed for the thus-obtained model (Fig. 5a) simulate the experimental X-band HYSCORE spectrum quite well (Fig. 5b). Similar to molecular complex 1, this indicates the correct positions of α-H atoms of a CH2tBu ligand in the optimized structure. Although 27Al couplings are not detected in the X-band HYSCORE spectrum, Q-band HYSCORE (Fig. 5c) provides the necessary information to determine the 27Al hyperfine couplings by least squares fitting (Fig. S10‡). Together with the calculated 14N hyperfine couplings, they fit reasonably to the Q-band HYSCORE spectrum (Fig. 5c, green). This allows us to consider the obtained model (Fig. 5a) as an explicit structure of Ti(III) surface species observed in our EPR studies. Therefore, we confirm that grafting of 1 onto the Al2O3-700 surface yields a neutral Ti(III) alkyl species, namely [(AlsO)Ti(nacnac)(CH2tBu)], with the structure shown in Fig. 5a. The production of UHMWPE by 1@Al2O3-700 as well as by 1, together with a high polymerization activity of 1@Al2O3-700, is consistent with a predominant presence of Ti(III) neutral alkyl species on the Al2O3-700 surface after grafting.
 | ||
| Fig. 5 (a) Model for 1@Al2O3-700 species representing a neutral [(AlsO)Ti(nacnac)(CH2tBu)] complex based on agreement with HYSCORE spectra. (b) X-band HYSCORE spectrum of 1@Al2O3-700 (blue) and simulation (green), based on DFT calculations on the obtained model. (c) Q-band HYSCORE spectrum of 1@Al2O3-700 (blue) and simulation (green), based on DFT computed EPR parameters from the obtained model (a) for 14N, combined with EPR parameters for 27Al, obtained from a least squares fit of the experimental spectrum. The ridge on the anti-diagonal in the left quadrant is due to an echo crossing that was not fully suppressed by phase cycling. | ||
13C-labeled complex 1* was characterized by pulse EPR spectroscopy. It exhibits the same echo-detected EPR spectrum and, consequently, the same g tensor parameters as non-labelled complex 1 (Fig. 6a). Detection of the 13C couplings of the coordinating (13CH2tBu) ligands proved difficult. According to DFT calculations for the optimized structure of 1 (Fig. 4a), the 13C hyperfine tensors for both α-C atoms of (13CH2tBu) ligands of complex 1* show large couplings that are mostly isotropic (aiso = −20.51 MHz, adip = [1.26 0.54 –1.80] MHz and aiso = −21.69 MHz, adip = [1.52 0.50 –2.01] MHz for the two 13C nuclei). This leads to a low probability of the forbidden electron-13C-nuclear spin transitions, making direct observation of the 13C signals with ESEEM-based techniques (e.g. HYSCORE) difficult (in the Q band) or impossible (in the X band). Furthermore, strong 14N ESEEM modulations in both the X- and Q-band may suppress the 13C modulations due to a cross-suppression effect.51 For this reason, we used an alternative EPR methodology based on the recently developed hyperfine technique CHEESY-detected NMR (CHEESY = chirp echo EPR spectroscopy).52 This method is based on long selective hole burning pulses that drive forbidden transitions, similar to ELDOR-detected NMR, but the detection is based on broadband chirp echoes and subsequent Fourier transform. This leads to a multiplex advantage and, consequently, to higher sensitivity (see ESI Part 2.4‡ for more details). Indeed, the 13C signals were observed already in the 1D CHEESY-detected NMR spectrum of 1*, revealed in comparison with the same spectrum of non-labelled complex 1 where the 13C signals were not observed (Fig. 6b).
 | ||
| Fig. 6 (a) Q-band echo-detected EPR spectra of 1 (blue) and 1* (red), intensities normalized; the position of further measurements is indicated with an arrow. (b) 1D CHEESY-detected NMR spectra of 1 (blue) and 1* (red); the 13C signals are marked on the spectrum. (c) Q-band 2D CHEESY-detected NMR spectrum of 1; the 13C nuclear Zeeman frequency is shown by a red antidiagonal line. (d) Q-band 2D CHEESY-detected NMR spectrum of 1*; the 13C nuclear Zeeman frequency is shown by a red antidiagonal line along which cross peaks due to 13C hyperfine coupling are observed (marked with arrows). The gray area has not been recorded during the experiment in order to optimize sensitivity and the spectra were recorded at slightly different fields and frequencies. | ||
These 13C signals, observed at the orientation corresponding to g = 1.983 (Fig. 6a, marked with an arrow), are better resolved in two-dimensional HYSCORE-type CHEESY-detected NMR spectra (Fig. 6c and d), which are obtained by applying a selective π pulse with variable frequency before the HTA pulse (see ESI Part 2.4‡ for the details of CHEESY-detected NMR experiments). The comparison of the spectra for 1 (Fig. 6c) and 1* (Fig. 6d) reveals the peaks at (21, 5) MHz, corresponding to the signals of 13C in [Ti(nacnac)(13CH2tBu)2]. Based on the obtained spectra (Fig. 6b–d), the 13C hyperfine coupling was estimated to be aiso(13C) = 16 MHz. This allowed us to confirm the assignment by Q-band HYSCORE and Q-band Davies ENDOR53 (see Fig. S11‡), where the weak spectral signals corresponding to 13C hyperfine couplings were identified by comparison to the 1D- and 2D-CHEESY detected NMR spectra.
Next, a benzene solution of 1* was brought into contact with C2H4 (1000 equivalents) at 80 °C for 2 hours. After the reaction, the excess C2H4 was removed and EPR measurements of (1* + C2H4) were performed. The similarities in the echo-detected Q-band EPR spectra (Fig. 7a) of complex 1* before and after polymerization indicate similar g tensor parameters consistent with conservation of the symmetry of Ti(III). Using the same methodology as before, 1D CHEESY-detected NMR spectra before and after the polymerization were measured (Fig. 7b) at the same frequency and field positions, with an identical microwave resonator profile (Fig. S12‡). Although the absolute echo intensities for both samples may still be slightly different, the CHEESY-detected NMR signals, being essentially the ratio of the spectra with and without a high turning-angle pulse, can be considered a quantitative tool to probe the amount of EPR active nuclei in the Ti(III) coordination sphere before and after the reaction with C2H4.
 | ||
| Fig. 7 (a) Q-band echo-detected EPR spectra of 1* (red) and 1* + C2H4 (black), intensities normalized; the position of further measurements is indicated by an arrow. (b) Top: experimental 1D CHEESY-detected NMR spectra of 1* (red) and 1* + C2H4 (black); no normalization was applied. Bottom: simulated 1D CHEESY-detected NMR spectra of 1* (red) and 1 (black). 13C signals, corresponding to A(13C) = 16 MHz are marked with lines under the spectra; the combination signal of (13C + 14N) is marked with an asterisk. The difference between the spectra due to difference in 13C signal intensities is colored in blue. | ||
An obvious decrease of the 13C signal intensity is observed after polymerization (Fig. 7b, top) for all previously observed 13C lines of 1*, corresponding to aiso(13C) = 16 MHz, as well as for the combination signal of (13C + 14N). At the same time, the spectral lines, determined by 14N hyperfine and quadrupole couplings (e.g. double-quantum 14N signals around 14 MHz), are the same before and after polymerization both regarding their frequencies and intensities. Since the frequencies of these 14N signals are sensitive even to small changes in the structure and conformation of 1* (see Fig. S7‡) we can conclude that the structure of the Ti(III) coordination sphere experiences minimal change upon polymerization. These 14N signals were reasonably simulated with the calculated values for the previously estimated conformation of 1 (Fig. 4a) in the spectra both before and after polymerization of ethylene (Fig. 7b, bottom). Together with the observed decrease of the 13C signal intensity, this indicates that ligand exchange of 13CH2tBu to (CH2CH2)n−1(13CH2tBu) occurs with preservation of the initial structure and conformation of 1*. Indeed, the experimentally observed decrease of 13C signals is simulated well as a difference between 1D CHEESY-detected NMR simulations for labelled complex 1* and non-labelled complex 1 (Fig. 7b, bottom). To that end, the experimental aiso(13C) = −16 MHz, together with DFT computed adip(13C) parameters, was used for the simulation of the spectrum of 1* (see ESI Part 2.4‡ for the details of the simulation). The comparison of the simulated and experimentally observed decrease of the 13C signal intensity implies that probably not all the complex 1* present is affected by the ligand exchange, but only a part of it. This indicates that only a part of molecules of 1* acts as active centres of ethylene polymerization under the aforementioned reaction conditions. This is consistent with the presence of an induction period at the beginning of polymerization, revealed by changes in ethylene consumption (Fig. 2a). It is also consistent with the calculated energy barrier for the first olefin insertion (vide infra). An exact quantification is difficult without precise knowledge of the full 13C hyperfine tensor, which also affects line intensities.
Based on the discussed experimental results, we propose that olefin polymerization takes place via C2H4 insertion into the Ti(III)–C bond in the molecular system Ti(nacnac)(CH2R)2–1. Unfortunately, we were not able to study ethylene polymerization with 1@Al2O3-700 due to T2 electron spin relaxation times, which are ca. 6 times shorter for 1@Al2O3-700 than for 1. This limits the observation window length and, consequently, the resolution of CHEESY-detected NMR26 such that the separation of 13C, 14N and 27Al signals becomes rather uncertain. However, the molecular weight distributions of produced PE are quite close for both 1 and 1@Al2O3-700, with UHMWPE being produced in both cases. This indicates a similar type of active center and polymerization mechanism for both catalysts. As we determined a neutral [(AlsO)Ti(nacnac)(CH2tBu)] species to be present in 1@Al2O3-700, which is similar in terms of structure and electronic properties to molecular complex 1, we propose that ethylene polymerization on 1@Al2O3-700 proceeds via the same mechanism as for 1, i.e. through C2H4 insertion into the Ti(III)–C bond in [(AlsO)Ti(nacnac)(CH2tBu)].
Electronic structures and the polymerization mechanism for 1 and 1@Al2O3-700
Essentially, α-agostic C–H bonds are described35,54 as the donation of electrons from the filled molecular orbital corresponding to the C–H bond to a metal d-orbital of appropriate symmetry that is empty for d0 metals. This agostic interaction has been recently related to a metal–carbon bond acquiring a π (or alkylidene) character,35,55 which favors the olefin insertion process. The degree of this π character could be indirectly estimated from the deviation of the Ti–C–H angle from 109° towards ca. 90°. In order to estimate directly the π character of Ti(III)–C bonds of 1 and 1@Al2O3-700, a Natural Bond Orbital (NBO) analysis56 was performed, using the program NBO 7.0.57 The molecular orbital sets for NBO analyses were generated using ORCA 4 (ref. 42) with the same parameters for the DFT calculations as the ones used for the simulations of the HYSCORE spectra (PBE0 functional together with the def2-TZVPP basis set for Ti and Al atoms and EPR-II basis set for all other atoms). Given the good agreement between the measured and calculated HYSCORE spectra, this computational method describes the electronic structures of 1 and 1@Al2O3-700 with sufficient accuracy.
The NBO analysis revealed a natural orbital, related to a singly occupied molecular orbital (SOMO) of paramagnetic complex 1 (Fig. 8a, red and blue). Its shape correlates well with the calculated distribution of spin density in space (Fig. 8a, green), thus confirming the close relation of this natural orbital to the SOMO. This orbital is nearly axially symmetric, which is consistent with the experimentally observed axial symmetry of the g tensor.
 | ||
| Fig. 8 (a) A natural spin-α orbital, related to the SOMO of 1 (red for positive and blue for negative signs of the wavefunction, the same in all the panels), together with the spin density distribution (green). (b) Qualitative molecular orbital diagram, illustrating the formation of the π*(Ti–(CH2tBu)2) orbital being the SOMO component of 1. (c) Natural SOMO of 1 as visible from the (CH2tBu)2 side, showing a central d-type part and two carbon p-type parts (marked with arrows). (d) Natural SOMO of 1@Al2O3-700, showing a carbon p-type part (marked with arrows). | ||
The spatial distribution of the SOMO-related natural orbital of 1 includes four lobes of the p-type on two nitrogen atoms and two carbon atoms of CH2tBu ligands, all being in antiphase with the central d-type lobes (Fig. 8a and c). The part of this orbital, which includes the central d-type part and the two carbon p-type parts (Fig. 8c), can be understood as a product of interaction of the half-filled dxy-type Ti orbital and one of two degenerate filled π− orbitals of the (CH2tBu)2 fragment (Fig. 8b). This orbital features π* symmetry with respect to the Ti–C bond.
Therefore, the presence of the π* orbital, as part of the SOMO and delocalized between two CH2tBu ligands, reveals the existence of π bonding in Ti–(CH2tBu)2. This π interaction, although being weakened by the unpaired electron in the antibonding π* orbital (Fig. 8b), stabilizes the structure with the α-agostic C–H bonds for 1. Compared to metal d0 complexes35 this α-H agostic interaction involves a half-filled metal d orbital instead of an empty one, as revealed by the NBO analysis for the refined structure of 1. Such an interaction brings a π character into both Ti–C bonds of 1. This π character is also evidenced by the deviation of the natural hybrid orbital (NHO) on carbon from the Ti–C axis (θNHO–C–Ti = 15.0° and 14.4° for the two Ti–C bonds of 1) – for a pure σ-bond no deviation would be expected (0.0°).35
A natural orbital of similar type, including a p-type lobe on the carbon atom of the single CH2tBu ligand, is also found for 1@Al2O3-700 (Fig. 8d). This indicates the presence of a π* orbital and, consequently, a π interaction in the [Ti–(CH2tBu)] system. However, the π character acquired by the Ti–C bond of 1@Al2O3-700 is less pronounced compared to that of complex 1 (θNHO–C–Ti = 9.2° for 1@Al2O3-700). The described π interaction, which involves half-filled metal d orbitals, might be present for all paramagnetic transition metal alkyl complexes, provided that the corresponding half-filled d orbitals have appropriate symmetry.
It is noteworthy that for the model Ti(IV) cationic analogue of complex 1, namely [Ti(nacnac)(CH2tBu)2]+ (1+), the ethylene insertion reaction is predicted to be less favorable compared to the α-H abstraction process, as revealed by calculations on the optimized structure (Fig. 9a). The calculated energy barrier for the α-H abstraction in d0 complex 1+ is ΔH‡298 = 29.7 kcal mol−1 and ΔG‡298 = 30.3 kcal mol−1, being slightly less than the one calculated for d1 complex 1. This is consistent with a stronger degree of π character in the Ti–C bonds (θNHO–C–Ti = 23.5° and 17.5° for the two Ti–C bonds) of 1+. For ethylene insertion involving 1+, the TS barrier is ΔH‡298 = 28.3 kcal mol−1 and ΔG‡298 = 41.7 kcal mol−1 with respect to the initial reagents. Despite having a stronger degree of π character of the Ti–C bonds, the TS energy of ethylene insertion is strongly increased by ΔG = + 8.0 kcal mol−1 for the d0 complex compared to the analogous d1 complex. This indicates that the unpaired electron in a singly occupied molecular orbital of complex 1 plays an important role in its reactivity towards ethylene insertion, significantly lowering the TS energy for d1 active species compared to similar d0 species.
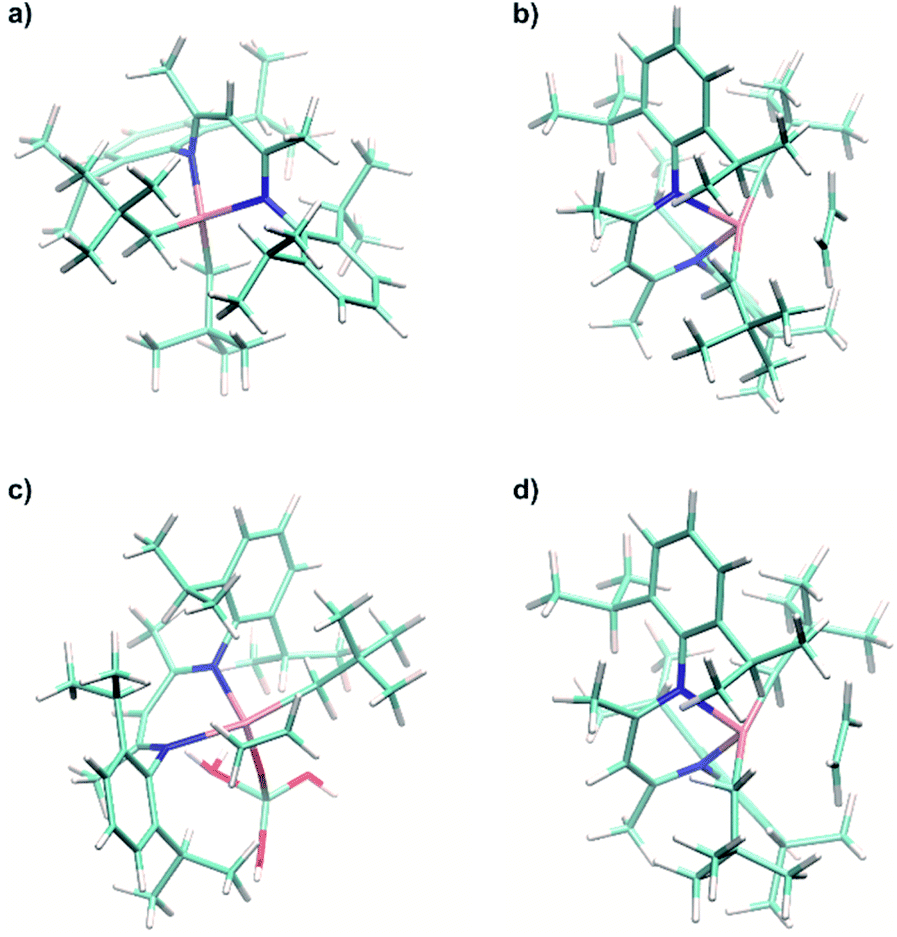 | ||
| Fig. 9 (a) Calculated structure of the model Ti(IV) complex [Ti(nacnac)(CH2tBu)2]+ (1+). (b) Calculated structure of π-ethylene complex 1⋯C2H4. (c) Calculated structure of π-ethylene complex 1@Al2O3-700⋯C2H4. (d) Calculated structure of model Ti(IV) π-ethylene complex 1+⋯C2H4. | ||
For the neutral supported alkyl species in 1@Al2O3-700, the overall energy barriers for C2H4 insertion into the Ti–C bond are ΔH‡298 = 16.7 kcal mol−1 and ΔG‡298 = 29.7 kcal mol−1 and thus both are lower than those found for molecular complex 1. This is consistent with the high polymerization activity of 1@Al2O3-700.
Ethylene insertion into the Ti–C bonds for both 1 and 1@Al2O3-700 follows the formation of π-ethylene complexes. The structures of the complexes 1⋯C2H4 (Fig. 9b) and [(AlsO)Ti(nacnac)(CH2tBu)(C2H4)] (1@Al2O3-700⋯C2H4, Fig. 9c) were obtained through DFT geometry optimizations along the TS imaginary modes (see ESI Part 2.6‡ for details of the optimization). These structures are close to those of the transition states for C2H4 insertion (see Fig. S13‡) with two CH2tBu ligands being trans to each other for complex 1⋯C2H4 and with the CH2tBu ligand being trans to the Ti–N bond of the nacnac ligand for complex 1@Al2O3-700⋯C2H4. In fact, for complex 1⋯C2H4, the calculated enthalpy and Gibbs free energy of formation with respect to the initial reagents (1 + C2H4) are ΔH0298 = 21.4 kcal mol−1 and ΔG0298 = 34.0 kcal mol−1 and very close to those of the TS (the corresponding TS energies are ΔH‡298 = 1.1 kcal mol−1 and ΔG‡298 = −0.3 kcal mol−1 with respect to the 1⋯C2H4 complex). The same is found for the 1@Al2O3-700⋯C2H4 π complex, with its CH2tBu ligand being trans to the Ti–N bond of the nacnac ligand. With ΔH0298 = 16.4 kcal mol−1 and ΔG0298 = 28.2 kcal mol−1 the energy barriers remaining to reach the TS are almost zero and calculated to be ΔH‡298 = 0.3 kcal mol−1 and ΔG‡298 = 1.5 kcal mol−1. Therefore, the energy cost for ethylene polymerization is mostly due to the initial formation of the π complexes followed by an almost barrier-less insertion, which is again in agreement with the observed induction period. The relatively high barrier of formation of the π-ethylene complex 1⋯C2H4 could be overcome by elevated ethylene pressure and increased temperature; this is consistent with the high temperatures and pressures (e.g. 80 °C, 7 bar) required for efficient polymerization on 1 (see Fig. 2a).
For the calculated Ti(IV) model cationic complex [Ti(nacnac)(C2H4)(CH2tBu)2]+ (1+⋯C2H4, Fig. 9d), the TS energy of ethylene insertion with respect to this complex is close to that of its d1 analogue 1⋯C2H4 (ΔH‡298 = 0.9 kcal mol−1 and ΔG‡298 = 2.4 kcal mol−1). Again, as soon as a π-ethylene complex is formed, the ethylene insertion into the Ti–C bond is nearly barrier-less; this behavior occurs in both d1 and d0 complexes. At the same time, while the structure is similar to that of the 1⋯C2H4 complex, the formation energy of 1+⋯C2H4 is much higher than that for 1⋯C2H4 (ΔH0298 = 27.4 kcal mol−1, ΔG0298 = 39.3 kcal mol−1). We thus further examine the π-ethylene complexes 1⋯C2H4, 1+⋯C2H4 and 1@Al2O3-700⋯C2H4via NBO analyses.
The degree of π character in Ti–C bonds of both the π complexes is further shown via the deviation of the corresponding NHO on C from the Ti–C axis, being θNHO–C–Ti = 31.3° for the shortened and 38.5° for the elongated Ti–C bonds of 1⋯C2H4, and θNHO–C–Ti = 26.1° for the Ti–C bond of 1@Al2O3-700⋯C2H4. For the Ti(IV) 1+⋯C2H4 complex, these values are θNHO–C–Ti = 25.6° and 39.7° for the two Ti–C bonds, being close to the ones for 1⋯C2H4. Therefore, a significant increase of the π character is observed for both d1 and model d0 π-ethylene complexes. These values are close to the ones previously found for the cationic Zr(IV) and Ti(IV) d0 complexes [Cp2MEt(C2H4)]+, with θNHO–C–Ti = 40.9° for M = Ti and θNHO–C–Ti = 40.4° for M = Zr.35 Note that all mentioned π-ethylene complexes, while having an enhanced degree of the π character in their Ti–C bonds, provide nearly barrier-less ethylene insertion with rather low calculated or reported35 TS energies. This indicates that ethylene insertion in neutral Ti(III) alkyl species or cationic Ti(IV) and Zr(IV) alkyls depends on the extent of the π character in Ti–C bonds. Therefore, a strong π character is a general reason for the facile ethylene insertion in d1 and d0 metal alkyl complexes after the coordination of C2H4.
It is noteworthy that for Ti(III) d1 complexes 1 and 1@Al2O3-700 “back donation” of the unpaired electron from a SOMO to the π* orbital of coordinated C2H4 is also involved in the insertion process. These two orbitals have a constructive overlap, as revealed by the NBO analysis for complex 1⋯C2H4via the overlap of negative parts of the corresponding natural orbitals (Fig. 10a). This results in some weakening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the C2H4 ligand (C–C distance is 1.344 Å compared to the calculated value of 1.333 Å for free ethylene), together with a population of the π*(C2H4) orbital (see ESI Part 2.7‡). The NBO energetic analysis reveals the stabilization effect, caused by the presence of a π*(C2H4) orbital in 1⋯C2H4, of 30.0 kcal mol−1. At the same time, the stabilization effect, caused by the presence of the π*(C2H4) orbital in the d01+⋯C2H4 complex, is 23.1 kcal mol−1. Therefore, the energy of the back donation of unpaired electron density in 1⋯C2H4 could be estimated through the difference of these two as a stabilization by 6.9 kcal mol−1, consistent with the difference of formation enthalpies ΔH0298 of 1⋯C2H4 and 1+⋯C2H4 of 6.0 kcal mol−1. This indicates that the back donation of the unpaired electron from the SOMO to the π* orbital of C2H4 is the main reason for relative stabilization of the π-ethylene complex 1⋯C2H4 compared to its d0 analogue 1+⋯C2H4. As the TS energies of ethylene insertion after the formation of π-ethylene complexes 1⋯C2H4 and 1+⋯C2H4 differ only by 2.7 kcal mol−1 (ΔG‡298 = −0.3 kcal mol−1 for 1⋯C2H4, compared to ΔG‡298 = 2.4 kcal mol−1 for 1+⋯C2H4), the principal reason for the lowering of the overall TS energy for d1 catalyst 1 by ΔG = 8.0 kcal mol−1 is the back donation of the unpaired electron to the π* orbital of C2H4.
C double bond of the C2H4 ligand (C–C distance is 1.344 Å compared to the calculated value of 1.333 Å for free ethylene), together with a population of the π*(C2H4) orbital (see ESI Part 2.7‡). The NBO energetic analysis reveals the stabilization effect, caused by the presence of a π*(C2H4) orbital in 1⋯C2H4, of 30.0 kcal mol−1. At the same time, the stabilization effect, caused by the presence of the π*(C2H4) orbital in the d01+⋯C2H4 complex, is 23.1 kcal mol−1. Therefore, the energy of the back donation of unpaired electron density in 1⋯C2H4 could be estimated through the difference of these two as a stabilization by 6.9 kcal mol−1, consistent with the difference of formation enthalpies ΔH0298 of 1⋯C2H4 and 1+⋯C2H4 of 6.0 kcal mol−1. This indicates that the back donation of the unpaired electron from the SOMO to the π* orbital of C2H4 is the main reason for relative stabilization of the π-ethylene complex 1⋯C2H4 compared to its d0 analogue 1+⋯C2H4. As the TS energies of ethylene insertion after the formation of π-ethylene complexes 1⋯C2H4 and 1+⋯C2H4 differ only by 2.7 kcal mol−1 (ΔG‡298 = −0.3 kcal mol−1 for 1⋯C2H4, compared to ΔG‡298 = 2.4 kcal mol−1 for 1+⋯C2H4), the principal reason for the lowering of the overall TS energy for d1 catalyst 1 by ΔG = 8.0 kcal mol−1 is the back donation of the unpaired electron to the π* orbital of C2H4.
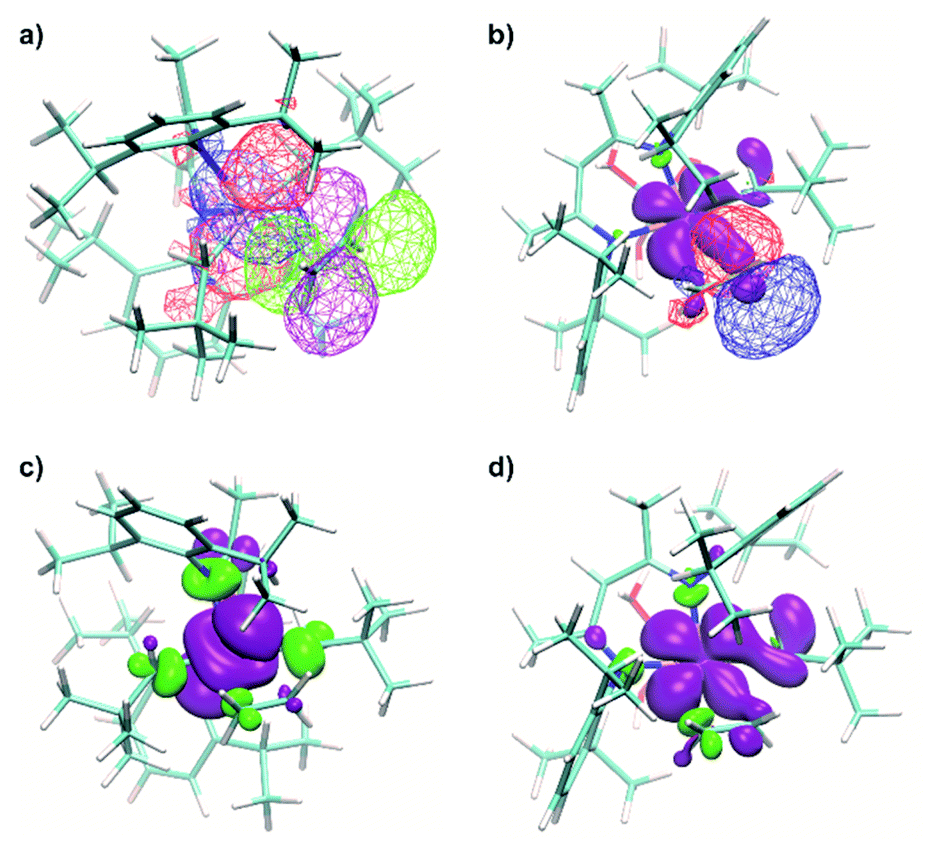 | ||
| Fig. 10 (a) Natural orbital, related to a SOMO (red for positive and blue for negative signs of the wavefunction), together with a natural orbital, related to π*(C2H4) (purple for positive and green for negative signs of the wavefunction) of 1⋯C2H4. (b) Calculated spin density distribution (purple for positive and green for negative, contour levels 0.2%), together with the natural orbital, related to π*(C2H4) (red for positive and blue for negative signs of the wavefunction), of the 1@Al2O3-700⋯C2H4 complex. (c) Calculated spin density distribution (purple for positive and green for negative signs, contour levels 0.1%) for the TS of ethylene insertion for 1. (d) Calculated spin density distribution (purple for positive and green for negative signs, contour levels 0.1%) for the TS of ethylene insertion for 1@Al2O3-700. The degree of delocalization of the unpaired electron could be estimated from the spin density distribution in the first coordination sphere of Ti, including ethylene. | ||
An even higher degree of back donation to π*(C2H4) is observed for the 1@Al2O3-700⋯C2H4 complex, since a strong spin density transfer from the initial SOMO to π*(C2H4) is revealed by the calculated spin density distribution (Fig. 10b). Similar to the molecular 1⋯C2H4 complex, this leads to a weakening of the CC double bond of the C2H4 molecule (C–C distance is 1.364 Å) and to the appearance of a bonding interaction between Ti in 1@Al2O3-700 and C2H4. In fact, NBO analysis, being one of the possible ways of representation of the electronic structure of 1@Al2O3-700⋯C2H4, shows a breaking of the π system of ethylene, followed by formation of a bonding set of natural orbitals, corresponding to a Ti–C(C2H4) bond (Fig. S14 and Table S2‡), and a partially occupied natural lone pair on the other carbon atom of C2H4, derived from π*(C2H4) (Fig. 10b). This indicates more favorable coordination of C2H4 to 1@Al2O3-700 where the stronger back donation in 1@Al2O3-700⋯C2H4 is caused by better orbital overlap between the initial SOMO and π*(C2H4). The better overlap between the SOMO and π*(C2H4) in the supported catalyst is mostly caused by the difference in geometry of the π-ethylene complexes induced by replacing one of the strong σ-donor alkyl ligands (CH2tBu) by a weaker O anionic surface ligand OAls. This is consistent with the lower formation energy of 1@Al2O3-700⋯C2H4 of ΔH0298 = 16.4 kcal mol−1 compared to ΔH0298 = 21.4 kcal mol−1 for complex 1 and with the ability of 1@Al2O3-700 to catalyze ethylene polymerization under milder conditions.
In fact, the unpaired electron in the 1@Al2O3-700⋯C2H4 complex appears to be strongly delocalized between the Ti d orbital, π*(C2H4) and σ*(Ti–CCH2tBu), as revealed by the spin density distribution (Fig. 10b) and the occupancies of the related natural orbitals (see Table S2‡). The same delocalization is also found in the structure of the TS of ethylene insertion for both 1 and 1@Al2O3-700, as revealed by the calculated spin density distributions (Fig. 10c and d). This delocalization appears to be stronger for the TS for 1@Al2O3-700 (Fig. 10d), while being weaker for the TS for 1 (Fig. 10c). It appears that the unpaired electron, while weakening the CC double bond of C2H4 and facilitating the formation of a Ti–C(C2H4) bond due to its presence at π*(C2H4), also favors the cleavage of the Ti–C bond of the CH2tBu ligand by occupation of the σ*(Ti–C) orbital before ethylene insertion. These factors together facilitate the C2H4 insertion into the Ti–C bond, in addition to the previously mentioned factor of its π character, which is consistent with a lowered energy of the TS of ethylene insertion in 1⋯C2H4, compared to 1+⋯C2H4, by ΔG = 2.7 kcal mol−1. We, therefore, propose that the mechanism of ethylene polymerization for 1 and 1@Al2O3-700 catalysts involves the delocalization of the unpaired electron in the TS (Scheme 2).
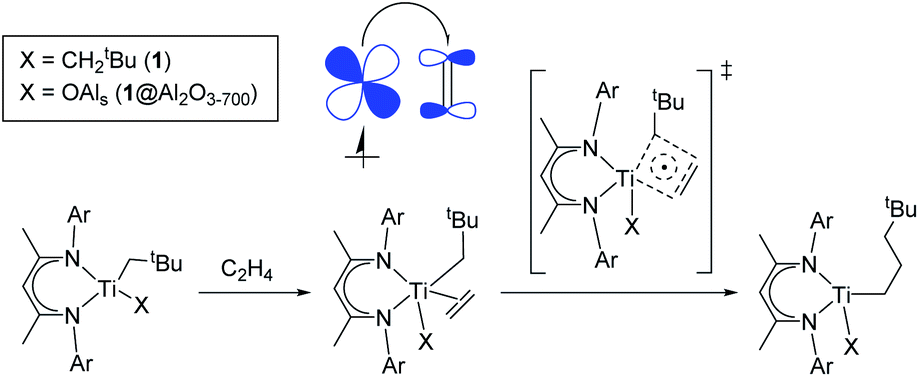 | ||
| Scheme 2 “Augmented” Cossee–Arlman mechanism of ethylene polymerization for 1 and 1@Al2O3-700. A partial electron transfer process from the SOMO to π*(C2H4) (“back donation”) is shown in the molecular orbital picture. | ||
In general, the mechanism of ethylene insertion in d1 Ti(III) alkyl complexes 1 and 1@Al2O3-700 is determined by two key factors: the π character in Ti–C bonds of (CH2tBu) ligands and the back donation of the unpaired electron. While the presence of the π character facilitates the insertion of ethylene into Ti–C bonds after its coordination, making the insertion in π-ethylene complexes nearly barrier-less at the TS, the back donation significantly lowers the formation energies of the π-ethylene complexes, which facilitates the overall reaction of C2H4 insertion. The delocalization of the unpaired electron in the TS structure, being noticeable for the systems with a high degree of back donation (i.e.1@Al2O3-700⋯C2H4), also has an effect on this reaction, favoring the cleavage of Ti–C and CC bonds and slightly lowering the TS barrier. We denote this process (Scheme 2) as an “augmented” Cossee–Arlman mechanism, being essentially a [2σ + 2π + d1] cycloaddition involving a partially alkylidenic σ(Ti–C) bond and a π(C2H4) bond together with a delocalized d1 electron.
The described delocalization of the unpaired electron is likely an important feature in d1 systems able to polymerize olefins, with a degree of the back donation (electron density transfer) that depends on the overlap of the SOMO and π*(C2H4), which in turn depends on the geometry of the system. For instance, a higher polymerization activity towards styrene polymerization was observed experimentally for Cp*Ti(OCH3)2/MAO and Cp2TiCl/MAO catalytic systems compared to Cp*Ti(OCH3)3/MAO and Cp2TiCl2/MAO, respectively.21 It is likely that the Ti(III) species, active towards styrene polymerization, show better performance compared to similar Ti(IV) systems due to a strong back donation (electron transfer), favored by the aromatic system of styrene. Therefore, under the same polymerization conditions, a d1 catalyst may be more active than a d0 catalyst of a similar structure. This finding also further suggests that Ti(III) alkyl species have competent electronic structures to act as efficient polymerization catalysts and may indeed be active species in the classical Ziegler–Natta heterogeneous catalysts.
Conclusions
In this work, we report the polymerization activity of molecular and the corresponding alumina-supported well-defined Ti(III) neutral alkyl species prepared via surface organometallic chemistry. Both of them were characterized and studied in detail by pulse EPR spectroscopy, combined with DFT calculations. This approach enabled us to identify the prevalent conformation of the molecular complex [Ti(nacnac)(CH2tBu)2] in a frozen toluene solution and to reveal the structure of the alumina-supported species that correspond predominantly to a neutral Ti(III) alkyl compound, i.e. [(AlsO)Ti(nacnac)(CH2tBu)]. To the best of our knowledge, these are the first examples of well-defined Ti(III) alkyl species able to efficiently polymerize ethylene, producing ultra-high molecular weight polyethylene. The ethylene insertion into the Ti(III)–C bond of [Ti(nacnac)(CH2tBu)2] was further evidenced by EPR hyperfine spectroscopy (CHEESY-detected NMR), using isotope-labeled Ti(nacnac)(13CH2tBu)2 in contact with C2H4.These Ti(III)-based polymerization pre-catalysts display α-agostic C–H bonds in their (CH2tBu) ligands, supporting the presence of π character in the corresponding metal–carbon bonds.35 Such a π character was further supported by DFT calculations via NBO analysis. It is noteworthy that the presence of the half-filled d1 Ti orbital does not prevent α-agostic C–H bonding. After coordination of C2H4, the degree of π character in the Ti–C bonds of (CH2tBu) ligands is significantly increased, which allows a nearly barrier-less insertion of C2H4 into the Ti–C bonds. Hence, the slow step is olefin coordination, consistent with the need for high pressure to carry out this reaction and with the observation of an induction period. The back donating interaction (electron transfer) between the SOMO and the π* orbital of C2H4 results in a significant lowering of the formation energies of π-ethylene complexes, which facilitates an overall reaction of ethylene insertion in these Ti(III) systems. Due to the back donation, the unpaired electron could be delocalized between the Ti d orbital and π*(C2H4) and σ*(Ti–C) orbitals in both π-ethylene complexes and transition states, which also lowers the energy barriers for ethylene insertion. All these factors, which combine to give an “augmented” Cossee–Arlman mechanism, facilitate the overall reaction of C2H4 insertion into the Ti–C bond, making the ethylene polymerization in d1 metal complexes potentially more efficient than that in d0 complexes of a similar structure under the same conditions.
This study shows that neutral d1 Ti alkyl complexes are competent in ethylene polymerization, being favored by a combination of the π character in the Ti–C bonds and the back donation of the unpaired electron. These findings lend further support to the notion that d1 Ti-alkyls are possible active sites in the heterogeneous Ziegler–Natta polymerization catalysts.
Author contributions
A. A. and F. A. contributed equally. A. A. carried out CW EPR and polymerization tests for 1, HYSCORE measurements, DFT computations, NBO analysis and wrote the manuscript. F. A. performed synthesis and characterization of 1, 1* and 1@Al2O3-700 and polymerization tests for 1@Al2O3-700. N. W. conducted CHEESY-detected NMR measurements. D. K. supervised the project. S. K. proposed the Ti(nacnac)(CH2tBu)2 complex as a stable Ti(III) alkyl compound to be tested for ethylene polymerization activity. G. J. and C. C. coordinated the project and provided guidance. All the authors revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. was supported by a SNF—ANR grant (Mr CAT 2-77275-15). We acknowledge David Trummer (ETHZ) and Gina Noh (ETHZ) for their assistance with the ethylene polymerization experiments; John Severn and Tigran Margossian (DSM) for SEC of the produced PE; Frank Krumeich (ETHZ) for SEM of 1@Al2O3-700; Christopher P. Gordon (ETHZ) and Erwin Lam (ETHZ) for discussions.Notes and references
- K. Ziegler, Bel. Pat., 533 362, 1953 CrossRef CAS; K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- G. Natta, P. Pino and G. Mazzanti, Ita. Pat., 537 425, 1954; G. Natta and F. Danusso, Stereoregular Polymers and Stereospecific Polymerizations, Pergamon Press, Oxford, 1967 Search PubMed.
- H. Sinn, W. Kaminsky, H.-J. Vollmer and R. Woldt, Angew. Chem., 1980, 92, 396–402 CrossRef CAS.
- R. F. Jordan, W. E. Dasher and S. F. Echols, J. Am. Chem. Soc., 1986, 108, 1718–1719 CrossRef CAS.
- W. Kaminsky, J. Chem. Soc., 1998, 9, 1413–1418 Search PubMed.
- S. Mecking, Macromol. Rapid Commun., 1999, 20, 139–143 CrossRef CAS.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS.
- T. J. Marks, Acc. Chem. Res., 1992, 25, 57–65 CrossRef CAS.
- O. Olabisi, M. Atiqullah and W. Kaminsky, J. Macromol. Sci., Polym. Rev., 1997, 37, 519–554 CrossRef.
- R. F. Jordan, J. Chem. Educ., 1988, 65, 285–289 CrossRef CAS.
- R. A. Collins, A. F. Russell, R. T. W. Scott, R. Bernardo, G. H. J. van Doremaele, A. Berthoud and P. Mountford, Organometallics, 2017, 36, 2167–2181 CrossRef CAS.
- Yu. V. Kissin, J. Catal., 2012, 292, 188–200 CrossRef CAS.
- E. Magni and G. Somorjai, Appl. Surf. Sci., 1995, 89, 187–195 CrossRef CAS.
- E. Magni and G. Somorjai, Catal. Lett., 1995, 35, 205–214 CrossRef CAS.
- T. I. Korányi, E. Magni and G. A. Somorjai, Top. Catal., 1999, 7, 179–185 CrossRef.
- T. Risse, J. Schmidt, H. Hamann and H.-J. Freund, Angew. Chem., Int. Ed., 2002, 41, 1517–1520 CrossRef CAS.
- S. Maurelli, E. Morra, S. Van Doorslaer, V. Busico and M. Chiesa, Phys. Chem. Chem. Phys., 2014, 16, 19625–19633 RSC.
- E. Morra, E. Giamello, S. Van Doorslaer, G. Antinucci, M. D'Amore, V. Busico and M. Chiesa, Angew. Chem., Int. Ed., 2015, 54, 4857–4860 CrossRef CAS.
- E. P. Moore, in Polypropylene handbook: polymerization, characterization, properties, processing, applications, Hanser-Gardner, Cincinnati, 1996, ch. 2, pp. 61–73 Search PubMed.
- G. Cecchin, G. Morini and F. Piemontesi, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Hoboken, 2000, vol. 26, pp. 502–554 Search PubMed.
- J. Schellenberg and N. Tomotsu, Prog. Polym. Sci., 2002, 27, 1925–1982 CrossRef CAS.
- F. Allouche, D. Klose, C. P. Gordon, A. Ashuiev, M. Wörle, V. Kalendra, V. Mougel, C. Copéret and G. Jeschke, Angew. Chem., Int. Ed., 2018, 57, 14533–14537 CrossRef CAS.
- N. Bahri-Laleh, M. Nekoomanesh-Haghighi and S. A. Mirmohammadi, J. Organomet. Chem., 2012, 719, 74–79 CrossRef CAS.
- P. H. M. Budzelaar, A. B. van Oort and A. G. Orpen, Eur. J. Inorg. Chem., 1998, 1998, 1485–1494 CrossRef.
- F. Basuli, B. C. Bailey, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2003, 125, 6052–6053 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef.
- D. G. H. Ballard, J. Polym. Sci., 1975, 13, 2191–2212 CAS.
- J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS.
- F. Allouche, K. W. Chan, A. Fedorov, R. A. Andersen and C. Copéret, Angew. Chem., Int. Ed., 2018, 57, 3431–3434 CrossRef CAS.
- M. Jezequel, V. Dufaud, M. J. Ruiz-Garcia, F. Carrillo-Hermosilla, U. Neugebauer, G. P. Niccolai, F. Lefebvre, F. Bayard, J. Corker, S. Fiddy, J. Evans, J.-P. Broyer, J. Malinge and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 3520–3540 CrossRef CAS.
- J. Joubert, F. Delbecq, P. Sautet, E. Le Roux, M. Taoufik, C. Thieuleux, F. Blanc, C. Coperet, J. Thivolle-Cazat and J.-M. Basset, J. Am. Chem. Soc., 2006, 128, 9157–9169 CrossRef CAS.
- M. M. Stalzer, M. Delferro and T. J. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS.
- B. D. Culver, H. Tafazolian and M. P. Conley, Organometallics, 2018, 37, 1001–1006 CrossRef.
- C. P. Nicholas, H. Ahn and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 4325–4331 CrossRef CAS.
- C. P. Gordon, S. Shirase, K. Yamamoto, R. A. Andersen, O. Eisenstein and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5867–5876 CrossRef.
- C. P. Gordon, C. Raynaud, R. A. Andersen, C. Copéret and O. Eisenstein, Acc. Chem. Res., 2019, 52, 2278–2289 CrossRef CAS.
- B. Rhers, A. Salameh, A. Baudouin, E. A. Quadrelli, M. Taoufik, C. Copéret, F. Lefebvre, J.-M. Basset, X. Solans-Monfort, O. Eisenstein, W. W. Lukens, L. P. H. Lopez, A. Sinha and R. R. Schrock, Organometallics, 2006, 25, 3554–3557 CrossRef CAS.
- S. M. Kurtz, in The UHMWPE handbook: ultra-high molecular weight polyethylene in total joint replacement, Academic Press, Cambridge, 2004, ch. 1, pp. 1–12 Search PubMed.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- J. P. Perdew and M. Ernzerhof, J. Chem. Phys., 1996, 105(22), 9982–9985 CrossRef CAS; C. Adamo and V. Barone, J. Chem. Phys., 1991, 110, 6158–6170 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 105104 CrossRef CAS; S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- N. Rega, M. Cossi and V. Barone, J. Chem. Phys., 1996, 105, 11060–11067 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS.
- K. H. Dahmen, D. Hedden, R. L. Burwell Jr and T. J. Marks, Langmuir, 1988, 4, 1212–1214 CrossRef CAS.
- G. Tosin, M. Delgado, A. Baudouin, C. Catherine, C. C. Santini, F. Bayard and J.-M. Basset, Organometallics, 2010, 29, 1312–1322 CrossRef CAS.
- G. Rao, A. B. Altman, A. C. Brown, L. Tao, T. A. Stich, J. Arnold and R. D. Britt, Inorg. Chem., 2019, 58, 7978–7988 CrossRef.
- S. Stoll, C. Calle, G. Mitrikas and A. Schweiger, J. Magn. Reson., 2005, 177, 93–101 CrossRef CAS.
- N. Wili and G. Jeschke, J. Magn. Reson., 2018, 289, 26–34 CrossRef CAS.
- G. Feher, Phys. Rev., 1956, 103, 834 CrossRef CAS.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395–408 CrossRef CAS.
- W. W. Lukens Jr, M. R. Smith III and R. A. Andersen, J. Am. Chem. Soc., 1996, 118, 1719–1728 CrossRef.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- E. D. Glendening, F. Weinhold and C. R. Landis, J. Comput. Chem., 2019, 40, 2234–2241 CAS.
- C. P. Gordon, K. Yamamoto, K. Searles, S. Shirase, R. A. Andersen, O. Eisenstein and C. Copéret, Chem. Sci., 2018, 9, 1912–1918 RSC.
- M. Besora, P. Vidossich, A. Lledós, G. Ujaque and F. Maseras, J. Phys. Chem. A, 2018, 122, 1392–1399 CrossRef CAS; L. Falivene, V. Barone and G. Talarico, Mol. Catal., 2018, 452, 138–144 CrossRef.
- G. Lanza, I. L. Fragalà and T. J. Marks, Organometallics, 2002, 21, 5594–5612 CrossRef CAS.
Footnotes |
| † We dedicate this work to the memory of Prof. Richard A. Andersen. |
| ‡ Electronic supplementary information (ESI) available: Details of experimental procedures and additional experimental data. See DOI: 10.1039/d0sc04436a |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |