
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the unique reactivity patterns of nickel/JoSPOphos manifold in the nickel-catalyzed enantioselective C–H cyclization of imidazoles†
Jian-Biao
Liu
 *a,
Xin
Wang
a,
Antonis M.
Messinis
b,
Xiao-Jun
Liu
a,
Rositha
Kuniyil
b,
De-Zhan
Chen
*a and
Lutz
Ackermann
*b
*a,
Xin
Wang
a,
Antonis M.
Messinis
b,
Xiao-Jun
Liu
a,
Rositha
Kuniyil
b,
De-Zhan
Chen
*a and
Lutz
Ackermann
*b
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, China. E-mail: jianbiaoliu.thu@gmail.com; chendzh@sdnu.edu.cn
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Göttingen 37077, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 4th November 2020
Abstract
The 3d transition metal-catalyzed enantioselective C–H functionalization provides a sustainable strategy for the construction of chiral molecules. A better understanding of the catalytic nature of the reactions and the factors controlling the enantioselectivity is important for rational design of more efficient systems. Herein, the mechanisms of Ni-catalyzed enantioselective C–H cyclization of imidazoles are investigated by density functional theory (DFT) calculations. Both the π-allyl nickel(II)-promoted σ-complex-assisted metathesis (σ-CAM) and the nickel(0)-catalyzed oxidative addition (OA) mechanisms are disfavored. In addition to the typically proposed ligand-to-ligand hydrogen transfer (LLHT) mechanism, the reaction can also proceed via an unconventional σ-CAM mechanism that involves hydrogen transfer from the JoSPOphos ligand to the alkene through P–H oxidative addition/migratory insertion, C(sp2)–H activation via σ-CAM, and C–C reductive elimination. Importantly, computational results based on this new mechanism can indeed reproduce the experimentally observed enantioselectivities. Further, the catalytic activity of the π-allyl nickel(II) complex can be rationalized by the regeneration of the active nickel(0) catalyst via a stepwise hydrogen transfer, which was confirmed by experimental studies. The calculations reveal several significant roles of the secondary phosphine oxide (SPO) unit in JoSPOphos during the reaction. The improved mechanistic understanding will enable design of novel enantioselective C–H transformations.
Introduction
The selective C–H functionalization has emerged as one of the most practical and powerful tools in molecular syntheses.1 Although major progress has been realized using the second and third row transition metals, recent developments have enabled many promising transformations that employ earth-abundant and less toxic 3d metals.2 In this context, nickel catalysts exhibit several unique properties3 and have served as an efficient alternative approach in a wide range of catalytic processes in the past few years.2b However, despite nickel being widely applied in numerous reactions, Ni-catalyzed enantioselective C–H activations have been rarely reported.4 The reactivity of nickel catalysts is highly sensitive to the substrates and ligands in most cases, resulting in narrow substrate scope and limited choices of viable chiral ligands.Imidazole derivatives are among the most significant heterocyclic structural moieties present in a broad variety of biologically active compounds.5 The construction and modification of imidazole derivatives, especially synthesis of imidazole compounds bearing stereocenters, are consequently of great interest and importance. In 2018, Ye and coworkers reported the enantioselective C–H cyclization of imidazoles with alkenes employing the Ni–Al bimetallic catalysis (Scheme 1a).6 A screening of a variety of chiral ligands indicated that the bis(t-butyl)phenyl-containing TADDOL-derived secondary phosphine oxide (SPO) ligand TADDOL–SPO was the optimal one. The TADDOL–SPO ligand enables the synthesis of polycyclic imidazoles with exclusive exo-selectivity and high enantiomeric excesses. In a very recent example, we reported a complementary approach that enables endo-selective hydroarylation of unactivated alkenes in the absence of organoaluminum reagents (Scheme 1b).7 Essential to the success of this reaction is the use of bidentate JoSPOphos ligand (L), whose applications were previously restricted to rhodium(I)-catalyzed hydrofunctionalizations.8 This simple catalytic system allows the synthesis of imidazole products containing chiral six-membered ring with high enantioselectivities.
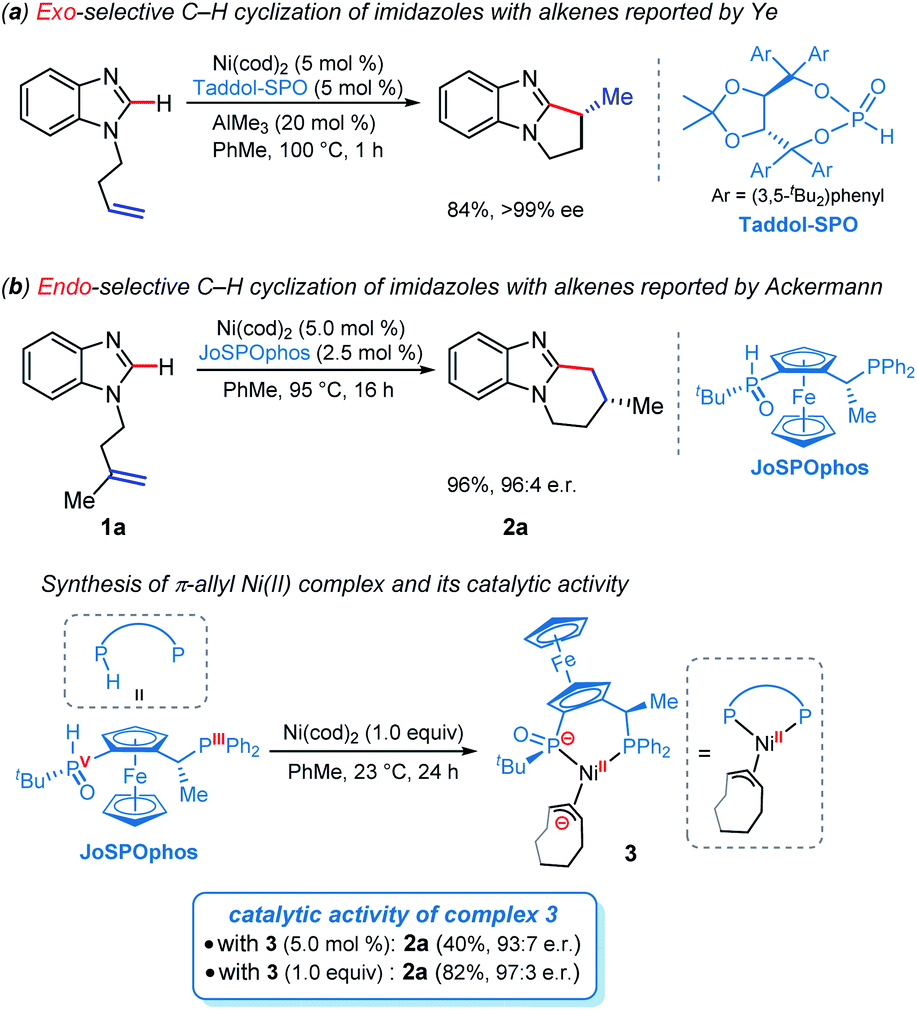 | ||
| Scheme 1 Ni-catalyzed intramolecular enantioselective C–H cyclization of imidazoles. | ||
Understanding the detailed mechanisms of Ni-catalyzed C–H functionalizations could guide the design of catalysts and the control of selectivity. However, the existing ligand exchanges and the potential of off-cycle intermediates lead to considerable complexities. To provide insights into the mechanism of the reaction in Scheme 1b, a well-defined η3–π-allyl nickel(II) complex 3 was isolated and tested for catalytic activity.7 This stable π-allyl nickel(II) complex was generated from precatalyst Ni(cod)2 (cod = 1,5-cyclooctadiene) and JoSPOphos via classical Ni-mediated chain-walking mechanism.9 Interestingly, complex 3 was found to be catalytically active in the enantioselective intramolecular alkylation of N-allylic benzimidazole 1a. Previous reports by Zimmerman and Montgomery have shown that the π-allyl complex is actually an off-cycle intermediate and its formation can drastically inhibit catalysis in the N-heterocyclic carbene–nickel-catalyzed hydroarylation.10 The experimentally observed reactivity of the π-allyl nickel(II) complex reported by us remains elusive.
Although the mechanism and selectivity of transition-metal-catalyzed hydroarylation of alkenes has been reported by several computational studies,11 the precise mode of C(sp2)–H activation12 and the origins of enantioselectivity remain unclear for the nickel/JoSPOphos system (Scheme 2). Herein, we report a detailed mechanistic study on the titled reaction by means of density functional theory (DFT) calculations. Our calculations show that both the nickel(II)-promoted σ-complex-assisted metathesis (σ-CAM)13 and the nickel(0)-catalyzed oxidative addition (OA) mechanisms are disfavored. Besides the common nickel(0)-catalyzed ligand-to-ligand hydrogen transfer (LLHT) mechanism,14 an unconventional nickel(0)-catalyzed σ-CAM mechanism is found to be also viable. The experimentally observed catalytic activity of π-allyl nickel(II) complex 3 can be rationalized by the regeneration of the nickel(0) complex via a stepwise hydrogen transfer from cod-derived π-allyl ligand to JoSPOphos, which subsequently promotes the nickel(0)/nickel(II) catalytic cycle. This new mechanistic scenario provides new insights into the unique reactivity patterns of the nickel/JoSPOphos catalytic system and new guidelines for future design of other asymmetric C–H transformation.
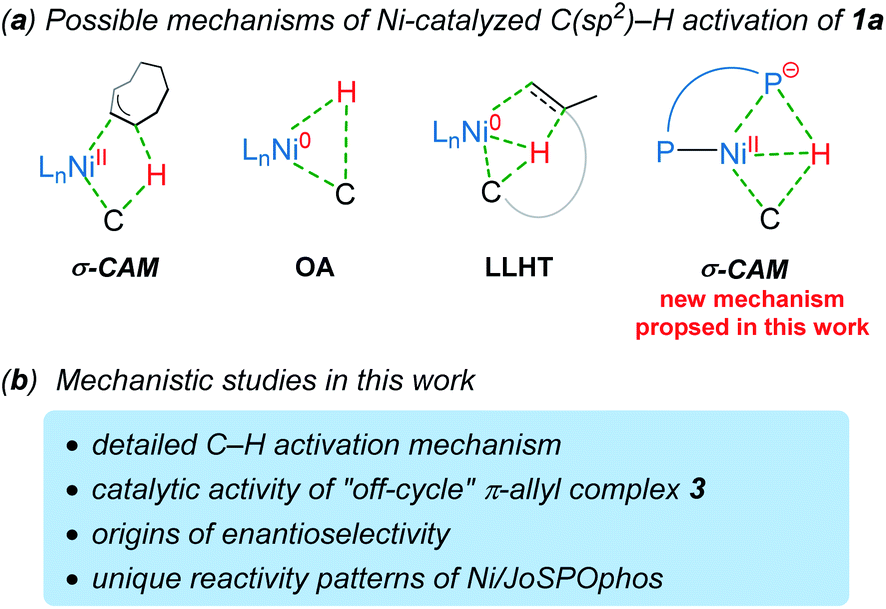 | ||
| Scheme 2 The plausible mechanisms of the Ni-catalyzed C(sp2)–H activation of 1a and mechanistic studies in this work. | ||
Computational details
Geometry optimizations were performed in solvent (toluene) at the B3LYP15-D3(BJ)16/6-31G(d,p)/LANL2DZ(Ni)17 level of theory with the corresponding effective core potential (ECP) on Ni. This computational method has been successfully applied to investigate various Ni-catalyzed C–H functionalizations.18 Frequency calculations were performed at the same theoretical level to verify the nature of the stationary points and to obtain the thermal Gibbs free energy corrections at 368.15 K. A correction to the harmonic oscillator approximation, as proposed by Grimme,19 was applied during the entropy calculations with a frequency cut-off of 50 cm−1. Considering the entropy in the solution phase is inadequately described due to the suppression of translational entropy upon moving from the gas phase to the solvent, the translational entropy in solution was estimated by the method of Whitesides.20 All the entropy corrections were finished by using the Goodvibes program.21 Intrinsic reaction coordinate (IRC) calculations were carried out to confirm that each transition state connected the corresponding reactant and product. To obtain more accurate electronic energies, single point calculations at B3LYP-D3(BJ)/def2-TZVP22 were carried out in the solvent. The continuum solvation model SMD23 was utilized to consider the bulk solvent effects. The correction caused by the different standard states in the gas phase and in solution was added to the free energies of all species.All the DFT calculations were conducted with the Gaussian 09 software.24 The natural population analysis (NPA) and localized orbital locator (LOL) analysis were performed by GenNBO 5.0 (ref. 25) and Multiwfn 3.6 (ref. 26) respectively, using the wavefunction obtained from B3LYP-D3(BJ)/6-31G(d,p)/LANL2DZ(Ni) level. The 3D diagrams of molecules were prepared by CYLview.27
Results and discussion
The four possible catalytic cycles
On the basis of previous investigations7,11 and our calculations, there are four plausible mechanisms of the Ni-catalyzed enantioselective C–H cyclization of imidazoles. As shown in Scheme 3, the σ-CAM mechanism, starting from the π-allyl nickel(II) complex 3, involves C(sp2)–H cleavage via σ-complex-assisted metathesis, migratory insertion and the second σ-complex-assisted metathesis. The OA mechanism starts from the nickel(0) complex 4 and proceeds via C(sp2)–H oxidative addition, migratory insertion and C–C reductive elimination to afford the final product and regenerate the active nickel(0) catalyst. The LLHT mechanism consists of C(sp2)–H activation via direct ligand-to-ligand hydrogen transfer from the imidazole to the alkene and the subsequent reductive elimination. This LLHT mechanism, originally proposed by Eisenstein and Perutz for hydrofluoroarylation of alkynes with Ni catalysts,14 is the more commonly proposed mechanism for Ni-catalyzed hydroarylation. Another unconventional σ-CAM mechanism, involving stepwise hydrogen transfer from JoSPOphos ligand to the alkene via P–H oxidative addition/migratory insertion, C(sp2)–H activation via σ-complex-assisted metathesis and reductive elimination, is proposed in this work.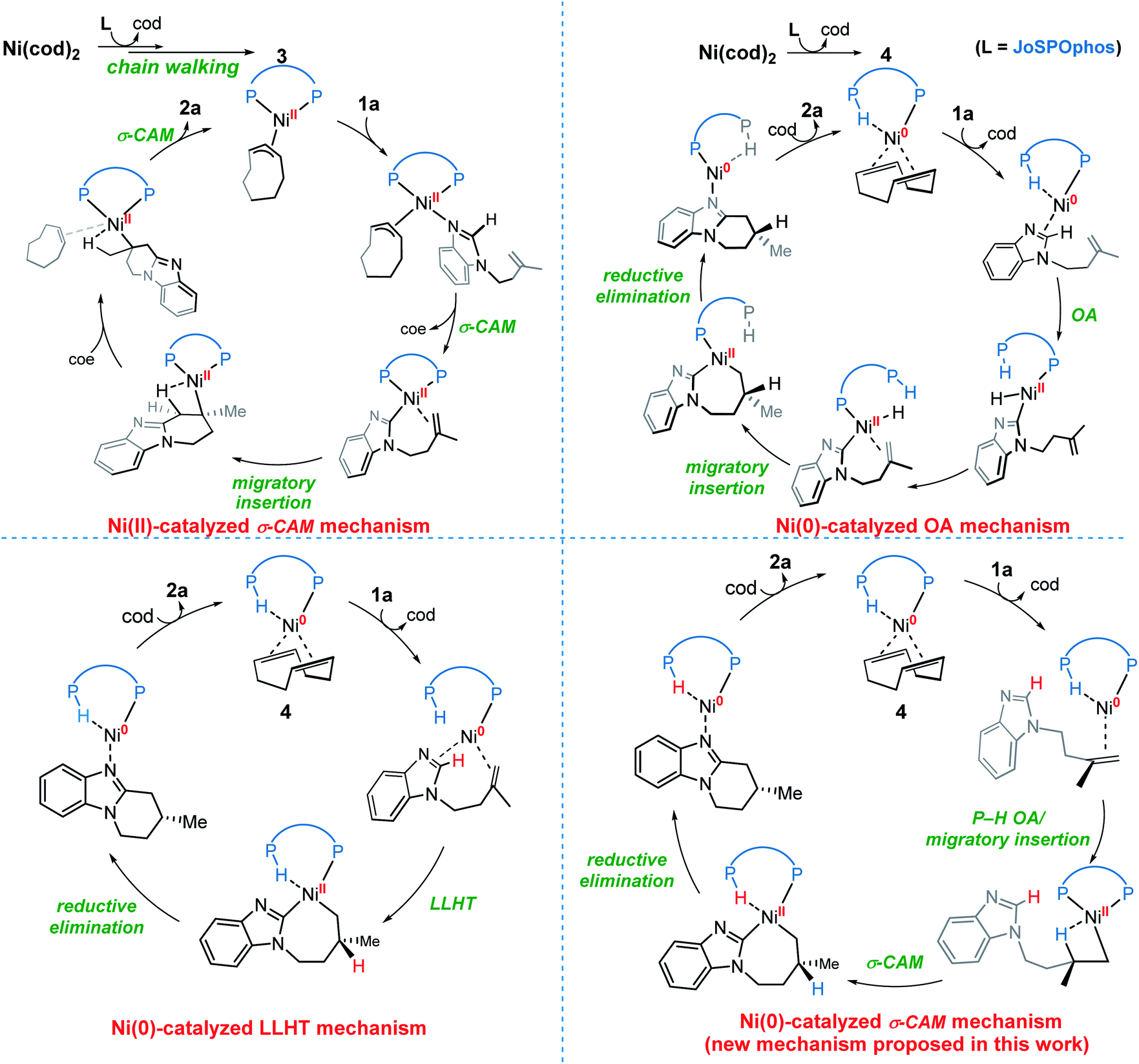 | ||
| Scheme 3 The four possible mechanisms of the Ni-catalyzed enantioselective C–H cyclization of imidazoles. | ||
Nickel(II)-catalyzed σ-CAM mechanism
We first investigated a catalytic cycle initiated by the π-allyl nickel(II) complex 3. The free energy profile for the nickel(II)-promoted hydroarylation is depicted in Fig. 1. All energies are presented with respect to complex 4, which is formed by ligand exchange of the precatalyst Ni(cod)2 with L. In complex 4, the bidentantate JoSPOphos ligand coordinates with the metal via the lone pair of the P(III) atom and the P(V)–H bond respectively. This complex is 3.1 kcal mol−1 more stable than the corresponding O-coordinated analog (see Scheme S1 and Table S1 in the ESI† for more details). We also considered the tautomeric equilibrium between the pentavalent phosphine oxide and the trivalent phosphinous acid.28 However, the tautomeric conversion is kinetically unfavorable for the JoSPOphos ligand (see Scheme S1 and Fig. S1 in the ESI†). For complex 4, the P–H bond becomes slightly longer by 0.04 Å after the formation of the agostic interaction. The agostic interaction between P–H with nickel(0) is further confirmed by Bader's atoms in molecules (AIM) analysis,29 which shows there exists a bond critical point between Ni and H atoms (Fig. 2). To the best of our knowledge, this kind of binding mode of JoSPOphos ligand has not been explored previously.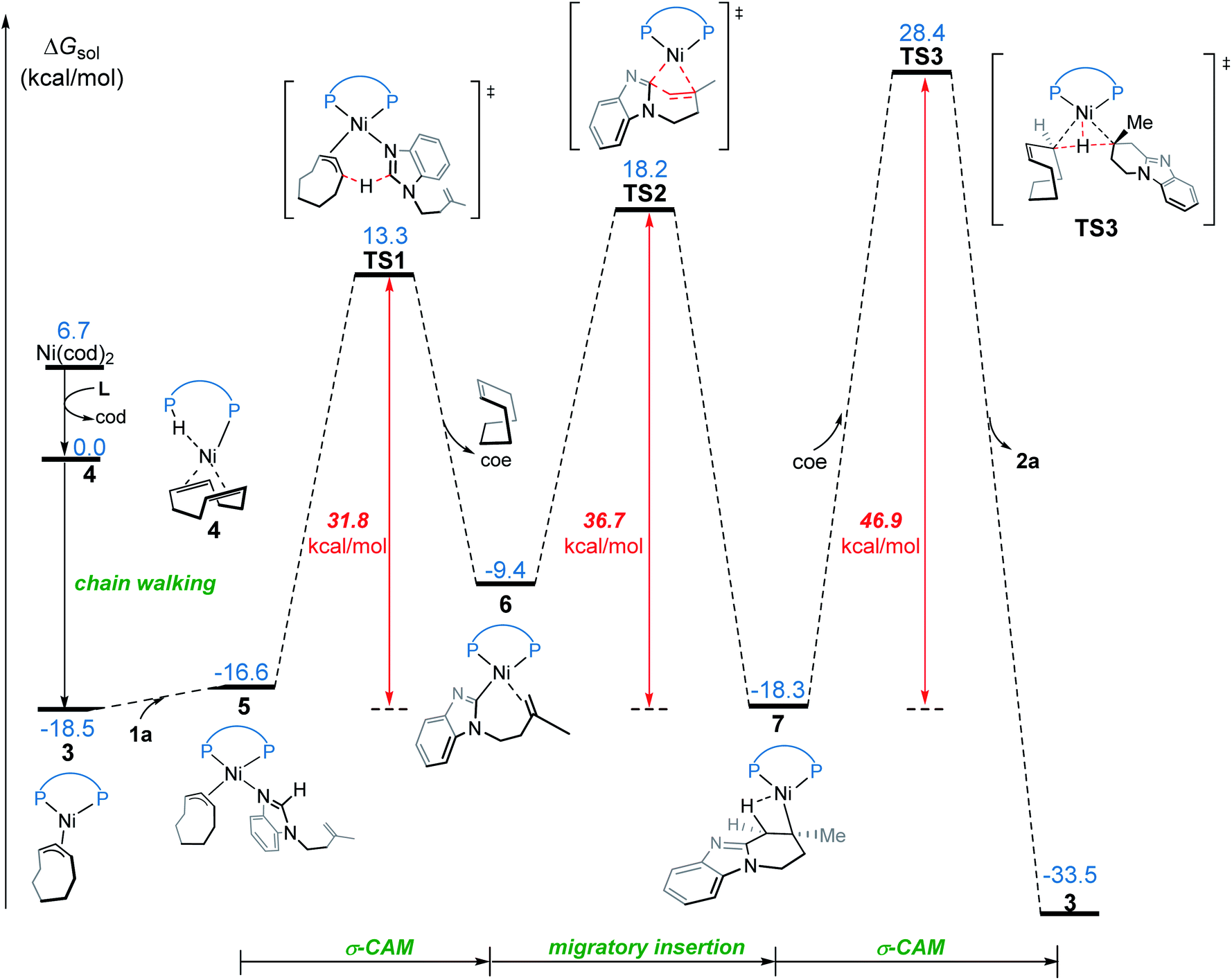 | ||
| Fig. 1 Free energy profile for nickel(II)-promoted hydroarylation via the σ-CAM mechanism. | ||
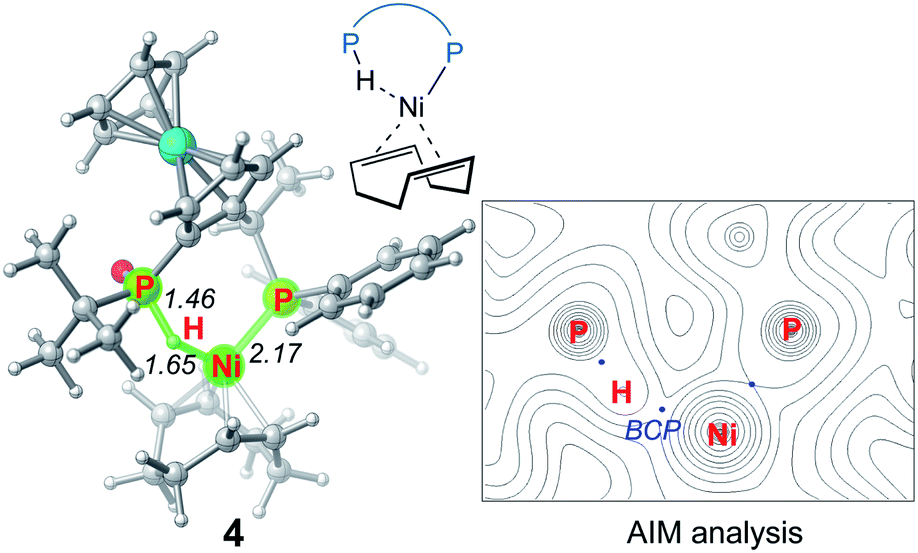 | ||
| Fig. 2 Optimized structure of 4 with selected bond distances in Å and corresponding AIM analysis. | ||
Complex 4 then undergoes facile P–H oxidative addition along with migratory insertion to form a nickel(II) intermediate, from which the traditional chain walking involving consecutive β-hydride elimination/migratory insertion occurs to yield the π-allyl nickel(II) complex 3 (Fig. S2 and S3 in the ESI†). As compared to the precatalyst Ni(cod)2, complex 3 is 25.2 kcal mol−1 more stable, explaining why it can thus be isolated in the experiment.7
From complex 3, coordination of the substrate 1avia N atom forms the four-coordinate complex 5. The subsequent σ-complex-assisted metathesis step with π-allyl as the base (TS1; see Fig. 3 for the optimized structure) requires 31.8 kcal mol−1, relative to 3. In the resulting deprotonated intermediate 6, the olefin moiety of the substrate is coordinated with the metal center to get prepared for the following cyclization. The olefin migratory insertion into the newly formed Ni–C bond takes place via four-membered transition state TS2, with a much higher barrier relative to 3.
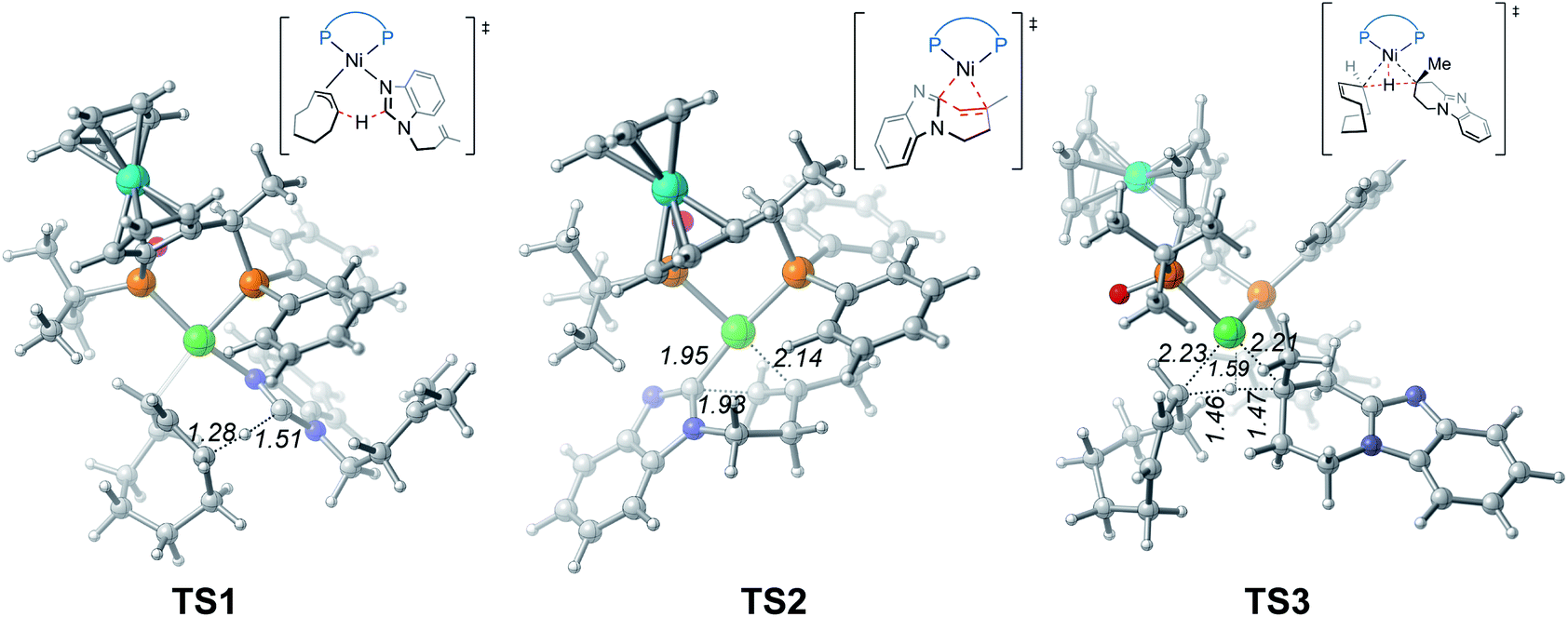 | ||
| Fig. 3 Optimized structures of transition states TS1, TS2 and TS3 with selected bond distances in Å. | ||
To complete the catalytic cycle, protonation is required for the cyclized intermediate 7 to liberate the final product. The corresponding C(sp3)–H bond cleavage occurs via σ-CAM transition state TS3, in which the proton is transferred from cyclooctene (COE) to the cyclized moiety. However, the σ-CAM process is highly disfavored due to the significantly higher barrier (TS3, ΔG‡ = 46.9 kcal mol−1 relative to 3). We also considered the proton abstraction from an additional substrate 1a, but the corresponding transition state is even higher in energy (see TS3′ in Fig. S4†). Overall, our calculations highlight that, while the step of σ-CAM C(sp2)–H activation is operative, this reaction pathway is kinetically unfavorable due to the considerably higher barrier of the succeeding steps, especially the second σ-CAM. The experimentally observed catalytic activity of nickel(II) complex 3 will be discussed later.
Nickel(0)-catalyzed OA mechanism
From complex 4, the substrate-ligand exchange with 1a can form several different substrate-coordinated complexes via either N- or alkene-coordination. As shown in Fig. 4, for the mono-imidazole complexes, the formations of η2-alkene coordination complex c1 and the N-coordinate complex c2 from 4 are endergonic by 2.3 and 7.2 kcal mol−1, respectively. Further coordination of the substrates results in various bisligated complexes, among which the Y-shape complex c3 is the most energetically favorable one. All the possible substrate-coordinated complexes are compared in Fig. S7 and Table S2 in the ESI.†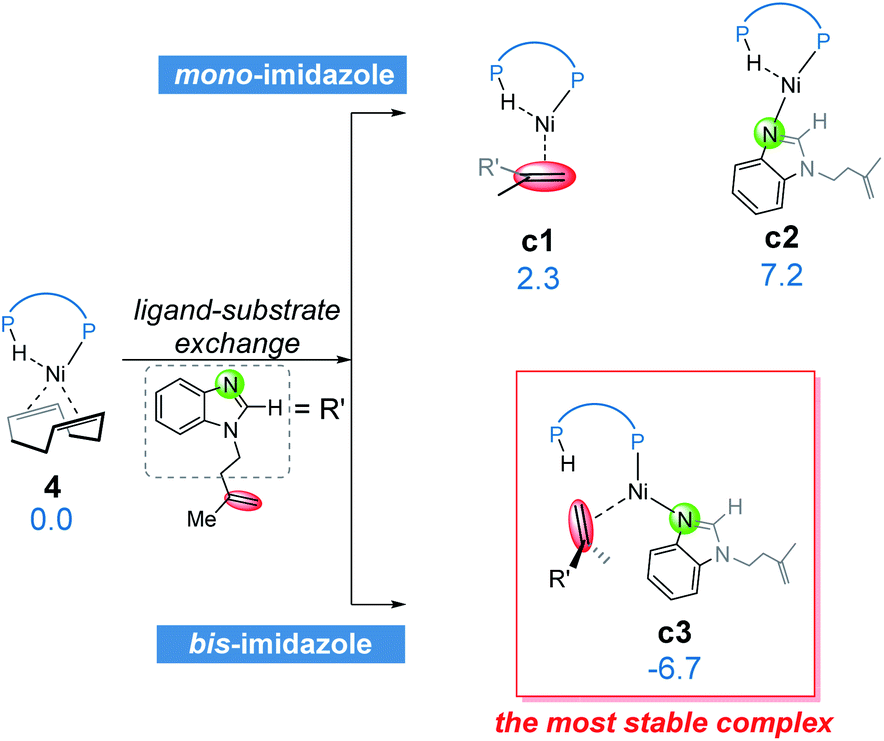 | ||
| Fig. 4 The possible ligand-substrate exchanges and representative substrate-coordinated complexes with energies in kcal mol−1. | ||
In the OA mechanism, dissociation of one substrate from catalyst c3 occurs first to generate intermediate 8, which is endergonic by 17.7 kcal mol−1 (Fig. 5). The C(sp2)–H oxidative addition then occurs via a three-membered transition state TS4, which requires a relatively higher barrier of 33.1 kcal mol−1. Subsequently, the resulting nickel hydride 9 undergoes consecutive isomerization to generate the η2-alkene coordination complex 11. This complex (11) undergoes migratory insertion viaTS5 to form the nickelacycle intermediate 12. The migratory insertion might also occur with intermediate 10, but this alternative reaction pathway (TS5′ in Fig. S8†) is less favorable as compared to TS5, probably due to the absence of agostic interaction. Intermediate 12 subsequently undergoes C–C reductive elimination viaTS6 to afford the product-coordinated complex 13. Product liberation of complex 13 releases the endo-product and regenerates the active catalyst c3. In the OA mechanism, the migratory insertion requires an activation free energy of 36.7 kcal mol−1 with respect to c3, which indicates this mechanism is infeasible under the experimental conditions.
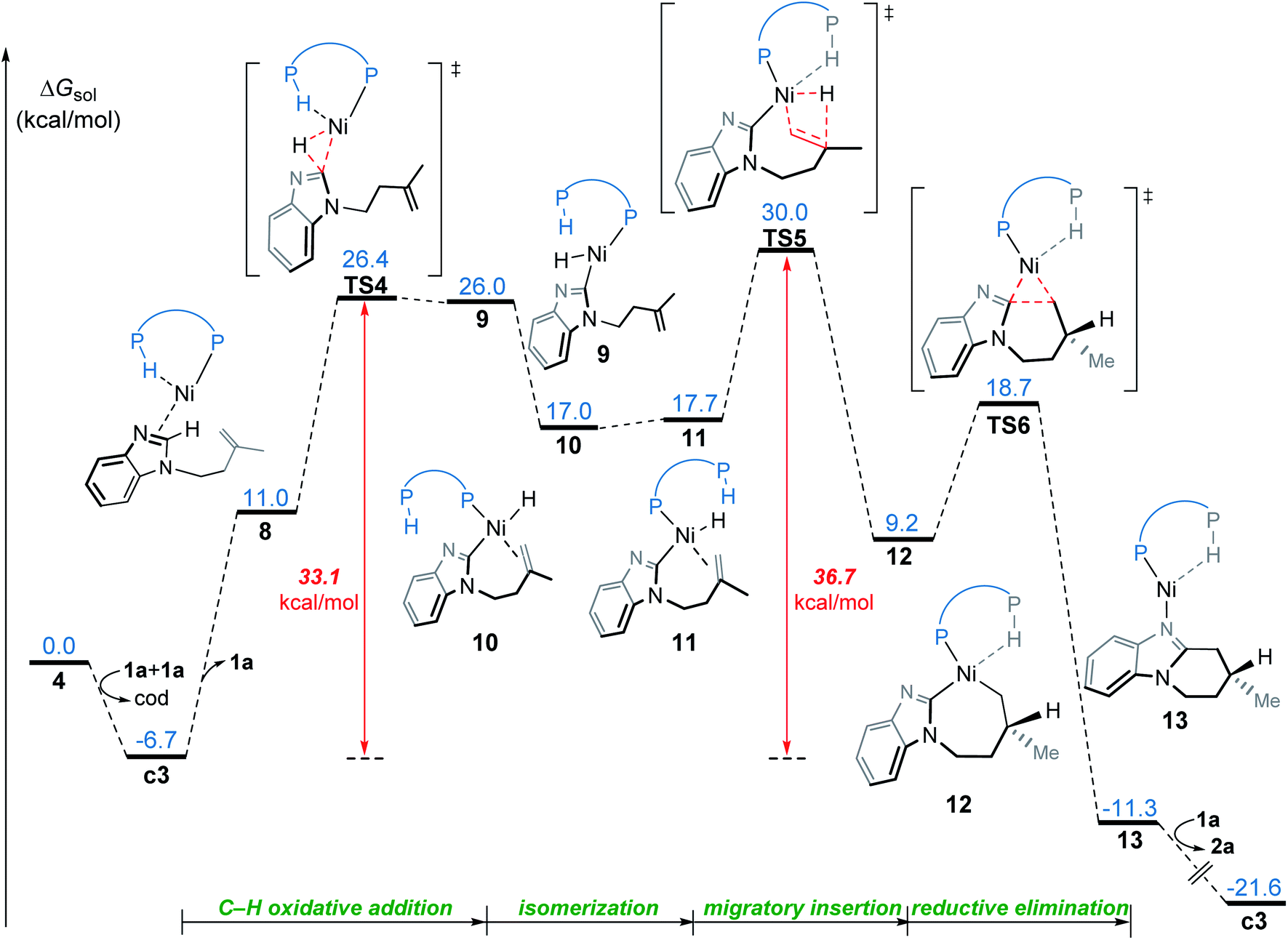 | ||
| Fig. 5 Free energy profile for nickel(0)-promoted hydroarylation via the OA mechanism. | ||
Nickel(0)-catalyzed LLHT mechanism
In the LLHT mechanism, the formation of the alkene-coordinated complex c7 from c3 is 14.7 kcal mol−1 endergonic (Fig. 6). In the three-coordinate complex c7, the C(sp2)–H bond becomes closer to the metal center and the agostic interaction between P–H with nickel(0) cannot be formed due to the steric repulsions between SPO and the benzoimidazolyl moiety in the substrate. Complex c7 then undergoes LLHT viaTS7,30,31 in which the oxidative addition of the C(sp2)–H bond is coupled with migratory insertion. The preference for LLHT mechanism for nickel catalyst can be attributed to the relatively small atomic radius of nickel and weak Ni(II)–H bond. To further investigate electronic structures of the transition state TS7, we performed additional NPA and LOL calculations. The LOL map in Fig. 7 clearly reveals that, although the Ni–H distance is relatively short at the transition state, the electron is localized around the migrating hydrogen instead of the middle regions between Ni and H atoms. The LOL picture is consistent with the calculated NPA charge of the transferring charge of H; the charge of H decreases significantly at the first-order saddle point.32 During the LLHT process, the NPA charge of Ni gradually increases, in agreement with the characteristic of oxidative addition.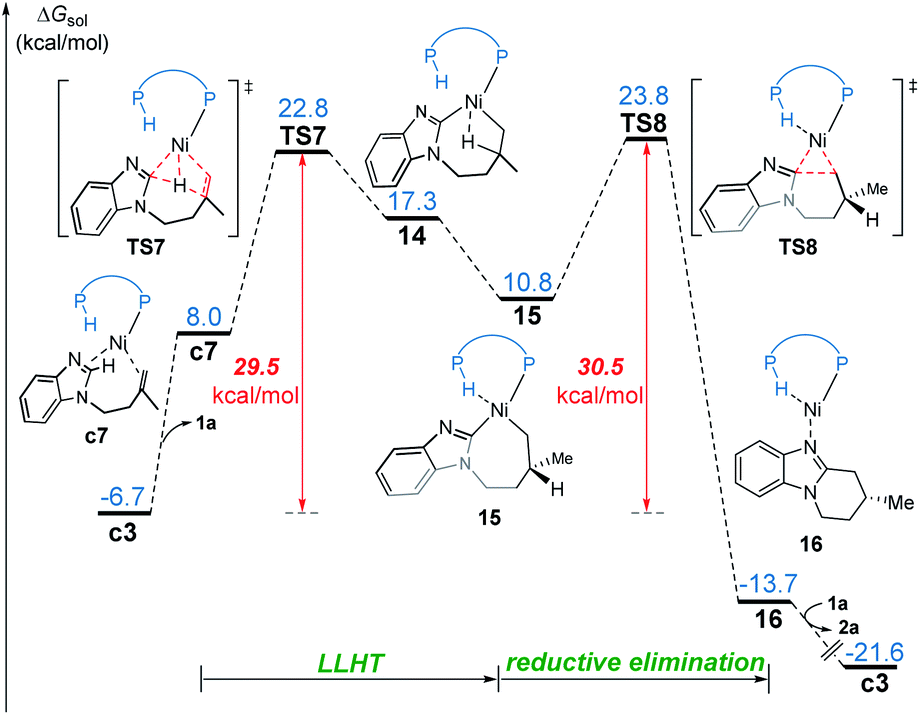 | ||
| Fig. 6 Free energy profile for nickel(0)-promoted hydroarylation via the LLHT mechanism. | ||
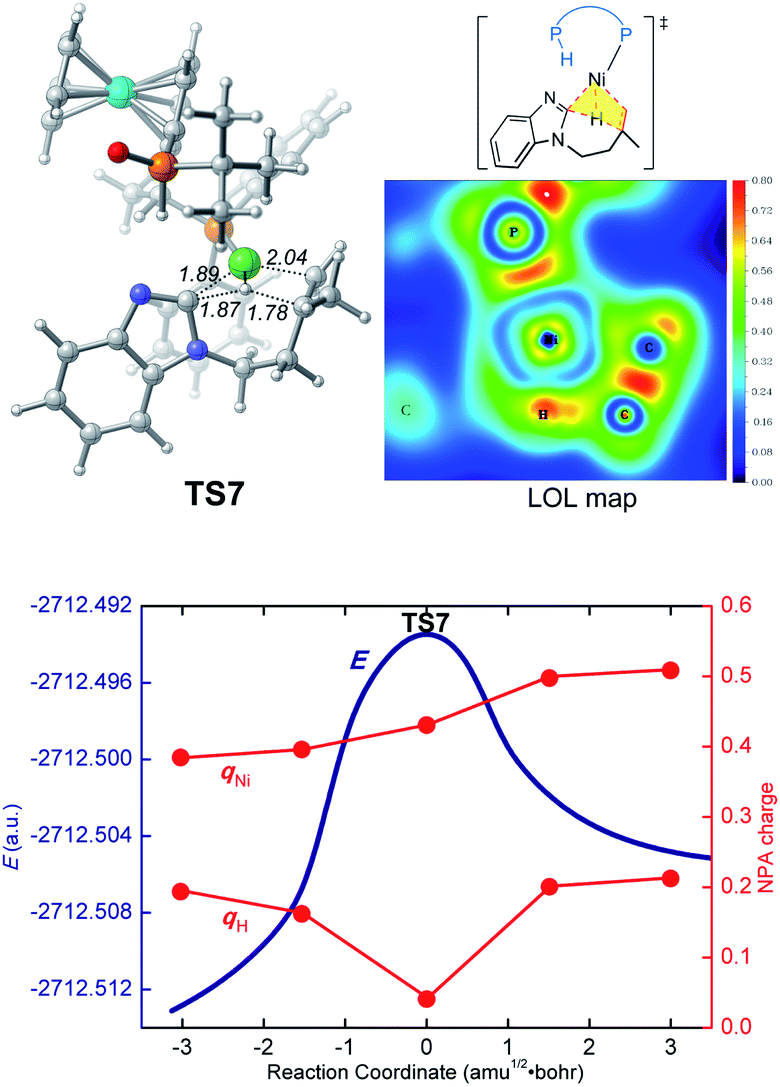 | ||
| Fig. 7 Optimized structure of the LLHT transition state TS7 with selected bond distances in Å, the LOL map, and the electronic energies (E) and NPA charges (q) along the intrinsic reaction coordinates. | ||
In intermediate 14, there exists a β-agostic interaction between nickel(II) with the newly formed C(sp3)–H bond. Prior to the following C–C reductive elimination, cleavage of the β-agostic interaction is required. From 14 to 15, the agostic interaction is transferred from C–H bond toward P–H bond, which is exergonic by 6.5 kcal mol−1. Intermediate 15 then undergoes irreversible reductive elimination viaTS8 to form the C–C bond, generating the stable product-coordinated complex 16. The reductive elimination barrier is only 1.0 kcal mol−1 higher than that of LLHT (TS8versusTS7). The rate-determining reductive elimination step is in good agreement with the experimentally-observed kinetic isotope effect (KIE) of kH/kD ≈ 1.1. Finally, the catalytic cycle was completed by ligand exchange between complex 16 and the substrate to liberate the desired product 2a and regenerate c3. Compared to the results of the OA pathway, the nickel(0)-promoted hydroarylation via the LLHT mechanism is much more favorable, which is consistent with previous results reported by Zhang on the Ni0/N-heterocyclic carbene-catalyzed C–H cyclization of pyridines with alkenes.11c
The reaction pathways for the formation of the other enantiomer of product are provided in Fig. S9 and S10.† Our calculations reveal that the LLHT transition state TS7′ leading to the (S)-product is 4.1 kcal mol−1 higher in energy than TS7 leading to the (R)-product (Fig. 8). Interestingly, in the pathway of the formation of (S)-product, the reductive elimination (TS8′ in Fig. S9†) becomes more facile and the LLHT is the rate-determining step. Finally, the overall barrier difference between the two competing pathways is 3.1 kcal mol−1, which results in a calculated enantiomeric ratio of 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 95 °C. This result is consistent with the experimentally observed ratio of 96:4.
:1 at 95 °C. This result is consistent with the experimentally observed ratio of 96:4.
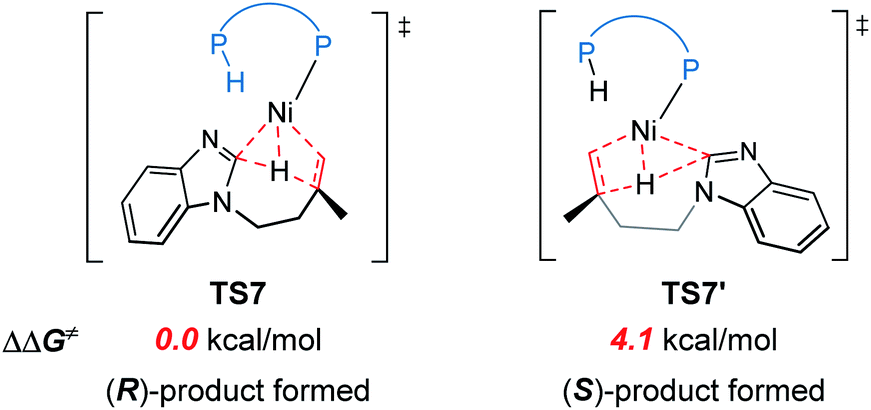 | ||
| Fig. 8 A comparison of the two competing LLHT transition states TS7and TS7′. | ||
A new type of σ-CAM mechanism
In the new σ-CAM mechanism, the σ-coordinated complex c1 first undergoes stepwise hydrogen transfer from the JoSPOphos to the alkene via the P–H oxidative addition/migratory insertion, generating the agostic intermediate 18 (Fig. 9). The c1 → 17 → 18 transformation is very facile, with a fairly low energy barrier. Interestingly, this process is rather similar to the migration of hydrogen to a cod ligand during the chain walking process in Fig. S2,† which indicates that this type of stepwise hydrogen transfer may be universal in the nickel(0)/JoSPOphos-catalyzed reactions that involve alkene or alkyne substrates. Rotation of the newly formed Ni–C bond in 18 results in the three-coordinate complex 19, which then undergoes σ-CAM viaTS11 to generate the nickelacycle intermediate 15. During this process, the hydrogen transfers from C(sp2)–H to the P(V) atom of JoSPOphos. Although the behavior of hydrogen transfer in σ-CAM transition state TS11 is similar to that in the LLHT transition state TS7, the changes in the formal oxidation state of Ni are obviously different. The metal is oxidized from Ni(0) to Ni(II) after the LLHT, while the formal oxidation state of Ni remains unchanged after the σ-CAM. To further illustrate this difference, we performed LOL and NPA calculations on TS11. As shown in Fig. 10, the LOL map clearly demonstrates the characteristic of the forming Ni–C and P–H bonds. In contrast to the results of LLHT (Fig. 7), the NPA charge of Ni remains almost constant during the σ-CAM, while the charge of the transferring H gradually decreases along the intrinsic reaction coordinates and becomes negative in intermediate 15. From intermediate 15, the subsequent elementary steps are the same with those in the LLHT mechanism. When comparing the two different modes of C–H activation in Fig. 6 and 9, the energy difference between the two transition states (TS7versusTS11) is only 0.4 kcal mol−1, indicating the LLHT mechanism and the new σ-CAM mechanism both being favorable under the experimental conditions. | ||
| Fig. 9 Free energy profile for nickel(0)-promoted hydroarylation via the σ-CAM mechanism. | ||
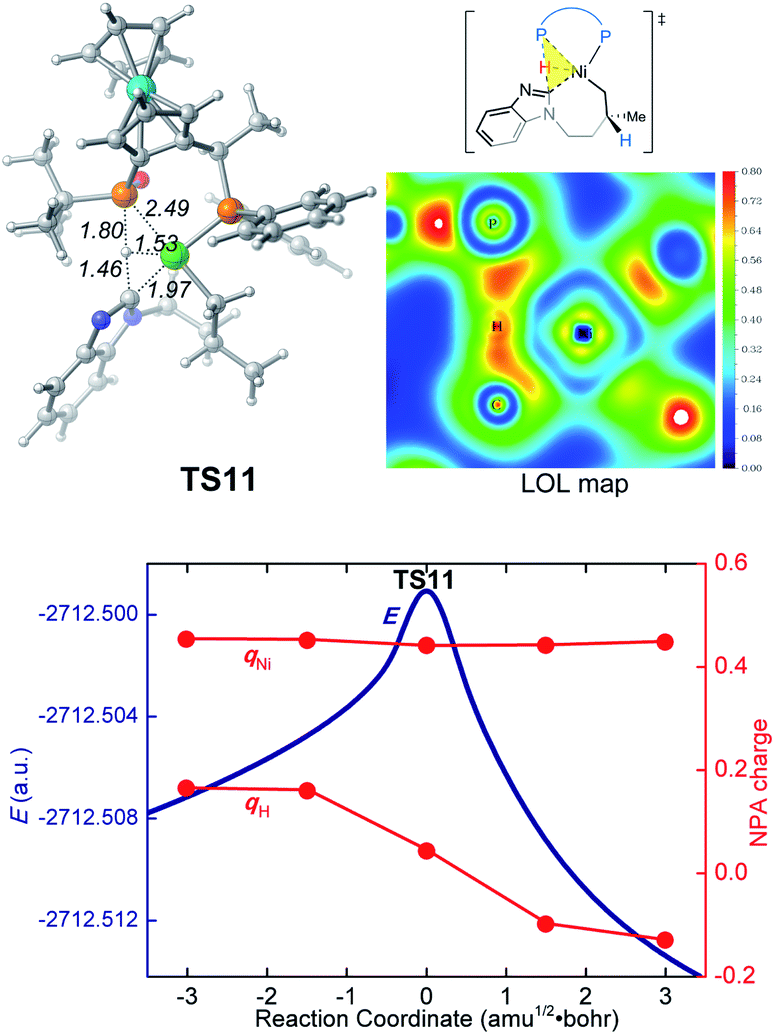 | ||
| Fig. 10 Optimized structure of TS11 with selected bond distances in Å, the LOL map and the electronic energies (E) and NPA charges (q) along the intrinsic reaction coordinates. | ||
On the basis of the above results of nickel(0)-catalyzed hydroarylation via the new σ-CAM mechanism, the coordination of alkene with nickel(0) through the Re- or Si-face and the subsequent migratory insertion of alkene into the Ni–H bond lead to the overall enantioselectivity. The insertion by the Re- or Si-face and the following σ-CAM and reductive elimination will lead to final (R)- or (S)-product, respectively (Fig. 11a and b). The free energy profile for the competing pathway that results in the (S)-product is given in Fig. S11.† Our calculated results reveal that the stepwise hydrogen transfer during the formation of (S)-product is also very facile. Finally, the enantiomeric ratio is determined by the rate-determining reductive elimination. The reductive elimination transition state TS8′′ leading to the (S)-product is 2.3 kcal mol−1 less stable than TS8 leading to the (R)-product (Fig. 11b). This activation energy difference corresponds to a calculated enantiomeric ratio of 96:4 at 95 °C. This result agrees indeed well with the experimentally observed ratio of 96:4.7
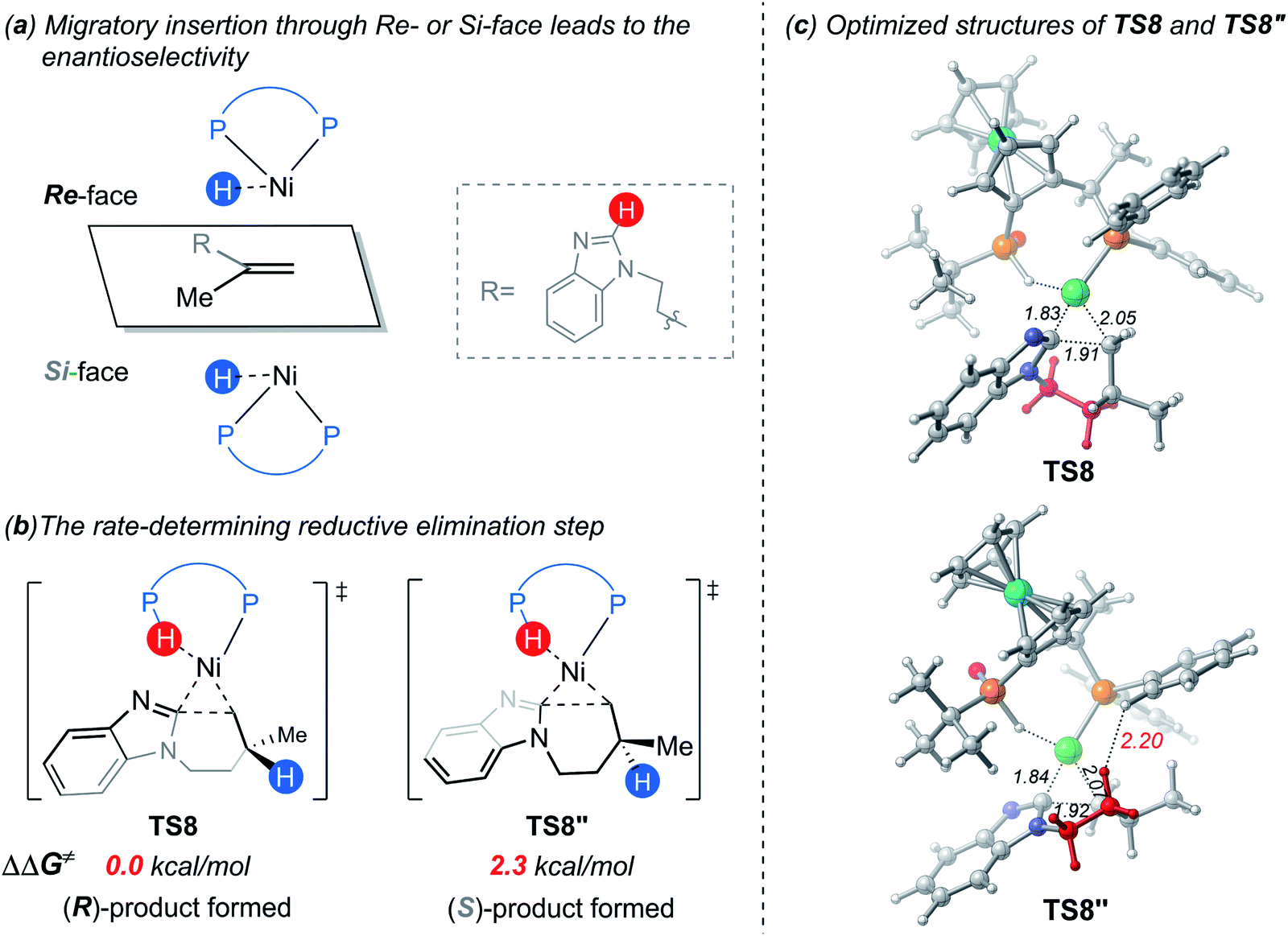 | ||
| Fig. 11 (a) Migratory insertion by the Re- or Si-face of the substrate; (b) the two competing reductive elimination transition states; (c) the optimized structures of TS8 and TS8′′ with selected bond distances in Å. | ||
In experiment, introducing different substituents on the two P atoms of JoSPOphos were found to affect the enantiomeric ratio, which highlights the importance of the interactions between the ligand and the substrate. To further understand the origins of the enantioselectivity, we examined the optimized structures of the two key reductive elimination transition states (Fig. 11c). A remarkable structural difference between TS8 and TS8′′ represents the orientation of dimethylene group in the substrates. In TS8, the dimethylene group points away from JoSPOphos, thus small steric repulsions are expected between the ligand and the substrate. In contrast, in the unfavorable TS8′′, the dimethylene motif points towards the ligand, resulting in relatively stronger steric repulsions between methylene group of the substrate and the bulky phenyl group in the ligand, as manifested by the shorter H⋯H distance (2.20 Å). Therefore, the more significant ligand-substrate steric repulsions in TS8′′ contribute to its relatively higher energy, eventually leading to the observed enantioselectivity.
Overall mechanism of the nickel-catalyzed hydroarylation
Our calculations reveal that the most favorable pathway involves the nickel(0) catalyst, while the nickel(II)-promoted reaction is not operative due to the significantly higher barrier of the last σ-CAM step (Fig. 1). This result prompted us to perform further experimental mechanistic investigations into the observed catalytic activity of the π-allyl nickel(II) complex 3. To this end, careful gas chromatographic GC-FID analysis of the solution resulting from the stoichiometric reaction between complexes 3 and 1a demonstrated the formation of 1,3-cyclooctadiene (1,3-cod) at the end of catalysis (Fig. S12†). On the basis of this experimental observation, a mechanism of the regeneration of nickel(0) catalyst c3 from nickel(II) complex 3 is proposed and shown in Fig. 12. The π-allyl nickel(II) complex 3 first isomerizes to the unstable agostic intermediate 20 that subsequently undergoes facile β-hydride elimination/reductive elimination to form the 1,3-cod-coordinated nickel(0) complex 22. From the thus-formed complex 22, ligand exchange with the substrate 1a regenerates the nickel(0) catalyst.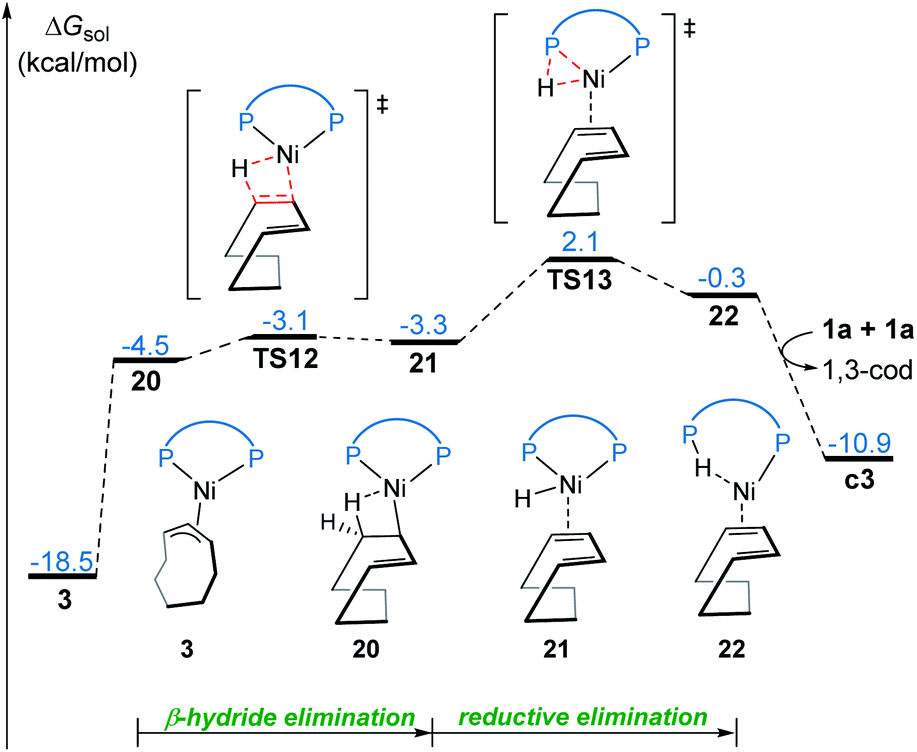 | ||
| Fig. 12 Free energy profile for the regeneration of the nickel(0) catalyst from 3. | ||
On the basis of the above mechanistic studies, the overall mechanism of Ni-catalyzed hydroarylation is summarized in Scheme 4. From the precatalyst Ni(cod)2, the ligand exchange generates the agostic complex 4. Further ligand-substrate exchange delivers the bisligated complex c3. From the catalyst c3, there exist two favorable mechanistic pathways to generate the nickelacycle intermediate B. In Path a, intermediate B is generated by the widely proposed LLHT mechanism. In Path b, facile P–H oxidative addition and migratory insertion occur to form the agostic nickel(II) intermediate A. Subsequently, a new type of σ-CAM occurs to generate intermediate B. Finally, the rate-determining C–C reductive elimination proceeds irreversibly to yield the final hydroarylation product.
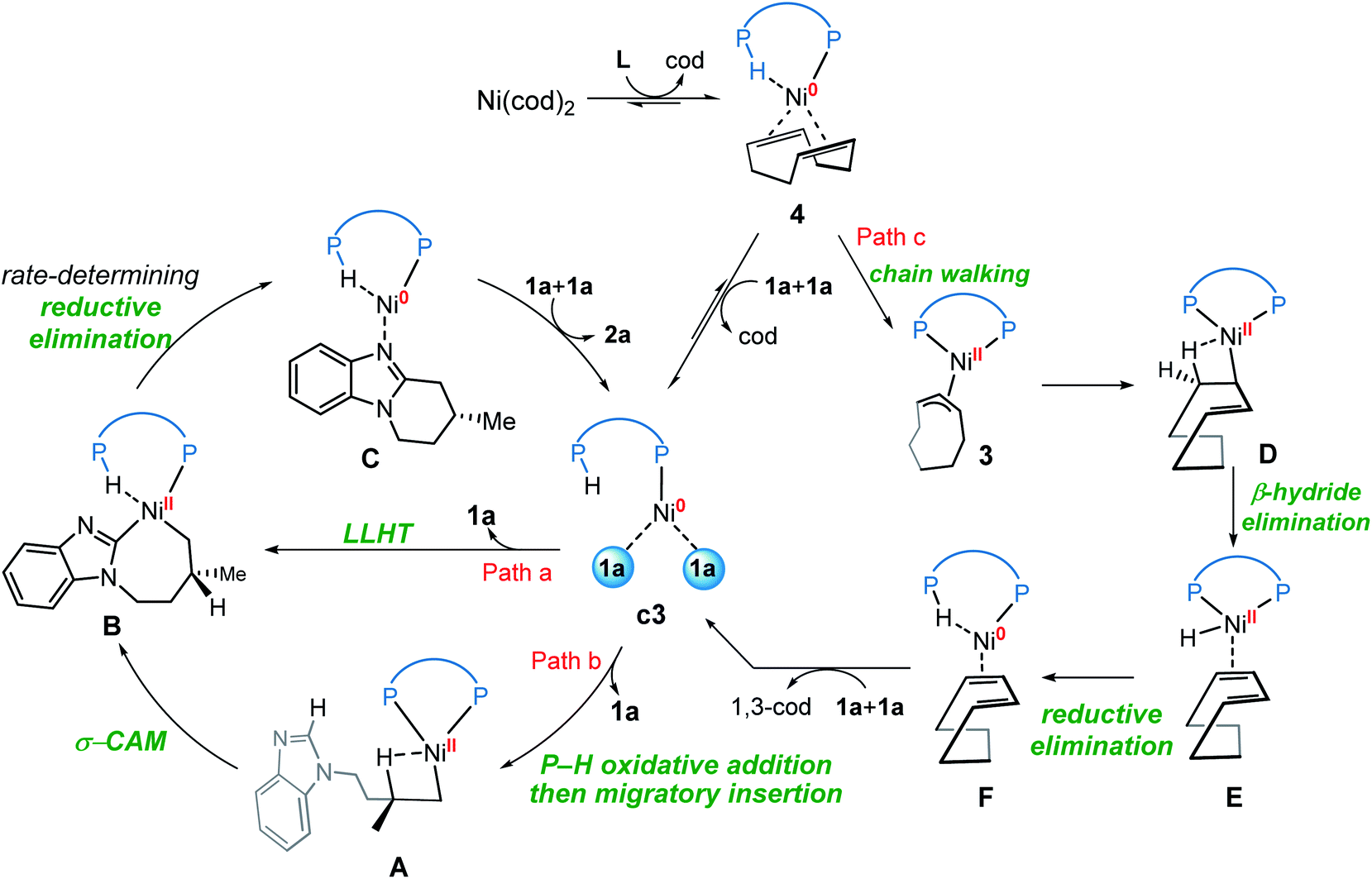 | ||
| Scheme 4 Overall mechanism of the nickel-catalyzed hydroarylation. | ||
From complex 4, a competing cod-mediated pathway can occur (Path c), ultimately resulting in the formation of the off-cycle π-allyl nickel(II) complex 3. The formation of 3 occurs via traditional chain walking mechanism, and is highly exergonic. Our calculations reveal that the π-allyl complex 3 is not the active catalyst. Through a sequential β-hydride elimination/reductive elimination, the catalyst resting state c3 is regenerated to initiate the product-forming pathway. Experimental mechanistic investigations provide additional support for this proposed mechanistic rationale. Furthermore, the overall mechanism indicates that increase in the amount of free cod ligand will facilitate the formation of off-cycle resting state 3 and impede reactivity of nickel(0) catalyst, which is consistent with the experimental observation that addition of free cod resulted in a deceleration of catalysis.
Concerning the reactivity of nickel catalysts, the relatively lower reduction potential and electronegativity leads to facile oxidative addition of nickel(0). Our computational results further disclose several unique reactivity patterns of the combination of nickel with JoSPOphos during the C–H activation with hydroarylation of alkenes (Scheme 5). The SPO can coordinate to nickel via an agostic interaction between P–H bond and the metal center, which is a somewhat more stable than the commonly proposed O-coordination. A facile hydrogen transfer between the SPO and the alkene through either a two-step P–H oxidative addition and migratory insertion or a concerted reverse LLHT (Fig. S5†) is observed for the nickel/JoSPOphos catalysis manifold. This reactivity mode is largely due to the relatively weak P–H bond and a facile oxidative addition at nickel(0). This manifold is expected to be facile for alkyne substrates likewise. Interestingly, the anion of SPO can function as a hydrogen acceptor, thus promoting the C(sp2)–H activation via an unconventional σ-CAM mechanism.
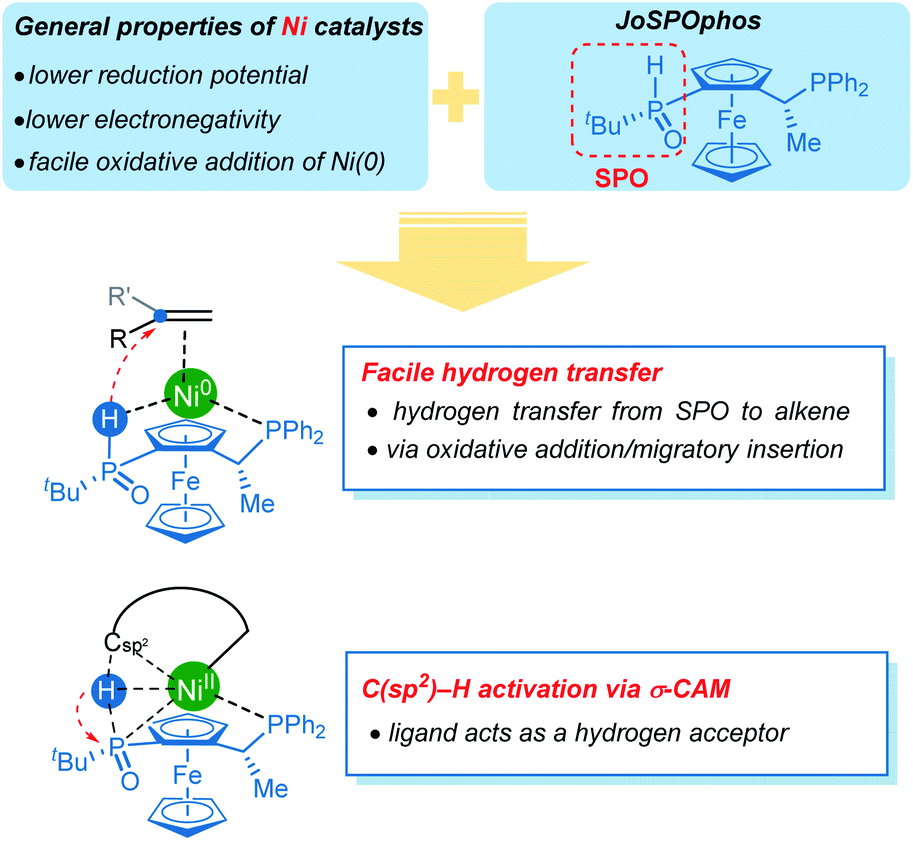 | ||
| Scheme 5 Summary of the reactivity of nickel/JoSPOphos catalytic system. | ||
Conclusions
In summary, the reaction mechanisms and origins of enantioselectivity of Ni-catalyzed intramolecular C–H cyclization of imidazoles were investigated by using DFT calculations. The calculations establish that the resting state of the catalyst is the nickel(0) complex rather than a π-allyl nickel(II) complex. In addition to the typically accepted LLHT/reductive elimination pathway, an unexpected nickel(0)/nickel(II) catalytic cycle comprising P–H oxidative addition, migratory insertion, C(sp2)–H activation via σ-CAM, and C–C reductive elimination is also favorable. Importantly, our computational results based on this new type of σ-CAM mechanism can reproduce rather well the experimentally observed enantioselectivity. In this new mechanism, the enantiomeric ratio is ultimately determined by the rate-determining reductive elimination steps. The different steric repulsions between methylene of the substrate and the methyl and phenyl groups of JoSPOphos ligand are responsible for the selectivity. Our calculations also show that the catalytic cycle initiated by the π-allyl nickel(II) complex is disfavored. The nickel(II) complex can be converted into the on-cycle nickel(0) complex via a sequential β-hydride elimination/reductive elimination, which was confirmed by experimental studies.The combined computational and experimental results provide a deeper understanding of the Ni-catalyzed enantioselective C–H cyclization, including the significant roles of the SPO unit in JoSPOphos. The SPO facilitates the coordination with the metal center via agostic interaction and the occurrence of hydrogen transfer and σ-CAM steps over the course of the reaction. We anticipate that this improved understanding will help to expand the scope of application of the nickel/JoSPOphos manifold, and provide further insights for the development of novel enantioselective C–H transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Natural Science Foundation of Shandong Province (ZR2019YQ11), the DFG (SPP 1807), and National Natural Science Foundation of China (NSFC No. 21601110) are gratefully acknowledged.Notes and references
- For selected reviews on C–H activation, see: (a) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS; (e) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 13, 8710–8753 CrossRef; (f) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (g) Q. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC; (h) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS.
- (a) K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu and E. A. Pidko, Chem. Rev., 2019, 119, 2453–2523 CrossRef CAS; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (c) Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS; (d) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS.
- (a) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- (a) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS; (b) L. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef.
- (a) I. Ali, M. N. Lone and H. Y. Aboul-Enein, MedChemComm, 2017, 8, 1742–1773 RSC; (b) Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef CAS.
- Y. Wang, S. Qi, Y. Luan, X. Han, S. Wang, H. Chen and M. Ye, J. Am. Chem. Soc., 2018, 140, 5360–5364 CrossRef CAS.
- J. Loup, V. Müller, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1749–1753 CrossRef CAS.
- For representative examples, see: (a) D. Berthold and B. Breit, Org. Lett., 2018, 20, 598–601 CrossRef CAS; (b) X. Wu, Z. Chen, Y. Bai and V. M. Dong, J. Am. Chem. Soc., 2016, 138, 12013–12016 CrossRef CAS; (c) A. M. Haydl, K. Xu and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 7149–7153 CrossRef CAS; (d) H. Landert, F. Spindler, A. Wyss, H. Blaser, B. Pugin, Y. Ribourduoille, B. Gschwend, B. Ramalingam and A. Pfaltz, Angew. Chem., Int. Ed., 2010, 49, 6873–6876 CrossRef CAS.
- A. J. Nett, J. Montgomery and P. M. Zimmerman, ACS Catal., 2017, 7, 7352–7362 CrossRef CAS.
- A. J. Nett, W. Zhao, P. M. Zimmerman and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 7636–7639 CrossRef CAS.
- (a) H. Shao, Y. Wang, C. W. Bielawski and P. Liu, ACS Catal., 2020, 10, 3820–3827 CrossRef CAS; (b) T. Sergeieva, T. A. Hamlin, S. Okovytyy, B. Breit and F. M. Bickelhaupt, Chem.–Eur. J., 2020, 26, 2342–2348 CrossRef CAS; (c) X. Zhao, X. Ma, R. Zhu and D. Zhang, Chem.–Eur. J., 2020, 26, 5459–5468 CrossRef CAS; (d) Y. Lang, M. Zhang, Y. Cao and G. Huang, Chem. Commun., 2018, 54, 2678–2681 RSC; (e) M. Zhang, L. Hu, Y. Lang, Y. Cao and G. Huang, J. Org. Chem., 2018, 83, 2937–2947 CrossRef CAS; (f) G. Huang and P. Liu, ACS Catal., 2016, 6, 809–820 CrossRef CAS.
- For selected reviews on the mechanisms of C–H activation, see: (a) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS; (b) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS; (c) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS; (e) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS.
- J. Guihaumé, S. Halbert, O. Eisenstein and R. N. Perutz, Organometallics, 2012, 31, 1300–1314 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909–9920 CrossRef CAS; (b) C. Ji and X. Hong, J. Am. Chem. Soc., 2017, 139, 15522–15529 CrossRef CAS; (c) S. Zhang, B. L. H. Taylor, C. Ji, Y. Gao, M. R. Harris, L. E. Hanna, E. R. Jarvo, K. N. Houk and X. Hong, J. Am. Chem. Soc., 2017, 139, 12994–13005 CrossRef CAS.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS.
- M. Mammen, E. I. Shakhnovich, J. M. Deutch and G. M. Whitesides, J. Org. Chem., 1998, 63, 3821–3830 CrossRef CAS.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000Research, 2020, 9, 291 Search PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, GenNBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- (a) A. Christiansen, C. Li, M. Garland, D. Selent, R. Ludwig, A. Spannenberg, W. Baumann, R. Franke and A. Börner, Eur. J. Org. Chem., 2010, 2010, 2733–2741 CrossRef; (b) L. Ackermann, Isr. J. Chem., 2010, 50, 652–663 CrossRef CAS; (c) L. Ackermann, Synthesis, 2006, 1557–1571 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- We also considered the possibility of the direct formation of intermediate 15 through a transition state by keeping P–H agostic interaction intact. However, we failed to locate the corresponding LLHT transition state due to the steric repulsions between SPO and the benzoimidazolyl.
- Similar transition states have been previously reported for other transition-metal catalysts, see: (a) S. Ding, L. Song, Y. Wang, X. Zhang, L. W. Chung, Y. Wu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 5632–5635 CrossRef CAS; (b) L. Song, S. Ding, Y. Wang, X. Zhang, Y. Wu and J. Sun, J. Org. Chem., 2016, 81, 6157–6164 CrossRef CAS; (c) B. E. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, ACS Catal., 2016, 6, 7536–7546 CrossRef CAS.
- X.-J. Liu, Y. Tian, H. Cui and H. Fan, Chem. Commun., 2018, 54, 7912–7915 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational and experimental results, calculated energies and imaginary frequencies, and Cartesian coordinates of optimized structures. See DOI: 10.1039/d0sc04578k |
| This journal is © The Royal Society of Chemistry 2021 |