
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArylation of gem-difluoroalkenes using a Pd/Cu Co-catalytic system that avoids β-fluoride elimination†‡
Kedong
Yuan
 a,
Taisiia
Feoktistova
b,
Paul Ha-Yeon
Cheong
*b and
Ryan A.
Altman
*c
a,
Taisiia
Feoktistova
b,
Paul Ha-Yeon
Cheong
*b and
Ryan A.
Altman
*c
aTianjin Key Laboratory of Advanced Functional Porous Materials, Institute for New Energy Materials & Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, P. R. China
bDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, OR, USA. E-mail: cheongh@oregonstate.edu
cDepartment of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: raaltman@purdue.edu
First published on 25th November 2020
Abstract
PdII/CuI co-catalyze an arylation reaction of gem-difluoroalkenes using arylsulfonyl chlorides to deliver α,α-difluorobenzyl products. The reaction proceeds through a β,β-difluoroalkyl–Pd intermediate that typically undergoes unimolecular β-F elimination to deliver monofluorinated alkene products in a net C–F functionalization reaction. However to avoid β-F elimination, we offer the β,β-difluoroalkyl–Pd intermediate an alternate low-energy route involving β-H elimination to ultimately deliver difluorinated products in a net arylation/isomerization sequence. Overall, this reaction enables exploration of new reactivities of unstable fluorinated alkyl–metal species, while also providing new opportunities for transforming readily available fluorinated alkenes into more elaborate substructures.
Introduction
Due to the intrinsic small size and high electronegativity, incorporation of fluorine at specific positions of bio-relevant molecules can improve pharmacokinetic, pharmacodynamic and physicochemical properties, thus facilitating the drug discovery process.1 For instance, replacing benzylic CH2 units with CF2 significantly influences metabolic properties, and strategies that incorporate fluorine directly into these positions aid in accessing the next generation of therapeutic candidates.2Recently, tremendous effort has been devoted to develop diverse reactions for accessing fluorinated drug-like substructures. One important strategy exploits fluorinated synthons, such as gem-difluoroalkenes, as valuable and readily-accessible building blocks for further functionalization.3 Relative to non-fluorinated alkenes, gem-difluoroalkenes show distinct reactivity trends:4 (i) reactions typically occur at the electron-deficient gem-difluorinated carbon to deliver α-functionalized products (Scheme 1), (ii) anionic intermediates typically decompose via β-F elimination to generate mono-defluorinated products (Scheme 1A), (iii) organometallic intermediates also decompose via β-F elimination (Scheme 1B).5–7 In contrast, transition metal catalysed reactions of gem-difluoroalkenes that avoids β-F elimination are extremely rare.7 Such a process would require an alternate reaction pathway to avoid β-F elimination and deliver difluoroalkyl substructures (Scheme 1C). Further, a convergent preparation would complement traditional and harsh deoxyfluorination reactions of ketones that might generate this substructure.8
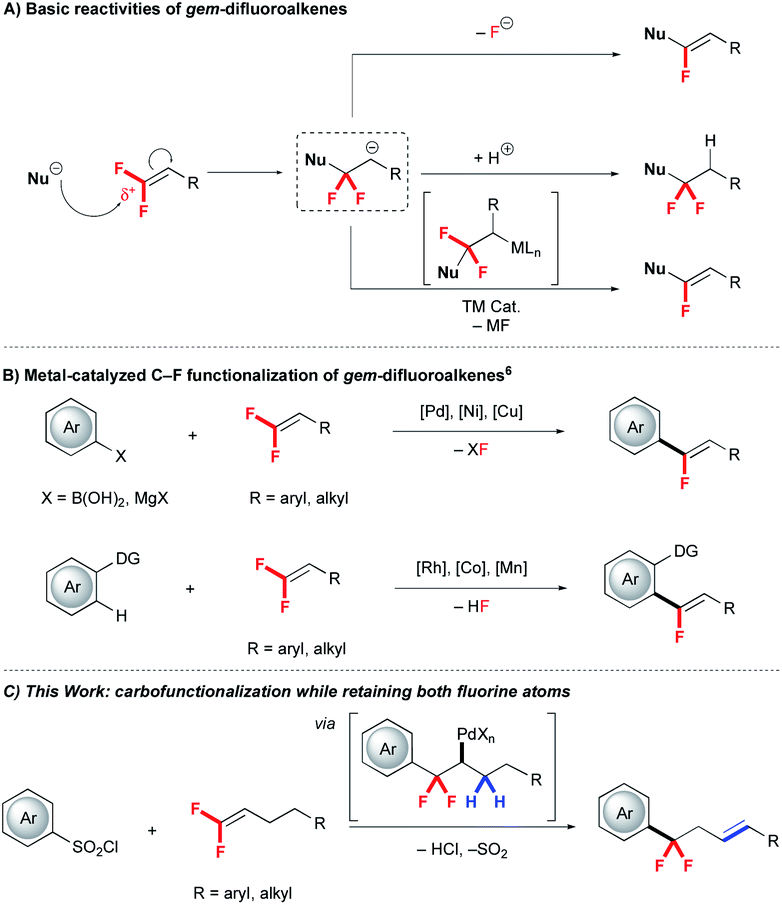 | ||
| Scheme 1 Reactivity of gem-difluoroalkenes. | ||
To avoid β-F elimination, we sought to offer an alternate route for the α,α-difluoroalkyl metal intermediate. Specifically, we hypothesized that β-H elimination might outcompete β-F elimination and deliver products containing both fluorine atoms. In practice, we exploited arylsulfonyl chlorides (ArSO2Cl) as readily available aryl reagents that show complementary reactivity and functional group tolerance relative to aryl-halides in cross-coupling and C–H functionalization reactions.9 These ArSO2Cl generate aryl radicals in the presence of CuI salts at high temperature10 that might avoid formation of anionic intermediates. Combined, these features inspired us to explore the unique reactivity of ArSO2Cl and gem-difluoroalkenes using a Pd/Cu-based system. Herein, we report a Pd/Cu co-catalyzed arylation-isomerization of gem-difluoroalkenes that avoids β-F elimination.
Results and discussion
Optimization of reactions
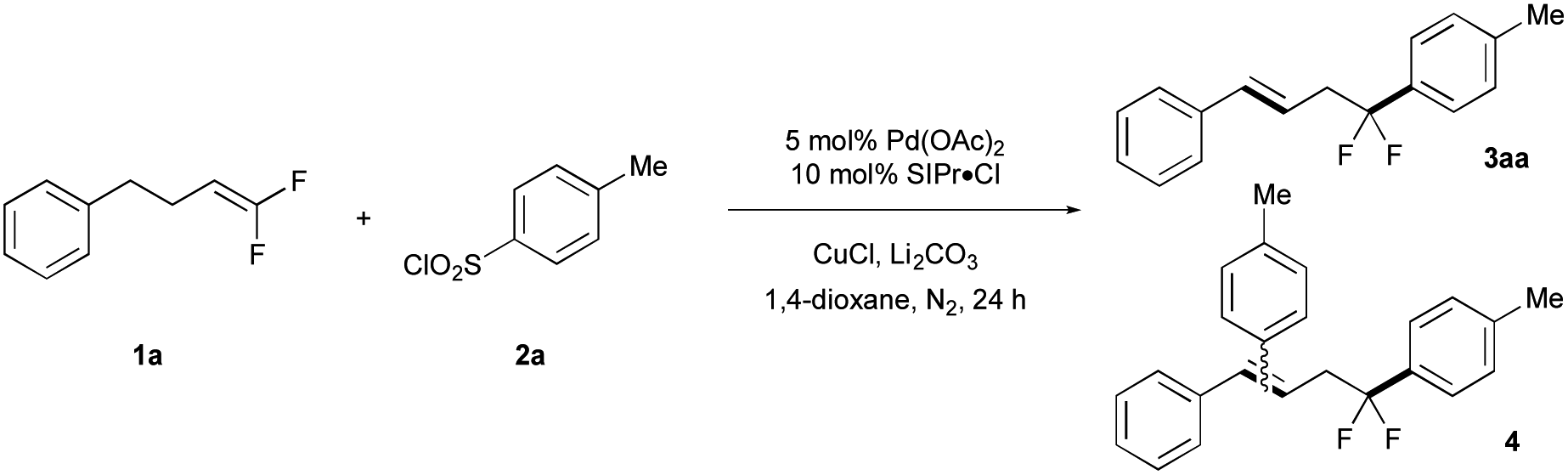
Optimal reaction conditions were identified by evaluating the cross coupling of gem-difluoroalkene 1a and ArSO2Cl (2a) to generate difluorobenzyl product 3aa (see ESI Tables 1–7‡). Ultimately, a system of Pd(OAc)2/CuCl/Li2CO3 was essential for generating the desired product (Table 1, entry 1), as removal of any individual component drastically decreased the yield of product (entries 2–4). In this reaction, use of an excess of 1a suppressed the formation of side products 4, which likely arose from Heck-arylation of alkene 3aa. Notably, the reaction proceeded well even without ligands (entry 5), though use of an NHC ligand (SIPr·Cl) reduced the yields of side products. Eventually, optimized conditions of 5 mol% Pd(OAc)2, 10 mol% SIPr·Cl, stoichiometric CuCl and Li2CO3 in refluxing 1,4-dioxane coupled 1a (2.25 equiv.) and 2a in 72% isolated yield (entry 6). Under these conditions, the difluorobenzyl group of 3aa did not decompose to a monofluoroalkene, even at high temperature (120 °C).|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Conv. (%) | Yield 3aab (%) |
| a Conditions: 1a (0.45 mmol), 2a (0.20 mmol), Pd(OAc)2 (0.010 mmol), SIPr·Cl (0.020 mmol), CuCl (0.24 mmol), Li2CO3 (0.40 mmol), 1,4-dioxane (0.50 mL, 0.40 M), N2, reflux for 24 h. Yields were determined by GC analysis using dodecane (20 μL) as internal standard. b Mixture of diarylation products (<10%) were observed. Isolated yields are given in parentheses. c 1-(2-Chloro-1,1-difluoro-4-phenylbutyl)-4-methylbenzene and mono-defluorinated arylation products were observed. d Reaction was performed based on 2a (0.50 mmol) in 0.33 M solution of 1,4-dioxane. SIPr·Cl = 1,3-bis[2,6-bis(1-methylethyl)phenyl]-1H-imidazolium chloride. | |||
| 1 | None | 100 | 78 (68) |
| 2 | No Pd(OAc)2 | <3 | 0 |
| 3c | No CuCl | 100 | 16 |
| 4 | No Li2CO3 | 100 | 0 |
| 5 | No SIPr·Cl | 100 | 75 (63) |
| 6d | Reaction on 0.5 mmol, 0.33 M | 100 | 76 (72) |
Evaluation of substrate scope
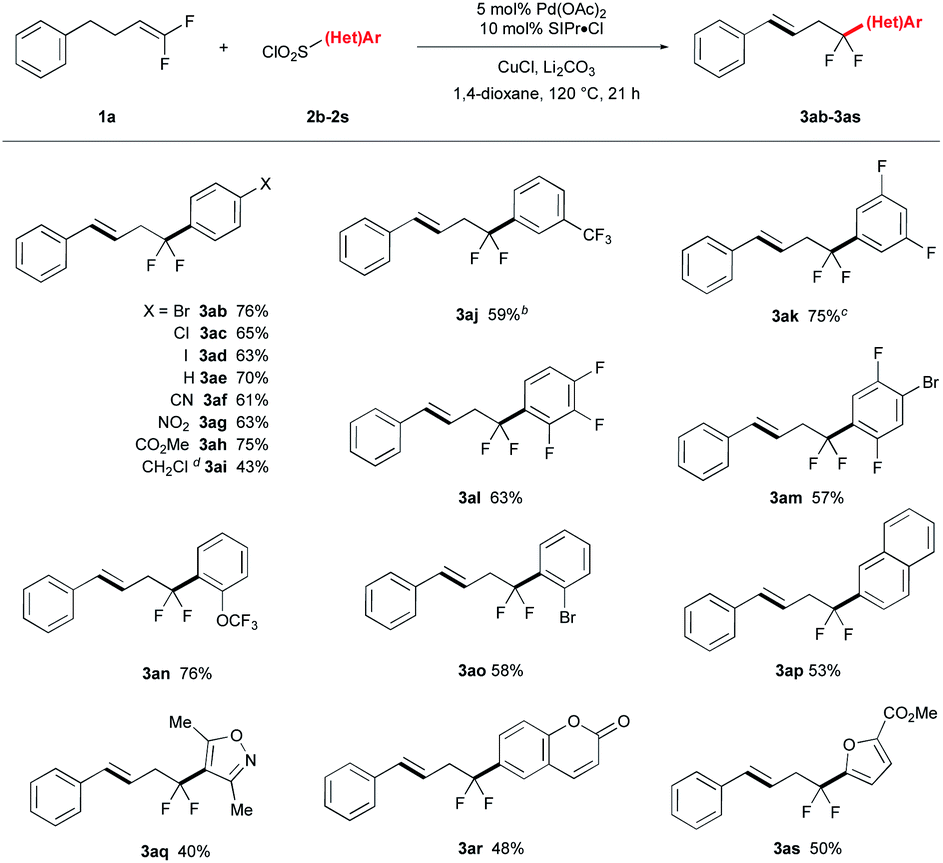
The reaction conditions tolerated a broad scope of ArSO2Cl bearing many important functional groups (Table 2). A variety of para-substituted ArSO2Cl were tolerated, including halogenated groups that are not tolerated by many Pd-catalyzed coupling reactions (I, Br, Cl; 3ab–3ad) and electron withdrawing groups (CN, NO2, CO2Me; 3af–3ah). Interestingly, benzyl chloride product 3ai was formed in moderate yield from reaction of 4-(bromomethyl)benzenesulfonyl chloride and 1a through an extra halogen exchange step. This benzyl electrophile would be useful for further synthetic elaboration. Fluorinated ArSO2Cl reacted smoothly to give the corresponding arylation product in good yields (3aj–3an). Ortho-substituted ArSO2Cl coupled effectively (3al–3ao), and heteroarylsulfonyl chlorides were tolerated albeit with reduced yields of product (3aq–3as). Notably, reactions of electron-rich (e.g. OMe, SMe, NHAc) and N-heteroaryl (pyridine, imidazole, pyrazole, quinoline) sulfonyl chlorides reacted in lower yield or with poor selectivity due to competing defluorination. The reaction proceeded on well on larger scales, with 3aj obtained on 5 mmol scale without decreasing the reaction yield.| a Conditions: 1a (0.875 mmol), 2 (0.50 mmol), Pd(OAc)2 (0.025 mmol), SIPr·Cl (0.050 mmol), CuCl (0.60 mmol), Li2CO3 (1.0 mmol), 1,4-dioxane (1.5 mL), N2, reflux for 21 h; isolated yields. b Reaction performed on 5.0 mmol scale of 2j. c Li2CO3 (3.0 equiv.). d Start from 4-(bromomethyl)benzenesulfonyl chloride. |
|---|
|
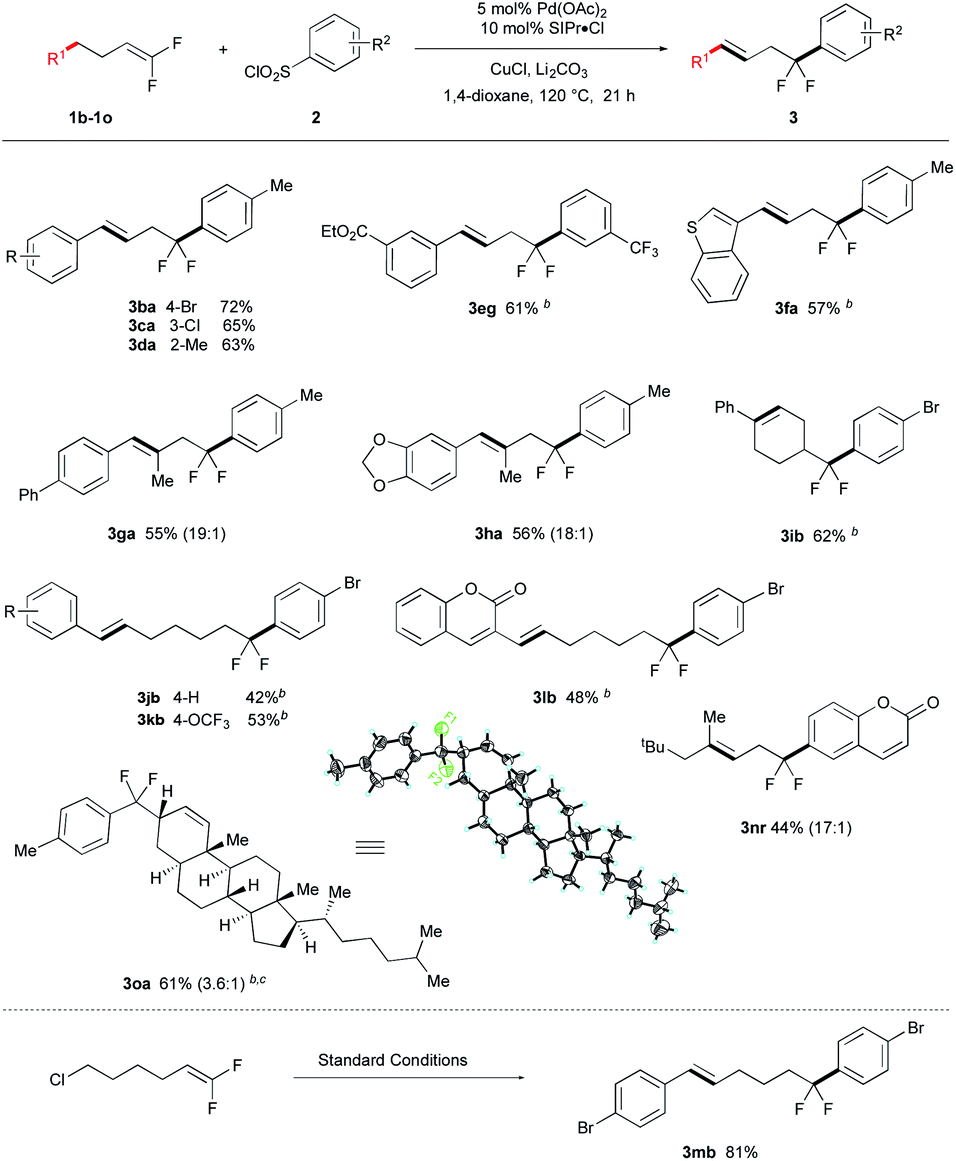
The catalytic system also coupled various aryl-substituted gem-difluoroalkenes (Table 3) and afforded 3ba–3da, 3eg and 3fa in good yields. Reaction of alkyl-substituted gem-difluoroalkenes gave trisubstituted akenes 3ga, 3ha and 3ib in good stereoselectivity. Extension the aliphatic carbon chain slightly decreased the yields (3jb–3lb), though these reactions required additional β-hydride elimination/reinsertion steps to produce the energetically favoured products. Notably, the reaction of cholesterol derivative 1o afforded coupled product 3oa in 61% yield as a mixture of diastereomers (3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), of which the relative stereochemistry was determined by X-ray crystallography (CSD: q79h).§ Finally, the reaction of 6-chloro-1,1-difluoro-hex-1-ene with 2b afforded diarylation product 3mb, which presumably proceed via a sequence involving arylation-isomerization-arylation (see figure inset).
:1), of which the relative stereochemistry was determined by X-ray crystallography (CSD: q79h).§ Finally, the reaction of 6-chloro-1,1-difluoro-hex-1-ene with 2b afforded diarylation product 3mb, which presumably proceed via a sequence involving arylation-isomerization-arylation (see figure inset).
| a Conditions: 1a (0.875 mmol), 2 (0.50 mmol), Pd(OAc)2 (0.025 mmol), SIPr·Cl (0.050 mmol), CuCl (0.60 mmol), Li2CO3 (1.0 mmol), 1,4-dioxane (1.5 mL), N2, reflux for 21 h; isolated yields; selectivity was determined by 19F NMR and GC analysis of crude mixture. b 1b, 1.75 equiv. c 110 °C, reflux for 38 h. X-ray structure of 3oa provided. |
|---|
|
Mechanistic investigations
A combination of computational and experimental mechanistic studies (see below) and previous literature,10–12 support a mechanism involving PdII/PdIII intermediates (Fig. 1). The cycle begins with PdII coordinating to the gem-difluoroalkene. Then a combination of the PdII catalyst, CuCl and Li2CO3 activate the ArSO2Cl to generate Ar˙, which combines with PdII to generate a PdIII–Ar intermediate. β-Migratory insertion of the Ar group into the gem-difluoroalkene would provide a PdIII–alkyl intermediate. The PdIII–alkyl intermediate undergoes β-H elimination preferentially over β-F elimination to generate alkene-coordinated PdIII–H species,14 and subsequent hydride insertion/elimination transfers the alkene to the thermodynamically stable position, thus delivering the product that retains both fluorine atoms.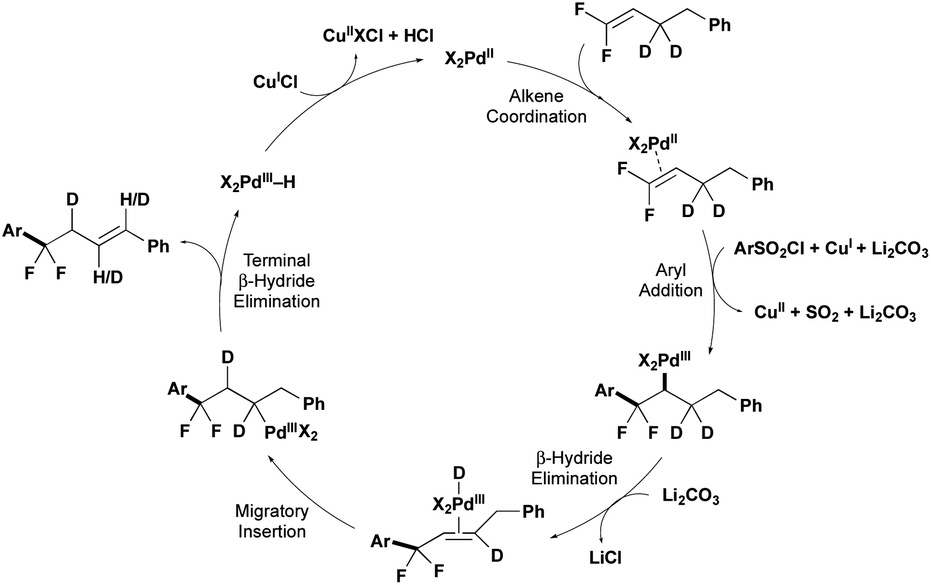 | ||
| Fig. 1 Plausible mechanism for the arylation of gem-difluoroalkenes. | ||
Experimental data supports early steps of the proposed cycle. First, the PdII precatalyst, Cu salt, and Li2CO3 are all required to activate the ArSO2Cl, as the absence of any one of these components provides low conversion of ArSO2Cl (2a) to generate Ar–Cl and homocoupling products (Scheme 2A, Table 1, entry 4; see Table ESI-4‡ for more details). This activation contrasts previous CuI-catalyzed reactions of ArSO2Cl that generated Ar˙ in the absence of PdII or PdII/CO32− additives.10a,b Second, decomposition of ArSO2Cl generates ArSO2˙ and subsequently Ar˙ intermediates, as evidenced by the generation of BHT adducts in both the full reaction (Scheme 2B) and half reaction (Scheme 2C). From this stage, the combination of the Ar˙, PdII catalyst, and gem-difluoroalkene could presumably react by multiple pathways (Scheme 2D; see Fig. S1‡ for more details). According to computations using density functional theory (DFT)-B3LYP-D3BJ/6-31G* & LANL2DZ/PCM (1,4-dioxane) at 120 °C, the lowest energy pathway involves a barrierless addition of the Ar˙ to PdII to generate a PdIII–Ar intermediate and subsequent β-migratory insertion of the Ar group into the gem-difluoroalkene. In contrast, antarafacial carbopalladation of the difluoroalkene is higher in energy by 17.1 kcal mol−1, while direct addition of Ar˙ to the uncoordinated gem-difluoroalkene to generate an unstabilized alkyl radical is 40.3 kcal mol−1 higher in energy.6a,13 Of note, the Pd catalyst plays a key role in generating the unfavorable C–C bond. Specifically, while the disfavored radical attack onto the difluoroalkene (either with or without coordination to PdII) would form the new C–C bond through the arene σ-system, the PdIII–Ar/β-migratory insertion pathway generates the new C–C bond through hybrid orbitals from the arene's π-system (see Fig. S1‡ for more details).
 | ||
| Scheme 2 Mechanistic experiments to support activation of ArSO2Cl, presence of Ar˙, and β-hydride elimination. | ||
Experimental and computational experiments also confirm that β-hydride elimination can outcompete β-fluoride elimination. As evidenced by the deuterium-scrambling reaction of deuterated substrate 1q, the reaction involves a Pd-mediated β-H elimination/reinsertion process that walks the alkene away from the difluorobenzyl moiety (Fig. 2A).15 Computations provided additional insight into these competing processes. Overall comparison of PdIII and PdII mechanisms reveals that the operative mechanism involves PdIII (see Fig. S2‡): (1) β-H elimination for PdIII is lower in energy than for PdII by 25.1 kcal mol−1; (2) similarly, β-F elimination is favored for PdIII over PdII by 37.1 kcal mol−1. Interestingly, when comparing PdIII- vs. PdII-based processes, β-H elimination is consistently favored over β-F elimination for PdIII- and PdII-based mechanisms by 2.5 and 14.5 kcal mol−1, respectively. Overall, for the operative PdIII mechanism, β-H elimination is favored over β-F elimination by 2.5 kcal mol−1 (Fig. 2B). We also evaluated whether the chemoselectivity is influenced by the homobenzylic and benzylic positions of the H and F atoms by computing the elimination processes for a hypothetical substrate on which the H atoms are benzylic and F atoms are homobenzylic (see Fig. S2–S4‡ for more details). In all cases, β-H elimination is markedly preferred over β-F elimination, suggesting that the conjugation effect of the benzylic or the homobenzylic positions are not sufficiently strong to reverse the selectivity. To elucidate the origins of β-H/F elimination selectivity, distortion–interaction analysis revealed that (Fig. 2B): (1) the interaction energies were almost identical in both processes (ca. −54 kcal mol−1); (2) the PdII catalyst was slightly more distorted at the transition state for the favoured β-H elimination (4.7 vs. 1.9 kcal mol−1); however, (3) the substrate was significantly more distorted at the transition state for the disfavoured β-F elimination (42.7 vs. 36.5 kcal mol−1), suggesting that the C–F bond is much stronger than the C–H bond (Fig. 2B). These results support the hypothesis that the selectivity arises from strong preference for breaking C–H bond vs. C–F bond.
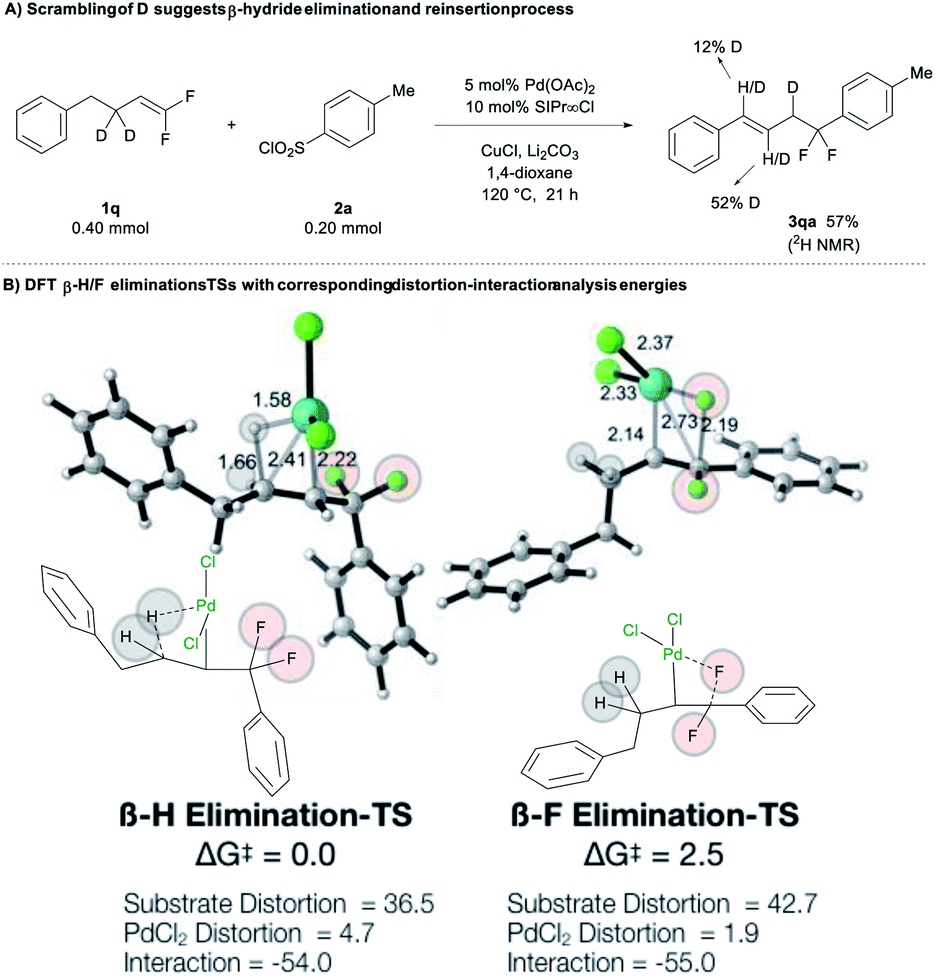 | ||
| Fig. 2 Calculated β-H/F elimination transition states (TSs) with corresponding distortion-interaction analysis energies. Distances in Å and energies in kcal mol−1. | ||
Conclusions
In summary, a PdII/CuI co-catalyzed cross-coupling reaction of gem-difluoroalkenes and ArSO2Cl react in a net arylation/isomerization sequence that demonstrated good functional group tolerance with respect to both components and provided products bearing the “CF2” motif at the benzylic position, which would block radical processes that might activate this position. DFT and mechanistic experiments indicate that Pd plays two key roles in the reaction, first by facilitating the formation of a challenging C–C bond, and second by reacting through a β-H elimination process, which overcomes the favoured metal-mediated β-F elimination process and delivers products bearing both fluorine atoms. These findings should enable the discovery of many complementary reactions for accessing a broad spectrum of fluoroalkyl substructures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institutes of Health (R35 GM124661) for supporting this work. NMR was provided by NIH Shared Instrumentation Grants S10OD016360 and S10RR024664, NSF Major Research Instrumentation Grants 9977422 and 0320648, and NIH Center Grant P20GM103418. We thank Dr Victor Day for X-ray analysis and NSF-MRI grant CHE-0923449 for the X-ray diffractometer and software. We thank Mr Bo Han for helping prepare NMR files. K. Y. acknowledges support from National Natural Science Foundation of China (21702145). PHYC is the Bert and Emelyn Christensen professor of OSU, and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family, and the National Science Foundation (NSF, CHE-1352663). TF acknowledges Summer Fellowship Award from the Department of Chemistry at OSU.Notes and references
- (a) H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS; (c) P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527 CrossRef CAS.
- (a) J. B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS; (b) P. Xu, S. Guo, L. Wang and P. P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955 CrossRef CAS; (c) Y. L. Xiao, Q. Q. Min, C. Xu, R. W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837 CrossRef CAS; (d) X. Li, J. Zhao, Y. Wang, J. Rong, M. Hu, D. Chen, P. Xiao, C. Ni, L. Wang and J. Hu, Chem.–Asian J., 2016, 11, 1789 CrossRef CAS; (e) Y. Ohtsuka and T. Yamakawa, J. Fluorine Chem., 2016, 185, 96 CrossRef CAS; (f) L. An, Y. L. Xiao, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6921 CrossRef CAS.
- (a) C. Liu, H. Zeng, C. Zju and H. Jiang, Chem. Commun., 2020, 56, 10442 RSC; (b) S. Koley and R. A. Altman, Isr. J. Chem., 2020, 60, 313 CrossRef CAS; (c) X. X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375 CrossRef CAS; (d) H. J. Tang, L. Z. Lin, C. Feng and T. P. Loh, Angew. Chem., Int. Ed., 2017, 56, 9872 CrossRef CAS; (e) J. Hu, X. Han, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 13342 CrossRef CAS; (f) J. Hu, Y. Zhao and Z. Shi, Nat. Catal., 2018, 1, 860 CrossRef CAS.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS; (b) D. L. Orsi and R. A. Altman, Chem. Commun., 2017, 53, 7168 RSC; (c) D. L. Orsi, B. J. Easley, A. M. Lick and R. A. Altman, Org. Lett., 2017, 19, 1570 CrossRef CAS.
- For review see: T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390 CrossRef CAS.
- (a) X. Lu, Y. Wang, B. Zhang, J. J. Pi, X. X. Wang, T. J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS; (b) R. T. Thornbury and F. D. Toste, Angew. Chem., Int. Ed., 2016, 55, 11629 CrossRef CAS; (c) W. Dai, H. Shi, X. Zhao and S. Cao, Org. Lett., 2016, 18, 4284 CrossRef CAS; (d) L. Zhou, C. Zhu, P. Bi and C. Feng, Chem. Sci., 2019, 10, 1144 RSC; (e) W. Dai, J. Xiao, G. Jin, J. Wu and S. Cao, J. Org. Chem., 2014, 79, 10537 CrossRef CAS; (f) P. Tian, C. Feng and T. P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef; (g) D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209 CrossRef CAS; (h) L. Kong, X. Zhou and X. Li, Org. Lett., 2016, 18, 6320 CrossRef CAS.
- J. Liu, J. Yang, F. Ferretti, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 4690 CrossRef CAS.
- (a) M. Hudlický, Org. React., 1988, 35, 513 Search PubMed; (b) T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199 CrossRef CAS.
- ArSO2Cl as coupling reagents, see: (a) S. R. Dubbaka and P. Vogel, J. Am. Chem. Soc., 2003, 125, 15292 CrossRef CAS; (b) S. R. Dubbaka and P. Vogel, Chem.–Eur. J., 2005, 11, 2633 CrossRef CAS; (c) K. Yuan and H. Doucet, Chem. Sci., 2014, 5, 392 RSC; (d) L. Loukotova, K. Yuan and H. Doucet, ChemCatChem, 2014, 6, 1303 CAS; (e) R. Jin, K. Yuan, E. Chatelain, J.-F. Soulé and H. Doucet, Adv. Synth. Catal., 2014, 356, 3831 CrossRef CAS.
- (a) Y. Amiel, J. Org. Chem., 1971, 36, 3697 CrossRef CAS; (b) X. Zeng, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 17638 CrossRef CAS; (c) X. Li, D. Liang, W. Huang, H. Zhou, Z. Li, B. Wang, Y. Ma and H. Wang, Tetrahedron, 2016, 72, 8442 CrossRef CAS.
- X. Zhao and V. M. Dong, Angew. Chem., Int. Ed., 2011, 50, 932 CrossRef CAS.
- High-valent palladium in catalysis, see: (a) K. Muñiz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef; (b) P. Sehnal, R. Y. K. Taylor and L. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS; (c) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS.
- (a) G. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 205 CrossRef CAS; (b) G. Maestri, M. Malacria and E. Derat, Chem. Commun., 2013, 49, 10424 RSC; (c) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111 CrossRef CAS.
- For PdIII species, see: (a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS; (b) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303 CrossRef CAS . DFT calculation for C–Cl bond formation from PdIII, see:; (c) R. Jagadeesan, G. Sabapathi, J. Madhavan and P. Venuvanalingam, Inorg. Chem., 2018, 57, 6833 CrossRef CAS.
- H. T. Zhao, A. Ariafard and Z. Y. Lin, Organometallics, 2006, 25, 812 CrossRef CAS.
Footnotes |
| † Dedication in Memoriam, James D. White, for his many contributions to organic synthesis. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05192f |
| § The crystal structure for 3oa can be found in the Cambrige Crystallographic Data Centre under code q79h. |
| This journal is © The Royal Society of Chemistry 2021 |