DOI:
10.1039/D0SC04444J
(Edge Article)
Chem. Sci., 2021,
12, 4570-4581
Short-chain reactive probes as tools to unravel the Pseudomonas aeruginosa quorum sensing regulon†
Received
13th August 2020
, Accepted 28th January 2021
First published on 28th January 2021
Abstract
In recent years, the world has seen a troubling increase in antibiotic resistance among bacterial pathogens. In order to provide alternative strategies to combat bacterial infections, it is crucial deepen our understanding into the mechanisms that pathogens use to thrive in complex environments. Most bacteria use sophisticated chemical communication systems to sense their population density and coordinate gene expression in a collective manner, a process that is termed “quorum sensing” (QS). The human pathogen Pseudomonas aeruginosa uses several small molecules to regulate QS, and one of them is N-butyryl-L-homoserine lactone (C4-HSL). Using an activity-based protein profiling (ABPP) strategy, we designed biomimetic probes with a photoreactive group and a ‘click’ tag as an analytical handle. Using these probes, we have identified previously uncharacterized proteins that are part of the P. aeruginosa QS network, and we uncovered an additional role for this natural autoinducer in the virulence regulon of P. aeruginosa, through its interaction with PhzB1/2 that results in inhibition of pyocyanin production.
Introduction
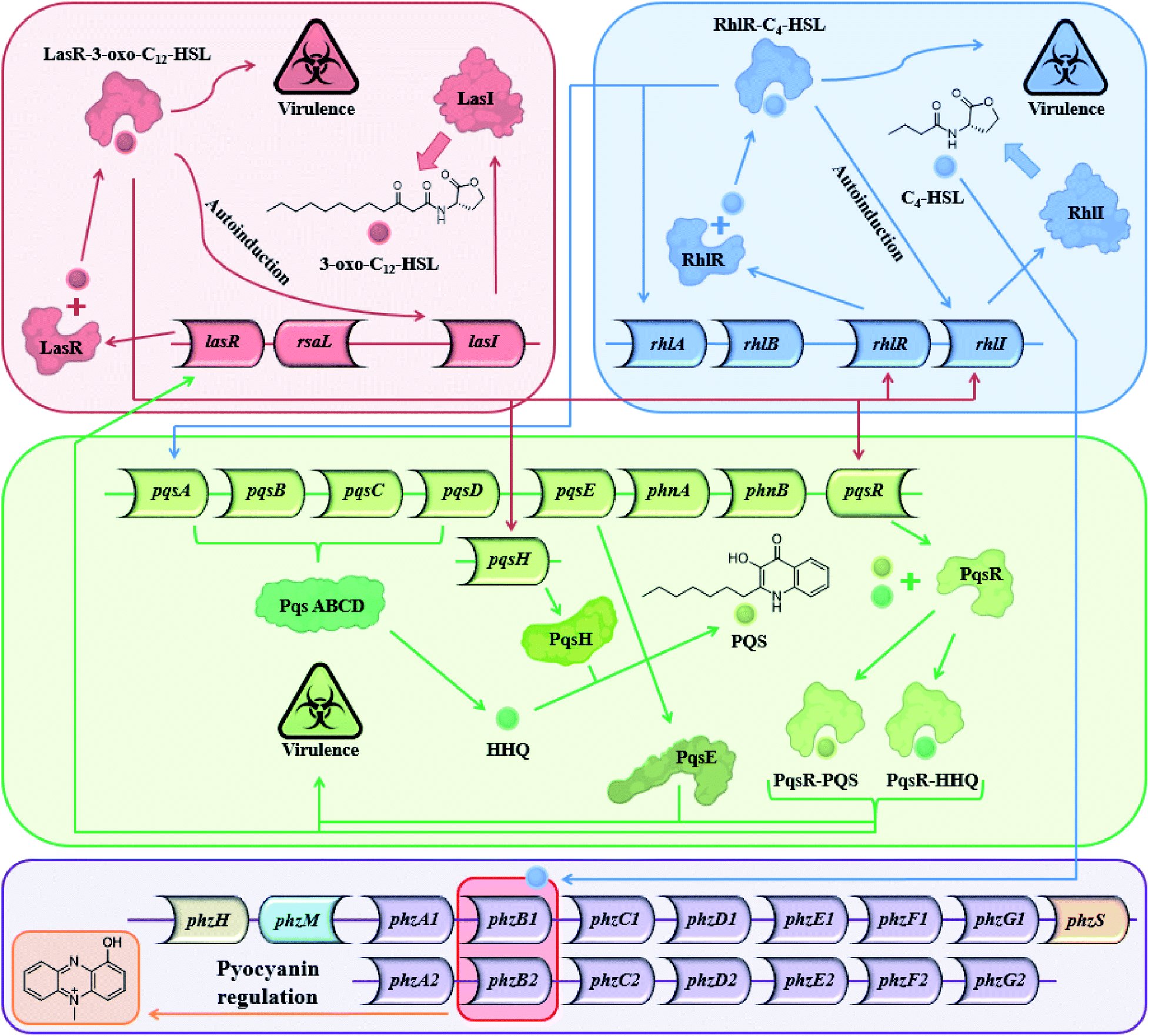
Most Gram-positive and Gram-negative bacteria use a type of cell to cell communication called QS in which bacteria produce, secrete and respond to small signaling molecules called autoinducers.1–3 These compounds help bacteria sense the bacterial population density during the growth of the culture. With the increase in the bacterial population, the concentration of the autoinducers increases, and at a certain population threshold (quorum), the bacteria start to act collectively and begin to express QS-regulated genes. The ensuing phenotypic changes help the bacteria to adapt to environmental challenges.4,5 QS is involved in the regulation of virulence factors, development of genetic competence, transfer of conjugative plasmids, sporulation, antimicrobial peptides, secondary metabolite synthesis and symbiosis with animals, bacteria and plants,6 both in Gram-positive and Gram-negative bacteria.5 Among the QS-regulated factors one can find bioluminescence in Vibrio fischeri, pyocyanin and biofilm production in Pseudomonas aeruginosa,7–9 pneumolysin and polysaccharide capsule production in Streptococcus pneumonia,10etc. In P. aeruginosa, a Gram-negative pathogen, the QS systems are regulated by two LuxI–LuxR homolog systems,11 the long-chain AHL-based, LasI–LasR system, and the short-chain AHL-based RhlI–RhlR system. In addition to the AHL-based systems, P. aeruginosa uses the PQS system, and in recent years, additional signals have been proposed to contribute to the complexity of the large cellular network of QS systems in P. aeruginosa.12 The Las and the Rhl systems regulate P. aeruginosa gene expression and are organized hierarchically. In the Las system, the LasR receptor protein responds to N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL) autoinducer which is synthesized by the LasI synthase protein. The Rhl system responds to N-butyryl-L-homoserine lactone (C4-HSL) autoinducer, which is synthesized by the RhlI synthase protein. C4-HSL is thought to bind to the RhlR receptor protein. The LasR–3-oxo-C12-HSL complex regulates the expression of lasI and induces a positive feedback loop through the up-regulation of 3-oxo-C12-HSL synthesis by the Las system. In addition, the expression of rhlI and rhlR is also regulated by the LasR–3-oxo-C12-HSL complex, placing the Las system higher up the hierarchy compared with the Rhl system.13 However, under certain conditions, the Las system is secondary to the Rhl system.14 The PQS (2-heptyl-3-hydroxy-4-quinolone) signal system is dependent on, and regulated by the balanced production of 3-oxo-C12-HSL and C4-HSL, and is thus linked with the Las and Rhl QS systems.15 In addition, PQS itself plays a role in the regulation and expression of virulence genes. From a biochemical perspective, the Rhl QS sub-system is the least well-understood of the three main QS signaling pathways and here we show another possible path for this system (Fig. 1).
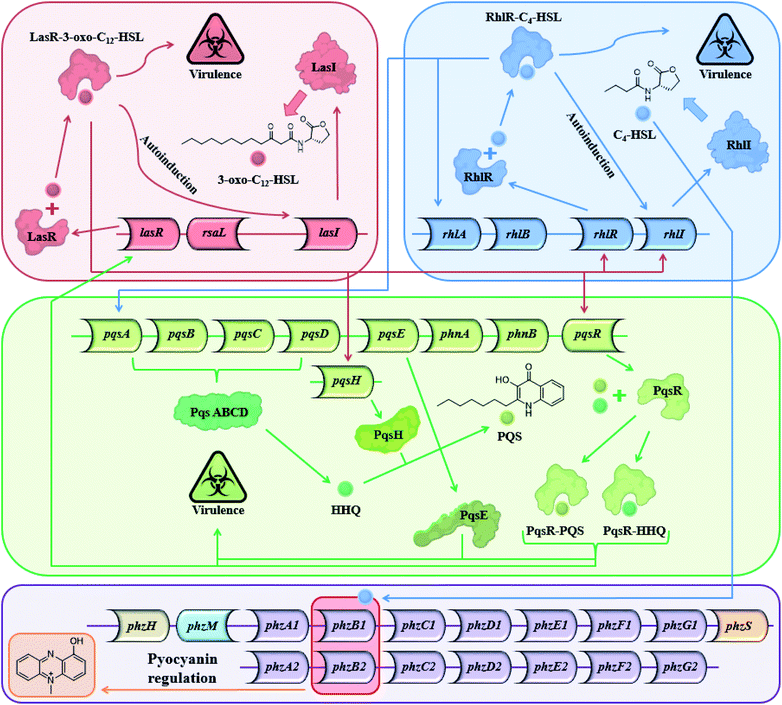 |
| | Fig. 1 The three main QS systems of P. aeruginosa and their signal molecules: N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL), N-butyryl-L-homoserine lactone (C4-HSL), and 2-heptyl-3-hydroxy-4-quinolone (PQS) signals which correspond to the Las, Rhl and PQS systems respectively – and a potential connection to the pyocyanin regulon. | |
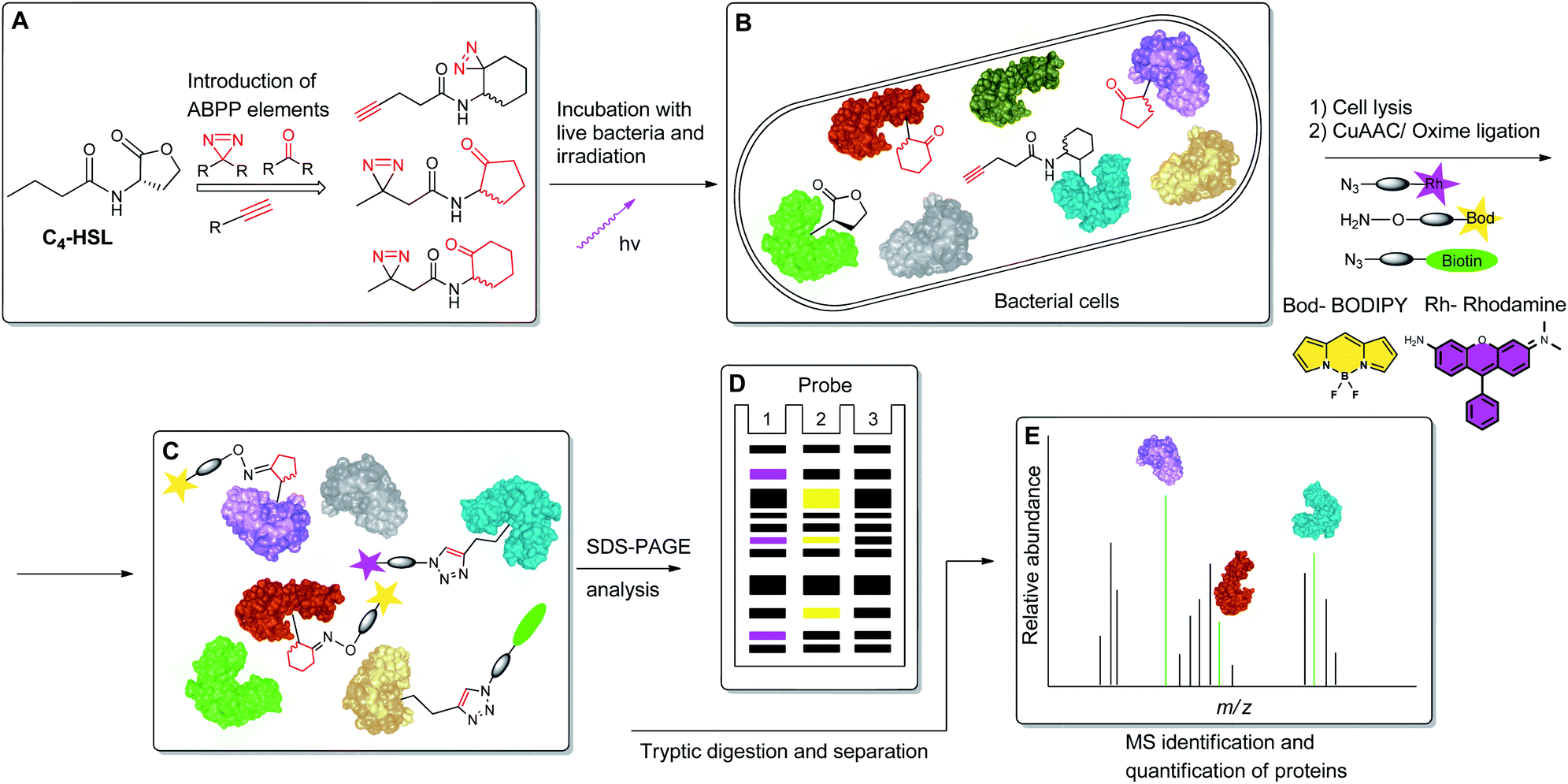
To begin to access the C4-HSL interactome, we used a powerful method termed Activity-Based Protein Profiling (ABPP), to help gain a deeper understanding of how C4-HSL impacts on cell physiology. This method has become a major tool for the identification and functional characterization of individual proteins in complex proteomes.16,17 We have synthesized a series of probes based on the scaffold of C4-HSL and introduced to the molecular structure a diazirine photoreactive moiety and ketone or alkyne analytical handles for oxime ligation or copper-mediated azide–alkyne coupling (CuAAC) reactions, respectively. We evaluated the biomimetic activity of these analogues with reporter strains, and with the use of the most suitable probe, we proceeded to label intact bacteria. After cell lysis and reaction with rhodamine azide, we evaluated the labeled proteome by SDS-PAGE. Finally, using a biotinylated tag, we captured the C4-HSL interactome by affinity chromatography and analyzed the labeled proteins by mass spectrometry. The resulting positive hits were evaluated through assays with defined transposon mutants and recombinant proteins, revealing a previously unknown connection between several proteins and the P. aeruginosa virulence regulon18 (Fig. 2).
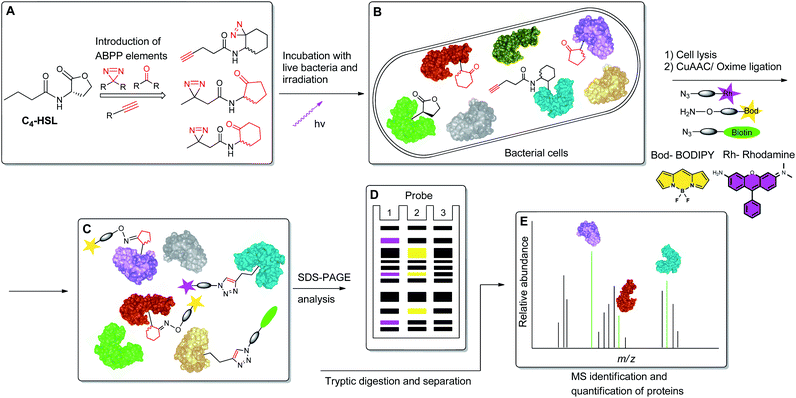 |
| | Fig. 2 Schematic representation of the ABPP methodology used in this study. (A) Structure-based modification of C4-HSL and incorporation of ABPP elements. (B) Incubation of bacteria with probes, followed by irradiation. (C) Application of CuAAC and oxime ligation reactions. (D) SDS-PAGE separation analysis. (E) Purification and LC-MS/MS analysis of target proteins. | |
Experimental
General methods
For flash chromatography, Sigma-Aldrich silica gel, high-purity grade, pore size 60 Å, 230–400 mesh particle size, 40–63 μm was used. TLC was performed using Merck TLC silica gel 60 F254 glass plates (20 × 20 cm), detected by UV (λ = 254 nm), or stained using potassium permanganate and heated at 150 °C. All compounds MS analyses were performed with LCQ Fleet mass spectrometer (Thermo Fisher Scientific) with an ESI source. Spectra were collected in positive ion mode and analyzed by Xcalibur software (Thermo Fisher Scientific). For LC-MS (ESI) analyses, a Surveyor Plus HPLC System (Thermo Fisher Scientific) was used, equipped with a Luna 5 μm C18(2), 100 Å, (150 × 4.6 mm) column at a flow rate of 0.5 mL min−1, using a mobile phase gradient of H2O containing 0.1% FA (solvent A) and acetonitrile containing 0.1% FA (solvent B). 1H and 13C NMR spectra were recorded on a Bruker DPX-400 MHz spectrometer. Chemical shifts are reported in δ ppm relative to TMS or residual solvent peak. Multiplicities were reported as singlet (s), broad singlet (br s), doublet (d), doublet of doublet (dd), doublet of doublet of doublet (ddd), doublet of triplet of doublet (dtd), triplet (t), triplet of doublet (td), quintet (quint), sextet (sext), multiplet (m) and broad multiplet (br m). NMR data were analyzed using MestReNova 6.0.2. Optical density and luminescence measurements were performed on a Varioskan Flash Spectral Scanning Multimode Plate Reader (Thermo Fisher Scientific) and SpectraMax M2 Microplate reader (Molecular Devices). EC50 values were calculated using GraphPad prism 9.0.0 and confirmed as reported.19 Protein concentrations were determined with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). UV irradiation was performed on TFX-20 LM 365/312 nm UV table (100% intensity). Cell lysates were prepared by sonication with a Sonics VCX-750 Vibra-Cell Ultra Sonic processor with a 3 mm probe. SDS-PAGE was separated with BIO-RAD Mini-PROTEAN Tetra system and PowerPac Basic power supply. Fluorescence readouts were obtained using an ImageQuant Fuji LAS4000 scanner. Quantitative ABPP analysis was performed on LTQ Orbitrap XL ETD (Thermo Fisher Scientific)/NanoLC-2D (Eksigent) and analyzed with MaxQuant 1.6.0.16.
Materials
All chemicals were obtained from Sigma Aldrich, Acros organics, Alfa Aesar, Bio-Lab, J.T. Baker, and Fluka unless otherwise noted, and were used without additional purification. Solvents were used as received from DAEJUNG, Bio-Lab, and J.T. Baker.
Strain and cell culture conditions
All bacterial strains were cultured from cryo stocks (maintained in 50% glycerol at −80 °C) and grown in LB (BACTO™) medium or LB agar (BACTO™). Wild-type P. aeruginosa strain PAO1 was grown in LB medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl) for liquid bacterial cultures, and on LB agar medium (LB with 1.5% agar) for solid bacterial cultures. To monitor Rhl signaling activity, P. aeruginosa double mutant, PAO-JP2 (ΔlasI/rhlI) carrying pKD-rhlA (encoding the rhlA promoter fused to the luxCDABE luminescence reporter gene20) was grown in LB medium and on LB agar supplemented with 300 μg mL−1 of trimethoprim (Sigma). P. aeruginosa transposon mutants, PW4328 and PW4329, were obtained from the University of Washington PAO1 transposon mutant library21,22 and grown in LB medium and on LB agar supplemented with 5 μg mL−1 of tetracycline (Fluka). Verification of transposon insertion was done via PCR as described (http://www.gs.washington.edu/labs/manoil/libraryindex.htm). Escherichia coli BL21 (DE3) carrying pRARE2 and phzB1 cloned into the expression vector, pET-19m, was grown in LB medium supplemented with 25 μg mL−1 of chloramphenicol (Sigma Aldrich) and 50 μg mL−1 carbenicillin disodium salt (CHEM-IMPEX INT'L INC).
Growth and luminescent activity assays
The P. aeruginosa Rhl reporter strain (PAO-JP2 pKD-rhlA), was inoculated into 20 mL of fresh LB medium in a 50 mL PP sterile conical centrifuge tube (Greiner Bio-One) and incubated aerobically at 37 °C with 180 rpm shaking overnight. The cultures were grown to late stationary phase and were diluted into fresh LB to OD600 of 0.05. Flat-bottomed, white polystyrene, 96-well, microtiter plates (Greiner Bio-One) were prepared with the desired range of probe concentrations (0–1 mM) from 10 mM DMSO stock solutions and the diluted culture was added to obtain a bacterial OD600 of 0.025. The plates were then incubated in a Varioskan Plate Reader for a period of 15 h at 37 °C with good aeration (vigorous agitation). During this time, luminescence and OD600 measurements were made at 20 min intervals. The luminescence and OD600 data were plotted against probe concentration, and EC50 values were calculated. For agonist control experiments, a final concentration of 10 μM C4-HSL was used.
Pyocyanin production assays
Triplicate samples of wild-type P. aeruginosa strain PAO1, and the otherwise isogenic phzB2 transposon mutants, PW4328 and PW4329, were incubated separately with good aeration in 50 mL PP sterile conical centrifuge tubes, each containing 15 mL of LB medium. The bacteria were grown overnight at 37 °C with shaking at 180 rpm to late stationary phase. Following this, 10 μL of the overnight-grown cultures (OD600 ≈ 1.0) were inoculated in triplicate into 25 mL LB in 250 mL autoclaved Erlenmeyer flasks. Various concentrations of C4-HSL, (0–100 μM, in DMSO) were added to each flask. The highest concentration (1% v/v) of DMSO (with no C4-HSL) employed in the dose–response experiment, was added to the control flasks. The bacteria were incubated aerobically for 24 h at 37 °C with shaking at 180 rpm. OD600 measurements were taken and pyocyanin was extracted directly from bacterial cultures as follows: five mL of bacterial culture and 3 mL of chloroform were added to a 15 mL PP sterile conical centrifuge tube (Greiner Bio-One). The mixture was agitated to extract the pyocyanin and clarified by centrifugation at 4 °C, 3220 × g for 10 min. The organic and aqueous phases were separated, and 2 mL of the greenish-blue chloroform layer was transferred to a new 5 mL glass test tube. This was mixed with 0.3 mL HCl (0.2 M) which gave the pyocyanin-containing aqueous layer a pinkish color. An aliquot (150 μL) of the aqueous layer from each sample was transferred to a TC Microwell 96F Nunclon D SI plate and the absorption was measured at 520 nm. HCl (0.2 M) was used as blank and subtracted from the absorption of the extracted pyocyanin samples.
His6-TEV-PhzB1 expression
A single bacterial colony of E. coli BL21 (DE3) carrying pRARE2 and phzB1::pET-19m strain was inoculated into 10 mL of fresh LB medium supplemented with 25 μg mL−1 chloramphenicol (Sigma Aldrich) and 50 μg mL−1 carbenicillin disodium salt (CHEM-IMPEX INT'L INC) in a 50 mL PP sterile conical centrifuge tube. The culture was incubated with good aeration at 37 °C overnight. Five mL of this starter culture was used to inoculate 800 mL of fresh LB medium supplemented with 25 μg mL−1 chloramphenicol and 50 μg mL−1 carbenicillin disodium salt in a 2 L sterile Erlenmeyer flask, and the culture was once again grown at 37 °C with good aeration on a rotary shaker (180 rpm) until OD600 reached 0.5–1. Protein expression was induced by addition of IPTG (ACROS ORGANICS) to a final concentration of 1 mM. Following induction, the temperature was lowered to 16 °C and growth (with vigorous aeration, shaking at 180 rpm) was continued overnight. The bacterial pellet expressing recombinant protein was collected by sedimentation (3220 × g, 15 min, 4 °C, or room temperature). The supernatant was discarded and the cell pellet was washed with ice-cold 1× phosphate-buffered saline solution (PBS). The washed cell pellet was resuspended in ice-cold 5 mL of lysis buffer (50 mM Tris–HCl, 300 mM NaCl, 10 mM imidazole, 2 μL mL−1 protease inhibitor cocktail (Merck), pH 8.0) and transferred to 15 mL PP sterile conical centrifuge tube. The bacterial suspension was sonicated for 7.5 min in fifteen 30 s pulses with 60 s rest between runs at 40% amplitude setting while maintaining the sample on ice. The cell debris was removed by sedimentation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 15 min, 4 °C, or room temperature), and the lysate was filtered through a 0.22 μm membrane filter (Millex®).
000 × g, 15 min, 4 °C, or room temperature), and the lysate was filtered through a 0.22 μm membrane filter (Millex®).
Purification of His6-TEV-PhzB1 protein using immobilized metal affinity chromatography (IMAC)
An aliquot (1.6–2 mL) of Ni-NTA agarose slurry (QIAGEN) was added to a 1 × 10 cm glass column (Glass Econo-Column®) and the resin was allowed to settle for a few minutes. The column was equilibrated with 10 resin-bed volumes of lysis buffer. The cleared lysate was applied to the equilibrated column and the flow rate was adjusted to 0.5–1 mL min−1. The column was rinsed with wash buffer (50 mM Tris–HCl, 300 mM NaCl, 20 mM imidazole, pH 8.0) until the eluate A280 had returned to baseline. The bound protein was eluted in elution buffer (50 mM Tris–HCl, 300 mM NaCl, 250 mM imidazole, glycerol (5% v/v), 5 mM β-mercaptoethanol, pH 8.0) and 1 mL fractions were collected. The A280 of the fractions was measured. Fractions containing protein were pooled and analyzed by SDS-PAGE (4% stacking and 12% resolving gel). The purified protein samples were dialyzed in Snakeskin Dialysis tubing 7000 MWCO (Thermo scientific) against 2 L of stirred PBS at 4 °C. After 8 h, the reservoir buffer was changed (again, 2 L of PBS) and dialysis continued overnight. The dialyzed protein sample was divided into aliquots in clear PP tubes (BIOLOGIX®) and frozen in liquid nitrogen. The frozen samples were stored at −80 °C.
In situ proteome oxime ligation
PAO-JP2 (pKD-rhlA) was inoculated into 200 mL of LB media in a sterile Erlenmeyer flask. The culture was grown overnight at 37 °C with good aeration to late stationary phase and harvested at 3220 × g at 4 °C. The pellet was washed and resuspended twice with fresh LB, then sedimented once more. The pellet was resuspended in 10 mL of fresh LB media. Aliquots of 1 mL were transferred to clear 15 mL PP sterile conical centrifuge tubes, and the bacteria were incubated in the presence of probe 13 (1, 0.5, 0.1, 0 mM) for 4 h. The cells were then pelleted for 10 min at 3220 × g (4 °C) and resuspended in 1 mL of PBS. The cell suspension was irradiated at 365 nm for 3.5 min. The samples were then mixed with 3 μL of EDTA-free protease inhibitor cocktail (Merck) and sonicated for a total of 2 min in twelve 10 second pulses with 45 s rest between runs at 40% amplitude setting. Protein concentrations were determined as was mentioned above. For the subsequent ligation reactions, the reaction mixtures (100 μL final volume) contained 0.5 mg mL−1 protein, 10 mM aniline, 100 mM sodium acetate buffer (pH 5.1), and 700 nM aminooxy bodipy.23 After 12 h at room temperature with vigorous mixing (shaking at 1000 rpm), the reactions were quenched and precipitated by the addition of methanol/chloroform/water mixture (2 reaction volumes of methanol, 1/2 reaction volume of chloroform and 1 reaction volume of H2O). The precipitate was washed once with ice-cold methanol to remove unreacted aminooxy bodipy and other reagents. The precipitate was pelleted and the supernatant was removed. The proteins were resuspended in the same reaction volume (100 μL) of 1× SDS sample buffer and heated for 10 min at 95 °C. The redissolved proteins were resolved by SDS-PAGE (4% stacking and 10% separating gels) alongside a protein sizing ladder (3 μL SMOBIO PM2500 Excel Band 3-color Regular Range Protein Marker). Fluorescent bands in the gels were revealed using a flatbed scanner and Cy3 filter. Following this, the gels were stained with Coomassie Brilliant Blue R-250, destained, and scanned in non-fluorescent mode.
In situ Cu1+-mediated azide–alkyne coupling (CuAAC) reactions
PAO-JP2 (pKD-rhlA) was inoculated into 200 mL of LB media in a sterile Erlenmeyer flask. The culture was grown overnight at 37 °C with good aeration to late stationary phase and harvested at 3220 × g (4 °C). The pellet was washed and resuspended twice with fresh LB. The pellet was then resuspended in 10 mL of fresh LB media. Aliquots of 1 mL were transferred to clear 15 mL PP sterile conical centrifuge tubes, and the bacteria were incubated in the presence of probe 12 (1, 0.5, 0.1, 0 mM) for 4 h. The cells were then pelleted for 10 min at 3220 × g (4 °C) and resuspended in 1 mL of PBS. The cell suspension was irradiated at 365 nm for 3.5 min. The samples were then mixed with 3 μL of EDTA-free protease inhibitor cocktail (Merck) and sonicated for a total of 2 min in twelve 10 s pulses with 45 s rest between runs, and 40% amplitude setting. Protein concentrations were determined as mentioned above. CuAAC reactions (200 μL total reaction volume (100 μL proteome + 100 μL click reagents)) contained final reagent concentrations of 0.5 mg mL−1 protein, 100 μM of rhodamine azide,24,25 100 μM TBTA (Sigma-Aldrich), 1 mM CuSO4 (Sigma-Aldrich) and 1 mM TCEP (Sigma-Aldrich) were performed. After 1 h at room temperature with vigorous mixing (shaking at 1000 rpm), the reactions were quenched and precipitated by the addition of 2 reaction volumes of methanol, 1/2 reaction volume of chloroform and 1 reaction volume of H2O. The resulting protein pellet was washed once with ice-cold methanol and pelleted. The pelleted proteins were resuspended in the same reaction volume (100 μL) of 1× SDS sample buffer, heated for 10 min at 95 °C, and analyzed as described previously.
Concentration-dependent His6-TEV-PhzB1 labeling
Purified His6-TEV-PhzB1 protein samples were thawed, and the protein concentration was measured. Labeling reactions were carried out in 1.5 mL transparent PP tubes (Greiner Bio-One) in a final volume of 100 μL. Samples adjusted to contain 0.5 mg mL−1 of His6-TEV-PhzB1 in PBS, were incubated in the presence of probe 12 (dissolved in DMSO) at the indicated concentrations of 0–50 μM for 45 min at room temperature, with vigorous mixing (shaking at 600 rpm). During this time, the samples were wrapped in aluminium foil to shield them from light. DMSO was used as control and never exceeded 2% final concentration. The reaction mixtures were UV irradiated at 365 nm for 30 s. After irradiation was completed, the reactions were quenched by the addition of 500 μL of acetone (−20 °C). The reaction mixtures were placed in the dark at −20 °C for 20 min and the precipitated protein was pelleted (20000 × g, 10 min, 25 °C). The supernatant was removed and the pellets were washed with 500 μL of cold acetone (−20 °C). The washed pellets were air-dried and redissolved in 2.5% w/v aqueous SDS. The samples were heated at 95 °C and vigorously vortexed to ensure complete dissolution for 5 min. CuAAC reactions (100 μL total reaction volume) contained a final concentration of 50 μM rhodamine azide, 50 μM TBTA (Sigma-Aldrich), 0.5 mM CuSO4 (Sigma-Aldrich), and 0.5 mM TCEP–HCl (Combi Blocks) were performed. After 1 h of vigorous mixing (shaking at 1000 rpm) at room temperature, the proteins were precipitated with 500 μL of cold acetone (−20 °C), pelleted, and the pellets were washed once with cold acetone. The resulting pellets were air-dried and resuspended in 100 μL SDS sample buffer and analyzed as described previously.
The specificity of labeling towards His6-TEV-PhzB1
His6-TEV-PhzB1, bovine serum albumin (BSA, Chem Cruz) and lysozyme (Sigma-Aldrich) were each prepared in PBS and subjected to the labeling reaction as described above for purified His6-TEV-PhzB1. All final protein concentrations in the reaction mixtures were adjusted to 0.5 mg mL−1 and probe 12 concentration fixed at 20 μM.
Labeling of His6-TEV-PhzB1 in the presence of competing C4-HSL
His6-TEV-PhzB1 protein samples were labeled as described above except that the reactions contained 0–1000 μM C4-HSL (dissolved in DMSO) and a fixed concentration (20 μM) of probe 12. The probe was added last and the labeling reaction was allowed to proceed for 5 min, after which, 8 s UV irradiation (365 nm) was performed and the proteins were analyzed by SDS-PAGE as described previously.
In vitro quantitative ABPP
Wild-type P. aeruginosa strain PAO1 was inoculated into 200 mL of LB media in a sterile Erlenmeyer flask. The culture was grown with good aeration (shaking at 180 rpm) at 37 °C overnight to late stationary phase. The cells were pelleted at 3220 × g (4 °C). The pellet was resuspended and washed three times with PBS. The final pellet, after washing, was frozen in liquid nitrogen. When required, the pellet was resuspended in 10 mL of PBS supplemented with EDTA-free protease inhibitor cocktail (Merck) (3 μL per mL of resuspended cells) and sonicated for 2 min in twelve 10 s pulses with 45 s rest between runs, at 40% amplitude setting. The protein concentration was determined. For the ABPP, two sets of triplicate reaction mixtures were set up in 1.5 mL transparent PP tubes. The mixtures contained 500 μL of bacterial protein extract, adjusted to 1 mg mL−1. One group was incubated in the presence of probe 12 (dissolved in DMSO) at a final concentration of 200 μM and the other group in the presence of C4-HSL (dissolved in DMSO) at a final concentration of 10 μM. After 1 h of incubation at room temperature, the samples were irradiated at 365 nm for 1.5 min and CuAAC reagents were added to make a final volume of 1 mL. The final concentration of CuAAC reagents in the reaction mixtures was 50 μM biotin azide, 50 μM TBTA (Sigma-Aldrich), 0.5 mM CuSO4 (Sigma-Aldrich) and 0.5 mM TCEP (Sigma-Aldrich). After 1 h at room temperature with vigorous agitation (shaking at 1000 rpm), the reaction mixtures were transferred to 15 mL transparent PP sterile conical centrifuge tube and the proteins were precipitated with methanol/chloroform/H2O (2 reaction volumes of methanol 1/2 reaction volume of chloroform and 1 reaction volume of H2O). The precipitated proteins were pelleted and washed once with cold methanol. The protein pellet was resuspended in 500 μL of 6 M urea (Sigma-Aldrich) and TCEP was introduced to a final concentration of 10 mM. The mixture was incubated for 0.5 h at 37 °C with shaking at 180 rpm, followed by the addition of IAA (Sigma-Aldrich) to a final concentration of 55 mM. The mixtures were left shielded from light at room temperature for 0.5 h with gentle agitation on a flat-bed shaker. Finally, 5 mL of PBS containing 0.2% of SDS was added to each of the tubes, thereby diluting the urea but retaining the labeled proteins in soluble form. In parallel, 100 μL aliquots of streptavidin-agarose bead suspension (Merck Novagen) were introduced to 1.5 mL transparent PP tubes. The beads were washed three times with PBS before use, and the overburden of supernatant was removed. For affinity enrichment, the washed beads were introduced to the SDS-solubilized reaction mixtures and the resulting suspensions were incubated for 1.5 h at room temperature with gentle agitation on a flat-bed shaker. The beads were then gently pelleted (1400 × g) for 3 min, and the overburden of liquid was decanted off. The beads were then washed with 5 mL of 0.2% SDS and re-pelleted. This was repeated three times. Following this, the beads were washed (three times) with 5 mL PBS, followed by three washes in pure water. The beads from each reaction were then transferred to new low-binding 1.5 mL transparent PP tubes (Eppendorf) and pelleted for 2 min at 1400 × g. The supernatant was removed and replaced with 200 μL of 2 M urea (Sigma-Aldrich) in PBS containing 1 mM CaCl2 (J.T. Baker) and 10 ng μL−1 trypsin (Promega). The mixture was incubated overnight at 37 °C with shaking at 180 rpm. Following this, the beads were pelleted at 1400 × g for 2 min and the supernatant (containing the released tryptic peptides) from each reaction mixture was transferred, separately, to a new low-binding 1.5 mL transparent PP tubes. The pelleted beads were washed once with 100 μL PBS and the supernatant was pooled to the corresponding reaction mixture containing the tryptic peptides. Each tryptic peptide mixture was supplemented with (5% final) HPLC grade formic acid (FA, Bio-Lab) and the samples were purified and desalted using StageTips (see below). Finally, the mixtures were dried in a speed-vac and stored at −20 °C until LC-MS/MS analysis. Data processing was performed with MaxQuant and peptides were identified with the Uniprot PAO1 proteome. Carbamidomethylation of cysteine residues was considered as a fixed modification and methionine oxidation (M) and acetyl (protein N-terminal) were treated as variable modifications. Otherwise, the default settings in the software were applied. For probe 12 and C4-HSL (control) triplicates, the experimental data were analyzed with Microsoft Excel and proteins represented by >1 peptide were filtered and quantified for all replicates.
StageTips preparation and desalting
StageTips were prepared according to a modified protocol26 with C18 solid-phase extraction disc (3M Empore). Between 4–6, 1 mm cut discs were loaded into 200 μL pipette tips. The discs were packed tightly with the help of a truncated needle, and washed once with MeOH, twice with 80% ACN containing 0.5% FA (Bio-Lab) and four times with H2O containing 0.5% FA (50 μL each buffer). The tips were spun at low speed (40 × g for 2 min) between the washes, allowing the C18 column to stay wet with a thin layer of liquid after each spin. The tryptic peptides from each sample were loaded in two consecutive batches onto each StageTip, and the tips were desalted once with 50 μL of H2O containing 0.5% FA. Peptides were eluted from the StageTips into 1.5 mL low-binding transparent PP tubes by the addition of four successive 50 μL aliquots of 80% ACN containing 0.5% FA. For subsequent MS analysis, the samples were resuspended (with sonication in a water bath) in 10 μL of H2O containing 0.5% FA.
Results and discussion
Probe design and structure–activity relationship studies
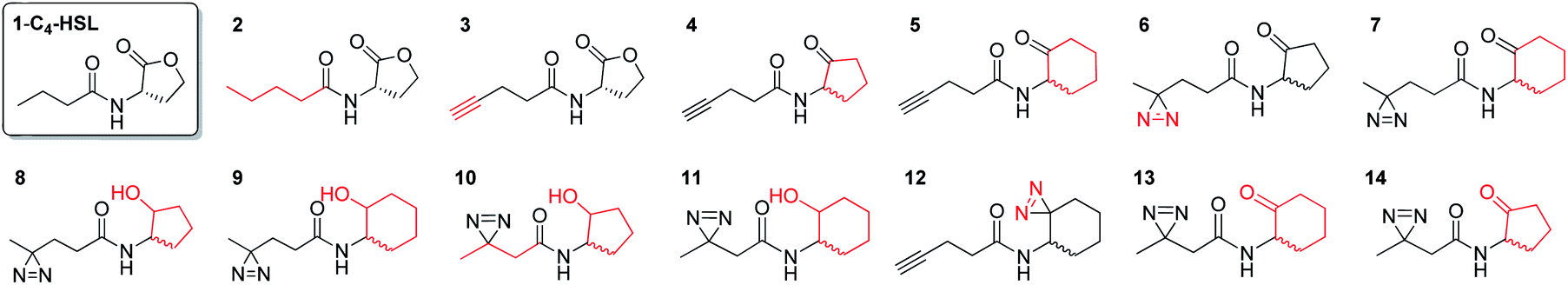
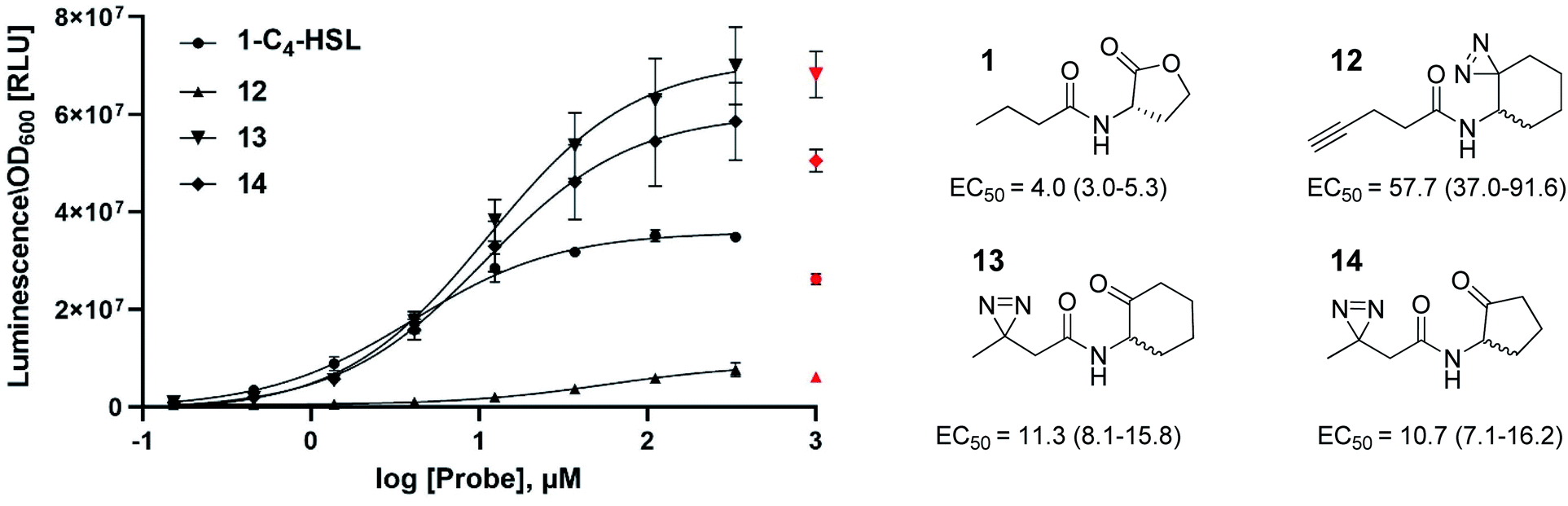
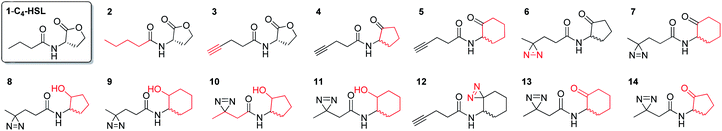
In this study, we aimed to determine which proteins in the P. aeruginosa proteome interact with the C4-HSL autoinducer. This way, we hoped to reveal potentially novel enzymes/proteins that might be regulated by C4-HSL. We synthesized molecular probes based on the C4-HSL structure, harboring handles that allow for bioorthogonal reactions. In all luminescent bioassays, we employed PAO-JP2 (pKD-rhlA). PAO-JP2 contains deletions in the lasI and rhlI genes, and therefore cannot synthesize endogenous 3-oxo-C12-HSL and C4-HSL (respectively). However, the chromosomally-encoded rhlR gene is intact in this mutant, enabling it to sense exogenously-added C4-HSL. In addition, the pKD-rhlA plasmid borne in this strain, is a reporter for RhlR activity. Here, the RhlR-regulated rhlA promoter is fused upstream of a cluster of lux genes, enabling facile monitoring of C4-HSL and C4-HSL agonist/antagonist activities through bioluminescence. We introduced the different structural probes to identical cultures of PAO-JP2 (pKD-rhlA) and compared their ability to stimulate lux gene expression with the natural autoinducer C4-HSL (Table 1). This allowed us to assess which probes best mimic the activity of the natural autoinducer. In terms of probe design, we adhered to three main principles of ABPP: (a) the affinity-based structural element should be minimally modified compared with the natural ligand, (b) the reactive group that allows for covalent attachment of the probe to the binding site on the target protein should be small and not overly reactive (here, we chose to employ the diazirine moiety, which, upon irradiation at 365 nm, forms a reactive carbene capable of binding covalently to the nearest carbon or heteroatom), and (c) the analytical “handle” assisting in visualization, separation, and analysis of the bound proteins should be minimally-reactive with any of the functional moieties naturally present in proteins. The “handles” we chose to employ in the current study were the alkyne group (which has been successfully used in copper-assisted azide–alkyne coupling (CuAAC)) and a ketone group for aniline-catalyzed oxime ligation reactions.27 The analysis of the probe activities is shown in Fig. S29–S32.† As outlined in Table 1, we evaluated differences in aliphatic chain length, diazirine position, ketone position, and ring size. In the first analogue that we synthesized, 2, we substituted the four-carbon chain of C4-HSL with a five-carbon chain. This change slightly increased the bioactivity of the compound. Based on this result, we decided to append the alkyne “handle” to the end of the aliphatic chain of C4-HSL. The resulting compound (3) showed increased bioactivity as well. We next tested whether replacement of the lactone with a ketone moiety was feasible. This was done in order to prevent possible lactonolysis of the probes.28 However, for both five and six-membered ring analogues this led to a large decrease in bioactivity (compounds 4 and 5). Next, we examined how the introduction of a diazirine moiety affected probe bioactivity. The diazirine moiety had strong impact on bioactivity compared to the alkyne containing probes (compounds 6 and 7). Interestingly, we observed that the reduced precursors of 6, 7, 13, and 14 (containing hydroxyl moieties in the ring, 8–11) also exhibited bioactivity compare to the corresponding ketones, indicating that small changes in the polar head group can be tolerated (notably, compounds 8 and 9 exhibited low activation levels of the system but still display activation of Rhl with low EC50 values). These combined observations led us to focus on three final probe designs; in compound 12, we introduced the diazirine moiety into the six-carbon ring and the alkyne moiety into the side chain, and in compounds 13 and 14, shorter-chain analogues of 7 and 6, respectively, we introduced the diazirine moiety into the side chain, and the ketone moiety into the ring. As all three ABPP probes, 12, 13 and 14 could potentially be used to label the P. aeruginosa proteome, we carried out a deeper quantitative analysis of their ability to activate the Rhl QS system (Fig. 3) and compare them to C4-HSL bioactivity as control.29 Interestingly 13 and 14 showed more robust activation of the Rhl system than the natural autoinducer C4-HSL. Compound 12 displayed lower maximal activation than 13 and 14, and lower potency (EC50 of 57.7 μM vs. 11.3 and 10.7 μM, respectively). However, based on these data we concluded that all three probes (12, 13 and 14) are capable of activating the Rhl QS system (albeit, to differing extents) and are able to mimic the natural autoinducer C4-HSL, at least in terms of RhlR-dependent activation of rhlA transcription. Based on these efficacy data, and taking into account the anticipated selectivity of the ligation reactions (CuAAC and oxime ligation), we chose to further examine compounds 12 and 13 in order to identify novel C4-HSL-binding proteins. Compound 12 was selected as the presence of an alkyne analytical handle enables a CuAAC click reaction with an azide-containing fluorophore or affinity tag, and 13 was selected due to its ability to activate rhl strongly, and the presence of a ketone moiety which can be used in oxime ligation reactions.
Table 1 Synthetic probes and their EC50 values compared to the relative value induced by exogenously-added C4-HSL, the natural autoinducer of the Rhl QS in P. aeruginosa. EC50 values correspond to the agonism portion of the curve
|
|
| Agonism |
| Compounds (probes) |
EC50 [μM] |
95% CI [μM] |
Compounds (probes) |
EC50 [μM] |
95% CI [μM] |
|
1-C4-HSL
|
4.0 |
3.0 to 5.3 |
8
|
7.2 |
3.1 to 16.5 |
|
2
|
2.9 |
2.2 to 3.7 |
9
|
9.7 |
6.0 to 15.7 |
|
3
|
4.6 |
3.3 to 6.2 |
10
|
69.1 |
54.4 to 88.2 |
|
4
|
25.1 |
21.7 to 29.1 |
11
|
41.9 |
33.1 to 53.2 |
|
5
|
22.1 |
18.3 to 26.8 |
12
|
57.7 |
37.0 to 91.6 |
|
6
|
110.7 |
88.9 to 138.1 |
13
|
11.3 |
8.1 to 15.8 |
|
7
|
75.0 |
53.9 to 104.2 |
14
|
10.7 |
7.1 to 16.2 |
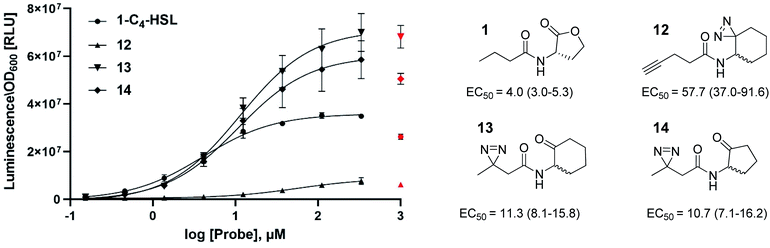 |
| | Fig. 3 Concentration dependent (0–1000 μM) activation of RhlR in PAO-JP2 (pKD-rhlA) by the reactive probes. Final activation curves are shown for probes 12, 13 and 14, compared to the natural autoinducer C4-HSL activation. EC50 values represented as EC50 [μM] (95% CI). All compounds were tested in triplicates and data represent the average ± SD of three independent experiments per probe. Three parameter fits were used to calculate EC50 values. Values for the highest concentrations were excluded from the curve fitting process for all compounds, since for most compounds 1 mM of probe resulted in lower activation of the Rhl system, most likely caused by membrane perturbation. | |
Rhodamine azide and bodipy aminooxy fluorescent dye labeling of P. aeruginosa PAO-JP2 (pKD-rhlA)
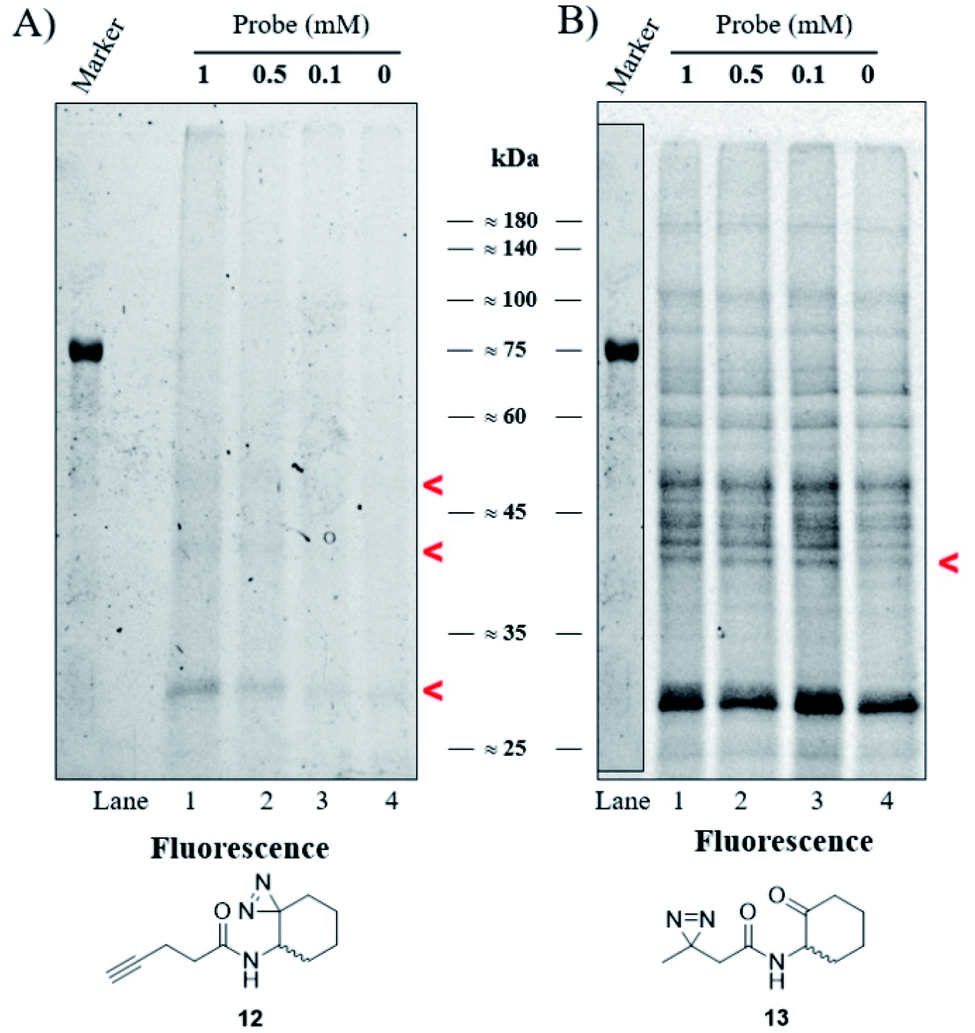
Our goal was to identify and characterize proteins that might play a hitherto unrecognized role in the P. aeruginosa QS response. Based on the fact that C4-HSL is membrane permeable,30 we used a two-step ABPP approach31 in which the C4-HSL based probes 12 and 13, which contain a diazirine reactive group and an alkyne (12) or ketone (13) analytical handle, were incubated at varying concentrations with cultures of P. aeruginosa PAO-JP2 (pKD-rhlA). The cells were washed, resuspended in PBS, irradiated, homogenized and then CuAAC click or oxime ligation reactions were performed. Fig. 4A, B and S33† show the results of the photo labeling experiments with probe 12 and probe 13. The fluorescent scans of the proteins resolved after SDS-PAGE show a clear difference in the selectivity of protein labeling between the two reactions, CuAAC with rhodamine azide and the oxime ligation with bodipy aminooxy and the two probes; probe 13 (or the fluorescent bodipy aminooxy tag) labels the P. aeruginosa proteome much less selectively due to the poor selectivity of the oxime ligation reaction, whereas probe 12 appears less reactive, but labeling is ultimately more specific due to the more selective CuAAC reaction. In the latter case, two weakly-labeled bands were observed with masses of ≈50 kDa and ≈42 kDa, and a more strongly-labeled band of mass ≈29 kDa.
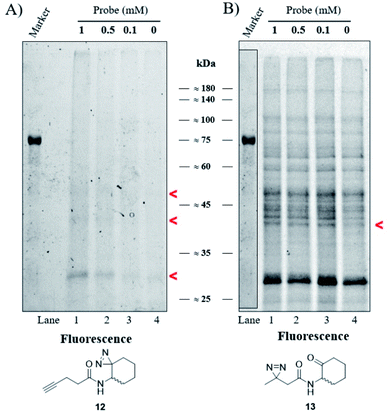 |
| | Fig. 4 Labeling of P. aeruginosa. (A) Fluorescent scan of rhodamine azide labeled PAO-JP2 (pKD-rhlA) proteins resolved by SDS-PAGE. (B) Fluorescent scan of bodipy aminooxy labeled proteins resolved by SDS-PAGE. Marker band positions in image (B) are copied from the marker band positions in gel (A). | |
Proteome analysis and protein identifications
After these initial labeling and optimization experiments, we repeated the labeling experiments with probe 12 but instead of conjugation with rhodamine azide, we now used biotin azide in order to isolate the labeled proteins. Wild-type P. aeruginosa strain PAO1 was grown to late stationary phase and harvested. The cell lysate was prepared and incubated separately with probe 12 and with C4-HSL (as negative control). The samples were then irradiated with UV light at 365 nm and subjected to CuAAC reactions with biotin azide. The biotinylated proteins were captured with streptavidin agarose beads and subjected to tryptic digestion, as described in the Experimental section. The tryptic digests were analyzed by LC-MS/MS and the proteins were quantified and identified using MaxQuant. Proteins that were identified in the negative controls were subtracted from the positive probe 12 experimental results. Although many proteins were identified in these experiments, only a few were seen in all seven replicates (Table 2), and among these are the virulence-related phenazine biosynthetic proteins, PhzB1 and PhzB2. These enzymes, which are almost identical at a sequence level, are expressed of different operons and comprise part of the pyocyanin biosynthesis pathway.32,33 Pyocyanin is a major virulence factor in P. aeruginosa, and is used by the pathogen to establish successful colonization in new niches. Another virulence-related protein that was identified is the putative transcriptional regulatory protein AlgP. AlgP, together with algR and algQ, is a key factor in the transcription of the biosynthesis gene algD. This protein is a key component of the production machinery for exopolysaccharide alginate, a critical virulence factor in P. aeruginosa. The alginate is associated with respiratory infections in cystic fibrosis patients and it is essential for adhesion and anti-phagocytosis and contributes to the persistence of the bacteria.34,35 Another labeled protein that was identified is a putative ClpA/B protease ATP-binding subunit. The ClpA and ClpB proteins belong to the Clp/Hsp100 ATPase family and function as chaperone proteins. Both ClpA and ClpB are involved in protein folding and stabilization; ClpA is a regulatory subunit in the ClpAP protease, which participates in the degradation of protein aggregates. ClpB is known to be required for colonization and in-host survival by several bacterial pathogens.36,37 We also identified an uncharacterized protein (Q9HU11/PA5178), which, after BLAST analysis, was found to contain a domain belonging to the LysM family of peptidoglycan-binding proteins. LysM homologs are present in a large number of proteins in both prokaryotes and eukaryotes, although the majority of LysM-homologs are bacterial peptidoglycan hydrolases. Several LysM domain-containing proteins from bacterial pathogens such as Staphylococcus aureus, Listeria monocytogenes, Streptococcus agalactiae are known virulence factors, involved in cell separation/autolysis and adherence to human cells, etc.38
Table 2 Protein identification. The most relevant protein hits (by score) that were identified by ABPP experiments with probe 12
| Uniprot |
Gene |
Protein |
Mass (Da) |
| Q9I659 |
PA0459 |
ClpA/B protease ATP-binding subunit |
94175 |
| O69753 |
PA4211 |
Phenazine biosynthesis protein PhzB1 |
18888 |
| Q9S508 |
PA1900 |
Phenazine biosynthesis protein PhzB2 |
19028 |
| Q9HU11 |
PA5178 |
Uncharacterized protein (peptidoglycan-binding protein, LysM) |
15461 |
| P15276 |
PA5253 |
Transcriptional regulatory protein AlgP |
34491 |
PhzB2 transposon mutant evaluation assays
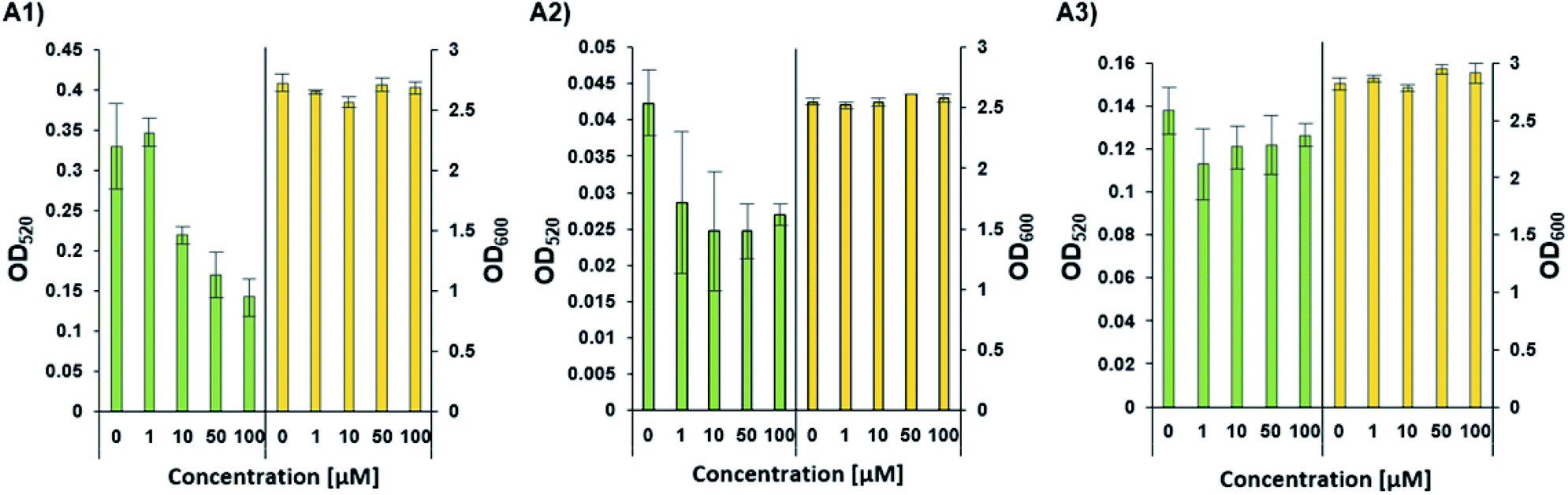
Phenazines are redox-active, heterocyclic compounds with distinctive coloration. They serve as both soluble electron mediators, and as virulence factors. They are secreted by a variety of different bacterial species, although the position of various substituents on the phenazine ring system determines the toxicity of the phenazine toward prokaryotic and eukaryotic organisms. In P. aeruginosa the biosynthesis of phenazines is carried out by enzymes expressed from two almost identical, but differentially regulated operons (phzA1B1C1D1E1F1G1 and phzA2B2C2D2E2F2G2). The enzymes encoded by PhzA-G convert chorismic acid to phenazine-1-carboxylic acid, and the latter is then converted to phenazine-1-carboxamide, 1-hydroxyphenazine and 5-methylphenazine-1-carboxylic acid betaine through the action of enzymes encoded by the unlinked genes phzH, phzS, and phzM, respectively. In a downstream reaction, 5-methylphenazine-1-carboxylic acid betaine is transformed by PhzS into pyocyanin. Pyocyanin is a strongly-colored blue phenazine and a potent P. aeruginosa virulence factor.32,39 Pyocyanin catalyzes the formation of reactive oxygen species, which can inhibit microbial growth40 and induce rapid apoptosis of human neutrophils.41 In people with cystic fibrosis, pyocyanin inhibits the ciliary function of respiratory epithelial cells and alters the host immune and inflammatory response.42 It can also stimulate the release of prostaglandin (prostacyclin), disrupt calcium homeostasis and inactivate the antioxidant enzyme, catalase.43 The synthesis of pyocyanin is ultimately regulated by several different QS systems in P. aeruginosa (among them, the PQS, and indirectly the Las and Rhl systems). In the Rhl system, the N-butyryl-L-homoserine lactone autoinducer binds to RhlR, which activates the transcription of pyocyanin biosynthesis genes.7 In addition, through a coordinated production of 3-oxo-C12-HSL and C4-HSL, the production of the PQS (2-heptyl-3-hydroxy-4-quinolone) signal is regulated, and this, in turn, affects the regulation and expression of virulence genes of the Rhl system, and thus, the production of pyocyanin.44 The PhzB2 protein has been suggested to be a major contributor to the synthesis of phenazine-1-carboxylic acid (PCA), the precursor of pyocyanin.45 To examine whether C4-HSL might influence pyocyanin production, we compared the effect of adding exogenous C4-HSL to cultures of wild-type P. aeruginosa strain PAO1 and cultures of two transposon mutants (PW4328 and PW4329) which both lack functional PhzB2 (Fig. 5). While we can observe a C4-HSL concentration-dependent inhibition of pyocyanin production in wild-type P. aeruginosa strain PAO1, this inhibitory effect is less pronounced in strain PW4328 and is almost absent in strain PW432s9 (Fig. 5A1, A2 and A3 resp. and Fig. S34†). This C4-HSL concentration-dependent inhibition of pyocyanin production indicates that in high concentrations of C4-HSL there appears to be a “switch off” effect on pyocyanin production, a phenomenon that could be linked to energy and resource-saving mechanisms in the bacteria in order to enable non-virulent survival in the host environment. Importantly, while the relatively high C4-HSL concentrations, at which we observe a significant decrease in pyocyanin production, is generally not reached in planktonic cultures, in biofilms, the local concentrations of autoinducers can be much higher (>600 μM).46 To our knowledge, this is the first indication that direct interaction between C4-HSL and factors of the pyocyanin biosynthetic machinery may enable P. aeruginosa to down-regulate the synthesis of its major virulence factor in a population-density dependent manner. Most likely P. aeruginosa has additional tools in its arsenal of factors that enable the correct and beneficial assessment of its environment under changing conditions, such as the other C4-HSL binding proteins that we identified (LysM, AlgP and ClpA/B), and this is the subject of ongoing investigations.
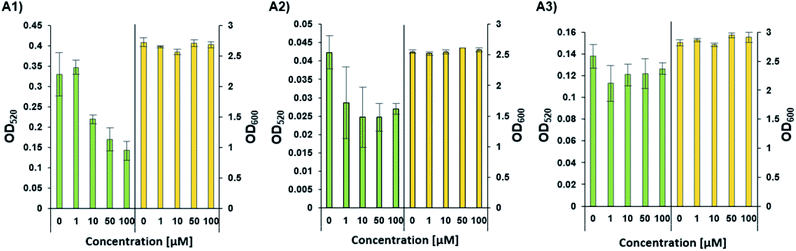 |
| | Fig. 5 The concentration-dependent response of pyocyanin production (OD520) and growth (OD600) by wild-type and different mutants of P. aeruginosa. (A1) Pyocyanin production by wild-type P. aeruginosa strain PAO1. (A2 and A3) Pyocyanin production by PhzB2 transposon insertion mutants PW4328 and PW4329 respectively. Strains were evaluated in the presence of increasing C4-HSL concentrations (0–100 μM). All strains were tested in triplicate and data represent the average ± SD of three independent experiments. | |
Cu1+-Mediated azide–alkyne coupling (CuAAC) reactions; bioorthogonal labeling of PhzB1
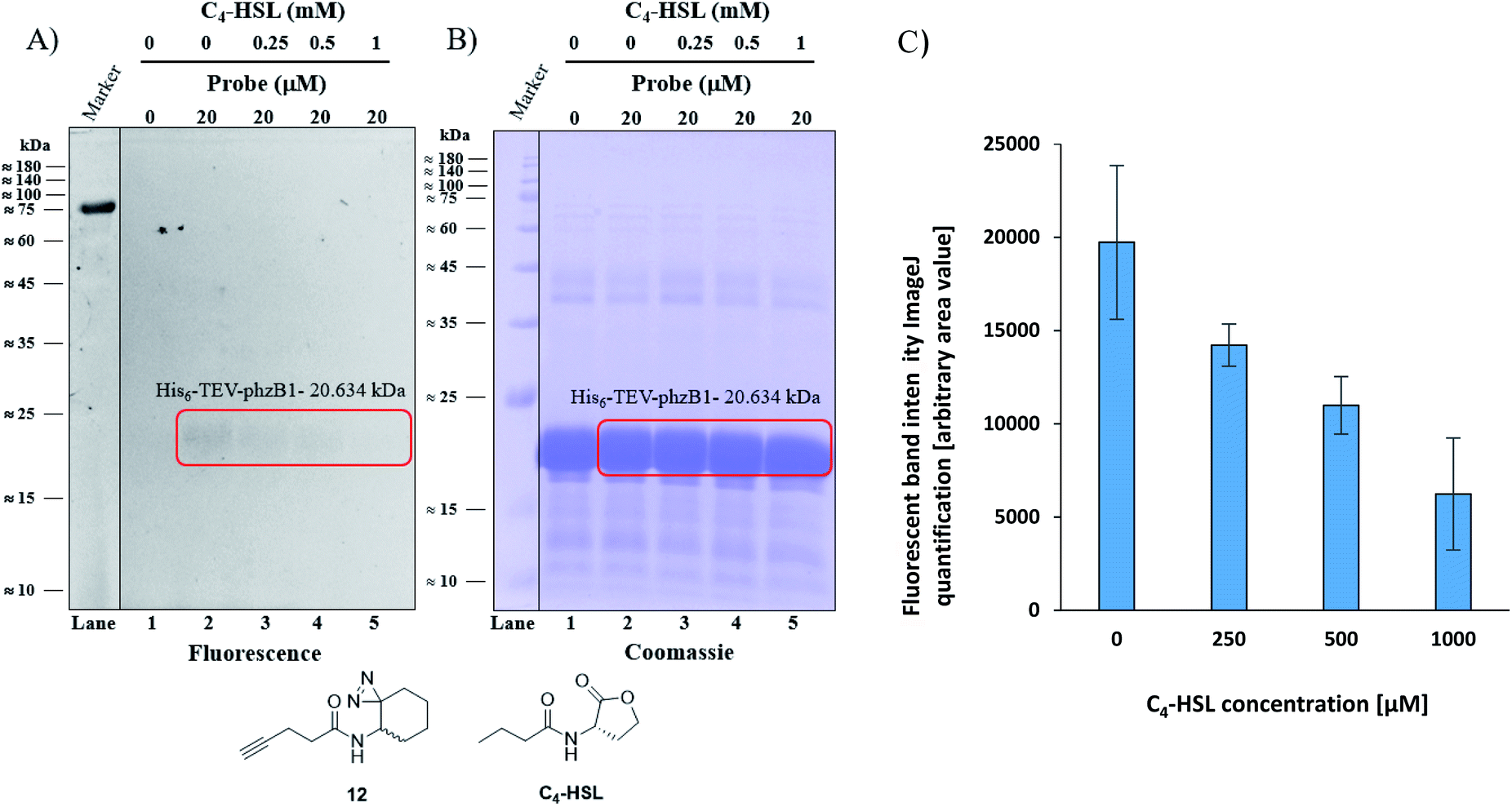
As mentioned above, P. aeruginosa has two almost identical operons, the phzA1-G1 (phz1) and phzA2-G2 (phz2), with ∼98% similarity at the nucleotide level.47,48 In P. aeruginosa, PCA is modified to yield bioactive phenazine derivatives by the enzymes PhzH, PhzS, and PhzM.49 PhzB1 is one of the enzymes that are responsible for the conversion of chorismic acid, an intermediate product of the shikimate pathway,50,51 to PCA and PDC via various phenazine intermediates with the help of the other mainly involved enzymes PhzA/B, PhzD, PhzE, PhzF, and PhzG, the crystal structures of which are known.52 The general role of PhzB in the phenazine biosynthesis is to catalyze the condensation of two precursor molecules of 6-amino-5-oxocyclohex-2-ene-1-carboxylic acid (AOCHC, see Fig. S39†) to form the tricyclic phenazine (HHPDC). This tricyclic phenazine intermediate can undergo enzymatic/nonenzymatic transformations to yield DHPCA and DHPDC, the precursors of more than 150 structural derivatives of strain-specific phenazines.53,54 To directly investigate the labeling of PhzB1 by the C4-HSL mimic, probe 12, we expressed recombinant His6-TEV-PhzB1 in E. coli Rosetta strain BL21 (DE3). The purified His6-TEV-PhzB1 protein migrated as a single band with a molecular mass of ≈20 kDa (Fig. S35B and C†) which corresponds to the predicted molecular mass of the protein based on the DNA sequence. The purified His6-TEV-PhzB1 protein was used to assess compound 12 and C4-HSL binding in vitro. First, we established how much probe 12 was required to label His6-TEV-PhzB1 in vitro (Fig. S36A and B†). Based on these data, we chose to work with 20 μM of probe 12 in subsequent experiments. This was a trade-off between the concentration required to give an intense fluorescent signal, and ensuring that the probe concentration would be comparable with the physiological range of C4-HSL concentration in vivo. Having selected the optimal concentration of probe 12 to use, we proceeded to assess the specificity of labeling towards His6-TEV-PhzB1. Probe 12 was incubated with His6-TEV-PhzB1 in the presence of BSA and lysozyme as control proteins (Fig. S37A and B†). Probe 12 efficiently labeled the ≈20 kDa His6-TEV-PhzB1 protein (lanes 2, 4 and 6) with much lower labeling of the BSA control (corresponding to the band with a molecular mass ≈60–62 kDa). No labeling of the lysozyme control (≈14 kDa) was observed. We conclude that probe 12 preferentially interacts with the His6-TEV-PhzB1 protein. We next examined whether C4-HSL would compete with probe 12 and block the labeling of His6-TEV-PhzB1. This was indeed the case (Fig. 6 and S38†); increasing concentrations of C4-HSL led to markedly lower labeling of the protein by probe 12 (Fig. 6A lanes 2–5 and S38A, lanes 1–4†). Quantitation of the fluorescent band intensity (Fig. 6C) confirmed this observation. From these experiments we conclude that C4-HSL binds to PhzB1, and currently we examine in more detail the mechanisms underlying the consequent reduction in pyocyanin biosynthesis (see phenazine biosynthetic pathway, Fig. S39†).52
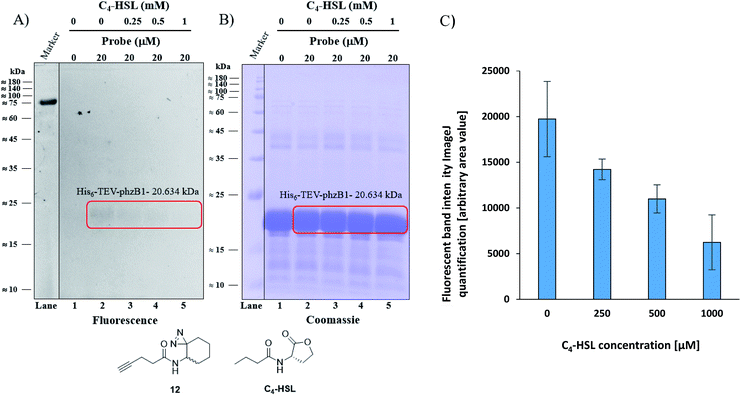 |
| | Fig. 6 Competitive labeling of PhzB1. (A) Competitive labeling of purified His6-TEV-PhzB1 with a fixed probe 12 concentration of 20 μM and increasing concentrations (0–1000 μM) of competing C4-HSL. (B) The corresponding Coomassie-stained gel image. (C) Quantitation (of band intensities) of two competitive labeling experiments of His6-TEV-PhzB1 ((A) and Fig. S38A†). Data represent the average ± SD of two independent experiments. | |
Conclusion
Using a C4-HSL-based ABBP strategy, we report here on the identification and validation of PhzB1/2 as C4-HSL binding proteins, and we propose that binding of the autoinducer to PhzB1/2 may result in a decrease of pyocyanin production. As such, this binding event may serve an important role in providing information to the bacteria regarding their population density. Both PhzB1 and PhzB2 are engaged in C4-HSL-induced phenazine biosynthesis, and upon an increase of population density, when P. aeruginosa reaches its quorum, a strong increase in production of pyocyanin can be observed. At higher population densities, however, this production is reduced, a phenomenon that can be mimicked by the addition of increasing concentrations of C4-HSL to growing P. aeruginosa cultures. Our studies show that C4-HSL binds PhzB1/2 and inhibits pyocyanin production, and while at lower concentrations this autoinducer activates the Rhl QS system, resulting in the production of virulence factors, the interaction with PhzB1/2 at higher concentrations could provide a negative feedback loop to this system. We surmise that this mechanism enables the bacteria to correctly sense and adjust their population density-dependent behaviour, such that the optimal amount of energy is spent on virulence factor production when needed, and these efforts can be reduced when the population has reached an optimal density.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank Aviad Mandaby for assisting with proteomics procedures and data analysis, Mark Karpasas for mass spectrometry support, Alexander Aronovich for PCR analysis, and Amira Rudy for NMR analysis assistance. We also acknowledge Benjamin Cravatt for valuable advice and discussions. This work was supported by the European Research Council (ERC Starting Grant 240356). Martin Welch was supported by a short-term EMBO fellowship.
Notes and references
- W. R. J. D. Galloway, J. T. Hodgkinson, S. D. Bowden, M. Welch and D. R. Spring, Chem. Rev., 2011, 111, 28–67 CrossRef CAS.
- G. J. Lyon and R. P. Novick, Peptides, 2004, 25, 1389–1403 CrossRef CAS.
- K. Winzer and P. Williams, Int. J. Med. Microbiol., 2001, 291, 131–143 CrossRef CAS.
- R. Czajkowski and S. Jafra, Acta Biochim. Pol., 2009, 56, 1–16 CrossRef CAS.
- B. M. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef.
- B. A. Jamuna and R. V. Ravishankar, Compr. Rev. Food Sci. Food Saf., 2011, 10, 184–193, DOI:10.1111/j.1541-4337.2011.00150.x.
- G. W. Lau, D. J. Hassett, H. Ran and F. Kong, Trends Mol. Med., 2004a, 10, 599–606 Search PubMed.
- R. M. Donlan, Emerging Infect. Dis., 2002, 8, 881–890 CrossRef.
- C. M. Miyamoto, Y. H. Lin and E. A. Meighen, Mol. Microbiol., 2000, 36, 594–607 CrossRef CAS.
- A. M. Mitchell and T. J. Mitchell, Clin. Microbiol. Infect., 2010, 16, 411–418 CrossRef CAS.
- W. C. Fuqua, S. C. Winans and E. P. Greenberg, J. Bacteriol., 1994, 176, 269–275 CrossRef CAS.
- J. Lee, J. Wu, Y. Deng, J. Wang, C. Wang, J. Wang, C. Chang, Y. Dong, P. Williams and L. H. Zhang, Nat. Chem. Biol., 2013, 9, 339–343 CrossRef CAS.
- P. Williams and M. Camara, Curr. Opin. Microbiol., 2009, 12, 182–191 CrossRef CAS.
- M. A. Welsh and H. E. Blackwell, Cell Chem. Biol., 2016, 23, 361–369 CrossRef CAS.
- E. Papaioannou, P. D. Utari and W. J. Quax, Int. J. Mol. Sci., 2013, 14, 19309–19340 CrossRef.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS.
- W. P. Heal, T. H. Dang and E. W. Tate, Chem. Soc. Rev., 2011, 40, 246–257 RSC.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS.
- B. Alexander, D. J. Browse, S. J. Reading and I. S. Benjamin, J. Pharmacol. Toxicol. Methods, 1999, 41, 55–58 CrossRef CAS.
- K. Duan and M. G. Surette, J. Bacteriol., 2007, 189, 4827–4836 CrossRef CAS.
- M. A. Jacobs, A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond and R. Levy,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14339–14344 CrossRef CAS.
- K. Held, E. Ramage, M. Jacobs, L. Gallagher and C. Manoil, J. Bacteriol., 2012, 194, 6387–6389 CrossRef CAS.
- J. Rayo, N. Amara, P. Krief and M. M. Meijler, J. Am. Chem. Soc., 2011, 133, 7469–7475 CrossRef CAS.
- Y. Haibo, X. Yi and G. Haiying, Org. Lett., 2012, 14, 2014–2017 CrossRef.
- A. Desvergne, Y. Cheng, S. Grosay-Gaudrel, X. Marechal, M. Reboud-Ravaux, E. Genin and J. Vidal, J. Med. Chem., 2014, 57, 9211–9217 CrossRef CAS.
- J. Rappsilber, M. Mann and Y. Ishihama, Nat. Protoc., 2007, 2, 1896–1906 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS.
- E. A. Yates, B. Philipp, C. Buckley, S. Atkinson, S. R. Chhabra, R. E. Sockett, M. Goldner, Y. Dessaux, M. Camara and H. Smith,
et al.
, Infect. Immun., 2002, 70, 5635–5646 CrossRef CAS.
- J. P. Pearson, L. Passador, B. H. Iglewski and E. P. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1490–1494 CrossRef CAS.
- J. P. Pearson, C. Van Delden and B. H. Iglewski, J. Bacteriol., 1999, 181, 1203–1210 CrossRef CAS.
- L. I. Willems, H. S. Overkleeft and S. I. van Kasteren, Bioconjugate Chem., 2014, 25, 1181–1191 CrossRef CAS.
- P. Nadal Jimenez, G. Koch, J. A. Thompson, K. B. Xavier, R. H. Cool and W. J. Quax, Microbiol. Mol. Biol. Rev., 2012, 76, 46–65 CrossRef.
- S. Shuang, C. Bo, J. Zi-Jing, Z. Lian, F. Yun-Ling, T. Chitti, R. Giordano and H. Ya-Wen, Mol. Microbiol., 2017, 104, 931–947 CrossRef.
- V. Deretic and W. M. Konyecsni, J. Bacteriol., 1990, 172, 5544–5554 CrossRef CAS.
- V. Deretic, N. S. Hibler and S. C. Holt, J. Bacteriol., 1992, 174, 824–831 CrossRef CAS.
- O. E. Petrova and K. Sauer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16690–16695 CrossRef CAS.
- M. Zolkiewski, T. Zhang and M. Nagy, Arch. Biochem. Biophys., 2012, 520, 1–6 CrossRef CAS.
- G. Buist, A. Steen, J. Kok and O. P. Kuipers, Mol. Microbiol., 2008, 68, 838–847 CrossRef CAS.
- D. V. Mavrodi, R. F. Bonsall, S. M. Delaney, M. J. Soule, G. Phillips and L. S. Thomashow, J. Bacteriol., 2001, 183, 6454–6465 CrossRef CAS.
- M. Z. El-Fouly, A. M. Sharaf, A. A. M. Shahin, H. A. El-Bialy and A. M. A. Omara, J. Radiat. Res. Appl. Sci., 2015, 8, 36–48 CrossRef CAS.
- L. Allen, D. H. Dockrell, T. Pattery, D. G. Lee, P. Cornelis, P. G. Hellewell and M. K. Whyte, J. Immunol., 2005, 174, 3643–3649 CrossRef CAS.
- H. Liang, J. Duan, C. D. Sibley, M. G. Surette and K. Duan, J. Med. Microbiol., 2011, 60, 22–34 CrossRef CAS.
- G. W. Lau, H. Ran, F. Kong, D. J. Hassett and D. Mavrodi, Infect. Immun., 2004b, 72, 4275–4278 Search PubMed.
- L. A. Gallagher, S. L. McKnight, M. S. Kuznetsova, E. C. Pesci and C. Manoil, J. Bacteriol., 2002, 184, 6472–6480 CrossRef CAS.
- S. Sun, L. Zhou, K. Jin, H. Jiang and Y. W. He, Sci. Rep., 2016, 6, 30352 CrossRef CAS.
- T. S. Charlton, R. de Nys, A. Netting, N. Kumar, M. Hentzer, M. Givskov and S. Kjelleberg, Environ. Microbiol., 2000, 2, 530–541 CrossRef CAS.
- S. Higgins, S. Heeb, G. Rampioni, M. P. Fletcher, P. Williams and M. Cámara, Front. Cell. Infect. Microbiol., 2018, 8, 1–13 CrossRef.
- D. V. Mavrodi, T. L. Peever, O. V. Mavrodi, J. A. Parejko, J. M. Raaijmakers, P. Lemanceau, S. Mazurier, L. Heide, W. Blankenfeldt and D. M. Weller,
et al.
, Appl. Environ. Microbiol., 2010, 76, 866–879 CrossRef CAS.
- D. A. Recinos, M. D. Sekedat, A. Hernandez, T. S. Cohen, H. Sakhtah, A. S. Prince, A. Price-Whelan and L. E. P. Dietrich, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19420–19425 CrossRef CAS.
- D. H. Calhoun, M. Carson and R. A. Jensen, J. Gen. Microbiol., 1972, 72, 581–583 CrossRef CAS.
- M. McDonald, D. V. Mavrodi, L. S. Thomashow and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 9459–9460 CrossRef CAS.
- W. Blankenfeldt and J. F. Parsons, Curr. Opin. Struct. Biol., 2014, 29, 26–33 CrossRef CAS.
- E. G. Ahuja, P. Janning, M. Mentel, A. Graebsch, R. Breinbauer, W. Hiller, B. Costisella, L. S. Thomashow, D. V. Mavrodi and W. Blankenfeldt, J. Am. Chem. Soc., 2008, 130, 17053–17061 CrossRef CAS.
- N. Guttenberger, W. Blankenfeldt and R. Breinbauer, Bioorg. Med. Chem., 2017, 25, 6149–6166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Abbreviations; synthetic procedures (Schemes S1–S14); 1H-NMR; 13C-NMR; LC-MS; biological evaluation results; growth and luminescence assays; strain activity comparison-pyocyanin production; SDS-PAGE images of labeling and purification experiments; phenazine biosynthetic pathway (Fig. S1–S39) (PDF). Pyocyanin; luminescence results; competitive labeling of His6-TEV-PhzB1; proteomics-experiments 1–7 and final proteomic results (XLSX). See DOI: 10.1039/d0sc04444j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Josep
Rayo
a,
Josep
Rayo