
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced sampling molecular dynamics simulations correctly predict the diverse activities of a series of stiff-stilbene G-quadruplex DNA ligands†
Michael P.
O'Hagan‡
 a,
Susanta
Haldar‡
ab,
Juan C.
Morales
c,
Adrian J.
Mulholland
*ab and
M. Carmen
Galan
*a
a,
Susanta
Haldar‡
ab,
Juan C.
Morales
c,
Adrian J.
Mulholland
*ab and
M. Carmen
Galan
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: adrian.mulholland@bristol.ac.uk; m.c.galan@bristol.ac.uk
bCentre for Computational Chemistry, School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
cInstituto de Parasitología y Biomedicina “López Neyra” Consejo Superior de Investigaciones Científicas (CSIC), PTS Granada, Avenida del Conocimiento 17, 18016 Armilla, Granada, Spain
First published on 26th November 2020
Abstract
Ligands with the capability to bind G-quadruplexes (G4s) specifically, and to control G4 structure and behaviour, offer great potential in the development of novel therapies, technologies and functional materials. Most known ligands bind to a pre-formed topology, but G4s are highly dynamic and a small number of ligands have been discovered that influence these folding equilibria. Such ligands may be useful as probes to understand the dynamic nature of G4 in vivo, or to exploit the polymorphism of G4 in the development of molecular devices. To date, these fascinating molecules have been discovered serendipitously. There is a need for tools to predict such effects to drive ligand design and development, and for molecular-level understanding of ligand binding mechanisms and associated topological perturbation of G4 structures. Here we study the G4 binding mechanisms of a family of stiff-stilbene G4 ligands to human telomeric DNA using molecular dynamics (MD) and enhanced sampling (metadynamics) MD simulations. The simulations predict a variety of binding mechanisms and effects on G4 structure for the different ligands in the series. In parallel, we characterize the binding of the ligands to the G4 target experimentally using NMR and CD spectroscopy. The results show good agreement between the simulated and experimentally observed binding modes, binding affinities and ligand-induced perturbation of the G4 structure. The simulations correctly predict ligands that perturb G4 topology. Metadynamics simulations are shown to be a powerful tool to aid development of molecules to influence G4 structure, both in interpreting experiments and to help in the design of these chemotypes.
Introduction
G-quadruplexes (G4) are secondary nucleic acid structures formed by sequences rich in guanine.1–3 They are composed of stacked arrangements of G-tetrads, square-planar arrangements of four guanine bases that are stabilized by Hoogsteen hydrogen bonding and coordination to a monovalent metal cation such as potassium4 or sodium.5 Of particular interest are G4s found at chromosome telomeres and in oncogene promoters, as these have been proposed as therapeutic targets in the treatment of human cancers.3,6,7 Ligands that target these structures with high specificity are much in demand as tools to validate biological and therapeutic hypotheses and as targets for the development of novel therapies. The precise structural definition and dynamic nature of G4 folding suggest a wide range of potential applications in the design of functional systems.8,9 Small molecule ligands have the potential to regulate G4 structures as well as bind to preformed structures.9 However, prediction of a molecule's ability to influence G4 folding remains elusive, precluding the rational design of these chemotypes.Given the huge range of chemical space available for G4 ligand design, the significant investment of time and resources needed to synthesize new ligands, combined with the difficulty of predicting the binding mode and influence on DNA folding of a putative G4-binding chemotype from the chemical structure alone, there is a need for tools that allow the activity of potential ligand candidates to be predicted efficiently and reliably in silico. Automated docking and molecular dynamics (MD) simulations have been applied to examine G4 ligand binding affinity and binding modes.10–15 However, ligand binding and associated conformational perturbation of the macromolecule occur on timescales usually beyond the capability of standard MD simulations, because these are typically long-timescale events, (i.e., rare events) in which several free energy basins may be separated by high energy barriers. Enhanced sampling simulation methods, such as metadynamics (MetaD), can overcome these barriers and sample multiple free energy minima on a rugged free energy surface.16 MetaD simulations are widely employed to investigate complex long timescale biomolecular events, and can allow the determination of the relevant free energy surface (FES) of the process of interest.17 MetaD simulations have been applied to study e.g. protein folding and unfolding,18,19 protein conformational behaviour and protein-ligand binding20–24 including in combination with docking to predict structures of protein ligand complexes,25 RNA folding,26 and the adsorption of DNA bases and small molecules onto surfaces.27–29 Moraca and co-workers have employed funnel-metadynamics30,31 to investigate the binding of berberine to G4 DNA,32 whilst Casini and colleagues studied the association of gold N-heterocyclic carbene complexes with G4 DNA.33 Both studies determined solution-phase association constants experimentally using fluorescence-based methods and found good agreement with the binding affinities predicted by the calculations. These simulations were undertaken from experimental (e.g. crystallographic) structures. Experimental structures of ligand/G4 complexes are only available in a small number of cases, useful for rationalizing experimental data but of limited use in the prediction of the behaviour of novel compounds. Therefore, the ability to predict structures of complexes of novel ligands, and to predict the ability of ligands to induce conformational perturbations would be of great benefit in the design of G4 ligands. Towards this end, Limongelli and co-workers previously applied MetaD to investigate the solution-phase binding mode of a G4 ligand (a benzothiazole derivative) for which less sophisticated computational methods did not fully describe the experimental data obtained by NMR spectroscopy.34 The enhanced sampling techniques revealed binding modes indicated by NMR that were not observed in standard MD simulations, though experimental data was not available to validate the binding affinity calculated from the free energy surface. The G4 model employed in that study was a symmetrical tetramolecular species. Unimolecular G4s are considered more physiologically relevant for targeting with ligands. Nonetheless, these promising results show the potential of MetaD to study G4 ligand binding. In the present study, we apply this approach to study ligand binding to a physiologically relevant unimolecular G4, which exhibits greater diversity in structural features and ligand binding sites (owing the presence of loop regions) as well as greater susceptibility to ligand-induced polymorphism. Furthermore, we test MetaD for predicting the diverse G4 binding behaviours of a series of related ligands, to assist ligand design.
Here, we present a synergistic application of MetaD simulations, circular dichroism and NMR spectroscopy to study the binding of a family of small molecules to G4 DNA. MetaD and MD predictions of structures, and ligand binding sites, are compared with solution-phase NMR and circular dichroism (CD) experiments. Our results reveal a striking correlation between the computational and experimental approaches and validate MetaD as a useful tool for predicting G4/ligand interactions, when the structure of the complex in question has not been experimentally determined previously. Moreover, the simulations correctly identify ligands that induce perturbations in G4 folded topology. Given the range of promising practical applications of G4 ligands, this combined simulation/experimental approach has significant potential as a tool to aid design of G4 ligands for biological and functional applications.
Results
Experimental approach and selection of G4/ligand models
We recently reported the design and synthesis of a novel family of stiff-stilbene G4 ligands (1–5, Fig. 1).35,36 The lead compound 1 displays high G4 thermal stabilization (ΔTm = 21 °C) and discrimination against duplex DNA (ΔTm = <1 °C), as well as interesting promise as the basis of anticancer and antiparasitic therapies, being toxic in the nanomolar range against HeLa cells whilst remaining 30-fold less toxic to non-tumoral mammalian cells. Through a variety of experimental approaches, we observed that the effect of ligand on G4 was sensitive to the nature of the ligand geometry, functionality and the nature of the G4 topology and metal cation. For example, whilst ligand 1 stabilized the native hybrid G4 fold in potassium-containing buffer, it triggered the disruption of the same sequence in sodium-rich conditions.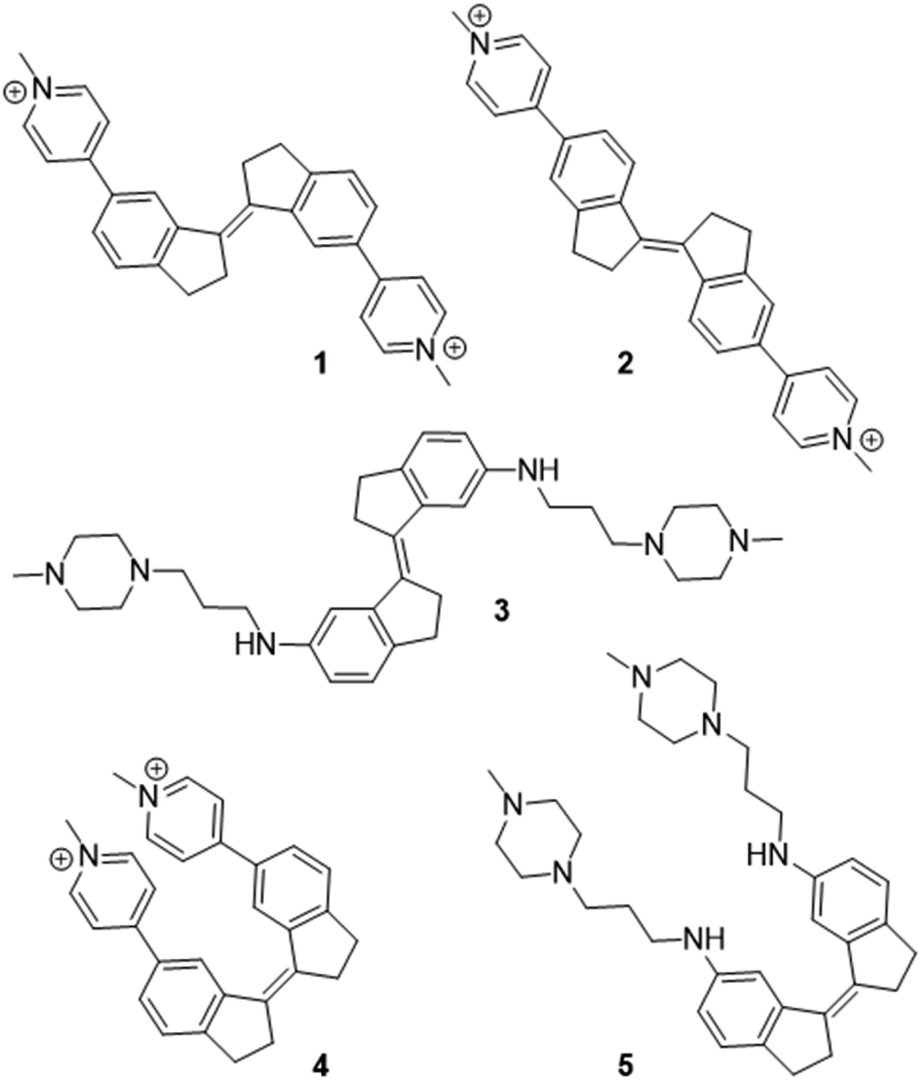 | ||
| Fig. 1 Stiff-stilbene G4 ligands studied in this work. | ||
Given the diverse activities obtained in this ligand series, the stiff-stilbene ligand family appears ideal to study as a test of the ability of MetaD simulations to predict G4 ligand activity more broadly. Indeed, we recently applied MetaD to rationalize the effect of ligand 1 on G4 DNA, which promotes G4 unfolding under sodium-rich conditions.36 The unfolding observed in experiment was corroborated by MetaD, but it was difficult to confirm ligand binding sites spectroscopically since the ligand-induced conformation perturbations resulted in severe line broadening and attenuation of the characteristic DNA proton resonances in the 1H-NMR spectra. Therefore, applying MetaD to related ligands, for which the folded G4 structure is preserved upon binding, allowed us to compare the binding site structures and binding affinity data with experiments.
We thus decided to investigate the binding of additional ligands (2–5) to G4 DNA in atomistic detail using MetaD and, in parallel, through solution-phase experiments to probe the structures of the G4/ligand complexes. Docking calculations, MD and well-tempered MetaD simulations were run using established protocols (see ESI† for full details).36 To provide experimental evidence for the association mode of ligands with G4, including effect on G4 folding topology and ligand binding sites, we employed a combination of CD spectroscopy and NMR spectroscopy. CD is a powerful technique to report on G4 secondary structure, with characteristic bands that indicate different types of folded structure.37 Such studies are important to determine whether the native G4 structure is preserved on ligand binding, or whether the ligand induces a conformational change in the oligonucleotide secondary structure.36 Meanwhile, NMR methods yield more detailed structural information on binding sites, e.g. through examination of ligand-induced chemical shift perturbations of the G4 resonances.38 Information from the experiment was used to test the simulation predictions. We note that the simulations were in no way fitted to or parameterized based on the experimental results.
Regarding the choice of G4 model: we required a system where ligand binding was evident using our chosen spectroscopic methods, and generated sufficiently simple spectra that could be readily interpreted for comparison to the simulations. In a previous study, we found that the ligands 1–5 interact more strongly with the hybrid (potassium) form of telomeric G4 than the antiparallel topology (formed in sodium-rich conditions).35 However, the presence of multiple conformations of the potassium sequence (telo23) in solution, coupled with overlapping signals and severe line-broadening during the NMR titration experiments (probably a result of intermediate exchange between species on the NMR timescale) made it difficult to extract reliable chemical shift perturbations that indicate binding hotspots. In the present study, we found that the NMR spectra of the telo22 antiparallel G4 model were much more straightforward to interpret (vide infra), with the weaker ligand binding moving the system towards the fast-exchange regime and allowing chemical shift perturbations to be followed with more confidence. Furthermore, the spectral assignment of key regions of the spectrum was straightforward by comparison of the 2D NOESY spectra with published data.5 The telo22 CD spectrum, 1D and 2D NMR spectra and keys to assignment of resonances discussed below are provided in the ESI (Fig. S1†).
Antiparallel telo22 is intercalated and disrupted by pyridinium stiff-stilbene ligands 1 and 2
In the absence of ligand, the CD spectrum of the antiparallel fold of telo22 is characterized by positive bands at 290 nm and 240 nm and a negative band at 260 nm (Fig. S1b†).37 We previously reported that ligand 1 unfolds this G4 system, as shown by both CD and NMR spectroscopy, and previous simulations indicated that this results from ligand intercalation and disruption of the hydrogen bonding network of the G-tetrads. This effect appeared to be much more pronounced for the related telo23 sequence: weaker structural perturbations were observed in NMR for telo22 than telo23. In the current study, we began by examining the binding of ligand 2 to telo22 G4, which differs from 1 only in the placement of the pyridinium substituents. Nonetheless, this modification confers a significantly different geometry and electronic structure, both factors that could affect the G4-ligand interaction.Docking of 2 to telo22 G4 DNA produced two high affinity poses. To test the stability of these poses, we performed unbiased MD simulations. The resulting dihedral distribution (Fig. 2a and ESI, Fig. S2†) obtained from the highest affinity docked pose shows three stable conformations (A, B and C) including the initial docked pose (Pose A, Fig. 2b). In this pose, the ligand binds to the major groove of G4 where it partially interacts with bases from the G-tetrads (G4, G8 and G9) and one of indane residues stacks with the T5 base. Pose B (Fig. 2b) has ligand 2 in the quadruplex groove with a slight change in ligand orientation. The ligand stacks with the T5 base and interacts with the G9 base in this pose, although an interaction with G10 is seen which results in loss of interaction with G8 compared to Pose A. As the simulation progresses, ligand 2 intercalates in the major groove, disrupting the hydrogen bonds in the G-tetrad (Pose C, Fig. 2b). In this pose, ligand 2 is intercalated between G2, G3 and G15 from the middle and lower G-tetrad. Following a further 500 ns simulation, the DNA backbone RMSD (Fig. 2c) begins to fluctuate between 3.0 to 5.0 Å, which suggests a loss of stability of the DNA fold due to rupturing of the G-tetrad hydrogen bonds. Ligand 2 was therefore expected to induce instability in G4 in a similar way to ligand 1. Analysis of the second docked pose is provided in the ESI (Fig. S3†); in this simulation, the ligand was found mainly to stack onto the residues of the lateral loops T18 and A7.
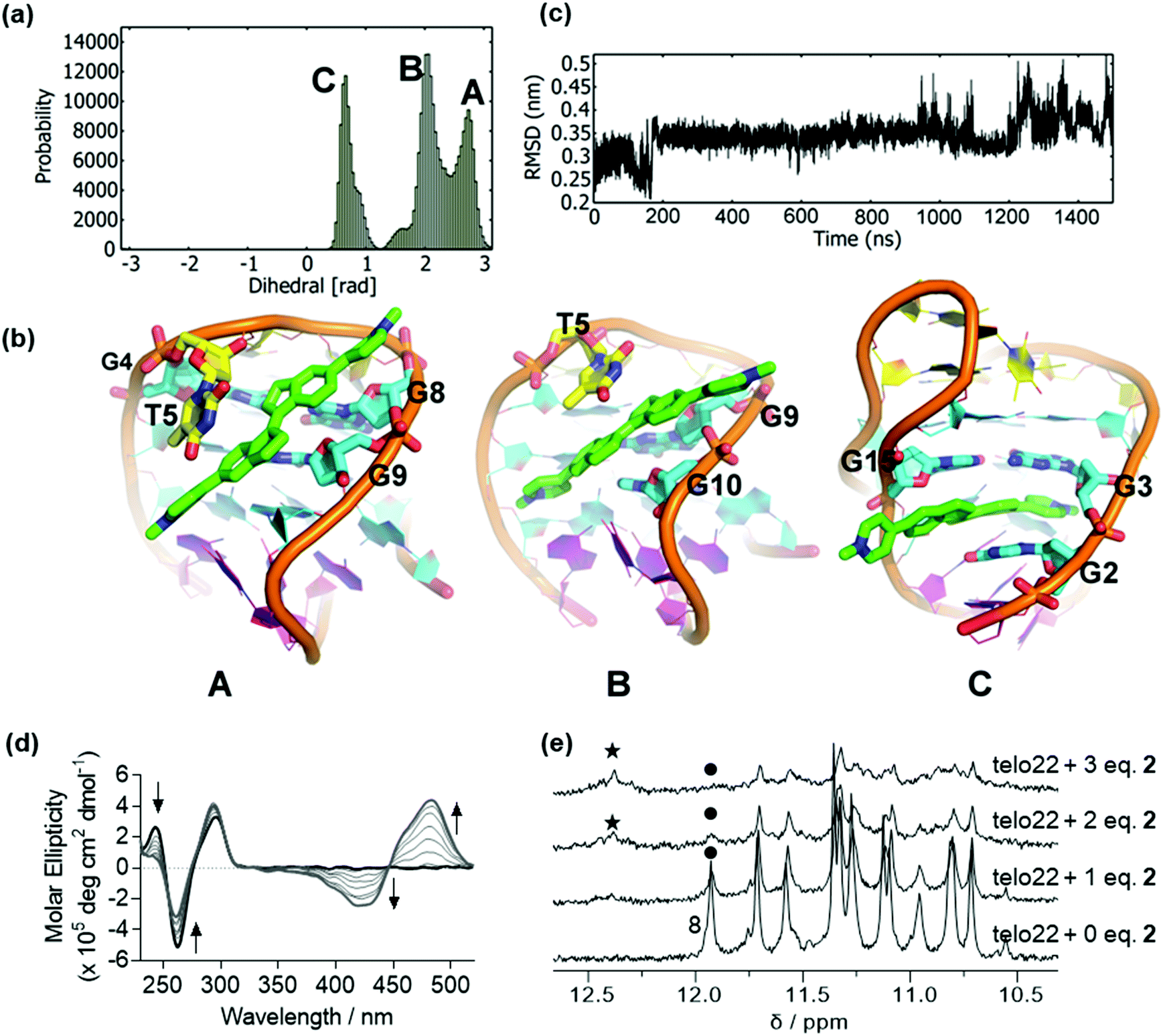 | ||
| Fig. 2 Binding of ligand 2 to antiparallel telo22 investigated by MD simulations, circular dichroism and NMR spectroscopy. (a) Dihedral angle distributions sampled in the MD simulations, (b) representations of the principal binding poses sampled in the MD simulations, key binding residues are indicated, (c) RMSD of the DNA backbone co-ordinates over the time course of the MD simulations, (d) circular dichroism spectra of telo22 (black trace) and increasing equivalents (light grey traces) of ligand 2 up to 7 eq. (dark grey trace), (e) NMR titration of telo22 with ligand 2, the attenuation of the G8 resonance is marked with a dot and the appearance of a new signal is marked with an asterisk. | ||
In agreement with the simulation results, the CD spectrum of telo22 in the presence of increasing concentrations of 2 shows a marked attenuation of the positive feature at 240 nm and negative feature at 260 nm corresponding to the G4 fold, along with a strong induced positive Cotton effect in the ligand region 350–550 nm (Fig. 2d). Such effects indicate disruption of the G4 structure (reduction in intensity of characteristic CD bands) that may arise upon intercalation. A small hypsochromic (blue) shift in the 295 nm positive dichroic band is also observed. Likewise, NMR titration (Fig. 2e) shows a marked decrease in intensity in the imino proton signals of telo22 and emergence of a new signal in the downfield (12.5 ppm) region of the spectrum, more characteristic of classical DNA base pairing rather than the Hoogsteen bonding found in G-tetrads, indicative of a degree of structural perturbation, as predicted by our MD simulations.
Antiparallel telo22 is stabilized by low concentrations of methylpiperazine ligand 3via binding to the top face of the G4, but the ligand induces misfolding at higher concentrations
Ligands of type 3, where the stiff-stilbene core is appended to flexible alkyl spacer terminated in a basic moiety (protonated at physiological pH) also bind appreciably to G4 DNA (ΔTm = 12 °C at 10 μM ligand concentration).35 However, the interaction is weaker than for pyridinium ligands 1 and 2, perhaps a result of the more flexible nature of the side chains which provide additional degrees of freedom in the unbound ligand.Fig. 3 depicts the binding conformations sampled in MD simulation of ligand 3 on the lowest-energy binding pose found in preliminary docking calculations. Two dominant conformations are sampled (Fig. 3a and ESI, Fig. S4†). The initial pose predicts the ligand to bind in the G4 major groove, interacting with G16 and G4 from the top G-quartet while one of the alkyl side-chains stacks with the A7, A19 and T6 bases (Pose A, Fig. 3b). However, MD simulation suggests that the ligand eventually slides towards the top face of the DNA, producing another binding conformation (Pose B, Fig. 3b). Here, ligand 3 binds mainly with the top face, external to the capping residues of the lateral loops, where both indane rings stack on the A19, A7 and T6 bases. Furthermore, a hydrogen bond is formed between the ligand tail and oxygen atom of the sugar phosphate backbone. The DNA secondary structure appeared stable during the simulation (Fig. 3c). MD simulation of a second binding pose also shows the ligand to be located in the major groove (ESI, Fig. S5†).
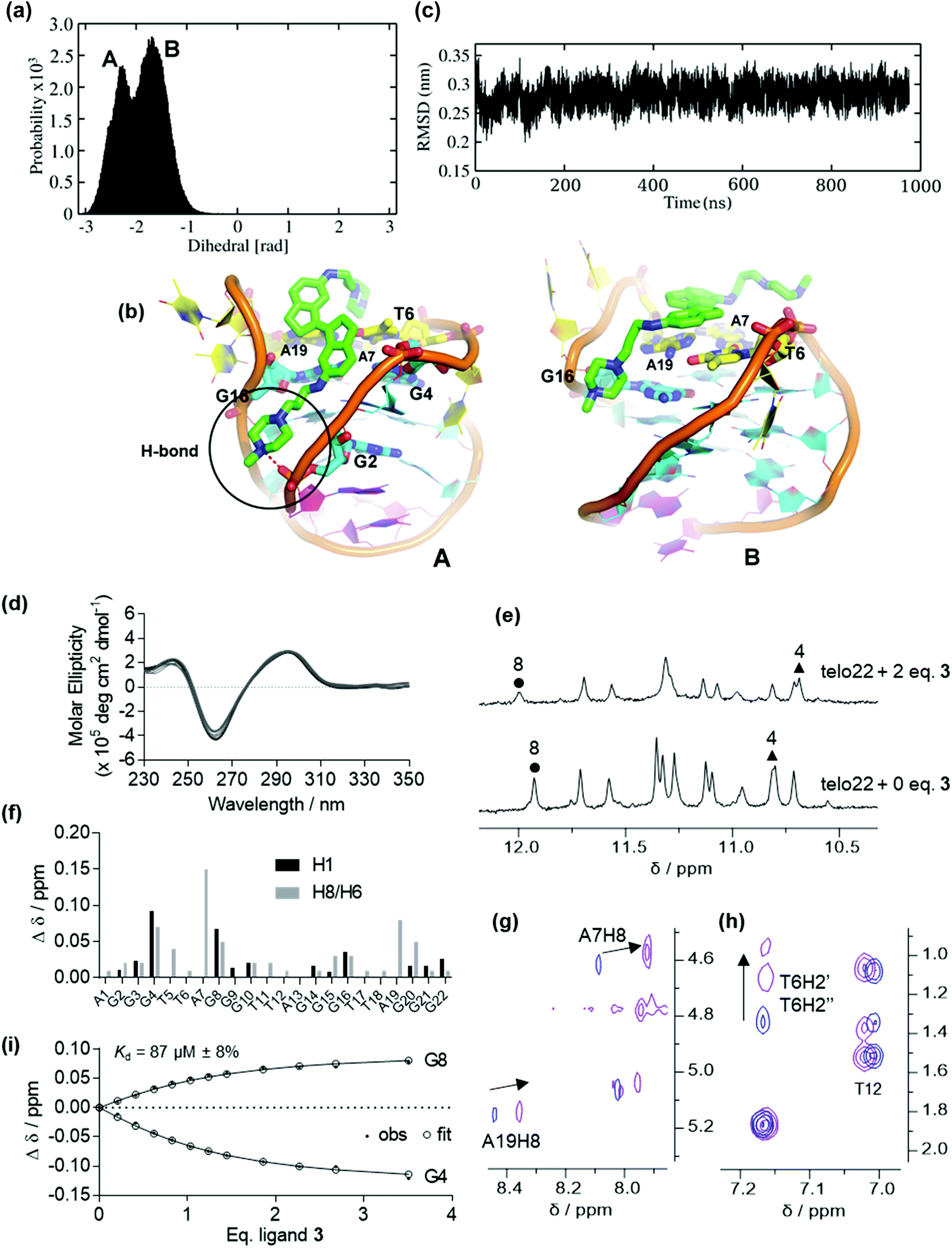 | ||
Fig. 3 Binding of ligand 3 to antiparallel telo22 investigated by MD simulations, circular dichroism and NMR spectroscopy. (a) Dihedral angle distributions sampled in the MD simulations, (b) representations of the principal binding poses sampled in the MD simulations, key binding residues are indicated, (c) RMSD of the DNA backbone co-ordinates over the time course of the MD simulations, (d) circular dichroism spectra of telo22 (black trace) and increasing equivalents (light grey traces) of ligand 3 up to 7 eq. (dark grey trace), (e) NMR titration of telo22 with ligand 3 showing shifts of key imino resonances, (f) changes in chemical shifts of imino an aromatic resonances of telo22 upon adding 2 eq. ligand 3 (g and h) partial NOESY spectra showing shifts of key (g) aromatic/anomeric (h) sugar aliphatic resonances, (i) estimation of Kd by fitting chemical shift perturbations to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. :1 binding model. | ||
Ligand 3 showed stable binding poses in MD simulation (unlike ligand 2), so we proceeded to run WTMetaD simulations on the end-stacked pose (see pose B in Fig. 3) in which 3 stacked on the A19 and A7 bases. The WTMetaD simulation was initiated from the equilibrium geometry after 20 ns from the unbiased MD simulation. Surprisingly, after approximately 250 ns of MetaD simulation, the G4 DNA appears unfolded (Fig. 4a). Initially, the G4 topology appears stable to ligand binding, and several binding and unbinding events are observed (0–50 ns). However, at approximately 50 ns, ligand 3 appears to intercalate between the G22 base and the remaining bases of the lower G-tetrad. From this position, both indane rings of ligand 3 appear to push towards the guanine bases of the middle G-tetrad and eventually break down the Hoogsteen hydrogen bonds between guanine bases of all G-tetrads (200 ns onwards) leading to disruption of the native structure.
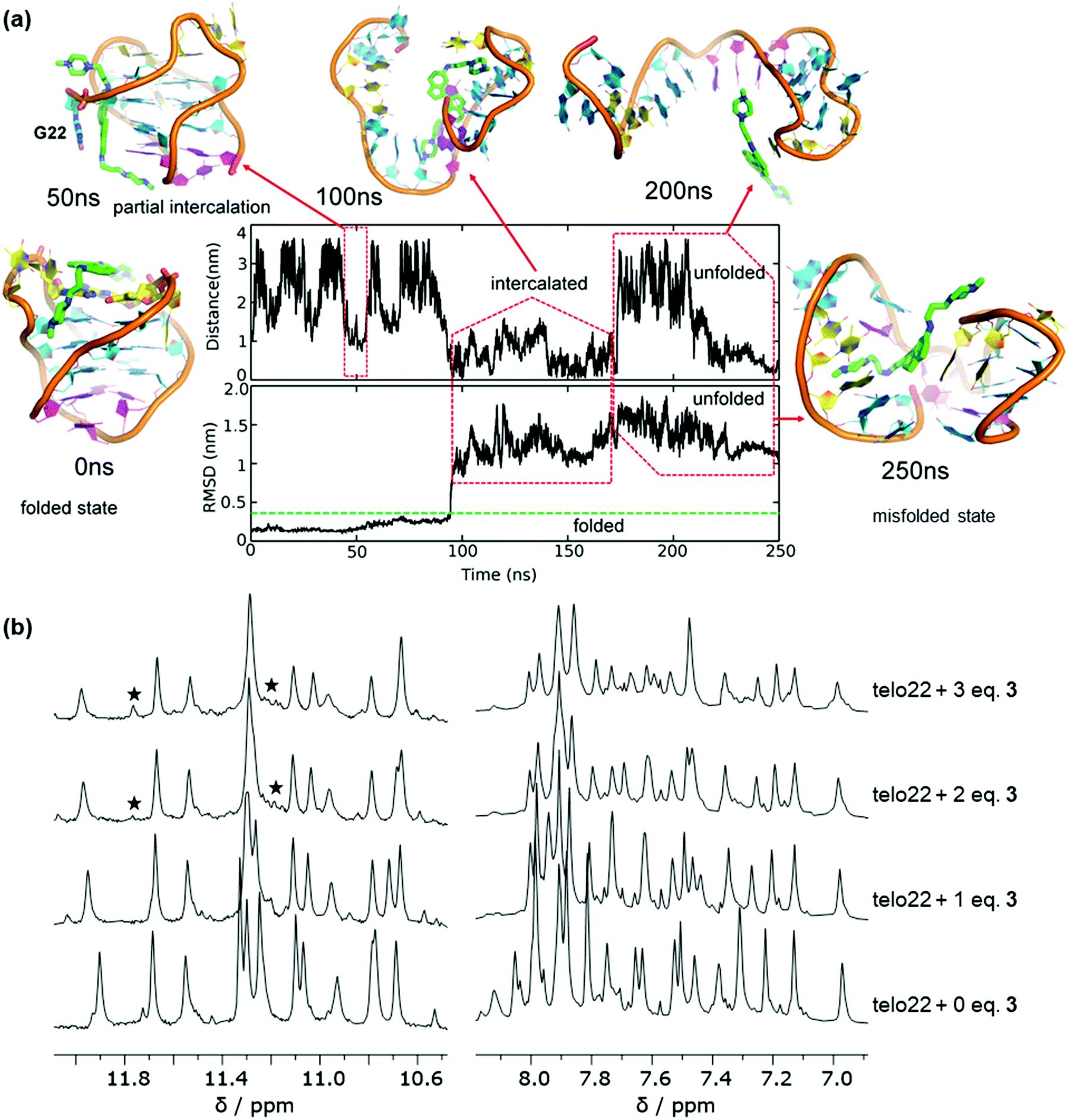 | ||
| Fig. 4 Effect of ligand 3 on antiparallel telo22 investigated by MetaD simulations and NMR spectroscopy. (a) MetaD calculations showing unfolding of telo22 by ligand 3via an intercalative mechanism, (b) comparison of 1H NMR spectra of telo22 with increasing equivalents of ligand 3 showing emergence misfolding conformation upon adding 3 equivalents of ligand 3, in particular, note the emergence of new signals in the imino region (marked with asterisks) indicating the presence of a misfolded species. | ||
With the simulation results in hand, we turned to examining the behaviour of ligand 3 by CD and NMR (Fig. 3 and ESI, Fig. S6 and S7†). No significant conformational change is observed at low ligand concentration by CD (Fig. 3d). In the NMR titration, the most significant chemical shift perturbations observed are for G4 and G8 (Fig. 3e and f). Such shifts probably arise from ring current effects and suggest the ligand is stacked onto the terminal G-tetrad, rather than external to the capping residues as observed by MD (vide supra). However, both simulation and experiment indicate that the ligand preferentially targets the top region of the G4. Notably, the H8 protons of A7 and A19 and the T6 H2′/H2′′ sugar protons are also perturbed (Fig. 3f–h), corresponding to residues found to be involved in binding by MD. In contrast, signals corresponding to the lower face of the G4 (e.g. T12, Fig. 3f and h) are comparatively unperturbed, indicating the ligand is unlikely to interact with this loop or the lower G-tetrad. Unfortunately, though the 2D NOESY spectra were helpful in assigning convoluted regions of the 1D titration spectra, no intermolecular DNA/ligand correlations could be observed.
On increasing the ligand stoichiometry to 3 equivalents, the aromatic region (7–8 ppm) of the spectrum appears more complex and an additional signals are observed in the imino (10–12 ppm) region (Fig. 4b). This suggests that ligand 3 is capable of perturbing the topology of the G4 at high concentrations and leading to the eventual emergence of misfolded states, perhaps through the mechanism suggested by MetaD (Fig. 4a), where initial intercalation leads to disruption of the hydrogen bond network. This result is particularly significant because, unlike for pyridinium ligand 2, topological perturbation is not detected in the standard MD simulations, demonstrating the importance of using MetaD as an enhanced sampling technique in order to obtain the full picture of the interaction mechanism of a ligand with G4 DNA by allowing the system to explore alternative regions of the free energy surface.
Antiparallel telo22 is stable to stiff-stilbene ligands 4 and 5 and ligands associate primarily with the top face of the G4
Previously, we observed that cis-ligands 4 and 5 interact with G4 DNA more weakly than their trans counterparts (1 and 3, respectively). This effect probably arises from the bent nature of the stilbene geometry in the cis isomers, hindering an optimal interaction with the G-tetrads. Here, we combine simulation and experiment to consider the G4 binding properties of stiff-stilbene isomers in greater detail. For pyridinium ligand 4 (the cis analogue of ligand 1), initial docking produced conformations in which the ligand is mainly bound on the top face of the DNA; the two lowest-energy poses were selected for further analysis. We subjected both poses to 1 μs MD simulations. For the lowest energy pose, a single stable but broad energy trough is found within the dihedral range of −0.5 to 1.5 rad, indicating that the ligand samples a range of binding conformations (Fig. 5a and ESI, Fig. S8†). Fig. 5b shows representative binding conformations sampled during the simulation of this pose. The RMSD of the DNA backbone is constant at ∼0.3 nm (3.0 Å), indicating that telo22 is stable during the unbiased MD simulation (Fig. 5c).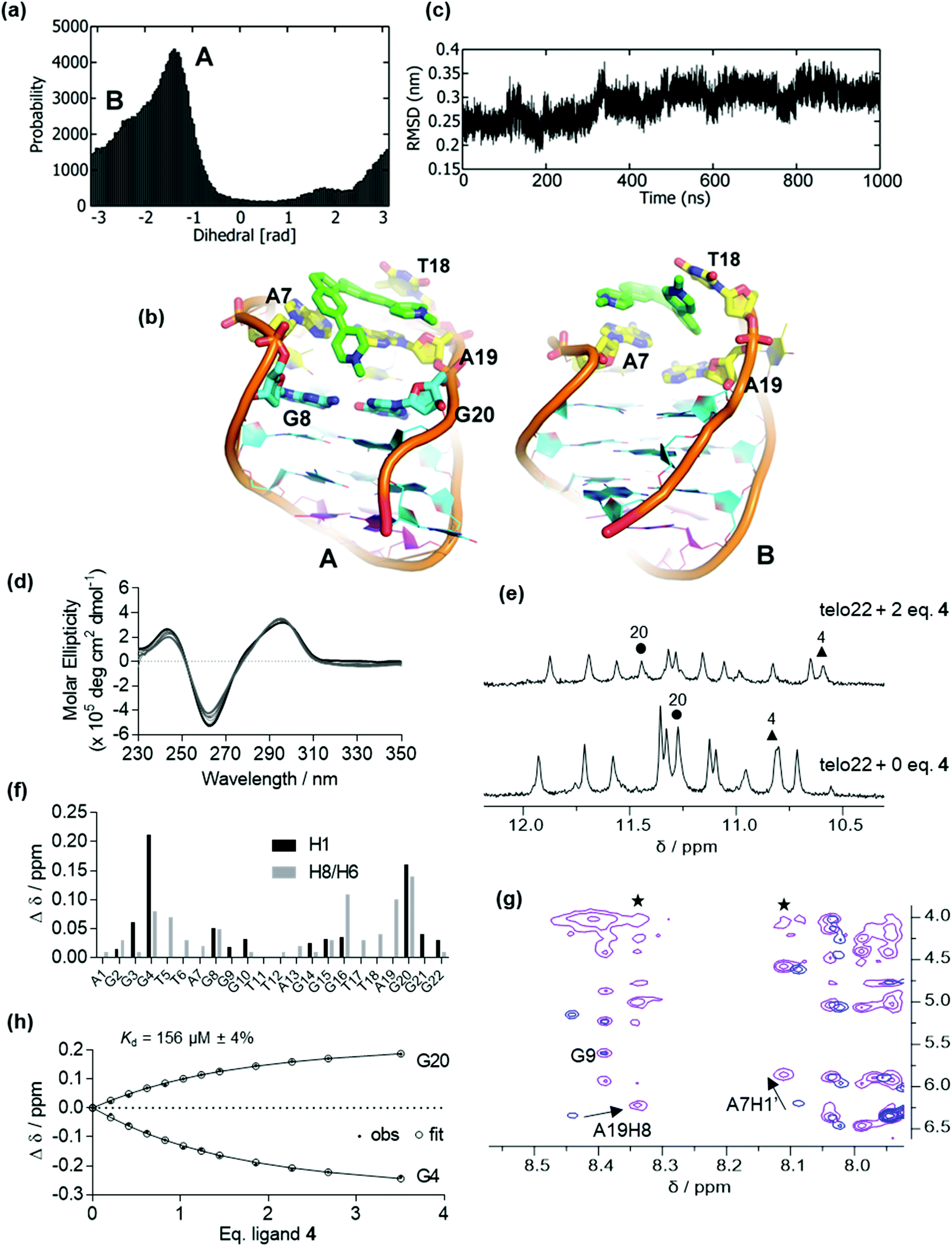 | ||
| Fig. 5 Binding of ligand 4 to antiparallel telo22 investigated by MD simulations, circular dichroism and NMR spectroscopy. (a) Dihedral angle distributions sampled in the MD simulations, (b) representations of the principal binding poses sampled in the MD simulations, key binding residues are indicated, (c) RMSD of the DNA backbone coordinates over the time course of the MD simulations, (d) circular dichroism spectra of telo22 (black trace) and increasing equivalents (light grey traces) of ligand 4 up to 7 eq. (dark grey trace), (e) NMR titration of telo22 with ligand 4 showing shifts of key imino resonances, (f) changes in chemical shifts of imino and aromatic resonances of telo22 upon adding 2 eq. ligand 4 (g) partial NOESY spectra showing shifts of key aromatic/anomeric resonances (h) estimation of Kd by fitting chemical shift perturbations to a 1:1 binding model. | ||
From these equilibrated structures, we performed WT-MetaD simulations of the binding of ligand 4 to telo22 G4. Unlike for ligands 1–3, which induce instability in the G4 fold under the experimental conditions, telo22 is stable in the presence of ligand 4 during MetaD simulations, which converge after ∼800 ns. Convergence was monitored by calculating the difference in the free energy between the bound and unbound state with time (ESI, Fig. S9†). The convergence of a binding/unbinding simulation can only be achieved when multiple re-crossing events from bound to unbound states, and vice versa, are seen. In our case, the exploration of the distance CV against time demonstrates multiple of these recrossing events (Fig. S10†). The two-dimensional FES was initially computed as a function of distance (D) and torsion (T) CVs (Fig. 6a). Four principal minima are observed: Basin A is the global minimum and it is more stable (by 1.5, 2.7, and 2.0 kcal mol−1, respectively) than basins B, C and D. In Basin A, the ligand interacts with the top face of the G4, sandwiched between T6, A7, A19 and T18. One of the central indane rings stacks with A7 in this pose as also observed in the MD simulation (Fig. 5b). The second indane ring stacks with the T18 base. Basin D is formed on the unbinding of ligand 4 from Basin A. In this binding mode, one of the pyridine rings is stacked with the A7 base, but both indane rings partially interact with the G8 and G20 bases which belong to the top G-tetrad.
 | ||
| Fig. 6 FES from MetaD simulations for binding of ligand 4 to telo22 G4. (a) FES expressed using d versus T collective variables; the inset shows the one dimensional potential of mean force calculated along the distance (D) collective variable showing the free energy difference between bound and unbound state; (b) FES expressed using DFA/POA(d.y) collective variables. The DFA/PA CVs show that ligand 4 binds only on the top. | ||
In order to sample Basin D, ligand 4 needs to slide and partially interact with the A7 base. We found a similar conformation on the MD simulation of Pose 2 (ESI, Fig. S11†) where ligand 4 interacts with A7, G8 and G20 bases. These results signify the binding of ligand 4 to involve the top G-tetrad formed by G4, G8, G16 and G20. In Basin B, ligand 4 is already exposed to the solvent, but partially interacts with the G4 by stacking on top of the T18 base. Taken together, these binding poses suggest significant entropic contributions to the binding free energy because in the bound state (Basin A) the ligand is sandwiched between DNA bases on both faces, leading to substantial desolvation. Meanwhile, intermediate binding poses reveal comparable stacking interactions (enthalpic contributions) but a greater entropic penalty due ordering of solvent molecules at the exposed ligand face. Since the ligand is solvent exposed, free rotation permits the formation of another energy minimum, Basin C, which is 1.2 kcal mol−1 less stable than Basin B since only one indane ring is responsible for binding following a 180° rotation of ligand relative to G4. A careful investigation suggests that going from minimum A to C via basin B and vice versa, ligand 4 is required to slide and rotate in order to find proper interactions with the aforementioned nucleic acid bases. Therefore, the binding/unbinding mechanism involves a state to state transition upon sliding, described as a hopping mechanism. To aid the reader in understanding the binding mode of ligand 4 to telo22, a movie showing the binding mechanism of the ligand to G4 DNA is included as part of the ESI.†
Though the D and T CVs describe the available binding modes to some degree, they are not sufficient to distinguish whether the ligand binds to the top face of the G4 through stacking, or by binding in the G4 groove (the MD simulations suggest binding to both of these regions). In order to capture all available binding modes, the FES was reconstructed using a reweighting algorithm introduced by Tiwary and Parinello.39 In this procedure, alternative representations of the FESs are created, based on different CVs which are not biased in the actual WT-MetaD simulation. Following the approach of Limongelli et al.,34 we chose another two CVs:POA (Position on the Axis) and DFA (Distance from Axis) (Fig. 6b). In this representation, the backbone of the DNA is aligned in the y-axis, and therefore, the POA CV is represented by the component of the distance along this axis (d.y); d.y and DFA axis are depicted in Fig. 6b. The DFA is taken as the distance from the centre of the d.y axis to the centre of mass of the heavy atoms of ligand 4.
This representation of the FES shows three stable minima, Basins A, B and D, as shown in Fig. 6b. This clearly shows all the binding modes are found within the CV regions of −1.0 to −1.7 nm (d,y) and 0 to ∼1.0 nm (DFA), corresponding to binding mostly on top of the G4 structure, as opposed to in the groove region. The region far from the d.y axis (DFA > 1.5 nm) representing the major groove does not appear to contain any free energy minima. These binding modes are depicted in different colours: Basin A (the global minimum) is represented in green and Basins B and D in orange and red, respectively. Basin C is not observed, because the POA/DFA representation of the FES lacks information concerning the orientation of the ligand 4 relative to the G4. Therefore, both combinations of CVs (D versus T and POA versus DFA) are needed in order to capture both the orientational effects and binding modes.
The stable binding of ligand 4 to telo22 predicted by MetaD (as opposed to the disruption of the G4 fold induced by trans counterpart 1) was also validated by the experiments (Fig. 5d–h and ESI, Fig. S12 and S13†). The CD spectrum of telo22 is virtually unperturbed on titration with ligand 4 (Fig. 5d) and the imino signals in the 1H NMR spectrum are also preserved (Fig. 5e). Binding residues can be inferred from the chemical shift perturbations (CSP) induced by ligand 4, most notably G20 and G4 imino resonances (corresponding to the top G-tetrad), the A7H1’ (anomeric) proton and the A19H8 (aromatic) proton (corresponding to residues in the lateral loop) (Fig. 5e–g). Furthermore, an intermolecular NOE correlation is observed between the ligand methyl group (δ = 4 ppm) and A19H8 and A7H1′ protons in the DNA ligand complex (asterisks, Fig. 5g). Resonances corresponding to other regions of the telo22 G4, especially the lower G-tetrad and diagonal loop, are comparatively unperturbed, indicating the ligand does not associate with these regions of the G4. These experimental observations support the binding modes identified by MetaD simulations, which also show binding to the top face of the G4, and the importance of the A7 and A19 bases in ligand binding.
Fitting of the chemical shift perturbations of G20 and G4 to a 1:1 binding model indicated a modest dissociation constant Kd = 156 μM (Fig. 5h), corresponding to a binding free energy of ΔGexpt = −5.3 kcal mol−1. The binding energy from the WT-MetaD free energy surface is ΔGcalc = −5.6 kcal mol−1 (Fig. 6a) in excellent agreement with experiment.
Finally, we examined the binding of ligand 5, the weakest affinity ligand identified in our previous study. Again, we ran WT-MetaD simulations on a stable ligand binding pose found from initial docking and MD simulations. In these simulations, the G4 was stable to the addition of ligand (unlike for its trans counterpart 3) and the simulation converged after ∼700 ns. As before, the FES was constructed as a function of D/T (Fig. 7) and POA/DFA CVs (ESI, Fig. S14†). Five consecutive minima are observed. Basins A, B and C are equally stable, whereas Basins D and E are less stable. In Basin A, ligand 5 stacks with A7 and A19, facing the major groove, while making a nonpolar interaction with the T17 base. The tail of ligand 5 is hydrogen bonded with a phosphate oxygen atom in the DNA backbone. In Basin B, ligand 5 is shifted slightly and sits on the top face of the DNA, stacked with A7 and T6 and again the ligand tail forms a hydrogen bond with the backbone. Basin C shows the ligand interacting primarily through the tail groups, with the indane residues exposed to solvent. Again, a hydrogen bond between the DNA backbone and one of the ligand tails is observed, with the other tail involved in stacking interactions with A7, A19 and T6 bases. Basin D is important because ligand 5 interacts with G20 base from the top G-quartet and also stacks with A7, while one of the indane rings stacks with the fraying base T17. Basin D is 1.3 kcal mol−1 less stable than Basin A. In Basin E, ligand 5 is oriented approximately 180° on the plane interacting with A7 base. The FES indicates that the association process of ligand 5 to telo22 G4 proceeds through initial formation of basin E followed by a 180° rotation on the plane to give Basin A.
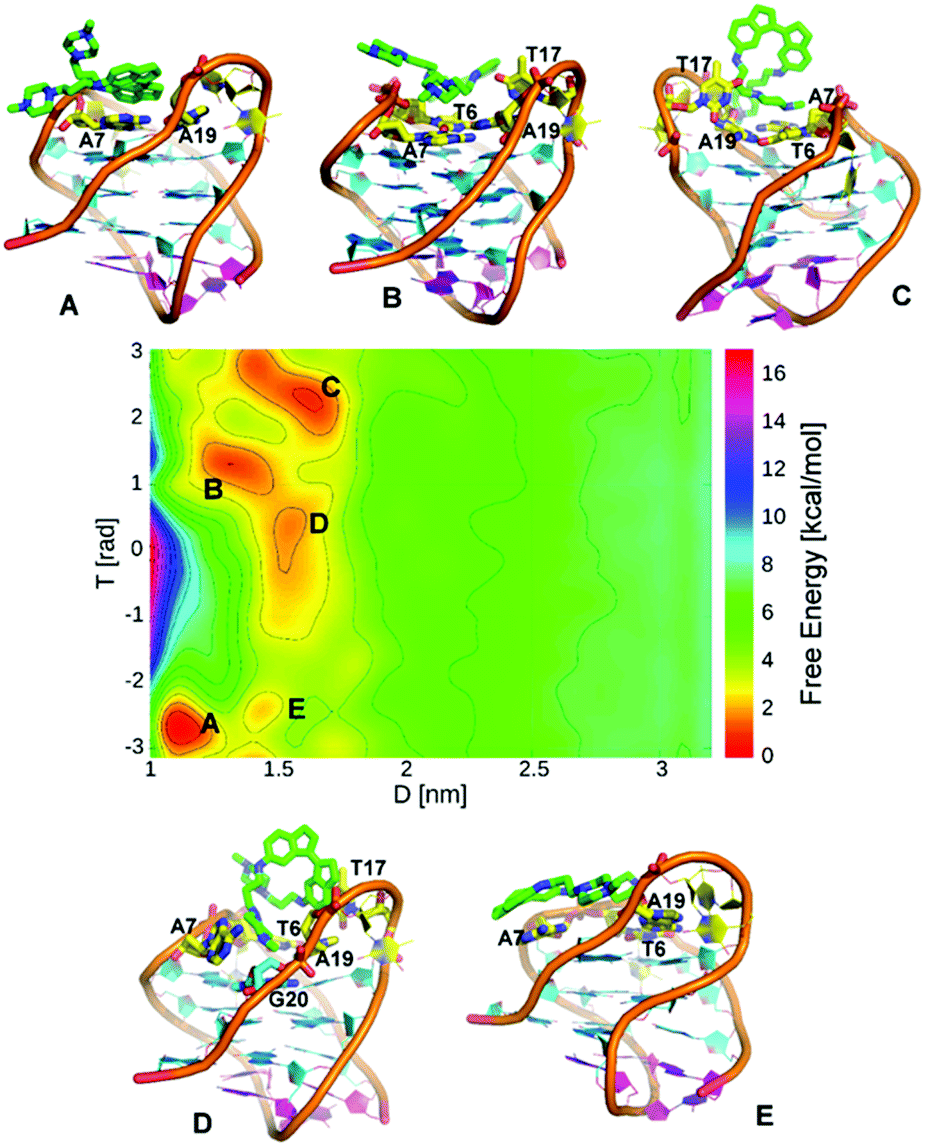 | ||
| Fig. 7 (A)–(E) show representative structures for each of the free energy minima. FES for binding of ligand 5 to telo22 G4 expressed using d versus T collective variables. | ||
The association of ligand 5 with telo22 was also investigated experimentally (ESI, Fig. S15 and S16†). We observed binding to be significantly weaker than for the trans counterpart (ligand 3), with the chemical shift perturbations being too weak to reliably extract a dissociation constant from the apparent binding isotherms. However, qualitatively, the binding site can be inferred to be similar to ligand 3, with the largest CSPs corresponding to A7, A19, G4 and G16 bases, corresponding to the top face of the G4 as observed in the MetaD simulations.
Conclusions
Design of selective G4 ligands remains a significant challenge, not least because of the polymorphism of these sequences, which can be perturbed upon binding of a small molecule. In this study, we assessed enhanced sampling simulations for predicting the effects of small molecules on G4 structures by detailed comparisons with experimental data. WT-MetaD simulations identify the main interaction sites of ligands with the target G4, and characterizes their interactions with specific DNA bases. In cases where ligands bind to the native G4 structure, this approach provides binding mechanisms and estimates of binding affinities at an affordable computational cost. Even more usefully, MetaD simulations also correctly predict the ability of certain ligands to induce structural perturbation in telo22 G4. The enhanced sampling simulations correctly predicted the diverse behaviours of a set of structurally related ligands that either perturb G4 folding (1–3) or bind to the existing topology without inducing such conformational perturbations (4–5). These results show that modelled structures (from docking and MD) provide useful starting points for MetaD simulations of G4/ligand interactions, showing that it is not necessary to use crystallographic structures of cognate ligand complexes in order to perform usefully predictive simulations. This is demonstrated by our tests against experiments on the DNA folding topology (CD spectroscopy) and ligand binding sites (NMR spectroscopy) in solution: the experimental results support the predicted binding models. The current work showcases the potential of MetaD as a predictive tool to explore ligand binding modes of novel chemotypes to G4 structures, and a gateway to understanding in more detail their possible effects on G4 folding dynamics. MetaD or other enhanced sampling biomolecular simulation methods16 can usefully contribute to integrated ligand design programmes, aiding and accelerating resource-intensive synthetic projects and experimental characterization of ligand activity at the molecular level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MPO thanks the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC EP/L015366/1, and the University of Bristol, for a PhD studentship. SH and AJM thank EPSRC for support [grant numbers EP/M015378/1 and EP/M022609/1. This work was carried out using the computational facilities of the Advanced Computing Research Centre, University of Bristol (http://www.bris.ac.uk/acrc/). JCMS thanks the Spanish Ministerio de Economía y Competitividad (Grant CTQ2015-64275-P). MCG thanks the European Research Council ERC-COG: 648239.Notes and references
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS.
- S. Neidle, J. Med. Chem., 2016, 59, 5987–6011 CrossRef CAS.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS.
- S. Neidle, FEBS J., 2010, 277, 1118–1125 CrossRef CAS.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS.
- F. Wang, X. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS.
- M. P. O'Hagan, J. C. Morales and M. C. Galan, Eur. J. Org. Chem., 2019, 4995–5017 CrossRef.
- S. Haider and S. Neidle, in Methods in molecular biology, Clifton, NJ, 2010, pp. 17–37 Search PubMed.
- S. Haider, J. Indian Inst. Sci., 2018, 98, 325–339 CrossRef.
- D.-L. Ma, T.-S. Lai, F.-Y. Chan, W.-H. Chung, R. Abagyan, Y.-C. Leung and K.-Y. Wong, ChemMedChem, 2008, 3, 881–884 CrossRef CAS.
- M. K. Si, S. K. Pramanik and B. Ganguly, Int. J. Biol. Macromol., 2019, 124, 1177–1185 CrossRef CAS.
- J. Luo, W. Wei, J. Waldispühl and N. Moitessier, Eur. J. Med. Chem., 2019, 168, 414–425 CrossRef CAS.
- R. C. Monsen and J. O. Trent, Biochimie, 2018, 152, 134–148 CrossRef CAS.
- D. J. Huggins, P. C. Biggin, M. A. Dämgen, J. W. Essex, S. A. Harris, R. H. Henchman, S. Khalid, A. Kuzmanic, C. A. Laughton, J. Michel, A. J. Mulholland, E. Rosta, M. S. P. Sansom and M. W. van der Kamp, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1393 Search PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS.
- F. Marinelli, F. Pietrucci, A. Laio and S. Piana, PLoS Comput. Biol., 2009, 5, e1000452 CrossRef.
- S. Haldar, F. Comitani, G. Saladino, C. Woods, M. W. van der Kamp, A. J. Mulholland and F. L. Gervasio, J. Chem. Theory Comput., 2018, 14, 6093–6101 CrossRef CAS.
- Q. Liao, Y. Kulkarni, U. Sengupta, D. Petrović, A. J. Mulholland, M. W. van der Kamp, B. Strodel and S. C. L. Kamerlin, J. Am. Chem. Soc., 2018, 140, 15889–15903 CrossRef CAS.
- F. L. Gervasio, A. Laio and M. Parrinello, J. Am. Chem. Soc., 2005, 127, 2600–2607 CrossRef CAS.
- V. Limongelli, M. Bonomi, L. Marinelli, F. L. Gervasio, A. Cavalli, E. Novellino and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5411–5416 CrossRef CAS.
- M. Bernetti, M. Masetti, M. Recanatini, R. E. Amaro and A. Cavalli, J. Chem. Theory Comput., 2019, 15, 5689–5702 CrossRef CAS.
- V. Limongelli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1455 CAS.
- A. J. Clark, P. Tiwary, K. Borrelli, S. Feng, E. B. Miller, R. Abel, R. A. Friesner and B. J. Berne, J. Chem. Theory Comput., 2016, 12, 2990–2998 CrossRef CAS.
- S. Haldar, P. Kührová, P. Banáš, V. Spiwok, J. Šponer, P. Hobza and M. Otyepka, J. Chem. Theory Comput., 2015, 11, 3866–3877 CrossRef CAS.
- S. Haldar, V. Spiwok and P. Hobza, J. Phys. Chem. C, 2013, 117, 11066–11075 CrossRef CAS.
- V. M. Miriyala, R. Lo, S. Haldar, A. Sarmah and P. Hobza, J. Phys. Chem. C, 2019, 123, 14712–14724 CrossRef CAS.
- V. Spiwok, P. Hobza and J. Řezáč, J. Phys. Chem. C, 2011, 115, 19455–19462 CrossRef CAS.
- V. Limongelli, M. Bonomi and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6358–6363 CrossRef CAS.
- S. Raniolo and V. Limongelli, Nat. Protoc., 2020, 15, 2837–2866 CrossRef CAS.
- F. Moraca, J. Amato, F. Ortuso, A. Artese, B. Pagano, E. Novellino, S. Alcaro, M. Parrinello and V. Limongelli, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2136–E2145 CrossRef CAS.
- D. Wragg, A. de Almeida, R. Bonsignore, F. E. Kühn, S. Leoni and A. Casini, Angew. Chem., Int. Ed., 2018, 57, 14524–14528 CrossRef CAS.
- F. S. Di Leva, E. Novellino, A. Cavalli, M. Parrinello and V. Limongelli, Nucleic Acids Res., 2014, 42, 5447–5455 CrossRef CAS.
- M. P. O'Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Chem.–Eur. J., 2020, 26, 6224–6233 CrossRef.
- M. P. O'Hagan, S. Haldar, M. Duchi, T. A. A. Oliver, A. J. Mulholland, J. C. Morales and M. C. Galan, Angew. Chem., Int. Ed., 2019, 58, 4334–4338 CrossRef.
- R. del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171–7175 CrossRef CAS.
- R. I. Mathad and D. Yang, in Methods in molecular biology, Clifton, NJ, 2011, pp. 77–96 Search PubMed.
- P. Tiwary and M. Parrinello, J. Phys. Chem. B, 2015, 119, 736–742 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Descriptions of materials and methods (experimental and computational procedures); supplementary figures; supplementary references (PDF). Video showing the binding mechanism of ligand 4 to telo22 (MP4). See DOI: 10.1039/d0sc05223j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |