
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA [3Cu:2S] cluster provides insight into the assembly and function of the CuZ site of nitrous oxide reductase†
Lin
Zhang
 a,
Eckhard
Bill
b,
Peter M. H.
Kroneck
c and
Oliver
Einsle
*a
a,
Eckhard
Bill
b,
Peter M. H.
Kroneck
c and
Oliver
Einsle
*a
aInstitut für Biochemie, Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, 79104 Freiburg im Breisgau, Germany. E-mail: einsle@biochemie.uni-freiburg.de
bMax-Planck-Institut für Chemische Energiekonversion, Stiftstr. 34-36, D-45470 Mülheim an der Ruhr, Germany
cFachbereich Biologie, Universität Konstanz, 78457 Konstanz, Germany
First published on 15th January 2021
Abstract
Nitrous oxide reductase (N2OR) is the only known enzyme reducing environmentally critical nitrous oxide (N2O) to dinitrogen (N2) as the final step of bacterial denitrification. The assembly process of its unique catalytic [4Cu:2S] cluster CuZ remains scarcely understood. Here we report on a mutagenesis study of all seven histidine ligands coordinating this copper center, followed by spectroscopic and structural characterization and based on an established, functional expression system for Pseudomonas stutzeri N2OR in Escherichia coli. While no copper ion was found in the CuZ binding site of variants H129A, H130A, H178A, H326A, H433A and H494A, the H382A variant carried a catalytically inactive [3Cu:2S] center, in which one sulfur ligand, SZ2, had relocated to form a weak hydrogen bond to the sidechain of the nearby lysine residue K454. This link provides sufficient stability to avoid the loss of the sulfide anion. The UV-vis spectra of this cluster are strikingly similar to those of the active enzyme, implying that the flexibility of SZ2 may have been observed before, but not recognized. The sulfide shift changes the metal coordination in CuZ and is thus of high mechanistic interest.
Introduction
Nitrous oxide (N2O) is an inert, odorless and non-toxic gas that nevertheless acts as a greenhouse agent with a global warming potential exceeding that of carbon dioxide (CO2) by a factor of 300.1 Its atmospheric concentration is on a steady rise, at a rate of 0.2–0.3% per year,1,2 which led to its designation as the most significant ozone-depleting substance of the 21st century.3 The biological reduction of N2O is catalyzed exclusively by nitrous oxide reductase (N2OR) as the ultimate step of the bacterial metabolic pathway of denitrification.2 N2OR is a periplasmic metalloprotein of 130 kDa that contains two copper centers, CuA and CuZ, in each monomer. Midpoint reduction potentials and spectroscopic properties of the two centers are distinct and provide detailed information about the respective electronic state of the sites.4–6 The three-dimensional structures of N2OR from Marinobacter hydrocarbonoclasticus (PDB 1QNI),7Paracoccus denitrificans (PDB 1FWX),8Achromobacter cycloclastes (PDB 2IWF),9Pseudomonas stutzeri (PDB 3SBQ),10 and Shewanella denitrificans (PDB 5I5M)11 were reported, providing structural details for various states of the copper centers. The dimeric N2OR forms a tight head-to-tail arrangement that juxtaposes the CuA site of one monomer with the CuZ site of the other, placing the two sites in sufficient proximity (10 Å) to form the composite active site of the enzyme at each dimer interface.7,8 CuA is a binuclear mixed-valent [Cu1.5+:Cu1.5+] site12,13 liganded by one methionine, one tryptophan, two cysteines and two histidines that is found in a very similar form in many respiratory heme:copper oxidases,14 and it mediates one-electron transfer at a midpoint potential of approximately +260 mV vs. SHE.6,15,16 When isolated under anoxic conditions – a state referred to as ‘form I’17 – the CuZ site that binds N2O is a tetranuclear [4Cu:μ4-S:μ2-S] cluster coordinated by seven histidines, as has been demonstrated for M. hydrocarbonoclasticus N2OR18 and P. stutzeri N2OR.10 The CuZ site seems to be prone to decomposition, losing sulfide ligand SZ2 to yield a [4Cu:μ4-S] site designated CuZ* in an enzyme referred to as form II.19,20 Notably, a conclusion of the present discussion will be that these terms require re-definition. In other structural analyses, CuZ sites were modelled to contain either a μ2-bridging water molecule, two terminal waters or an iodide anion that acted as an inhibitor, adding to a lack of clarity regarding the relevant active forms of this site.7–9,20To date, understanding of the biogenesis of CuZ remains fragmentary, but a series of maturation factors involved in the process have been identified.5 Interestingly, the apoenzyme is exported as a folded dimer via the Tat pathway, although the entire maturation of the metal sites occurs in the periplasm.21 Here the dedicated metallochaperone NosL, a lipoprotein, binds Cu+ and delivers it to apo-N2OR,22 and a recent study has suggested its role to be in particular for CuZ site assembly.23
In addition, the ABC transporter NosFY in conjunction with the periplasmic NosD protein is required to provide a sulfur species to the periplasm in order to complete CuZ maturation.17 We recently established a recombinant system that included all essential genes nosRZDFYLX for N2OR production, able to generate functional enzyme containing both CuA and CuZ sites in E. coli,24 facilitating mutagenesis studies of the key residues coordinating the copper sites.
Results and discussion
Properties of the seven histidine variants of CuZ
In the present study we focus on understanding the role of the seven ligands of the CuZ site, namely H129, H130, H178, H326, H382, H433 and H494 of P. stutzeri N2OR (PsN2OR). All histidine residues were individually mutated to alanine. The corresponding variants were purified (Fig. S1†), followed by spectroscopic characterization and the determination of three-dimensional structures by X-ray crystallography (Table S1†).Not unexpectedly, six of the seven variants of the coordinating histidine residues, namely H129A, H130A, H178A, H326A, H433A and H494A only showed spectral properties confirming the presence of the CuA site (Fig. S2†), with absorption maxima at 485 nm, 525 nm and 795 nm, although the relative occupancy of CuA was found to differ (Table S2†). However, the H382A variant showed UV-vis spectra that were nearly identical to those of wild-type N2OR (Fig. 1). The oxically isolated H382A was of blue colour, with partially oxidized copper sites as indicated by comparing the spectra of sample as isolated (Fig. 1D, blue) and ascorbate-reduced (Fig. 1E, blue). After oxidation with ferricyanide the protein underwent a colour change to purple, with absorption maxima at 533 nm and 780 nm and two prominent shoulders at 485 nm and 625 nm (Fig. 1D, purple). These features do not merely reflect a mixed-valent CuA site, but correspond to the properties of a wild-type form I N2OR (Fig. 1A),6,25,26 except for the more pronounced shoulder peak at 625 nm. The selective reduction of CuA by ascorbate resulted in two distinct bands at 535 nm and 625 nm (Fig. 1E, blue). We have earlier described this two-peak spectrum of CuZ as a signature of the [4Cu:2S] form I, with the 625 nm maximum originating from SZ1 and the second peak, found at 562 nm in wild-type N2OR, as originating from SZ2,10 although a difference assignment attributing this transition to also originate from SZ1 was made by other.18 The spectrum of H382A N2OR thus indicated that – unlike in CuZ* – sulfide SZ2 was still present, but the shifted peak indicated a change in its chemical environment. Also, the intensity ratio of the two CuZ bands indicated an incomplete occupancy for SZ2. Intriguingly, further reduction with dithionite did not lead to a loss of either band (Fig. 1F, cyan), indicating the “CuZ” site in H382A was redox-inert. In wild-type N2OR, dithionite reduction leads to a single charge transfer peak at 650 nm (Fig. 1C), which was assigned to the reduction of CuZ from a [2Cu+:2Cu2+] state to a [3Cu+:1Cu2+] form.4,18,27 We then determined the N2O-reducing activity using reduced benzyl viologen as electron donor. H382A was not active in N2O reduction (Fig. S3F†). We also determined the specific activities for the other six variants, and while H129A, H130A, H326A, H433A and H494A were completely inactive as expected (Fig. S3†), H178A showed low activity, with a decrease in vmax to 0.03 ± 0.01 μmol N2O per min per mg, approximately 50-fold lower than that of wild-type enzyme,24 and KM (N2O) of 268 ± 24 μM (Fig. S3D†). This residual activity (validated by 3 replicates) might be originated from a small portion of H178A containing a functional CuZ site, but the overall occupancy of possibly only 2% given the 50-fold lower activity was too low to be observed in the UV-vis spectra and crystal structure (vide infra).
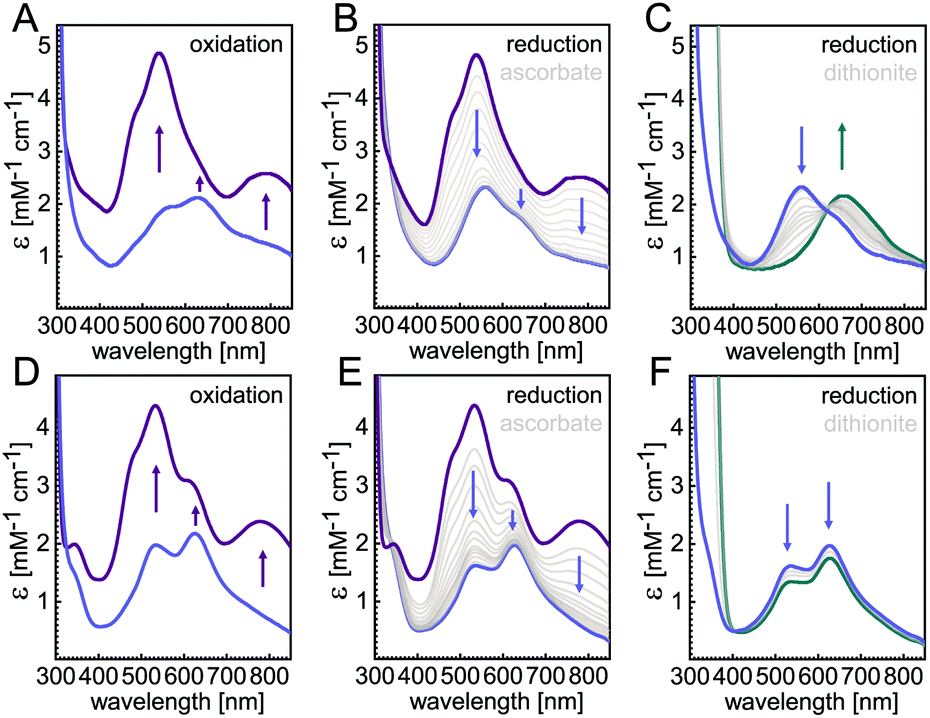 | ||
| Fig. 1 UV-vis spectra of WT PsN2OR (A–C) and variant H382A (D–F). As isolated, H382A (D) showed two absorption peaks at 535 nm and 625 nm (blue); upon oxidation with ferricyanide the spectra (purple) were similar to the wild type form I N2OR (A). (B and E) Selective reduction of CuA with ascorbate yielded a two-peak spectrum indicative of a [4Cu:2S] CuZ site (blue). Upon extended reduction with dithionite, the typical loss of the 535 nm band seen for WT PsN2OR (C) was not observed in the H382A (F) variant (cyan). | ||
We further proceeded to crystallize all variants and determined their three-dimensional structures to resolutions ranging from 1.67 Å to 1.49 Å (Table S3 and Fig. S4–S11†). As expected, the overall fold and dimeric structure of all variants remained unchanged,24 with root-mean-squared deviations for all atoms from wild-type N2OR at 0.34 Å (H129A), 0.28 Å (H130A), 0.14 Å (H178A), 0.48 Å (H326A), 0.11 Å (H382A), 0.32 Å (H433A), and 0.23 Å (H494A). Overall, the major structural difference was that the Ca2+-binding loop (N257–D273) was disordered in some of variants. This disorder seemed to correlate with the occupancy of the CuA site (Tables 1 and S2†), in line with an early report that the presence of Ca2+ ions was required for a stable insertion of the center.11
| Variant | Sidea | Ca2+-binding loop | CuZ | CuA–His583b | |
|---|---|---|---|---|---|
| a Side A shows CuA of chain A, Ca2+-binding loop and CuZ of chain B, which together form one active site; side B means the other way around. b CuA is present in all seven variants. ‘Bound’ indicates H583 is a ligand of CuA1, while ‘unbound’ means it is turned away. The given distances are between Cu1 and H583; see Table S4 for more details. c PDB accession code 6RL0. d Chain A of H382A shows weak density for Cu1 in the CuZ site. See Fig. S9B for more details. | |||||
| WTc | A | Ordered | [4Cu:2S] | Unbound | 3.74 Å |
| B | Ordered | [4Cu:2S] | Unbound | 3.44 Å | |
| H129A | A | Ordered | Empty | Bound | 2.51 Å |
| B | Disordered | 1 Zn2+ | Unbound | 3.46 Å | |
| H130A | A | Ordered | Empty | Unbound | 3.49 Å |
| B | Disordered | Empty | Unbound | 3.84 Å | |
| H178A | A | Ordered | Empty | Bound | 2.33 Å |
| B | Ordered | Empty | Bound | 2.46 Å | |
| H326A | A | Disordered | 1 Zn2+ | Unbound | 3.93 Å |
| B | Disordered | 1 Zn2+ | Unbound | 3.86 Å | |
| H382A | A | Ordered | [3Cu:2S] | Unbound | 3.69 Å |
| B | Ordered | [3Cu:2S]d | Unbound | 3.68 Å | |
| H433A | A | Ordered | Empty | Unbound | 3.47 Å |
| B | Disordered | Empty | Unbound | 3.85 Å | |
| H494A | A | Ordered | 1 Zn2+ | Bound | 2.82 Å |
| B | Disordered | 2 Zn2+ | Unbound | 3.29 Å | |
Although at different occupancies, the CuA site was present in all seven-variants (Table S2 and Fig. S4–S11†). The CuA ligands C618, W620, C622, H626, and M629 were in place to coordinate two copper ions, but the remaining ligand, H583, was in one of two possible conformations (e.g. Fig. S4†). In a ‘bound’ conformation, the Nδ atom of the imidazole moiety coordinated CuA1 at a distance of 2.5 Å, and the Nε atom formed a short hydrogen bond (2.7 Å) to residue D576 (Fig. S4A†). This is a state for CuA28 that is most commonly observed and was also found in N2OR of M. hydrocarbonoclasticus,7P. denitrificans,8 and A. cycloclastes,9 as well as in cytochrome c oxidases (PDB ID: 2CUA).29 In the second, ‘unbound’ state, however, the imidazole group of H583 was rotated away from CuA by approximately 135°, so that the Nδ atom now formed a hydrogen bond to residue S550, while the H-bond between Nε atom and D576 remained unchanged (Fig. S4B†). This histidine flip at CuA was previously reported for P. stutzeri N2OR,10 and we proposed a role in gating electron transfer from an external redox partner to CuA.20 In our previous study, H583 showed only partial ligation of CuA in either form I and II of recombinant PsN2OR.24 However, the conformational switch of H583 was randomly distributed among the variants (Table 1). Geometry changes of CuA were also observed between the two conformations of H583. The major difference was that the bond lengths of CuA2 to the sulfur atoms of C618 and C622 were about 0.1 Å longer when H583 was not a ligand (Table S4†).
The UV-vis spectra of the six variants H129A, H130A, H178A, H326A, H433A and H494A lacked the S → Cu charge transfer bands typically associated with the CuZ center (Fig. S2†). This is primarily indicative of an absence of sulfide, but the presence of histidine-coordinated Cu alone should also lead to similar N → Cu CT bands, albeit with lower intensity. Only the H326A variant might show such a peak at 630 nm (Fig. S2D†), while the other five gave no indication for the presence of Cu. Structural analysis revealed that in the variants H130A, H178A and H433A the CuZ site was only occupied by water molecules (Fig. S5, S6 and S10†). In the H129A, H326A and H494A variants, one or two zinc ions were instead found at the binding site for CuZ (Fig. S4, S7 and S11†), as confirmed by anomalous scattering data collected at the X-ray absorption K-edge of Zn (9700 eV). The presence of Zn2+ in the CuZ site can be rationalized by non-specific incorporation via a periplasmic zinc chaperone such as ZinT30 or ZraP,31,32 and the presence of multiple histidines, which are suitable ligands for zinc.33 The metal might occupy this site if the regular CuZ maturation pathway is dysfunctional. Note that in no case either a single or two Cu ions were observed. Since the maturation factors NosDFYL were present for the production of all N2OR variants in E. coli, copper (and sulfide) delivery should have been possible. The complete lack of Cu thus either means that each of the six histidines mutated here is essential for assembly, or that any site assembled without the full complement of ligands is unstable and the metal is quickly lost.
A [3Cu:2S] cluster in the H382A variant
The unexpected UV-vis spectra (Fig. 1D) were reflected in an unprecedented, only partially assembled [3Cu:μ3-S:μ-S] cluster at the CuZ site (Fig. 2B). In this cluster, Cu1 was absent, leaving SZ1 as a μ3-bridging sulfide ligating the remaining three copper ions. The histidine coordination of these was identical to native CuZ, in that Cu2 was coordinated by H129 and H178, Cu3 by H130 and H433, and Cu4 by H494. Furthermore, the second sulfide, SZ2, was still present as a ligand to Cu4, in spite of the absence of Cu1. It shifted position towards the nearby lysine K454, forming a hydrogen bond that stabilized the [3Cu:2S] cluster (Fig. 3B). As a consequence, the SZ1–Cu4–SZ2 bond angle in the [3Cu:2S] site increased by approximately 26° with respect to the one in the native, [4Cu:2S] CuZ. In contrast, the changes to the individual bond lengths in the cluster were insignificant (Fig. 3C).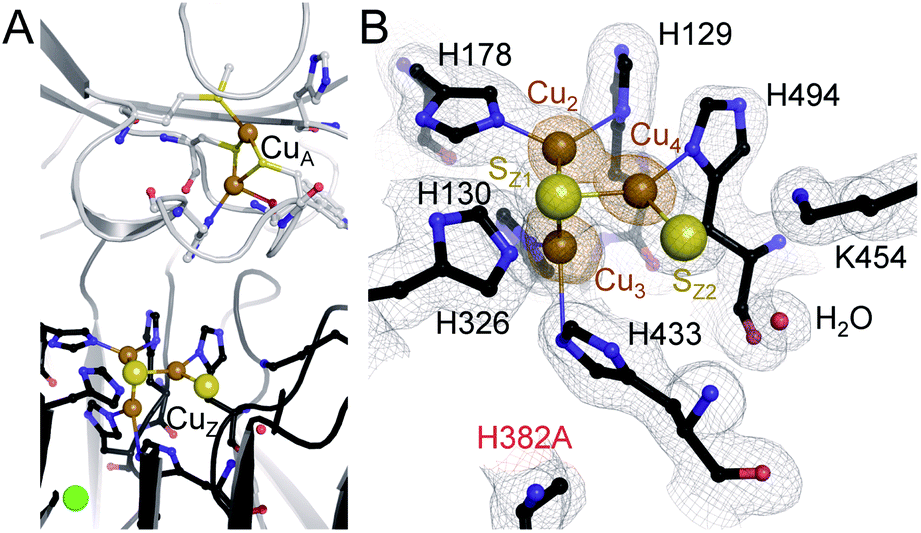 | ||
| Fig. 2 Characterization of the PsN2OR variant H382A. (A) The three-dimensional structure of H382A shows the presence of binuclear CuA site, while CuZ is an incomplete [3Cu:2S] cluster due to the loss of copper atom CuZ1. (B) Electron density map around the [3Cu:2S] site. The grey map is a 2Fo − Fc electron density maps contoured at the 1σ level, and anomalous difference Fourier maps (data collected at the X-ray absorption edge of Cu, 9050 eV) are shown in orange at the 6σ level, confirming the presence of three copper ions. | ||
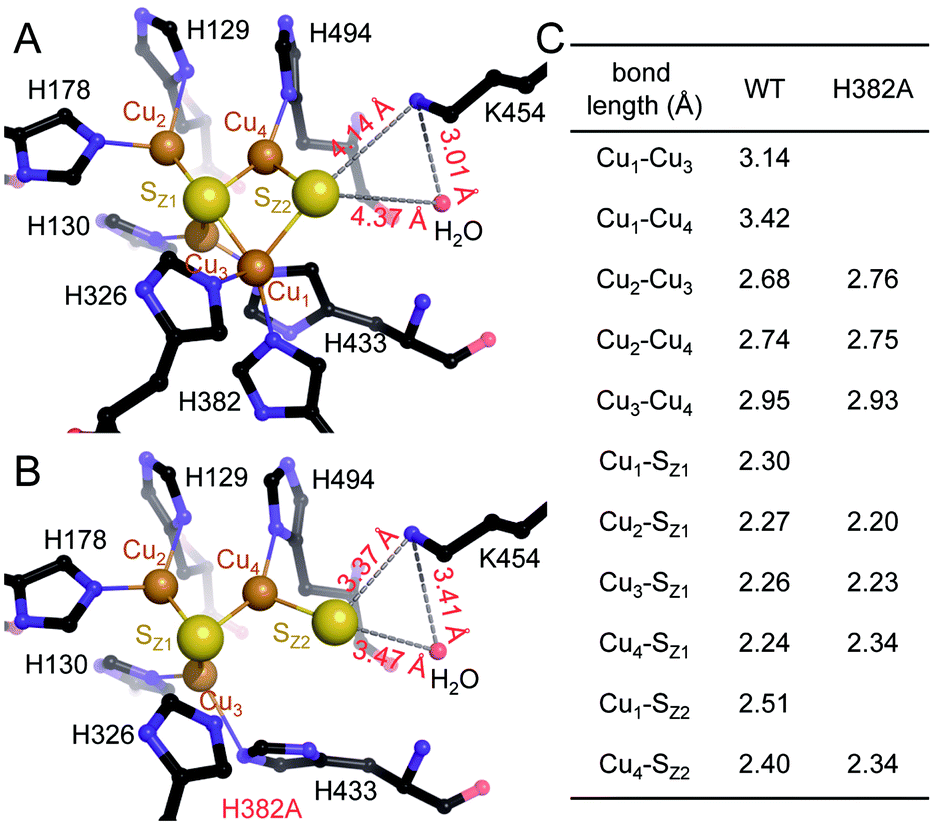 | ||
| Fig. 3 Structural comparison of CuZ site and [3Cu:2S] site. (A) Structure of holo-N2OR isolated from P. stutzeri (PDB code: 3SBQ) shows CuZ site in the [4Cu:2S] state coordinated by seven histidine residues. K454 is too far from SZ2 (4.14 Å) to form a bond. (B) In [3Cu:2S] site of variant H382A, residues H129, H130, H178, H326, H494 are in the same conformation as observed in the wild-type. However, H433 rotates by approximately 55° due to the space released by H382A. The distance between K454 and SZ2 is close enough to form a weak hydrogen bond (3.37 Å). (C) Comparison of bond lengths for CuZ and [3Cu:2S] sites. | ||
The X-band EPR spectra for both WT N2OR (Fig. 4A) and H382A variant (Fig. 4D) showed a similar 7-line hyperfine splitting pattern in the g‖ region originating from the mixed-valent [Cu1.5+:Cu1.5+] state of oxidized CuA,12,14 although the peak in the g‖ region of H382A was more pronounced. In the spectrum of WT PsN2OR, reduction with ascorbate selectively removed the contribution of CuA, leaving CuZ in a di-cupric [2Cu+:2Cu2+] state (Fig. 4B).4 The further addition of dithionite reduced CuZ to a [3Cu+:Cu2+] state (Fig. 4C).34 In [2Cu+:2Cu2+] CuZ, the two oxidized coppers couple antiferromagnetically,4,35 with a total spin of S = 0.21 The EPR signal shown in Fig. 4B was derived from residual [3Cu+:Cu2+] CuZ* that was described to have lost sulfide SZ2.4,6,34 Interestingly, EPR spectra of both ascorbate- and dithionite-reduced H382A showed a 4-line hyperfine splitting pattern in the g‖ region (g‖ = 2.18, A‖ = 66 G) (Fig. 4E and F), indicating the presence of a Cu(II) ion (S = 1/2, 63/65Cu I = 3/2) with its ground state in a 3dx2–y2-derived molecular orbital,36,37 and consistent with that of typical mononuclear type 1 (T1) copper as found in plastocyanin,37,38 azurin,39–41 cucumber basic protein,42 as well as copper-containing nitrite reductase (NiR),43–45 especially from fungal laccase46–48 and Fet3p,49,50 where Cu(II) is trigonal-planar coordinated by one cysteine and two histidine residues.
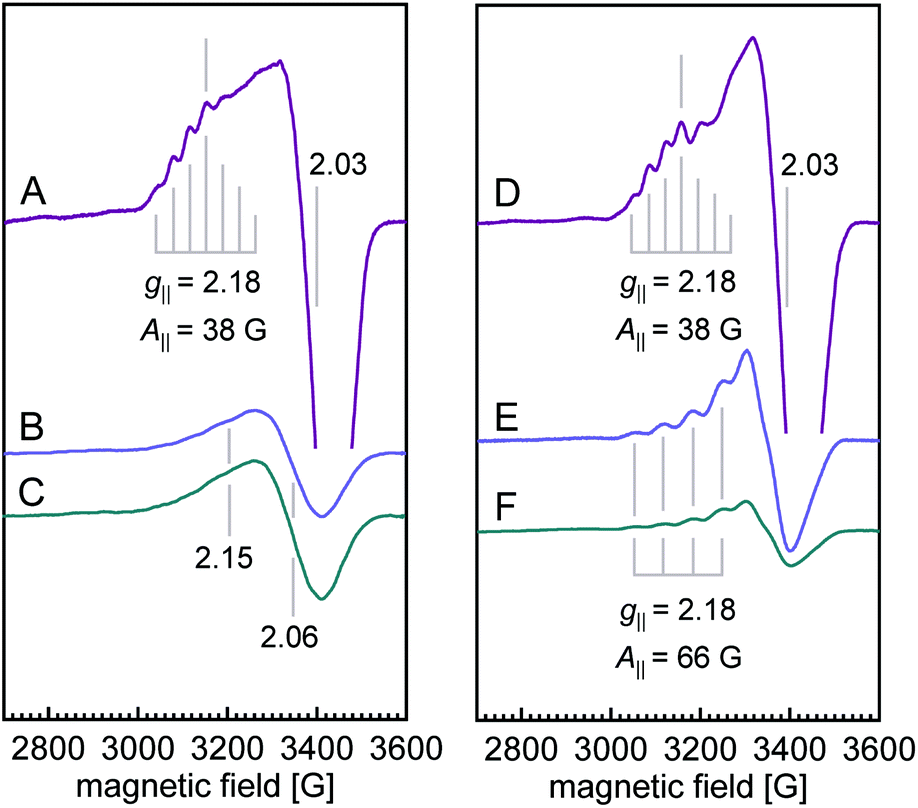 | ||
| Fig. 4 X-band CW EPR spectra of WT PsN2OR (A–C) and variant H382A (D–F). Spectra for ferricyanide-oxidized (purple), ascorbate-reduced (blue), and dithionite-reduced (cyan) samples are shown, respectively. The intensities are normalized to spins per protein. Temperature, 10 K; power, 0.2 mW; microwave frequency, 9.635 GHz; modulation amplitude, 7.46 G. | ||
Reduction with dithionite (Fig. 4F) reduced the signal intensity to about 1/3 of that for the ascorbate-reduced sample (Fig. 4E), and also destabilized the cluster, as shown by the broad peak in the g = 2.3–2.4 region (Fig. S12†). Therefore, the [3Cu:2S] cluster is very likely in a [2Cu+:Cu2+:2S2−]0 state, with Cu4 (Fig. 3B) as Cu(II), which is trigonally coordinated in the S(μ3-S)–N(His494)–S(μ2-S) plane.
Residue H326, the second ligand to Cu1 in the CuZ site (Fig. 3A), did not coordinate a metal in the [3Cu:2S] cluster (Fig. 3B). Thus, we expected the [3Cu:2S] cluster to be present in variant H326A as well. However, the H326A only contained a single Zn2+ (Fig. S7†), indicating the stabilizing effect of H326 and H382 to the CuZ site differs. We hypothesize that H326 is required already in the early stages of CuZ maturation, so that the site will not assemble if this histidine residue is mutated. H382, in contrast, only seems come into play once the entire cluster is assembled. Our data do not reveal whether CuZ is initially complete as a [4Cu:2S] cluster that is then prone to lose Cu1 in the absence of the support by H382. Residue K454 (K397 in the case of N2OR from M. hydrocarbonoclasticus) was proposed to play a role in the catalytic cycle of N2OR by providing protons.34,51,52 Our data show that it is also involved in the stabilization – and possibly assembly – of CuZ through hydrogen-bonding interactions (Fig. 3B).
Concluding remarks
Beyond these questions regarding the assembly of CuZin vivo, the [3Cu:2S] cluster in PsN2OR H382A also has interesting functional implications. In the variant, the cluster retains sulfide SZ2, but has it shifted towards residue K454, leading to UV/vis properties that fall between the forms I and II described earlier. Form II N2OR was proposed to contain a [4Cu:S] CuZ* center, requiring the loss of SZ2 as a prerequisite for reductive activation.4 Nevertheless the [4Cu:2S] CuZ state has been consistently isolated from cells grown under denitrifying conditions (i.e. after having turned over in vivo).4,6,10,18,52,53 The H382A variant may now help to reconcile these seemingly incompatible results, suggesting that SZ2 can indeed change its position from ligating Cu1 of CuZ to the nearby K454 (similar sulfur-shift mechanism was envisaged by Moura and Pauleta).53 This would leave both Cu1 and Cu4 with three remaining ligands, and thus with the opportunity to bind an additional exogenous ligand, the substrate N2O, in a 1,3-bridging fashion. This binding mode is similar to the one proposed by Moura and Solomon,52 but does not require dissociation of SZ2 in accordance with our structural data.24 It would also imply a binding mode of N2O that is very much compatible with the N2O binding site we observed at CuZ after pressurizing crystals of the enzyme with the substrate gas.10In particular, the UV-vis properties of the H382A variant highlight the fact that preparations of the enzyme that were typically assigned to a ‘form II’, or CuZ* species that implies a [4Cu:S] site may well be of a different nature. The loss of the charge transfer band at 550 nm that characterizes this form II may be rooted in the shift of SZ2 towards K454, without being fully lost form the cluster. This finding is also in line with the frequent observation of a less well-defined, elongate electron density feature at the Cu1–Cu4 edge of the CuZ cluster in different published and unpublished structures. Such features were frequently interpreted as two H2O ligands,9 or inspired an originally suggested binding mode for N2O54 that was, however, never observed experimentally. The present data now offers an alternative rationalization for the reported spectroscopic features that may eventually lead to a unified picture of the structural and functional features of the unique and enigmatic CuZ site.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the staff at beam lines X06SA and X06DA, Swiss Light Source, Villigen, CH, for excellent assistance with data collection, and Anne-Catherine Abel for help with the experiments. This work was supported by the European Research Council (grant 310656 to O. E.) and Deutsche Forschungsgemeinschaft (CRC 992, project no. 192904750).Notes and references
- K. Butterbach-Bahl, E. M. Baggs, M. Dannenmann, R. Kiese and S. Zechmeister-Boltenstern, Philos. Trans. R. Soc., B, 2013, 368, 20130122 CrossRef.
- A. J. Thomson, G. Giannopoulos, J. Pretty, E. M. Baggs and D. J. Richardson, Philos. Trans. R. Soc., B, 2012, 367, 1157–1168 CrossRef CAS.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS.
- E. M. Johnston, S. Dell'Acqua, S. Ramos, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 614–617 CrossRef CAS.
- S. R. Pauleta, S. Dell'Acqua and I. Moura, Coord. Chem. Rev., 2013, 257, 332–349 CrossRef CAS.
- T. Rasmussen, B. C. Berks, J. N. Butt and A. J. Thomson, Biochem. J., 2002, 364, 807–815 CrossRef CAS.
- K. Brown, M. Tegoni, M. Prudencio, A. S. Pereira, S. Besson, J. J. Moura, I. Moura and C. Cambillau, Nat. Struct. Biol., 2000, 7, 191–195 CrossRef CAS.
- K. Brown, K. Djinovic-Carugo, T. Haltia, I. Cabrito, M. Saraste, J. J. G. Moura, I. Moura, M. Tegoni and C. Cambillau, J. Biol. Chem., 2000, 275, 41133–41136 CrossRef CAS.
- K. Paraskevopoulos, S. V. Antonyuk, R. G. Sawers, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 2006, 362, 55–65 CrossRef CAS.
- A. Pomowski, W. G. Zumft, P. M. Kroneck and O. Einsle, Nature, 2011, 477, 234–237 CrossRef CAS.
- L. K. Schneider and O. Einsle, Biochemistry, 2016, 55, 1433–1440 CrossRef CAS.
- J. A. Farrar, F. Neese, P. Lappalainen, P. M. H. Kroneck, M. Saraste, W. G. Zumft and A. J. Thomson, J. Am. Chem. Soc., 1996, 118, 11501–11514 CrossRef CAS.
- D. R. Gamelin, D. W. Randall, M. T. Hay, R. P. Houser, T. C. Mulder, G. W. Canters, S. de Vries, W. B. Tolman, Y. Lu and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 5246–5263 CrossRef CAS.
- M. E. Llases, M. N. Lisa, M. N. Morgada, E. Giannini, P. M. Alzari and A. J. Vila, FEBS J., 2020, 287, 749–762 CrossRef CAS.
- C. L. Coyle, W. G. Zumft, P. M. H. Kroneck, H. Körner and W. Jakob, Eur. J. Biochem., 1985, 153, 459–467 CrossRef CAS.
- C. Carreira, M. M. C. Dos Santos, S. R. Pauleta and I. Moura, Bioelectrochemistry, 2020, 133, 107483 CrossRef CAS.
- W. G. Zumft and P. M. H. Kroneck, Adv. Microb. Physiol., 2007, 52, 107–227 CrossRef CAS.
- E. M. Johnston, S. Dell'Acqua, S. R. Pauleta, I. Moura and E. I. Solomon, Chem. Sci., 2015, 6, 5670–5679 RSC.
- W. G. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- L. K. Schneider, A. Wüst, A. Pomowski, L. Zhang and O. Einsle, Met. Ions Life Sci., 2014, 14, 177–210 CAS.
- S. R. Pauleta, M. S. P. Carepo and I. Moura, Coord. Chem. Rev., 2019, 387, 436–449 CrossRef CAS.
- M. A. McGuirl, J. A. Bollinger, N. Cosper, R. A. Scott and D. M. Dooley, J. Biol. Inorg Chem., 2001, 6, 189–195 CrossRef CAS.
- S. P. Bennett, M. J. Soriano-Laguna, J. Bradley, D. A. Svistunenko, D. J. Richardson, A. J. Gates and N. E. Le Brun, Chem. Sci., 2019, 10, 4985–4993 RSC.
- L. Zhang, A. Wüst, B. Prasser, C. Müller and O. Einsle, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12822–12827 CrossRef CAS.
- S. Dell'Acqua, S. R. Pauleta, J. J. Moura and I. Moura, Philos. Trans. R. Soc., B, 2012, 367, 1204–1212 CrossRef.
- J. M. Charnock, A. Dreusch, H. Körner, F. Neese, J. Nelson, A. Kannt, H. Michel, C. D. Garner, P. M. Kroneck and W. G. Zumft, Eur. J. Biochem., 2000, 267, 1368–1381 CrossRef CAS.
- P. Chen, I. Cabrito, J. J. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 10497–10507 CrossRef CAS.
- P. M. H. Kroneck, J. Biol. Inorg Chem., 2018, 23, 27–39 CrossRef CAS.
- P. A. Williams, N. J. Blackburn, D. Sanders, H. Bellamy, E. A. Stura, J. A. Fee and D. E. McRee, Nat. Struct. Biol., 1999, 6, 509–516 CrossRef CAS.
- A. I. Graham, S. Hunt, S. L. Stokes, N. Bramall, J. Bunch, A. G. Cox, C. W. McLeod and R. K. Poole, J. Biol. Chem., 2009, 284, 18377–18389 CrossRef CAS.
- I. Petit-Hartlein, K. Rome, E. de Rosny, F. Molton, C. Duboc, E. Gueguen, A. Rodrigue and J. Coves, Biochem. J., 2015, 472, 205–216 CrossRef.
- C. A. Blindauer, Chem. Commun., 2015, 51, 4544–4563 RSC.
- I. Dokmanic, M. Sikic and S. Tomic, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 257–263 CrossRef CAS.
- S. Ghosh, S. I. Gorelsky, S. DeBeer George, J. M. Chan, I. Cabrito, D. M. Dooley, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 3955–3965 CrossRef CAS.
- S. I. Gorelsky, S. Ghosh and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 278–290 CrossRef CAS.
- E. I. Solomon and R. G. Hadt, Coord. Chem. Rev., 2011, 255, 774–789 CrossRef CAS.
- E. I. Solomon, R. K. Szilagyi, S. D. George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS.
- J. M. Guss and H. C. Freeman, J. Mol. Biol., 1983, 169, 521–563 CrossRef CAS.
- H. B. Gray, B. G. Malmström and R. J. P. Williams, J. Biol. Inorg Chem., 2000, 5, 551–559 CrossRef CAS.
- H. Nar, A. Messerschmidt, R. Huber, M. Vandekamp and G. W. Canters, J. Mol. Biol., 1991, 218, 427–447 CrossRef CAS.
- C. M. Groeneveld, R. Aasa, B. Reinhammar and G. W. Canters, J. Inorg. Biochem., 1987, 31, 143–154 CrossRef CAS.
- L. B. LaCroix, D. W. Randall, A. M. Nersissian, C. W. G. Hoitink, G. W. Canters, J. S. Valentine and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 9621–9631 CrossRef CAS.
- L. B. LaCroix, S. E. Shadle, Y. N. Wang, B. A. Averill, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1996, 118, 7755–7768 CrossRef CAS.
- K. Olesen, A. Veselov, Y. W. Zhao, Y. S. Wang, B. Danner, C. P. Scholes and J. P. Shapleigh, Biochemistry, 1998, 37, 6086–6094 CrossRef CAS.
- H. Iwasaki, S. Noji and S. Shidara, J. Biochem., 1975, 78, 355–361 CrossRef CAS.
- A. E. Palmer, D. W. Randall, F. Xu and E. I. Solomon, J. Am. Chem. Soc., 1999, 121, 7138–7149 CrossRef CAS.
- A. Messerschmidt and R. Huber, Eur. J. Biochem., 1990, 187, 341–352 CrossRef CAS.
- D. M. Dooley, J. Rawlings, J. H. Dawson, P. J. Stephens, L. E. Andreasson, B. G. Malmström and H. B. Gray, J. Am. Chem. Soc., 1979, 101, 5038–5046 CrossRef CAS.
- T. E. Machonkin, L. Quintanar, A. E. Palmer, R. Hassett, S. Severance, D. J. Kosman and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 5507–5517 CrossRef CAS.
- A. B. Taylor, C. S. Stoj, L. Ziegler, D. J. Kosman and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15459–15464 CrossRef CAS.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2018, 54, 1097–1100 RSC.
- E. M. Johnston, C. Carreira, S. Dell'Acqua, S. G. Dey, S. R. Pauleta, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 4462–4476 CrossRef CAS.
- C. Carreira, R. F. Nunes, O. Mestre, I. Moura and S. R. Pauleta, J. Biol. Inorg Chem., 2020, 25, 927–940 CrossRef CAS.
- S. Ghosh, S. I. Gorelsky, P. Chen, I. Cabrito, J. J. G. Moura, I. Moura and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 15708–15709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05204c |
| This journal is © The Royal Society of Chemistry 2021 |