
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on the reactivity of nucleophilic radiohalogens with arylboronic acids in water: access to an efficient single-step method for the radioiodination and astatination of antibodies†
Marion
Berdal
 a,
Sébastien
Gouard
a,
Romain
Eychenne
ab,
Séverine
Marionneau-Lambot
ae,
Mikaël
Croyal
cd,
Alain
Faivre-Chauvet
ae,
Michel
Chérel
af,
Joëlle
Gaschet
a,
Jean-François
Gestin
*a and
François
Guérard
*a
a,
Sébastien
Gouard
a,
Romain
Eychenne
ab,
Séverine
Marionneau-Lambot
ae,
Mikaël
Croyal
cd,
Alain
Faivre-Chauvet
ae,
Michel
Chérel
af,
Joëlle
Gaschet
a,
Jean-François
Gestin
*a and
François
Guérard
*a
aUniversité de Nantes, CNRS, Inserm, CRCINA, F-44000 Nantes, France. E-mail: francois.guerard@univ-nantes.fr
bArronax GIP, Saint-Herblain, France
cCRNH-O, Mass Spectrometry Core Facility, F-44000 Nantes, France
dNUN, INRA, CHU Nantes, UMR 1280, PhAN, IMAD, CRNH-O, F-44000 Nantes, France
eDepartment of Nuclear Medicine, CHU Nantes, Nantes, France
fICO-René Gauducheau, Saint-Herblain, France
First published on 23rd November 2020
Abstract
Easy access to radioiodinated and 211At-labelled bio(macro)molecules is essential to develop new strategies in nuclear imaging and targeted radionuclide therapy of cancers. Yet, the labelling of complex molecules with heavy radiohalogens is often poorly effective due to the multiple steps and intermediate purifications needed. Herein, we investigate the potential of arylboron chemistry as an alternative approach for the late stage labelling of antibodies. The reactivity of a model precursor, 4-chlorobenzeneboronic acid (1) with nucleophilic iodine-125 and astatine-211 was at first investigated in aqueous conditions. In the presence of a copper(II) catalyst and 1,10-phenanthroline, quantitative radiochemical yields (RCYs) were achieved within 30 minutes at room temperature. The optimum conditions were then applied to a CD138 targeting monoclonal antibody (mAb) that has previously been validated for imaging and therapy in a preclinical model of multiple myeloma. RCYs remained high (>80% for 125I-labelling and >95% for 211At-labelling), and the whole procedure led to increased specific activities within less time in comparison with previously reported methods. Biodistribution study in mice indicated that targeting properties of the radiolabelled mAb were well preserved, leading to a high tumour uptake in a CD138 expressing tumour model. The possibility of divergent synthesis from a common modified carrier protein demonstrated herein opens facilitated perspectives in radiotheranostic applications with the radioiodine/211At pairs. Overall, the possibility to develop radiolabelling kits offered by this procedure should facilitate its translation to clinical applications.
1. Introduction
Astatine-211 is a radionuclide of high interest in nuclear medicine for targeted α-therapy, a promising modality for the treatment of disseminated cancers.1,2211At emits one high energy α particle (5.7 or 7.4 MeV) per decay, with an intermediate half-life of 7.21 h making it compatible with a broad spectrum of carrier compounds pharmacokinetics, from small organic molecules to heavy intact antibodies. Reports over the past two decades have shown its promising efficacy on the eradication of isolated cancer cells or small cell clusters with limited toxicity at preclinical and clinical stages.3–6 On the other hand, several radioisotopes of iodine have long been considered of interest for therapeutic and imaging applications, the most representative being iodine-131 (β− and γ emitter, t1/2 = 8 days), iodine-123 (γ emitter, t1/2 = 13.2 h), iodine-124 (β+ emitter, t1/2 = 4.18 days) and iodine-125 (γ and Auger electrons, t1/2 = 59.5 days).7Astatine and iodine are both halogens and neighbours in the periodic table and, therefore, they exhibit a number of similar properties. Consequently, most reactions known for radioiodine are applicable similarly with astatine. Radiolabelling approaches have long been limited to nucleophilic halogen (or isotope) exchange, and to the highly popular electrophilic halodestannylation.8,9 These reactions are mainly used in order to form halo(hetero)aryl compounds, sp2 carbon–halogen bonds being preferred for stability reasons over sp3 carbon. Alternatively, promising nucleophilic strategies using the radiohalogen as a nucleophilic species (X−) were recently reported, based on aryliodonium salts or arylboronic acids/esters precursors that provided high radiochemical yields (RCYs) on a broad scope of substrates.10–12
The availability of efficient nucleophilic radiolabelling approaches are in several aspects desirable. First of all, they avoid the use of electrophilic species and oxidizing conditions required for their formation that may alter the substrate or lead to side reactions. Additionally, these approaches exhibit advantages in the practical point of view: on the one hand, iodine radioisotopes are available commercially as sodium iodide solutions, ready to be used in nucleophilic reactions without a reduction step. On the other hand, the nucleophilic At− species is much easier to produce since it is stable in reducing media over a broad pH range, whereas the electrophilic At+ species exists only in a small pH and redox potential domain.13 Better procedure robustness using nucleophilic astatine is thus expected in comparison with electrophilic approaches in which overoxidized non-reactive At species may negatively impact RCYs.
The first efficient approach for radiolabelling proteins with 211At, which remains also the most widely used so far, is a two-step procedure based on the radiosynthesis of an intermediate prosthetic group, N-[211At]succinimidylastatobenzoate ([211At]SAB), via electrophilic astatodestannylation of the corresponding aryltrialkylstannane precursor (Scheme 1a),14 similarly to the analogous procedure that had previously been developed for radioiodination.15 To overcome issues with the use of electrophilic astatine and to eliminate the need of toxic organotin agents, we recently revisited this approach using nucleophilic astatine or iodine and an aryliodonium salt precursor (Scheme 1b).16 On the other hand, a single-step strategy was developed by Lindegren et al., for which the precursor is grafted to the antibody prior to the radiolabelling, saving time and increasing RCYs compared to two-step strategies (scheme 1c).17,18 It is however still based on the previous electrophilic destannylation reaction and, although efficient for astatination, it cannot be applied for radioiodination since electrophilic radioiodine would react competitively with tyrosine and histidine residues, leading to suboptimal in vivo stability.19 This issue does not exists in the case of astatine since electrophilic astatine cannot react with tyrosines or histidines unless very drastic conditions are applied.20–22 Similarly, the one-step approach based on boron clusters which provides improved in vivo stability of radiolabelling, but can dramatically alter antibody biodistribution (Scheme 1d),23,24 is also not applicable to radioiodination since it requires the electrophilic halogen species that may, again, react with tyrosines and histidine residues.
 | ||
| Scheme 1 Strategies for radioiodination and astatination of antibodies. | ||
This incompatibility of use with radioiodine is a hurdle to the development of radiotheranostic antibodies based on the 211At/123,124I pairs. The concept of radiotheranostic pharmaceuticals is based on the use of a unique compound to perform both imaging and therapy,25,26 and is considered as a promising approach in personalized therapy.
In this context, it appears essential to develop late stage radiohalogenation approaches applicable to proteins with both iodine and astatine radioisotopes. This implies the use of nucleophilic halogens in order to avoid issues inherent to electrophilic approaches discussed above. Previously reported nucleophilic astatination reactions require elevated temperatures, which is not compatible with proteins.10,27,28
Interestingly, boron reagents have focused an increasing attention in radiolabelling chemistry over the past decade.29 The recently reported metal catalysed radiohalogenation of arylboron substrates appeared to us attractive to investigate as it was shown to be efficient at low temperature. However radiolabelling reactions were performed in organic solvents (acetone, methanol, acetonitrile) that are incompatible with antibodies that tolerate only aqueous conditions.12,30 A recent report on radiobromination showed that a substantial proportion of water (up to 50%) can be tolerated for this reaction although high temperatures were still used (80 °C).31
In this study, we aimed to reinvestigate radioiodination and astatination of arylboronic acids in water, at first on a model compound. We then applied the methodology to the 125I-radioiodination and 211At-astatination of an anti-CD138 monoclonal antibody (mAb) that is relevant for targeting multiple myeloma tumour cells. Our novel approach was then compared with the previously reported two-step approach in biodistribution studies with mice grafted with CD138 expressing tumours.
2. Results and discussion
2.1 Preliminary investigation with model compound
Before translation to a relevant biomacromolecule, the reactivity of iodide and astatide with the arylboronic acid functionality in water was probed and optimized with a simple aryl derivative, allowing for accurate analyses by reverse phase radio-HPLC that would not be possible with complex proteins. Thus, 4-chlorobenzeneboronic acid (1) was chosen as a model compound for its simplicity. The chloride in para position is moderately electron withdrawing (Hammet constant σ = 0.227) and may be considered as an average substituent in terms of electronic effects that may be found in functionalized compounds. The choice of the substituent, however, does not seem crucial for this preliminary set of experiments as little to no influence of electronic effects on the copper catalyzed radiohalogenation of arylboronic acids or esters was reported (unlike steric effects).12,30,32Before running the radiolabelling in water, we investigated the reaction in organic medium. We started with conditions similar to previous studies that showed Cu(pyridine)4(OTf)2 to be an efficient source of copper(II) for the Chan–Evans–Lam based reaction with radiohalogens.12,33 The reaction, conducted initially with a relatively high precursor concentration (25 mM) and equimolar amount of catalyst, for 30 min at room temperature, provided nearly quantitative RCYs with both 125I and 211At (Table 1). Interestingly, the same reaction conducted without catalyst led, as expected, to no radioiodination, whereas a substantial RCY (44%) was observed with [211At]NaAt. A nucleophilic process is unlikely to lead to this chemical conversion. Rather, our hypothesis is the possibility for the astatide anion to oxidize in the reaction medium into At+ that would then react electrophilically with the arylboronic acid group. This hypothesis is supported by previous studies having reported on the radioiodination of aryltrifluoroborate or arylboronic acid precursor using electrophilic radioiodide, in the presence or absence of a catalyst.34,35 We then probed the possibility to reduce the precursor concentration to a level acceptable for mAb radiolabelling. No drop in RCYs was observed when precursor concentration was decrease to 250 μM, a condition being chosen as it corresponds approximately, in terms of available arylboronic groups, to a solution of mAb concentrated at 6 mg mL−1 with a ratio of 6 arylboronic acids grafted per mAb.
|
|
|||||
|---|---|---|---|---|---|
| C(1) (mM) | C(Cu(OTf)2pyr4) (mM) | C(1,10-phenanthroline) (mM) | Solvent (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v) :v) |
RCY (%)a | |
| 125I | 211At | ||||
|
a Standard conditions: [125I]NaI or [211At]NaAt (0.5–1 MBq), 100 μL, 30 min, 23 °C, average of n = 3 runs.
b
n = 7.
|
|||||
| 25 | 0 | 0 | MeOH/H2O (9:1) |
0 | 44 ± 7b |
| 25 | 25 | 0 | MeOH/H2O (9:1) |
99 ± 1 | 99 ± 1 |
| 10 | 10 | 0 | MeOH/H2O (9:1) |
>99 | 98 ± 3 |
| 0.25 | 10 | 0 | MeOH/H2O (9:1) |
>99 | 99 ± 1 |
| 0.25 | 10 | 0 | H2O/MeOH (85:15) |
17 ± 2 | 74 ± 5 |
| 0.25 | 0 | 0 | H2O/MeOH (85:15) |
— | 0 |
| 0.25 | 10 | 10 | H2O/MeOH (85:15) |
85 ± 2 | >99 |
| 0.25 | 10 | 10 | H2O/DMF (85:15) |
91 ± 1 | 99 ± 1 |
| 0.25 | 10 | 10 | H2O/DMSO (85:15) |
88 ± 2 | 99 ± 1 |

We then switched to aqueous conditions, keeping 15% of methanol as co-solvent for dissolution of the precursor and catalyst. Divergent results were observed for [125I]iodide and [211At]astatide: whereas radioiodination RCY dropped to 17%, it remained at a relatively high level (74%) in the case of astatination. The first hypothesis to explain this difference was an electrophilic pathway with 211At, as in the assay performed in MeOH without catalyst. However, this hypothesis was ruled out as no radiochemical conversion was obtained when the radiolabelling was performed in water without copper salt.
The second hypothesis to explain this large discrepancy is the higher polarizability of astatide, which is consequently less deactivated by hydration than iodide in nucleophilic processes, the nucleophilicity of halogens being increasingly repressed by water from the heaviest to the lightest halogens.
In order to improve RCYs, we added 1,10-phenanthroline to the radiolabelling solution, this kind of ligands having already been reported by Zhang et al. and Reilly et al. to increase RCYs in organic medium.12,30 Excellent RCYs were reached again in this case (85% for radioiodination and a quantitative astatination). Radioiodination was further improved by changing methanol to DMF or DMSO to 91% and 88% RCY respectively, whereas astatination RCY remained quantitative. Since DMF was slightly better for radioiodination, and that it is known to be compatible with proteins (when used in reasonable proportions, below 20%), it was kept as a co-solvent for the continued study.
To come closer to radiolabelling conditions compatible with proteins that are generally stored and used in a buffer solution rather than in pure water, we investigated the reaction in relevant buffers (Table 2). All buffers were used at pH 7.4–7.5 in order to dissociate the effect of pH on the reaction from the nature of ions in solution. All tested conditions led to a significant decrease in radioiodination RCY, the worst salt being sodium citrate. This could be explained by chelation of copper(II) by citrates, limiting catalyst availability for the reaction. Chloride ions seem to be disadvantageous for the reaction as RCYs decreased in 0.9% NaCl solution. A hypothesis is the possible competition between chloride ions and iodide or astatide in the reaction. This can also explain in part the poor RCYs observed in PBS, as this solution contains a high concentration of chlorides as well. However, even lower RCYs were observed in PBS compared with NaCl, which may be attributed to the presence of phosphate ions that are known to cause copper precipitation.
| Solvent (v:v) |
RCYa (%) | |
|---|---|---|
| 125I | 211At | |
| a Standard conditions: (1) (0.25 mM), Cu(OTf)2pyr4 (10 mM), 1,10-phenanthroline (10 mM), [125I]NaI or [211At]NaAt (1–5 MBq), 30 min, 100 μL, 23 °C, n = 3. b Cu(OTf)2pyr4 (5 mM), 1,10-phenanthroline (5 mM). | ||
| 0.9% NaCl/DMF (85:15) |
38 ± 3 | 78 ± 13 |
| PBS/DMF (85:15) |
21 ± 2 | 31 ± 7 |
| Sodium acetate buffer 0.1 M pH 7.5/DMF (85:15) |
40 ± 4 | 84 ± 8 |
| Sodium citrate buffer 0.5 M pH 7.5/DMF (85:15) |
0b | — |
| Tris buffer 0.5 M pH 7.5/DMF (85:15) |
35 ± 8 | 47 ± 9 |
Even if no precipitation in PBS was observed in our assays, the interaction between phosphates and the copper catalyst is likely to have altered the reaction. NaCl solution and acetate buffer gave the best results, although moderate RCYs were obtained for radioiodination. However, we observed precipitation of a mAb in these buffers (the 9E7.4 IgG used in radiolabelling assays as discussed below) when the catalyst was added. This precipitation was attributed to the presence of copper(II) in solution since copper salts, and more generally polyvalent metallic ions are known to interact with proteins, leading to their precipitation or degradation.36,37 Whereas decreasing the copper catalyst or the mAb concentrations were optimizations that could be considered to limit this phenomenon, we choose to investigate the influence of the buffer nature on the protein precipitation. Among the tested buffers, only citrate and Tris prevented precipitation of the proteins. As radioiodination was inefficient in citrate buffer, Tris was selected as the buffer solution to investigate further. It has been previously shown that Tris (tris-(hydroxymethyl)aminomethane) can act as an efficient chelating agent for Cu2+via its three hydroxyl groups and its primary amine at pH ≥ 7, preventing free copper(II) to interact with proteins.38 Nonetheless, RCYs were moderate and not satisfying for the development of an efficient radiolabelling method. However, an investigation of the influence of pH showed quantitative RCYs from pH = 7 down to pH = 2 below which the catalyst precipitated (Fig. 1). This last round of optimization showed that arylboronic acid chemistry is extremely efficient for radioiodination and astatination in water and is a realistic approach for the late stage radiolabelling of proteins.
 | ||
Fig. 1 Influence of pH on the 125I-iodination ( ) and 211At-astatination ( ) and 211At-astatination ( ) of 4-chlorobenzeneboronic acid (1). RCYs determined by radio-HPLC analysis of the crude product. Standard conditions: (1) (0.25 mM), Cu(OTf)2pyr4 (10 mM), 1,10-phenanthroline (10 mM), [125I]NaI or [211At]NaAt (1–5 MBq), Tris buffer 0.5 M/DMF (85:15), 30 min, 100 μL, 23 °C, n ≥ 2. ) of 4-chlorobenzeneboronic acid (1). RCYs determined by radio-HPLC analysis of the crude product. Standard conditions: (1) (0.25 mM), Cu(OTf)2pyr4 (10 mM), 1,10-phenanthroline (10 mM), [125I]NaI or [211At]NaAt (1–5 MBq), Tris buffer 0.5 M/DMF (85:15), 30 min, 100 μL, 23 °C, n ≥ 2. | ||
2.2 Arylboronic acid conjugation to mAb and radiolabelling
In order to investigate the potential of this chemical approach on a relevant cancer targeting mAb, we chose the 9E7.4 mAb. This rat anti-mouse CD138 antibody developed in our laboratory has previously shown suitable properties for the study of targeted alpha therapy, including with 211At, or beta therapy and immunoPET imaging of multiple myeloma in a syngeneic animal model.39–42Bioconjugation of arylboronic acid groups with the 9E7.4 mAb was performed on amino groups of the lysine residues using 3-(succinimidyloxycarbonyl)phenylboronic acid (4) prepared from 3-boronobenzoic acid, providing aBA-9E7.4 (Scheme 2). Precursor (4) was chosen as it leads to the same radiolabelled mAbs than with the widely used two-step approach based on the conjugation of SIB or SAB to lysine groups, allowing direct comparison of methods. When 10 equivalents of (4) were used in the coupling step, we measured a ratio of 3.4 ± 0.6 arylboronic acid groups/mAb by mass spectrometry (see ESI Fig. S7†), a value that we considered as sufficient to perform radiolabelling while being low enough to expect preservation of protein pharmacokinetic properties.
 | ||
| Scheme 2 Strategy for late stage 125I-iodination and 211At-astatination of 9E7.4 mAb via arylboronic acid chemistry. | ||
First radiolabelling assays performed on aBA-9E7.4 using optimal conditions defined with the model compound (Tris buffer pH 6, with a reduction of the DMF ratio to 7.5% instead of 15% in order to lower the risk of protein denaturation), provided excellent results, with RCYs of 87 ± 1% and 94 ± 3% for 125I-iodination and 211At-astatination, respectively. In order to limit the risk of protein alteration or the presence of residual copper in the final radiopharmaceutical, we investigated further the possibility to reduce the catalyst concentration during the radiolabelling step, while keeping an equimolar amount of ligand. Radioiodination RCYs were observed to decrease steadily (from 87% to 58%) while catalyst concentration was decreased from 10 mM to 1.25 mM (Table 3). On the other hand, astatination RCYs remained unchanged (93–96%) even at the lowest concentration tested.
| Catalyst and ligand concentration (mM) | RCY (%) | |||
|---|---|---|---|---|
| 125I | 211At | |||
| 9E7.4-aBA | 9E7.4 | 9E7.4-aBA | 9E7.4 | |
|
a Standard conditions: modified 9E7.4-aBA or unmodified 9E7.4 (32 μM), Cu(OTf)2Pyr4, 1,10-phenanthroline, [125I]NaI or [211At]NaAt (1–5 MBq), 30 min, 23 °C in 0.5 M TRIS buffer/DMF (92.5:7.5), n ≥ 3. RCYs are based on the radio-TLC analysis of the crude product.
|
||||
| 10 | 87 ± 1 | 0.4 | 94 ± 3 | 62 ± 8 |
| 5 | 79 ± 4 | 1.2 | 93 ± 4 | 42 ± 5 |
| 2.5 | 73 ± 2 | — | 96 ± 1 | 32 ± 7 |
| 1.25 | 58 ± 8 | 0.3 | 93 ± 4 | 37 ± 12 |
We then investigated the non-specific binding of 125I and 211At to the mAb. For this, the unmodified mAb (not conjugated with the arylboronic acid precursor (4)) was submitted to the same radiolabelling conditions. In the case of radioiodination, non-specific binding to 9E7.4 was insignificant (≤1.2%) whereas astatination was high (62 ± 8% with 10 mM catalyst and ligand). 211At binding was reduced by half when using 5 mM catalyst and ligand concentration, but it could not be further reduced at lower concentrations. The binding of astatine on proteins is an already known phenomenon as discussed below. Since radioiodination RCY remained satisfyingly elevated (79 ± 4%) with 5 mM catalyst and ligand concentration, we selected this concentration for the rest of this study whereas it was set at 2.5 mM for astatination.
We then investigated the influence of the mAb concentration on the RCYs, the purposes being: (i) to probe the possibility to reduce 211At non-specific binding on the mAb and (ii) to optimize the specific activity (As) that can be achieved by this method. Initial conditions discussed above used the mAb at a concentration of 4.8 mg mL−1, which is the minimum needed for efficient radioiodination or astatination of this mAb by two-step approaches. This concentration may vary depending on mAbs.49 This relatively elevated concentration limits optimization of As. Radioiodination RCYs remained optimal (80 ± 9%) when aBA-9E7.4 concentration was decreased to 3.6 mg mL−1 (Fig. 2), and decreased progressively along with further concentration decrease. In the case of astatination, the mAb concentration could be further reduced to 1.8 mg mL−1 without significantly impacting the RCY (91 ± 2% vs. 96 ± 1% at the highest tested concentrations). Interestingly, non-specific binding of 211At on 9E7.4 also decreased with mAb concentration from 32 ± 7% at 4.8 mg mL−1 to 16 ± 4% at 1.8 mg mL−1 or even 6 ± 1% at 0.6 mg mL−1.
 | ||
Fig. 2 Influence of mAb concentration on the 125I-radioiodinationa of aBA-9E7.4 ( ) and 9E7.4 ( ) and 9E7.4 ( ) and 211At-astatinationb of aBA-9E7.4 ( ) and 211At-astatinationb of aBA-9E7.4 ( ) and 9E7.4 ( ) and 9E7.4 ( ). RCYs determined by radio-ITLC-SG analysis of the crude product. Standard conditions: 30 min, 100 μL, 23 °C, Tris buffer 0.5 M pH 6/DMF (92.5:7.5), n ≥ 2. aCu(OTf)2pyr4 (5 mM), 1,10-phenanthroline (5 mM), [125I]NaI (1–5 MBq) bCu(OTf)2pyr4 (2.5 mM), 1,10-phenanthroline (2.5 mM), [211At]NaAt (1–5 MBq). ). RCYs determined by radio-ITLC-SG analysis of the crude product. Standard conditions: 30 min, 100 μL, 23 °C, Tris buffer 0.5 M pH 6/DMF (92.5:7.5), n ≥ 2. aCu(OTf)2pyr4 (5 mM), 1,10-phenanthroline (5 mM), [125I]NaI (1–5 MBq) bCu(OTf)2pyr4 (2.5 mM), 1,10-phenanthroline (2.5 mM), [211At]NaAt (1–5 MBq). | ||
It is known that free astatine can bind to proteins as reported in previous studies. In particular At(I) species have been shown to bind weakly to proteins in the absence of free sulfhydryl group from cysteine at their surface. The bond formed is so weak that At dissociates extensively upon protein precipitation or when the protein is passed through a Sephadex gel during chromatographic analysis or purification.22 From results depicted in Fig. 2, the possibility that non-specific binding occurs competitively with expected astatodeboronation reaction was investigated. For this, we performed a size exclusion chromatographic purification (using a Sephadex based PD10 column) of both astatinated aBA-9E7.4 and unmodified 9E7.4 followed by a chromatographic analysis on ITLC-SG strips using MeOH as eluent of the purified products. Under these conditions, proteins precipitate at the deposit point whereas free astatine migrates with the solvent front. Chromatographic analyses showed that, after purification, activity was detected all along the ITLC-SG strip in the case of the unmodified [211At]9E7.4 due to the progressive release of weakly bound astatine from the mAb during the elution (Fig. S6†). Conversely, the radiochemical purity of [211At]aBA-9E7.4 was very high, most of the activity remaining at the bottom of the TLC (>99%). These results suggest that non-specific binding does not occur during 211At-astatination of the aBA-9E7.4, or that if occurring, it is removed during the purification step.
The similar chromatographic and precipitation behaviour of non-specifically astatinated 9E7.4 suggests a similar mode of weak binding than previously reported by Visser et al.22 The nature of this bond was recently reinvestigated by molecular modelling, suggesting the formation of covalent bonds between the AtO+ species and nitrogen and sulphur atoms on proteins.43
In order to investigate the potential impact of this approach in a pre-clinical research setting, we performed radioiodination and astatination of the mAb with the optimized conditions, at higher activities (≈30 MBq starting activity). Results were compared with the classical two-step approaches via electrophilic and nucleophilic labelling reported previously for this mAb.16 Our method led to significantly improved results regarding major aspects (Table 4): RCYs, activity yields and specific activities (due to the lower amount of mAb needed) were increased whereas the procedure time was decreased. The time aspect is of high importance with radionuclides with short half-life such as 211At. As the use of a copper catalyst can be a problem related to copper toxicity, we investigated the presence of residual Cu by Induced Coupled Plasma-Mass Spectrometry (ICP-MS) of the purified astatinated aBA-9E7.4. Results showed that the copper concentration was below the detection limit of 1 ppm.
| Method | RCYs (%) | Activity yieldc (MBq) | Specific activityc (MBq mg−1) | Duration time (min) | |||
|---|---|---|---|---|---|---|---|
| 125I | 211At | 125I | 211At | 125I | 211At | ||
| a [211At]SAB prepared from the tin precursor with N-chlorosuccinimide as oxidizing agent before the conjugation to the mAb (n = 2). b [125I]SIB or [211At]SAB prepared from the aryliodonium salt with sodium sulfite as reducing agent before conjugation to the mAb (n = 5). c Starting with 30 MBq. | |||||||
| Two-step electrophilica | — | 20–30 | — | 5.5 | — | 24.4 | 200 ± 10 |
| Two-step nucleophilicb | 43 | 53–57 | 12.9 | 13.2 | 57.3 | 58.7 | 140 ± 10 |
| This work | 80 | 56–68 | 24 | 16.1 | 66.7 | 119 | 90 ± 10 |
2.3 Immunoreactivity and precursor shelf-life
The immunoreactivity, which illustrates the proportion of protein in a batch that is able to bind to its target, was measured for both 125I-iodinated and 211At-astatinated 9E7.4. Immunoreactive fraction (IRF) of 90% and 88% were respectively obtained with 125I- and 211At-labelled 9E7.4. These results were similar to the unmodified 9E7.4 (IRF = 85%) or to the previously reported two-step labelling procedure (IRF = 86% for 125I-iodination and 211At-astatination),16 indicating an excellent preservation of the mAb.In addition, to check that the aBA-9E7.4 immunoconjugate can be stored in solution and used over time, we performed a series of radiolabelling assays with the same production batch stored in the radiolabelling buffer (Tris buffer at pH = 6) either at 4 °C or – 18 °C over more than a year (Fig. 3). High RCYs were maintained during the whole study (close to 80% with both radionuclides), which indicates that protodeboronation that may impact boronic acid compounds in water as reported previously does not occur significantly under investigated storage conditions.44,45 The integrity of the mAb was also verified via its immunoreactivity and IRFs of 85% and 90% for the conjugate stored at 4 °C and −18 °C were respectively measured after radioiodination, confirming that it was not altered by the storage conditions. These results suggest the possibility to develop storable radiolabelling kits containing the pre-modified mAb and the reagents needed, facilitating the production of labelled antibodies in a clinical setting.
 | ||
Fig. 3 Radiolabelling of aBA-9E7.4 with 125I after storage at 4 °C ( ) or – 18 °C ( ) or – 18 °C ( )a and 211At after storage at 4 °C ( )a and 211At after storage at 4 °C ( ) or −18 °C ( ) or −18 °C ( )b in Tris buffer 0.5 M at pH 6. Standard conditions: 30 min, 100 μL, 23 °C. aCu(OTf)2pyr4 (5 mM), 1,10-phenanthroline (5 mM), [125I]NaI (1–10 MBq) bCu(OTf)2pyr4 (2.5 mM), 1,10-phenanthroline (2.5 mM), Tris buffer 0.5 M pH 6/DMF (92.5:7.5), [211At]NaAt (1–5 MBq). RCYs determined by radio-ITLC-SG analysis of the crude product. )b in Tris buffer 0.5 M at pH 6. Standard conditions: 30 min, 100 μL, 23 °C. aCu(OTf)2pyr4 (5 mM), 1,10-phenanthroline (5 mM), [125I]NaI (1–10 MBq) bCu(OTf)2pyr4 (2.5 mM), 1,10-phenanthroline (2.5 mM), Tris buffer 0.5 M pH 6/DMF (92.5:7.5), [211At]NaAt (1–5 MBq). RCYs determined by radio-ITLC-SG analysis of the crude product. | ||
2.4 Biodistribution study
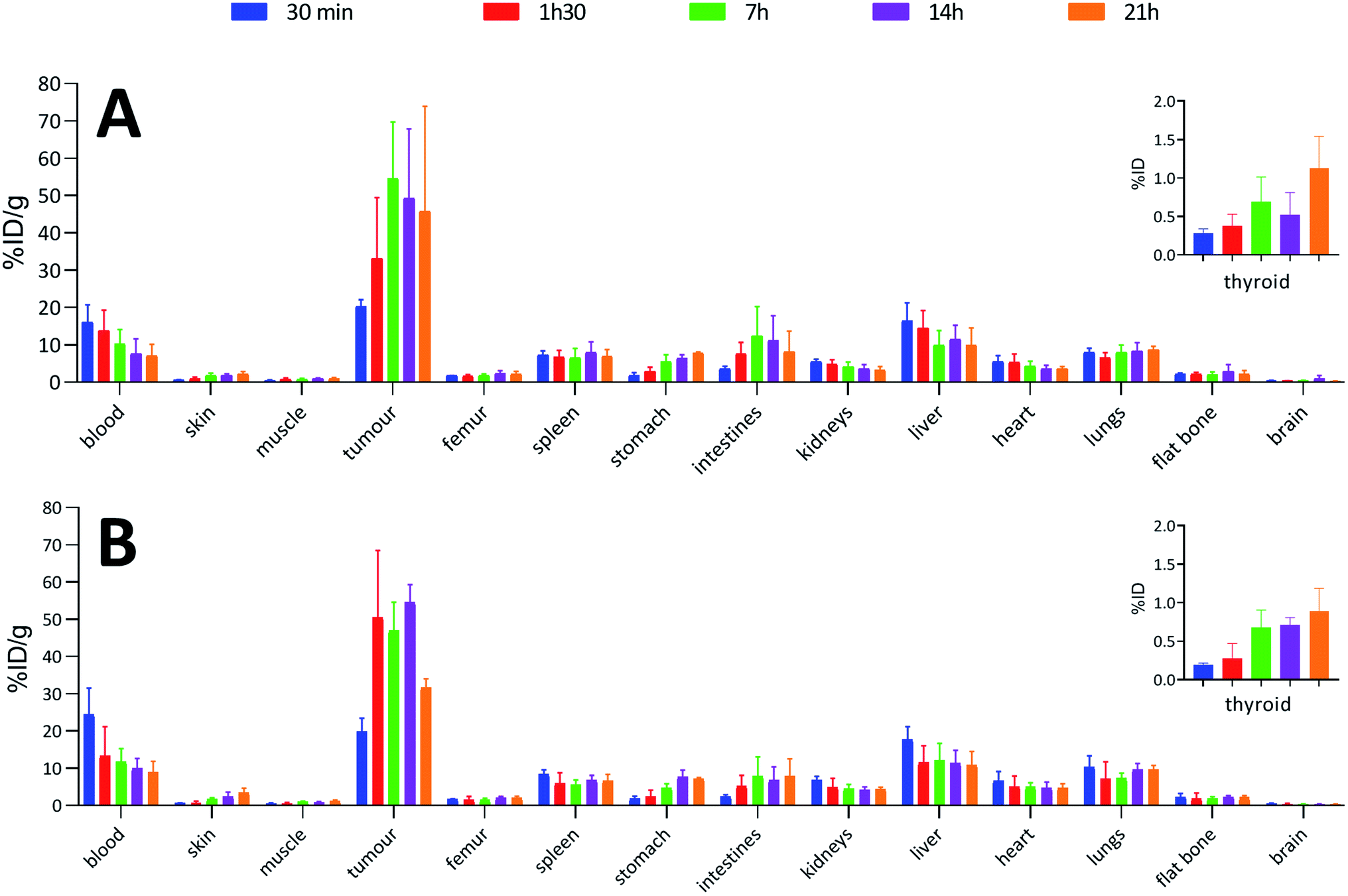
To confirm the viability of this technology for in vivo applications, biodistribution of both 125I-iodinated and 211At-astatinated aBA-9E7.4 was monitored over 21 h after intravenous injection on BALB/c mice grafted subcutaneously with the CD138 expressing MOPC-315 plasmacytoma cell line. Results were compared with the conventional two-step approach that consisted in the synthesis of [125I]SIB and [211At]SAB intermediates followed by conjugation to the mAb.16Overall, biodistribution data indicated that the pharmacokinetic profiles were globally preserved when using our single-step approach since % ID g−1 values were nearly identical (p > 0.05) for most major organs when comparing both radioiodination approaches (Fig. 4A and B) or both astatination approaches (Fig. 5A and B). For 125I-labelling, significant differences between both methods appeared in intestines at 1 h 30 (p = 0.0080) and 7 h (p = 0.0009) and in the liver at 1 h 30 (p = 0.0348) for which a significantly reduced uptake was visible using the single-step labelling approach. The only statistical difference with the astatination was for blood at 30 min (p = 0.0006), which was lower using the two-step approach.
 | ||
| Fig. 4 Biodistributions in BALB/c mice with subcutaneous tumours (MOPC-315) injected with: (A) 0.04 MBq of 125I-labelled 9E7.4 via the two-step method; (B) 0.04 MBq of 125I-labelled 9E7.4 via the arylboronic acid single-step method. | ||
 | ||
| Fig. 5 Biodistributions in BALB/c mice with subcutaneous tumours (MOPC-315) injected with: (A) 1 MBq of 211At-labelled 9E7.4 via the two-step method; (B) 1 MBq of 211At-labelled 9E7.4 via the arylboronic acid single-step method. | ||
In all cases, high uptake was observed in tumours. The significantly higher tumour uptake observed with the two-step 125I-labelling at 7 h (p < 0.0001) and 14 h (p < 0.0001) and with the one-step 211At-labelling at 1 h 30 (p = 0.0003) can be explained by the lower tumour weight of the mice in these groups. Indeed, radioimmunoconjugate uptake in the tumour appeared to be inversely correlated to the tumour weight (Fig. 6), which may be explained by the difference in vasculature between large and small tumours (unpublished observation). Large tumours are often poorly vascularized with high interstitial pressure leading to the development of necrotic tissues in their centre with a loss of CD138 expression.
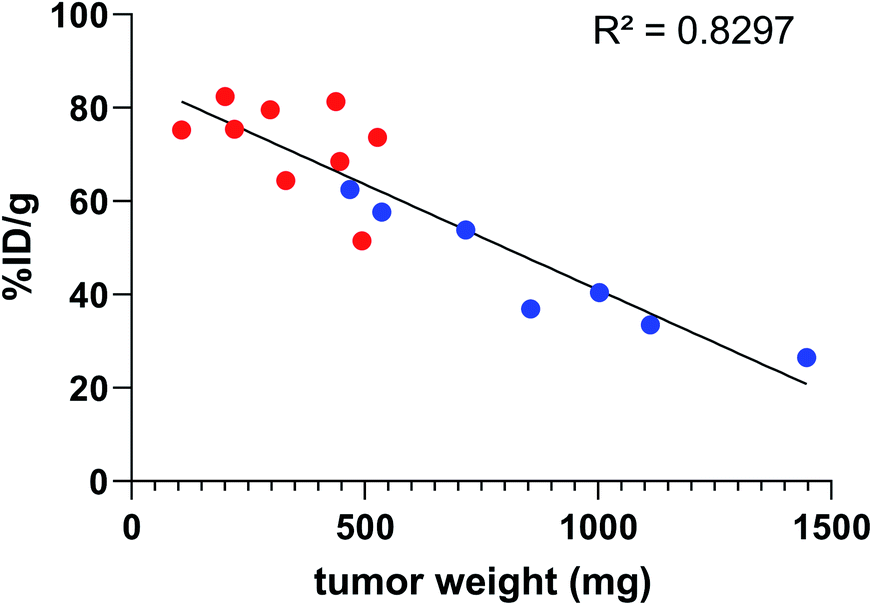 | ||
Fig. 6 Activity uptake in tumour compared to tumour weight 7 h and 14 h post injection of 9E7.4 125I-iodinated by the two-step method ( ) or the arylboronic acid single-step method ( ) or the arylboronic acid single-step method ( ). ). | ||
When comparing radioiodinated 9E7.4 with the astatinated analogue within the same radiolabelling strategy, both led to the same higher uptake in thyroid, stomach, spleen, lungs and kidneys with the 211At-immunoconjugates. This observation was expected and can be attributed to the progressive release of 211At from the carrier IgG in vivo since these organs are well-known targets of free astatine.46 The instability remained however moderate, with a relatively slow increase of uptake over time in these organs, and it was not impacted by the radiolabelling method employed (p > 0.05). Research for novel labelling strategies involving other bonds than aryl–astatine bonds with improved stability remains one of the biggest challenges, with a pressing need especially for applications with small, rapidly metabolized carrier compounds. In our case, the instability remained moderate, and if needed, this non-specific uptake may be reduced significantly by the use of appropriate blocking agents.47 We decided to not use them in our experiment to check the presence of free 211At, or 211At weakly bound to the IgG in the injected solution, which would rapidly result in a higher uptake in thyroid at early biodistribution points. Our results indicated a low 211At uptake in thyroid in the shortest time (<0.3% ID at 0.5 h), similarly to iodine, followed by a progressive increase in uptake with a difference visible only after 7 h. Consequently, the analysis of thyroid activity uptake excludes the hypothesis of residual non-specifically bound astatine discussed in previous sections, as it would have resulted in a rapid increase in activity uptake in this tissue.
3. Conclusions
We demonstrated herein the high reactivity of arylboronic acids with radioiodide and astatide in water at room temperature, suggesting their high potential for radiolabelling sensitive carrier bio(macro)molecules. The standard conditions set up on a simple model compound were straightforwardly translated to the radiolabelling of an anti-CD138 mAb relevant for the imaging and treatment of multiple myeloma. We showed that the new procedure, based on a nucleophilic single-step approach, outperformed previously reported methods in terms of RCYs, activity yields, specific activities and duration, while maintaining nearly identical radioimmunoconjugates biodistribution and high uptake on CD138 expressing tumours in mice. The first divergent radiosynthesis of a radioiodinated mAb and its 211At-labelled analogue, performed in a single step from a common pre-conjugated aryl precursor was reported, facilitating perspectives for radiotheranostic applications using radioiodide/211At pairs. Additionally, the long-term stability in solution of the mAb pre-modified by conjugation with arylboronic acids led to high RCYs over more than a year, suggesting the possibility to develop ready-to-use radiolabelling kits. Overall, the efficiency and simplification provided by this novel approach should facilitate the transfer of astatinated radiopharmaceuticals and their theranostic combinations into the clinic.4. Experimental section
4.1 Radiochemistry
:1 (0.05% TFA) and analysed by reverse phase HPLC.
4.2 Protein radiolabeling
:1) (5 μL), 100 mM 1.10-phenanthroline in DMF (5 μL), and [125I]NaI (10 μL, 1 to 33 MBq). After 30 min of incubation at room temperature, RCY was assessed by elution of an aliquot deposited on an ITLC-SG strip (MeOH as eluent), and analysis of the strip using a Cyclone phosphorimager scanner (PerkinElmer). Purification was performed by gel filtration on a Sephadex G-25 resin loaded column (PD-10, GE healthcare) using PBS as eluent, affording the purified radiolabelled antibody (6a) with a >99% radiochemical purity as assessed by ITLC-SG.
:1) (50 mM, 5 μL), 1.10-phenanthroline in DMF (50 mM, 5 μL), and [211At]NaAt (10 μL, 1 to 40 MBq). After 30 min of incubation at room temperature, RCY was assessed by ITLC-SG strip similarly to radioiodination. Purification was performed by gel filtration on a Sephadex G-25 resin loaded column (PD-10, GE healthcare) using PBS as eluent, affording the purified radiolabelled antibody (6b) with a >99% radiochemical purity as assessed by ITLC-SG.
4.3. In vivo studies
BALB/c mice were inoculated subcutaneously with 2 × 105 MOPC-315 plasmacytoma cells (ATCC® TIB-23™). When tumour size was between 200 and 1500 mm3, the animals were divided in 4 groups. Group A was treated with [125I]aBA-9E7.4 (40 kBq per mice, 20 μg in 100 μL), group B with [125I]9E7.4 radiolabelled via the two-step method (40 kBq per mice, 20 μg in 100 μL), group C with [211At]aBA-9E7.4 (1 MBq per mice, 20 μg in 100 μL), and group D with [211At]9E7.4 radiolabelled via the two-step method (1 MBq per mice, 20 μg in 100 μL).16 The mice were sacrificed by cervical dislocation in groups of 3 to 6 animals at 5 different time points post injection: 30 min, 1 h 30, 7 h, 14 h and 21 h. The selected tissues (blood, skin, muscle, tumour, femur, spleen, stomach, intestines, kidneys, liver, heart, lungs, flat bone, brain) were dissected, weighed and counted on a calibrated and normalized gamma-counter (PerkinElmer). For each organ, the percentage of injected dose per gram (% ID g−1) was calculated. The neck (thyroid) was not weighed but counted as well on the gamma counter for the determination of the % ID. No blocking agent were used in order to use organs uptake (thyroid, stomach, spleen, lungs, kidneys, liver) as indicator of free astatine.
Conflicts of interest
The authors declare the following competing financial interest: M. B., A. F.-C., J.-F. G. and F. G. have filed a provisional patent application relating to this work.Acknowledgements
This work was supported by grants from the French National Agency for Research called “Investissements d'Avenir”, Equipex Arronax-Plus (ANR-11-EQPX-0004), Labex IRON (ANR-11-LABX-18-01), ISITE NExT (ANR-16-IDEX-0007) and INCa-DGOS-Inserm_12558. The UTE facility (SFR Santé) and the radioactivity platform (SFR Santé) are thanked for the technical assistance.Notes and references
- M. Makvandi, E. Dupis, J. W. Engle, F. M. Nortier, M. E. Fassbender, S. Simon, E. R. Birnbaum, R. W. Atcher, K. D. John, O. Rixe and J. P. Norenberg, Target. Oncol., 2018, 13, 189–203 CrossRef.
- C. Parker, V. Lewington, N. Shore, C. Kratochwil, M. Levy, O. Lindén, W. Noordzij, J. Park and F. Saad, JAMA Oncol., 2018, 4, 1765–1772 CrossRef.
- F. Guérard, J.-F. Gestin and M. W. Brechbiel, Cancer Biother.Radiopharm., 2013, 28, 1–20 CrossRef.
- M. R. Zalutsky, D. A. Reardon, G. Akabani, R. E. Coleman, A. H. Friedman, H. S. Friedman, R. E. McLendon, T. Z. Wong and D. D. Bigner, J. Nucl. Med., 2008, 49, 30–38 CrossRef CAS.
- H. Andersson, E. Cederkrantz, T. Bäck, C. Divgi, J. Elgqvist, J. Himmelman, G. Horvath, L. Jacobsson, H. Jensen, S. Lindegren, S. Palm and R. Hultborn, J. Nucl. Med., 2009, 50, 1153–1160 CrossRef CAS.
- S. O'Steen, M. L. Comstock, J. J. Orozco, D. K. Hamlin, D. S. Wilbur, J. C. Jones, A. Kenoyer, M. E. Nartea, Y. Lin, B. W. Miller, T. A. Gooley, S. A. Tuazon, B. G. Till, A. K. Gopal, B. M. Sandmaier, O. W. Press and D. J. Green, Blood, 2019, 134, 1247–1256 CrossRef.
- T. Ferris, L. Carroll, S. Jenner and E. O. Aboagye, J. Labelled Compd. Radiopharm. DOI:10.1002/jlcr.3891.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153–163 RSC.
- L. Xie, M. Hanyu, M. Fujinaga, Y. Zhang, K. Hu, K. Minegishi, C. Jiang, F. Kurosawa, Y. Morokoshi, H. K. Li, S. Hasegawa, K. Nagatsu and M.-R. Zhang, J. Nucl. Med., 2020, 61, 242–248 CrossRef CAS.
- F. Guérard, Y.-S. Lee, K. Baidoo, J.-F. Gestin and M. W. Brechbiel, Chem.–Eur. J., 2016, 22, 12332–12339 CrossRef.
- L. Navarro, M. Berdal, M. Chérel, F. Pecorari, J.-F. Gestin and F. Guérard, Bioorg. Med. Chem., 2019, 27, 167–174 CrossRef CAS.
- S. W. Reilly, M. Makvandi, K. Xu and R. H. Mach, Org. Lett., 2018, 20, 1752–1755 CrossRef CAS.
- D.-C. Sergentu, D. Teze, A. Sabatié-Gogova, C. Alliot, N. Guo, F. Bassal, I. D. Silva, D. Deniaud, R. Maurice, J. Champion, N. Galland and G. Montavon, Chem.–Eur. J., 2016, 22, 2964–2971 CrossRef CAS.
- M. R. Zalutsky, P. K. Garg, H. S. Friedman and D. D. Bigner, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7149–7153 CrossRef CAS.
- M. R. Zalutsky and A. S. Narula, Int. J. Radiat. Appl. Instrum., Part A, 1987, 38, 1051–1055 CrossRef CAS.
- F. Guérard, L. Navarro, Y.-S. Lee, A. Roumesy, C. Alliot, M. Chérel, M. W. Brechbiel and J.-F. Gestin, Bioorg. Med. Chem., 2017, 25, 5975–5980 CrossRef.
- S. Lindegren, S. Frost, T. Bäck, E. Haglund, J. Elgqvist and H. Jensen, J. Nucl. Med., 2008, 49, 1537–1545 CrossRef CAS.
- E. Aneheim, A. Gustafsson, P. Albertsson, T. Bäck, H. Jensen, S. Palm, S. Svedhem and S. Lindegren, Bioconjugate Chem., 2016, 27, 688–697 CrossRef CAS.
- D. S. Wilbur, S. W. Hadley, M. D. Hylarides, P. G. Abrams, P. A. Beaumier, A. C. Morgan, J. M. Reno and A. R. Fritzberg, J. Nucl. Med., 1989, 30, 216–226 CAS.
- G. W. M. Visser, E. L. Diemer and F. M. Kaspersen, Int. J. Appl. Radiat. Isot., 1979, 30, 749–752 CrossRef CAS.
- G. W. M. Visser, E. L. Diemer and F. M. Kaspersen, Int. J. Appl. Radiat. Isot., 1980, 31, 275–278 CrossRef CAS.
- G. W. M. Visser, E. L. Diemer and F. M. Kaspersen, Int. J. Appl. Radiat. Isot., 1981, 32, 905–912 CrossRef CAS.
- D. S. Wilbur, M.-K. Chyan, D. K. Hamlin and M. A. Perry, Nucl. Med. Biol., 2010, 37, 167 CrossRef CAS.
- K. Fujiki, Y. Kanayama, S. Yano, N. Sato, T. Yokokita, P. Ahmadi, Y. Watanabe, H. Haba and K. Tanaka, Chem. Sci., 2019, 10, 1936–1944 RSC.
- H. Jadvar, X. Chen, W. Cai and U. Mahmood, Radiology, 2018, 286, 388–400 CrossRef.
- K. Ogawa, T. Takeda, K. Mishiro, A. Toyoshima, K. Shiba, T. Yoshimura, A. Shinohara, S. Kinuya and A. Odani, ACS Omega, 2019, 4, 4584–4591 CrossRef CAS.
- G. J. Meyer, K. Roessler and G. Stoecklin, J. Am. Chem. Soc., 1979, 101, 3121–3123 CrossRef CAS.
- G. J. Meyer, A. Walte, S. R. Sriyapureddy, M. Grote, D. Krull, Z. Korkmaz and W. H. Knapp, Appl. Radiat. Isot., 2010, 68, 1060–1065 CrossRef CAS.
- T. C. Wilson, T. Cailly and V. Gouverneur, Chem. Soc. Rev., 2018, 47, 6990–7005 RSC.
- P. Zhang, R. Zhuang, Z. Guo, X. Su, X. Chen and X. Zhang, Chem.–Eur. J., 2016, 22, 16783–16786 CrossRef CAS.
- D. Zhou, W. Chu, T. Voller and J. A. Katzenellenbogen, Tetrahedron Lett., 2018, 59, 1963–1967 CrossRef CAS.
- T. C. Wilson, G. McSweeney, S. Preshlock, S. Verhoog, M. Tredwell, T. Cailly and V. Gouverneur, Chem. Commun., 2016, 52, 13277–13280 RSC.
- M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751–7755 CrossRef CAS.
- G. W. Kabalka and M.-L. Yao, J. Organomet. Chem., 2009, 694, 1638–1641 CrossRef CAS.
- S. Webster, K. M. O'Rourke, C. Fletcher, S. L. Pimlott, A. Sutherland and A.-L. Lee, Chem.–Eur. J., 2018, 24, 937–943 CrossRef CAS.
- M. V. Dam, G. E. Wuenschell and F. H. Arnold, Biotechnol. Appl. Biochem., 1989, 11, 492–502 Search PubMed.
- Z. K. Glover, L. Basa, B. Moore, J. S. Laurence and A. Sreedhara, mAbs, 2015, 7, 901–911 CrossRef CAS.
- J. Nagaj, K. Stokowa-Soltys, E. Kurowska, T. Frączyk, M. Jeżowska-Bojczuk and W. Bal, Inorg. Chem., 2013, 52, 13927–13933 CrossRef CAS.
- N. Fichou, S. Gouard, C. Maurel, J. Barbet, L. Ferrer, A. Morgenstern, F. Bruchertseifer, A. Faivre-Chauvet, E. Bigot-Corbel, F. Davodeau, J. Gaschet and M. Chérel, Front. Med., 2015, 2, 76 Search PubMed.
- C. Bailly, S. Gouard, M. Lacombe, P. Remaud-Le Saëc, B. Chalopin, M. Bourgeois, N. Chouin, R. Tripier, Z. Halime, F. Haddad, A. Faivre-Chauvet, F. Kraeber-Bodéré, M. Chérel and C. Bodet-Milin, Oncotarget, 2018, 9, 9061–9072 CrossRef.
- C. Bailly, S. Gouard, F. Guérard, B. Chalopin, T. Carlier, A. Faivre-Chauvet, P. Remaud-Le Saëc, M. Bourgeois, N. Chouin, L. Rbah-Vidal, R. Tripier, F. Haddad, F. Kraeber-Bodéré, C. Bodet-Milin and M. Chérel, Int. J. Mol. Sci., 2019, 20, 2564 CrossRef CAS.
- S. Gouard, C. Maurel, S. Marionneau-Lambot, D. Dansette, C. Bailly, F. Guérard, N. Chouin, F. Haddad, C. Alliot, J. Gaschet, R. Eychenne, F. Kraeber-Bodéré and M. Chérel, Cancers, 2020, 12, 2721 CrossRef.
- F. Bassal, J. Champion, S. Pardoue, M. Seydou, A. Sabatié-Gogova, D. Deniaud, J.-Y. L. Questel, G. Montavon and N. Galland, Inorg. Chem., 2020, 59, 13923–13932 CrossRef CAS.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS.
- J. Spetz, N. Rudqvist and E. Forssell-Aronsson, Cancer Biother.Radiopharm., 2013, 28, 657–664 CrossRef CAS.
- R. H. Larsen, S. Slade and M. R. Zalutsky, Nucl. Med. Biol., 1998, 25, 351–357 CrossRef CAS.
- S. Lindegren, T. Bäck and H. J. Jensen, Appl. Radiat. Isot., 2001, 55, 157–160 CrossRef CAS.
- M. Bourgeois, F. Guérard, C. Alliot, M. Mougin-Degraef, H. Rajerison, P. Remaud-Le Saëc, J.-F. Gestin, F. Davodeau, M. Chérel, J. Barbet and A. Faivre-Chauvet, J. Labelled Compd. Radiopharm., 2008, 51, 379–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05191h |
| This journal is © The Royal Society of Chemistry 2021 |