
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe site-selectivity and mechanism of Pd-catalyzed C(sp2)–H arylation of simple arenes†
Daeun
Kim
ab,
Geunho
Choi
ab,
Weonjeong
Kim
b,
Dongwook
Kim
c,
Youn K.
Kang
 d and
Soon Hyeok
Hong
*a
d and
Soon Hyeok
Hong
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: soonhyeok.hong@kaist.ac.kr
bDepartment of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Republic of Korea
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
dDepartment of Chemical Energy Engineering, Sangmyung University, Seoul 03016, Korea
First published on 27th October 2020
Abstract
Control over site-selectivity is a critical challenge for practical application of catalytic C–H functionalization reactions in organic synthesis. Despite the seminal breakthrough of the Pd-catalyzed C(sp2)–H arylation of simple arenes via a concerted metalation–deprotonation (CMD) pathway in 2006, understanding the site-selectivity of the reaction still remains elusive. Here, we have comprehensively investigated the scope, site-selectivity, and mechanism of the Pd-catalyzed direct C–H arylation reaction of simple arenes. Counterintuitively, electron-rich arenes preferably undergo meta-arylation without the need for a specifically designed directing group, whereas electron-deficient arenes bearing fluoro or cyano groups exhibit high ortho-selectivity and electron-deficient arenes bearing bulky electron-withdrawing groups favor the meta-product. Comprehensive mechanistic investigations through a combination of kinetic measurements and stoichiometric experiments using arylpalladium complexes have revealed that the Pd-based catalytic system works via a cooperative bimetallic mechanism, not the originally proposed monometallic CMD mechanism, regardless of the presence of a strongly coordinating L-type ligand. Notably, the transmetalation step, which is influenced by a potassium cation, is suggested as the selectivity-determining step.
Introduction
The functionalization of aromatic compounds is a central topic in organic synthesis, which has great applicability.1 Among them, electrophilic aromatic substitution (EAS) has been widely used for the functionalization of arenes.2 Since the pioneering work of Friedel and Crafts,3 its site-selectivity has been well established, in which an electron-donating group (EDG) or halogen substituent directs an incoming electrophile (E+) to the ortho- and para-positions, whereas an electron-withdrawing group (EWG) directs it to the meta-position (Scheme 1a).2b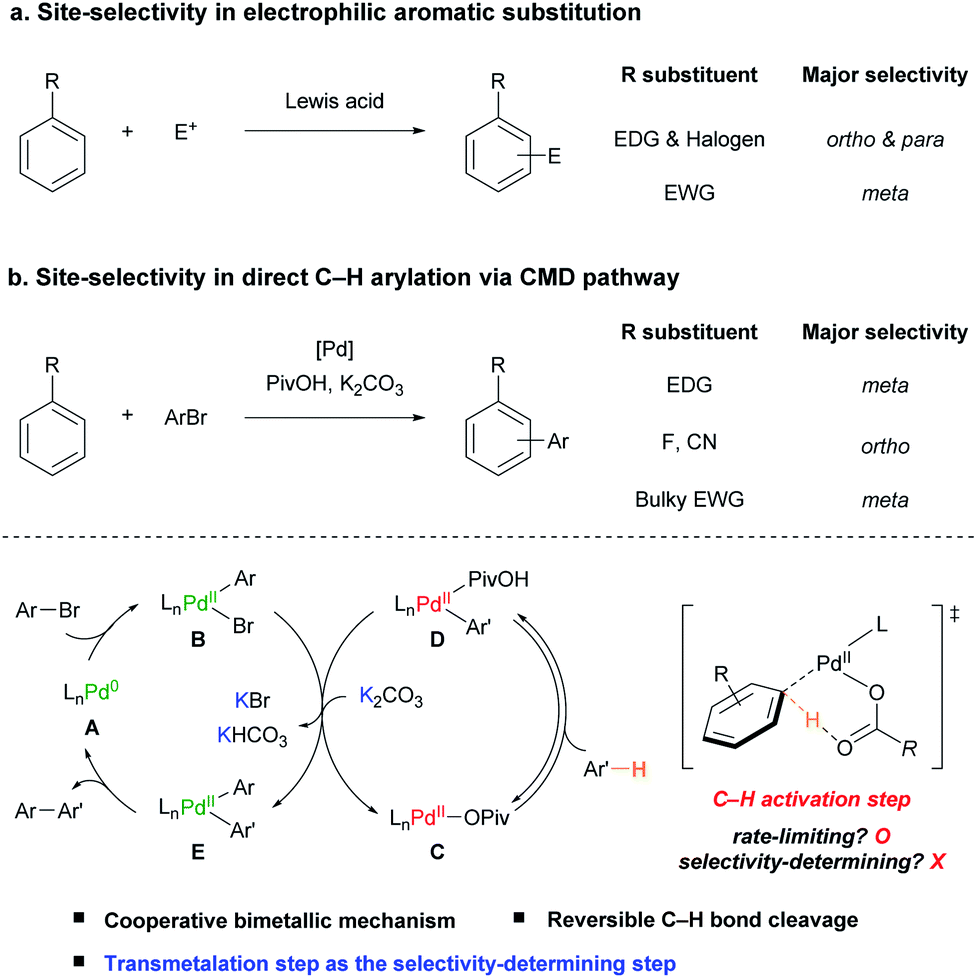 | ||
| Scheme 1 Direct C–H arylation of simple arenes and selectivity. | ||
Biaryls are one of the essential scaffolds found in many types of natural products, pharmaceuticals, and functional materials.4 Direct C(sp2)–H arylation is a powerful and attractive tool used to construct biaryls from arenes in an efficient manner because traditional cross-coupling methods require the preparation of prefunctionalized organometallic or main-group reagents.1b,c,4a,d,5 To date, significant advances have been achieved in the C–H arylation of heteroarenes5a,6 and electronically biased7 or chelation-assisted8 arenes to activate certain C–H bonds. However, these approaches limit the applicability of the reaction by requiring electronically activated substrates or additional synthetic operations, such as the installation and removal of a directing group to attain the target compound. Therefore, site-selective C–H arylation of unactivated, simple arenes is a fundamental challenge in catalysis to broaden the value of the reaction. Recently, site-selective C–H arylations of simple arenes have been achieved by elaborately designed strategies. For example, para-selective arylations of simple arenes have been reported using various arylating reagents such as diaryliodonium salts,9 arylsilanes,10 electron-poor arenes,11 and aryl boronic acids.4e,12 Inspired by the Catellani reaction,13 Yu and co-workers developed a meta-selective arylation reaction of simple arenes such as anisole and fluorobenzene with aryl iodide using a Pd/norbornene-based catalytic system to relay the palladium catalyst to the meta-position.14 The Larrosa group also reported a one-pot direct meta-selective arylation of simple arenes using a traceless directing group relay strategy utilizing CO2.4f,15 To develop a highly selective and efficient catalytic system, it is crucial to understand the underlying mechanism and factors that control the site-selectivity of the intrinsic palladium catalytic system.
During the past few decades, significant attention has been focused on investigating the underlying mechanism of Pd(II)-catalyzed C(sp2)–H bond cleavage. In 1985, Ryabov and co-workers proposed an electrophilic palladation pathway in the directed C–H activation of N,N-dimethylbenzylamine by Pd(OAc)2.16 Subsequently, Martinez,17 and Davies and Macgregor18 suggested that C–H bond cleavage proceeds via a concerted intramolecular proton abstraction by the acetate ligand. This concerted metalation–deprotonation (CMD) is regarded as an elegant pathway to cleave C–H bonds with a base ligand as the proton shuttle and has been extensively applied in various catalytic C–H functionalization reactions.4b,7d,19
In the pioneering work of Pd-catalyzed C–H arylation of arenes by the Fagnou group, the CMD pathway efficiently facilitates the aryl C–H bond activation step operating using the combination of a sub-stoichiometric amount of pivalic acid (PivOH) and K2CO3.20 Both the Brønsted acidity of the C–H bond and distortion–interaction analysis of the CMD transition state using density functional theory (DFT) studies have been adopted to predict the site-selectivity of the Pd-catalyzed C–H arylation of (hetero)arenes.21 However, the prediction was not well-matched with various simple arenes such as anisole (1b) and benzotrifluoride (1m). For example, although the ortho C–H bonds of 1b and 1m are the most acidic and reactive positions based on the current understanding of the CMD mechanism, the meta-isomers were observed as the major products in the previously reported studies.20–22
After the initial proposition of the Pd(II)-catalyzed monometallic CMD mechanism in the C–H arylation with aryl halides,17,23 Hartwig and co-workers experimentally uncovered that a cooperative bimetallic mechanism is operative in the C–H arylation of pyridine N-oxide catalyzed by Pd(OAc)2 and PtBu3.24 Gorelsky reported that the cooperative bimetallic mechanism involving two palladium species is favored using computational studies with phosphine-based Pd catalytic systems.25 Our group has also reported that a Pd-diimine catalyst facilitates the direct C–H arylation of benzene via a cooperative bimetallic mechanism with noticeably improved TONs.26 However, despite these advances, it is still questionable whether the suggested bimetallic pathway is generally operative even in the absence of a relatively strongly coordinating L-type ligand, such as phosphine or diimine. Moreover, although it is crucial to achieve the site-selective catalytic C–H arylation of simple arenes, there has been no focused study on understanding the site-selectivity of the reaction.
Herein, we investigate the scope and site-selectivity of the Pd-catalyzed C–H arylation reaction of simple arenes with aryl bromides (Scheme 1b). Comprehensive mechanistic studies have been conducted using a combination of extensive kinetic measurements and stoichiometric experiments with arylpalladium complexes to address the operating catalytic cycle and factors determining selectivity. Consequently, it is demonstrated that the applied Pd-based catalytic system works via a cooperative bimetallic mechanism involving two Pd catalytic species, which accounts for the observed counterintuitive site-selectivity trend by disclosing the transmetalation step as the selectivity-determining step.
Results and discussion
The reaction of ethoxybenzene 1a with 3-bromotoluene 2a in DMA was initially chosen to investigate both the reactivity and site-selectivity of the Pd-catalyzed direct C–H arylation (Table 1). The use of palladium acetate (Pd(OAc)2) and DavePhos, employed under Fagnou's condition,20 afforded the desired biaryl product 3aa in 54% yield with 69% meta-selectivity (entry 1). When using PtBu3 as a ligand, which is used for the arylation of pyridine N-oxide,24,27 Pd(OAc)2 exhibits similar reactivity to DavePhos (entry 2). Increased reactivities in the C–H arylation reaction were observed in the absence of a phosphine ligand, as reported by the Hartwig group, along with slightly increased meta-selectivity (entries 3 and 4).28 Changing the Pd precursor to PdCl2 enhanced the reactivity (75%, entry 5). To our delight, the use of Pd(CH3CN)2Cl2 further improved the reaction efficiency to 79%, which could be attributed to the improved solubility of the Pd precursor (entry 6). The previously reported Pd-diimine catalyst also displayed good reactivity (81%) with 68% meta-selectivity (entry 7).26 Increasing the catalyst loading (10 mol%) or the use of stoichiometric KOPiv instead of a combination of PivOH and K2CO3 resulted in a much lower yield (15% and 32%, respectively) accompanied by the homocoupling and hydrodebromination of 2a (entries 8 and 9).20 It is interesting to see that an appropriate choice of an acid and base rather than the Pd catalyst or ligand is essential to obtain high efficiency and selectivity (entries 10–12 and Tables S1–S3†).19b,29 The meta-selectivity was noticeably decreased when Rb2CO3, NaOPiv, or CsOPiv was used as the base instead of K2CO3 or KOPiv, suggesting that there may be a cation effect on the site-selectivity (entries 10–12). The use of other aryl halides including aryl chloride and iodide gave poor reactivities (0% and 4.4%, respectively), suggesting the involvement of halide anions in the catalytic cycle (entries 13 and 14). Control experiments indicated that a source of pivalate is essential for the reaction, which is consistent with previous reports (entry 15).20,26,29a|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalytic condition | Base | Yield (%) | Selectivity (o/m/p) | % selectivity (o/m/p) |
| a Reaction conditions: 2a (0.2 mmol), 1a (60 equiv.), [Pd] catalyst (3 mol%), ligand (3 mol%), PivOH (0.3 equiv.), base (2.5 equiv.) and DMA (1.2 mL) at 120 °C for 18 h; the total yield and regioselectivity (ortho/meta/para) were determined by GC analysis of the reaction mixture using dodecane as an internal standard. b Isolated yield. c [Pd] catalyst (10 mol%). d Without PivOH. diimine = (1E,2E)-N1,N2-bis(2,6-di(pentan-3-yl)phenyl)ethane-1,2-diimine. e With 3-chlorotoluene. f With 3-iodotoluene. | |||||
| 1 | Pd(OAc)2, DavePhos | K2CO3 | 54 | 1/7.2/2.3 | 9/69/22 |
| 2 | Pd(OAc)2, PtBu3–HBF4 | K2CO3 | 57 | 1/6.8/2.6 | 10/65/25 |
| 3 | Pd(OAc)2 | K2CO3 | 68 | 1/9.7/3.1 | 7/70/23 |
| 4 | Pd(OPiv)2 | K2CO3 | 67 | 1/10.0/3.0 | 7/71/22 |
| 5 | PdCl2 | K2CO3 | 75 | 1/10.0/3.1 | 7/71/22 |
| 6 | Pd(CH3CN)2Cl2 | K2CO3 | 79 (76b) | 1/10.2/3.2 | 7/71/22 |
| 7 | Pd(diimine)Cl2 | K2CO3 | 81 | 1/9.0/3.0 | 7/68/25 |
| 8c | Pd(CH3CN)2Cl2 | K2CO3 | 15 | 1/10.3/3.0 | 7/72/21 |
| 9d | Pd(CH3CN)2Cl2 | KOPiv | 32 | 1/6.2/2.0 | 11/68/21 |
| 10 | Pd(CH3CN)2Cl2 | Rb2CO3 | 75 | 1/5.7/2.9 | 10/59/31 |
| 11d | Pd(CH3CN)2Cl2 | NaOPiv | 1.3 | 1/1.6/1.2 | 26/43/31 |
| 12d | Pd(CH3CN)2Cl2 | CsOPiv | 4.3 | 1/1.4/1.8 | 31/45/24 |
| 13e | Pd(CH3CN)2Cl2 | K2CO3 | 0 | — | — |
| 14f | Pd(CH3CN)2Cl2 | K2CO3 | 4.4 | 1/2.0/4.0 | 14/29/57 |
| 15d | Pd(CH3CN)2Cl2 | K2CO3 | 0 | — | — |

With the optimized conditions in hand, the site-selectivity of the direct C–H arylation of various simple arenes was examined with 1-bromo-3,5-dimethylbenzene 2b as the coupling partner (Table 2). The major isomers are illustrated in Table 2 and the observed % site-selectivity is reported in parenthesis as (ortho/meta/para) or (α/β). To gain an idea of the correlation between the C–H bond acidity and site-selectivity in the transformation, proton affinity at every C-position in the various arenes was calculated (Table S17†); however, no consistent correlation was observed (Table 2). Various monosubstituted electron-rich arenes exhibit good reactivities, counter-intuitively providing the meta-isomer as the major product (>60% selectivity). In a similar manner to ethoxybenzene, anisole provided biaryl products 3bb in 58% yield with 67% meta-selectivity. In the case of alkylbenzenes, an almost statistical ratio of the meta- and para-isomers (∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was observed in excellent yields (3cb–3eb). N-Methylacetanilide provided biaryl products 3fb (>96%) with 62% meta-selectivity. Electron-neutral arenes also exhibited good efficiency. Benzene afforded biaryl 3gb in quantitative yield (>96%). Naphthalene provided 3hb (70%) in a 1.7:1 ratio favoring the α-arylated product. Various monosubstituted electron-deficient arenes exhibit higher efficiency even when using a lower amount of the arene (6 equiv.) or a shorter reaction time (3 h). Fluorobenzene provided biaryl products 3ib in 96% yield with 88% ortho-selectivity when using 6 equiv. of arene over 3 h. Similarly, when benzonitrile was subjected to the standard reaction conditions, the arylated products 3jb were obtained in 72% yield in favor of the ortho-isomer (69% selectivity). The ortho-selectivity observed using the electron-deficient arenes has been well acknowledged in the literature and attributed to both the most acidic ortho C–H bond and its lower distortion energy in the CMD transition state.21a,c However, we observed that the steric effect of the substituent can significantly affect the site-selectivity in the CMD catalytic system. Chlorobenzene, which is predicted to exhibit a similar trend to fluorobenzene,21c efficiently produced biaryls 3kb (95%) in favor of the meta-isomer (64% selectivity). (Trifluoromethoxy)benzene and benzotrifluoride gave products 3lb (64%) and 3mb (86%) with good meta-selectivities (61% and 71%, respectively). When using acetophenone as the arene substrate, α-arylation of the ketone occurred exclusively because the α-C(sp3)–H bond adjacent to the carbonyl group is much more acidic than the aryl C(sp2)–H bond (Scheme S1†).30 When pivalophenone was used to block the undesired C(sp3)–H arylation, 3nb was produced in 91% yield with 66% meta-selectivity. Nitrobenzene provided biaryl products 3ob (95%) with decreased meta-selectivity (44%) and an almost statistical distribution of regioisomers. The selectivity is significantly dependent on the solvents, as demonstrated in the ortho-arylation of nitrobenzene by the Fagnou group29b and our solvent screening results (Table S4†). The current method is well suited for disubstituted arenes and displays excellent regioselectivity. m-Xylene provided biaryl product 3pb (53%) as the sole isomer functionalized at the least sterically hindered meta-position with respect to the methyl substituent. Fluorotoluene and fluoroanisole resulted in products 3qb–3sb, which were exclusively functionalized adjacent to the fluoro group. In summary, the C–H arylation of electron-rich arenes exhibits meta-selectivity. High ortho-selectivity is observed in the case of electron-deficient arenes containing fluoro or cyano groups, as previously reported. However, arenes bearing large electron-withdrawing groups are arylated in favor of the meta-isomer rather than at the most acidic ortho C–H position.
:1) was observed in excellent yields (3cb–3eb). N-Methylacetanilide provided biaryl products 3fb (>96%) with 62% meta-selectivity. Electron-neutral arenes also exhibited good efficiency. Benzene afforded biaryl 3gb in quantitative yield (>96%). Naphthalene provided 3hb (70%) in a 1.7:1 ratio favoring the α-arylated product. Various monosubstituted electron-deficient arenes exhibit higher efficiency even when using a lower amount of the arene (6 equiv.) or a shorter reaction time (3 h). Fluorobenzene provided biaryl products 3ib in 96% yield with 88% ortho-selectivity when using 6 equiv. of arene over 3 h. Similarly, when benzonitrile was subjected to the standard reaction conditions, the arylated products 3jb were obtained in 72% yield in favor of the ortho-isomer (69% selectivity). The ortho-selectivity observed using the electron-deficient arenes has been well acknowledged in the literature and attributed to both the most acidic ortho C–H bond and its lower distortion energy in the CMD transition state.21a,c However, we observed that the steric effect of the substituent can significantly affect the site-selectivity in the CMD catalytic system. Chlorobenzene, which is predicted to exhibit a similar trend to fluorobenzene,21c efficiently produced biaryls 3kb (95%) in favor of the meta-isomer (64% selectivity). (Trifluoromethoxy)benzene and benzotrifluoride gave products 3lb (64%) and 3mb (86%) with good meta-selectivities (61% and 71%, respectively). When using acetophenone as the arene substrate, α-arylation of the ketone occurred exclusively because the α-C(sp3)–H bond adjacent to the carbonyl group is much more acidic than the aryl C(sp2)–H bond (Scheme S1†).30 When pivalophenone was used to block the undesired C(sp3)–H arylation, 3nb was produced in 91% yield with 66% meta-selectivity. Nitrobenzene provided biaryl products 3ob (95%) with decreased meta-selectivity (44%) and an almost statistical distribution of regioisomers. The selectivity is significantly dependent on the solvents, as demonstrated in the ortho-arylation of nitrobenzene by the Fagnou group29b and our solvent screening results (Table S4†). The current method is well suited for disubstituted arenes and displays excellent regioselectivity. m-Xylene provided biaryl product 3pb (53%) as the sole isomer functionalized at the least sterically hindered meta-position with respect to the methyl substituent. Fluorotoluene and fluoroanisole resulted in products 3qb–3sb, which were exclusively functionalized adjacent to the fluoro group. In summary, the C–H arylation of electron-rich arenes exhibits meta-selectivity. High ortho-selectivity is observed in the case of electron-deficient arenes containing fluoro or cyano groups, as previously reported. However, arenes bearing large electron-withdrawing groups are arylated in favor of the meta-isomer rather than at the most acidic ortho C–H position.
|
|
|---|
| a Reaction conditions: 2b (0.2 mmol), arene (60 equiv.), Pd(CH3CN)2Cl2 (3 mol%), PivOH (0.3 equiv.), K2CO3 (2.5 equiv.) and DMA (1.2 mL) at 120 °C for 18 h. All yields are isolated yields. The regioselectivity (ortho/meta/para or α/β) was measured by GC analysis of the reaction mixture using dodecane as an internal standard. b Arene (6 equiv.). c 3 h. d GC yield. e The C-position with the lowest proton affinity (calculated using G3MP2; see Table S17, ESI). |

|

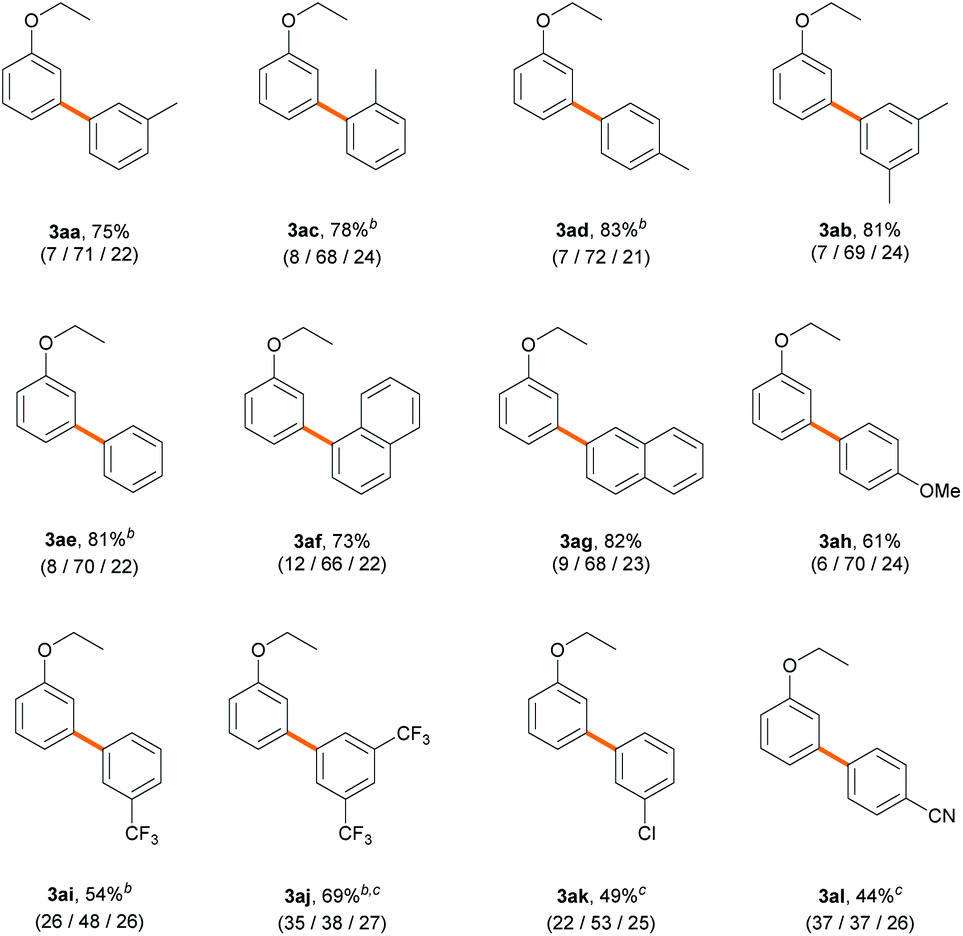
Next, we explored an array of aryl bromides as the coupling partner in the C–H arylation of ethoxybenzene (Table 3). A variety of electron-rich aryl bromides provided their corresponding biaryl products efficiently (61–83%) with good meta-selectivities (66–72%). Methyl- and dimethyl-substituted aryl bromides displayed good reactivities (3aa–3ad). Phenyl and naphthyl groups also gave good product yields (3ae–3ag). 4-Bromoanisole (2h), an aryl ether, exhibited slightly decreased reactivity (61%), but the meta-selectivity (70%) was maintained (3ah). Electron-deficient aryl bromides bearing trifluoromethyl, chloro, or cyano substituents also provided their corresponding biaryl products (3ai–3al) in moderate to good yields (44–69%). However, to our surprise, the presence of an electron-withdrawing substituent on the aryl bromide resulted in dramatically decreased meta-selectivities (37–53%), contrary to our expectation that the nature of aryl bromide would not significantly affect site-selectivity. Similar results were observed with stoichiometric amounts of ethoxybenzene (Scheme S2†). The results imply that the electronic properties of aryl bromide significantly affect the selectivity-determining step in the catalytic cycle.
|
|
|---|
| a Reaction conditions: aryl bromide (0.2 mmol), 1a (60 equiv.), Pd(CH3CN)2Cl2 (3 mol%), PivOH (0.3 equiv.), K2CO3 (2.5 equiv.) and DMA (1.2 mL) at 120 °C for 18 h; the total yields and regioselectivity (ortho/meta/para) were determined by GC analysis of the reaction mixtures using dodecane as an internal standard. b 48 h. c 140 °C. |
|
To understand general reactivity and selectivity trends mechanistically, the reaction rates were first compared with a variety of arenes (Scheme 2). As rationally expected in the rate-limiting proton-transfer CMD pathway,2a,21b,27 arenes with inductively withdrawing groups reacted faster than electron-rich arenes, which is a reversed tendency to the reactivity profiles obtained for electrophilic aromatic substitution reactions.2b
 | ||
| Scheme 2 Comparision of reaction rates. Combined yields of all isomers, determined by GC using dodecane as an internal standard, were used for kinetic plots. σp = Hammett constant. krel = relative reaction rate vs. k(R=H). | ||
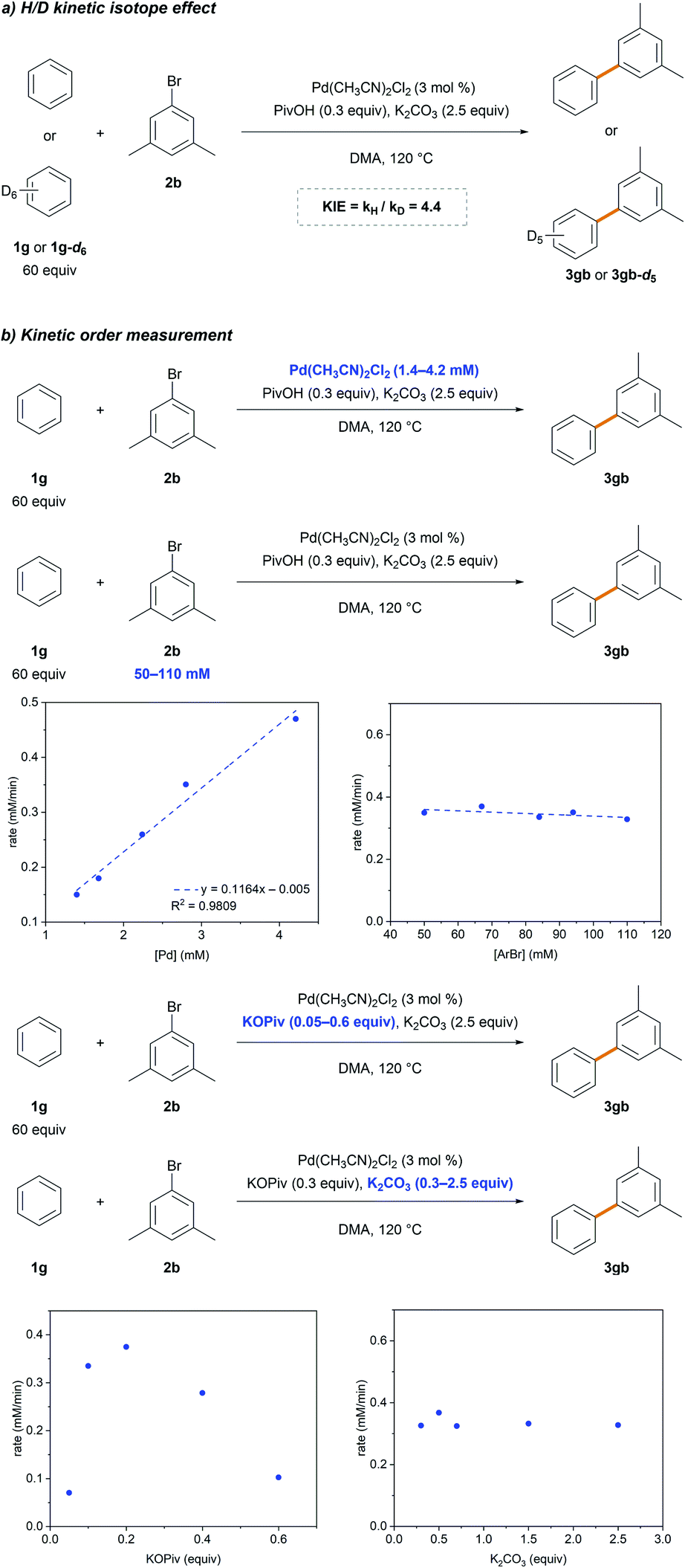
Standard kinetic isotope effect (KIE) experiments were conducted using benzene 1g and benzene-d61g-d6 to gain additional insight into the rate-limiting step (Scheme 3a). The observed large primary KIE (kH/kD = 4.4) suggests that the C–H bond cleavage step is involved in the rate-limiting step. This is in agreement with that previously reported,26,27 implying the involvement of C–H bond cleavage in the rate-limiting step. The dependence of the reaction rate on [Pd], [ArBr], KOPiv and K2CO3 was comprehensively investigated (Scheme 3b). A first-order dependence with respect to [Pd] and a zero-order with respect to [ArBr] and K2CO3 were observed. A positive dependence of the reaction rate on KOPiv was observed,31 but undesired homocoupling and hydrodebromination of 2b were observed using higher amounts of pivalate (>0.2 equiv.), which retarded the reaction (Table 1, entry 9). These results indicate that the C–H bond cleavage step follows the Pd-pivalate-promoted CMD pathway.
 | ||
| Scheme 3 Mechanistic studies. | ||
Following the initial mechanistic studies, a mechanistic outline involving Pd-pivalate-promoted C–H bond cleavage was constructed. For the Pd-catalyzed C–H arylation reaction, two pathways can be operative: (1) the originally proposed Pd(0/II) monometallic mechanism (Scheme 4, top pathway) or (2) a cooperative bimetallic mechanism (Scheme 4, bottom pathway), based on the literature precedent.20,24–26 As outlined in Scheme 4, the most distinguishable difference between the monometallic and cooperative bimetallic processes is the presence of a transmetalation step that only exists in the bimetallic mechanism. Stahl32 and Hong26 previously proposed that a Pd-catalyzed arylation reaction operated by a cooperative bimetallic pathway can display a first-order dependence at high [Pd], but second-order dependence at low [Pd], where the transmetalation step becomes the rate-limiting step. The second-order dependence can be more pronounced in the deuterated substrate because the slower rate of C–D bond cleavage significantly lowers [Pd–Ar] and thus, further decelerates the rate of the transmetalation step. In previous Pd-diimine catalyzed C–H arylations of simple arenes, we proposed that the rigid bidentate diimine ligand drives the cooperative bimetallic pathway because an open coordination site for the C–H bond of the arene is required to undergo the CMD process for the monometallic pathway to be operative. In regard to the current Pd catalytic system, we conjectured a monometallic CMD pathway in operation because there is no strongly coordinating ligand such as a diimine or electron-rich phosphine present. To verify the operative catalytic cycle, we investigated the dependence of the reaction rate at low concentrations of Pd(CH3CN)2Cl2 in benzene-d6 (Scheme 4c). To our astonishment, the kinetic study revealed second-order dependence at very low [Pd], strongly supporting that the reaction follows a cooperative bimetallic mechanism (Scheme 4b). Inspired by the results, we performed extensive kinetic studies to determine whether the cooperative bimetallic mechanism is generally operative. Second-order kinetics were consistently observed at low concentrations of Pd(OAc)2 in the presence of various phosphine ligands (DavePhos, PCy3, PPh3, and P(C6F5)3) (Fig. S14–S21†). Notably, the Pd(OAc)2-based catalytic system without any external ligand (Table 1, entry 3) also exhibits second order kinetics at very low concentrations of Pd(OAc)2 (Fig. S13†), indicating that the cooperative bimetallic pathway is generally working in the Pd-catalyzed arylation of simple arenes with aryl bromides under CMD conditions.
 | ||
| Scheme 4 Mechanistic hypothesis and kinetic studies. | ||
With evidence for the cooperative bimetallic mechanism in hand, we attempted to elucidate the origin of the observed site-selectivity. Meta-selectivity observed for many electron-rich and electron-deficient arenes (Table 2) was unexpected, counter-intuitive, and contrary to previous reports suggesting that the CMD step is both the rate-limiting and selectivity-determining step based on a monometallic mechanism.21a,33
Because the C–H bond cleavage step is independent of the oxidative addition of aryl bromides in the cooperative bimetallic mechanism, the dependence of selectivity on the electronic effect of aryl bromides with ethoxybenzene (Table 3) indicates that the C–H activation step is not the selectivity-determining step. The site-selectivity distribution was further investigated using electron-poor arenes, such as fluorobenzene (1i) and chlorobenzene (1k), in the C–H arylation with two electronically different aryl bromides (2b and 2j) under stoichiometric conditions (Scheme 5). In the case of highly ortho-selective fluorobenzene, the site-selectivity slightly changed when using 2b and 2j. In the arylation of chlorobenzene, the site-selectivity significantly varied from 46% meta-selectivity with 2b to 61% ortho-selectivity with 2j, supporting the validity of the hypothesis that the C–H activation step is not the selectivity-determining step.
 | ||
| Scheme 5 C–H arylation of fluorobenzene and chlorobenzene with two different aryl bromides. | ||
To scrutinize which elementary step is the selectivity-determining step, we first checked the reversibility of the oxidative addition and the C–H cleavage steps. A competition experiment was carried out using electronically different sets of aryl bromides (Scheme 6a). The reaction was conducted using 2e and electron-rich aryl bromide 2a, while the other reaction used 2e and electron-deficient aryl bromide 2i as the competing substrate. In the former reaction, similar yields of 3ae and 3aa were observed in a combined 54% yield. In contrast, only a trace amount of 3ae was produced in the presence of 2i, and 3ai was formed as the dominant product, albeit in a low yield (10%). If the oxidative addition was reversible, the coupling reaction with electron-richer aryl bromide 2e could occur with a reasonable yield despite the predominant oxidative addition of the electron-poor substrate 2i.34 Instead, the observed poor yields indicate that the oxidative addition step is irreversible. We hypothesized that electron-poor substrates exhibit poor yields, despite the predominant oxidative addition, due to the transmetalation step. The same trend of lower reactivity with electron-poor aryl bromides was observed when examining the substrate scope (Table 3, 2i–2l). Subsequently, deuterium exchange reactions were carried out with stoichiometric amounts of ethoxybenzene (1b) in the presence of D2O and benzene-d6 (3a-d6), respectively, to confirm the reversibility of the C–H bond cleavage step (Scheme 6b). Deuterium incorporations into the aryl group of 3ab (10.2 and 6.4%, respectively) via H/D scrambling were observed using 2H NMR spectroscopy. If the C–H bond cleavage by the Pd-pivalate species were irreversible, deuterium incorporation into 3ab would not occur. These results strongly support the reversibility of the C–H activation step. Therefore, the following steps, transmetalation or reductive elimination, may govern the site-selectivity of the reaction.
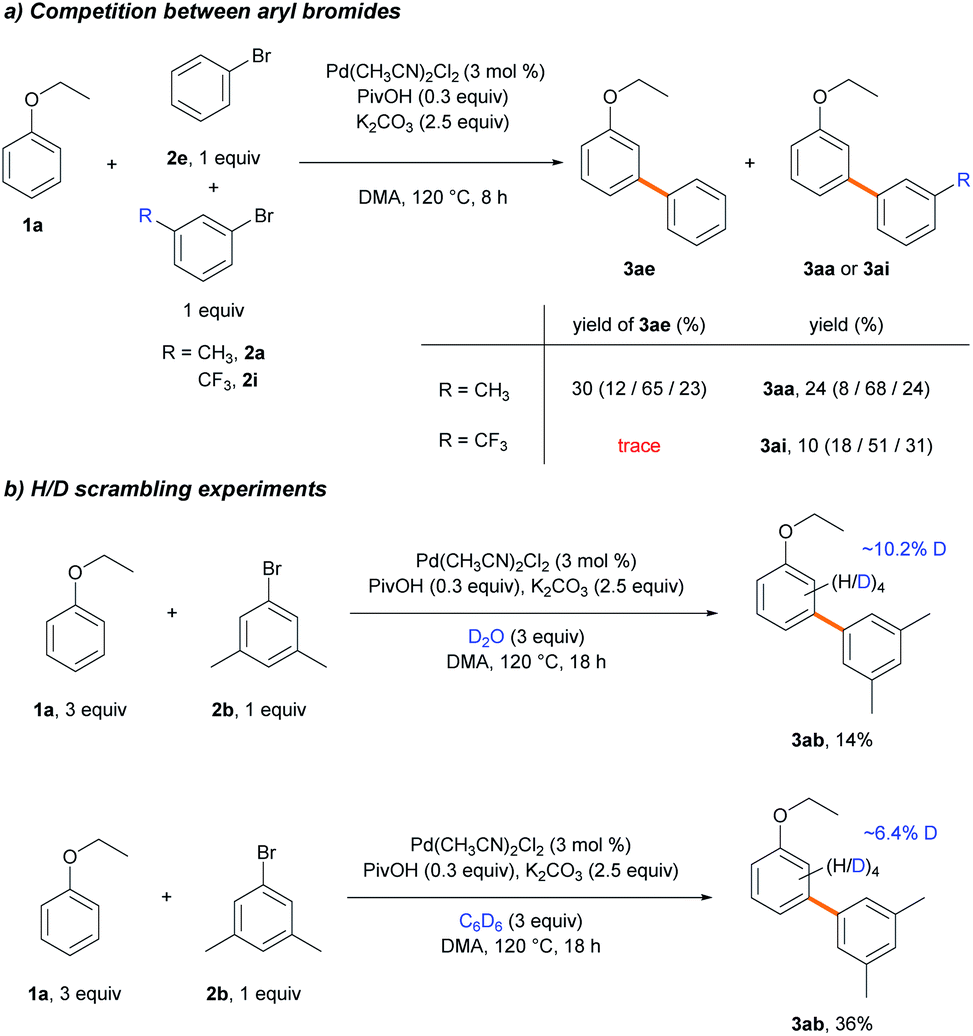 | ||
| Scheme 6 Competition experiments. | ||
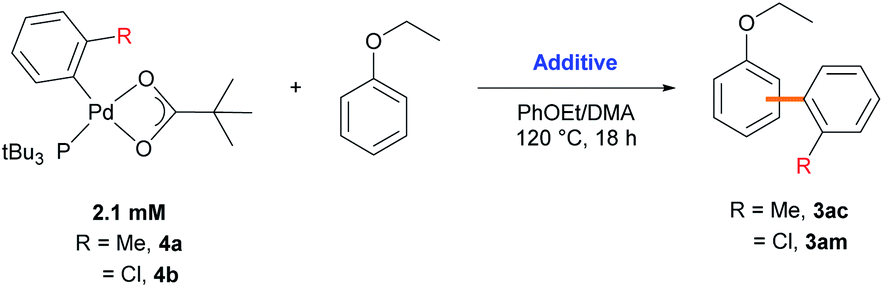
Stoichiometric reactions with arylpalladium complexes, which are analogous intermediates, were designed to affirmatively validate the cooperative bimetallic mechanism and further explore the selectivity-determining step. Two different arylpalladium pivalate complexes bearing either electron-rich or electron-poor aryl groups (4a and 4b) were prepared upon the reaction of Pd(dba)2 with PtBu335) in neat aryl iodide, followed by ligand exchange from iodide to pivalate using AgOPiv (Scheme 7).28 The solid-state structure of Pd complex 4b was confirmed by single-crystal X-ray diffraction analysis (CCDC 2000400). Complex 4a containing the o-tolyl group was subjected to stoichiometric reactions with ethoxybenzene in DMA (Table 4). The reaction did not provide the desired product 3ac in the absence of any additive (entry 1). The addition of PivOH, K2CO3, and a bromide source increased the reactivity up to 21%; however, the resulting meta-selectivity was significantly low (30 and 35%, entries 2 and 3). In contrast, when an additional Pd source, Pd(CH3CN)2Cl2, was added, the meta-selectivity increased (24% and 64%, entries 4 and 5) to a level similar to that observed under the standard conditions using PtBu3 (65% meta-selectivity, Table 1, entry 2). These results strongly suggest that meta-selectivity can be only achieved by the cooperative bimetallic mechanism. When complex 4b containing an o-chlorophenyl group was subjected to the stoichiometric reactions, the selectivities observed in the products 3am were completely different from those observed with 4a. A high ortho-selectivity (84%), albeit in a low yield (7%), was observed in the absence of an additional Pd complex (entry 6). Notably, when an additional Pd source was added, the ortho-selectivity significantly decreased (from 84 to 48%) with increased meta-selectivity (from 8 to 33%, entry 7). If the reaction would follow a monometallic mechanism, the reaction efficiency and selectivity would not vary significantly, regardless of the addition of an external Pd complex. These results consistently support that the reaction majorly operates via a cooperative bimetallic mechanism and the C–H bond cleavage step was not the selectivity-determining step because C–H activation is independent of the electronics of the aryl groups of the arylpalladium species under the cooperative bimetallic mechanism.
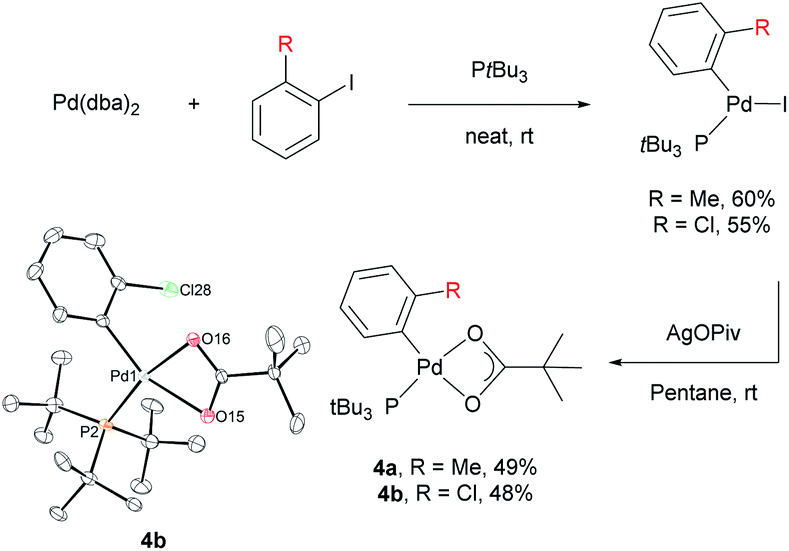 | ||
| Scheme 7 Synthesis of arylpalladium pivalate complexes. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Additive (equiv.) | Yield of 3ac or 3am (%) | Selectivity (o/m/p) | % selectivity (o/m/p) |
| a The total yield and regioselectivity (ortho/meta/para) were determined by GC analysis of the reaction mixture using dodecane as an internal standard. | |||||
| 1 | 4a | None | 0 | — | — |
| 2 | 4a | PivOH (20) and K2CO3 (160) | 0.6 | 1/0.7/0.7 | 42/30/28 |
| 3 | 4a | PivOH (20), K2CO3 (160), and nBu4NBr (10) | 21 | 1/1.3/1.4 | 27/35/38 |
| 4 | 4a | PivOH (20), K2CO3 (160), and Pd(CH3CN)2Cl2 (5) | 26 | 1/0.8/1.6 | 29/24/47 |
| 5 | 4a | PivOH (20), K2CO3 (160), nBu4NBr (10), and Pd(CH3CN)2Cl2 (5) | 65 | 1/6.2/2.6 | 10/64/26 |
| 6 | 4b | PivOH (20), K2CO3 (160), and nBu4NBr (10) | 7 | 1/0.1/0.1 | 84/8/8.0 |
| 7 | 4b | PivOH (20), K2CO3 (160), nBu4NBr (10), and Pd(CH3CN)2Cl2 (5) | 65 | 1/0.7/0.4 | 48/33/19 |
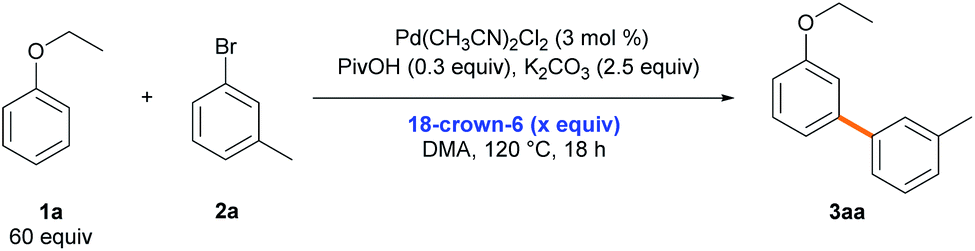
Notably, the effect of the cation in the carbonate base on the site-selectivity of the C–H arylation of ethoxybenzene (1a) was observed (Table 1, entry 10). The cation effect was further investigated upon the addition of 18-crown-6 under the standard reaction conditions.36 As the amount of 18-crown-6 increased, the meta-selectivity in the C–H arylation of ethoxybenzene gradually decreased (from 71 to 39%, Table 5). The same phenomenon was also observed in the C–H arylation of toluene (1c), excluding the effect of potential cation–ethoxybenzene interactions on the meta-selectivity (Table S17†). Amatore and Jutand demonstrated that the counterion of the base can affect the rate of transmetalation by coordinating the countercation to the Pd intermediate in Pd-catalyzed Suzuki–Miyaura cross-coupling reactions.37 Although we do not fully understand the Pd species involved in the transmetalation step, the results indicate that a potassium cation can affect the site-selectivity in the transmetalation process rather than the reductive elimination process.
|
|
||||
|---|---|---|---|---|
| Entry | 18-Crown-6 (x equiv.) | Yielda (%) | Selectivity (o/m/p) | % selectivity (o/m/p) |
| a Reaction conditions: 2a (0.2 mmol), 1a (60 equiv.), Pd(CH3CN)2Cl2 (3 mol%), PivOH (0.3 equiv.), K2CO3 (2.5 equiv.), 18-crown-6, and DMA (1.2 mL) at 120 °C for 18 h; the total yield and regioselectivity (ortho/meta/para) were determined by GC analysis of the reaction mixture using dodecane as an internal standard. | ||||
| 1 | — | 79 | 1/10.2/3.2 | 7/71/22 |
| 2 | 0.3 | 91 | 1/8.0/2.6 | 9/69/22 |
| 3 | 1 | 90 | 1/4.4/1.9 | 14/60/26 |
| 4 | 2.5 | 85 | 1/2.8/1.5 | 19/53/28 |
| 5 | 5 | 80 | 1/2.0/1.4 | 23/47/30 |
| 6 | 10 | 59 | 1/1.4/1.2 | 29/39/32 |
Reductive elimination is a common process in transition-metal catalysis and its mechanism is well understood.38 The reductive elimination of [Pd(II)]ArAr′ complexes to form biaryls has been thoroughly examined as a facile and irreversible process, particularly at our reaction temperature of 120 °C.39 It is less likely to be the selectivity-determining step of the reaction. To confirm it further, we conducted DFT modelling of the reductive elimination pathways to check whether the reductive elimination process theoretically produces the experimentally observed site-selectivity. C–H arylations of fluorobenzene and chlorobenzene using 2b were selected as model reactions for the computational studies. Scheme 5 shows fluorobenzene and chlorobenzene undergo the arylation reaction to produce biaryls 3ib (96%) with 88% ortho-selectivity and 3kb (32%) with 46% meta-selectivity, respectively. To compare with the experimental results, the reductive elimination processes for each isomeric position were calculated (Table 6 and Fig. S23†). The Gibbs free energy changes (ΔG), energy barriers (ΔG‡), and energetic differences (ΔΔG‡) are listed in Table 6. In the case of fluorobenzene, the reductive elimination at the meta-position is preferred over at the ortho-position, as indicated by the negative ΔΔG‡ value (−1.1 kcal mol−1). In the case of chlorobenzene, the para-position is the most reactive in the reductive elimination process with a negative ΔΔG‡ value of −1.7 kcal mol−1. The obtained theoretical selectivities, assuming the reductive elimination step is the selectivity-determining step, are completely different from the experimental results. Therefore, the possibility of the reductive elimination being the site-selectivity determining step is excluded using the computational investigations and the previous studies on the reductive elimination processes occurring in Pd-catalyzed biaryl cross-coupling reactions.39 Thus, we concluded that the transmetalation step is the selectivity-determining step based on the overall observations and control experiments. Due to the nature of the transition state and intermediate in the transmetalation step still being unknown,39a,b,41 attempts to predict site-selectivity using computational studies were unsuccessful.
|
|
||||||
|---|---|---|---|---|---|---|
| Arylation site | DFT calculateda | Experimental | ||||
| ΔG | ΔG‡ | ΔΔG‡b | % selectivity (o/m/p) | ΔΔG‡b | % selectivity (o/m/p) | |
| a Free energies in solution (kcal mol−1) obtained at the B3LYP-D3(IEFPCM)/SDD/6-311++G**//B3LYP-D3/LanL2DZ/6-31G** level of theory.40 b The value obtained using ΔG‡(ortho) − ΔG‡(position). A negative value indicates the lower energy barrier of the C-position than the ortho-position. Ar = 3,5-dimethylphenyl. | ||||||
| Fluorobenzene | ||||||
| Ortho | −5.0 | 13.3 | 0 | (18/70/12) | 0 | (88/10/2) |
| Meta | −8.1 | 12.2 | −1.1 | 1.3 | ||
| Para | −7.0 | 13.6 | 0.3 | 3.0 | ||
|
||||||
| Chlorobenzene | ||||||
| Ortho | −3.3 | 14.2 | 0 | (9/14/77) | 0 | (34/46/20) |
| Meta | −5.1 | 13.9 | −0.3 | −0.3 | ||
| Para | −6.8 | 12.5 | −1.7 | 0.4 | ||

A summary of the investigated mechanism is illustrated in Scheme 1. The Pd catalytic system without an external ligand operates via a cooperative bimetallic mechanism. One Pd–Ar species B is generated from the irreversible oxidative addition of the aryl bromide (ArBr) to Pd(0) A, while another Pd–Ar′ species D is generated from the rate-limiting C–H bond cleavage of the arene (Ar′H) by Pd(II)-pivalate species C. Both Pd species (B and D) undergo transmetalation to afford Pd(II)ArAr′ E and regenerate Pd(II)-pivalate species C. Importantly, the transmetalation step is disclosed to be the selectivity-determining step with the help of a potassium cation after the reversible C–H activation step. Finally, the reductive elimination of Pd(II)ArAr′ E affords the desired biaryl product and the initial Pd(0) species A, which then re-enters the catalytic cycle.
Conclusions
In conclusion, we have comprehensively investigated the scope and site-selectivity of the Pd-catalyzed direct C–H arylation of simple arenes using aryl bromides as a coupling partner. The site-selectivity of the C–H arylation reaction is affected by the steric effect of the substituents on the arene as well as C–H bond acidity. Generally, electron-rich arenes exhibit meta-selectivity. Although electron-deficient arenes bearing fluoro or cyano groups result in high ortho-selectivity, electron-deficient arenes bearing bulky electron-withdrawing groups produce the meta-isomer as the major product. Thorough mechanistic investigations, including kinetic studies, control experiments, and stoichiometric reactions with arylpalladium complexes, suggest that the reaction follows a cooperative bimetallic mechanism. Notably, the transmetalation step, which is influenced by a cation, is identified as the selectivity determining step, which refutes the previous understanding that the C–H bond cleavage step determines the site-selectivity in the reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jangwoo Koo for helping with the preliminary study, and Jungwon Kim and Geun Seok Lee for their help with the computational analyses. This work was supported by the National Research Foundation of Korea (NRF-2019R1A2C2086875 and NRF-2014R1A5A1011165, Center for New Directions in Organic Synthesis; NRF-2018R1D1A1B07049976) and the Korea Institute of Science and Technology Information with supercomputing resources including technical support (KSC-2018-CHA-0068 and KSC-2019-CRE-0182).Notes and references
- (a) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS.
- (a) R. Taylor, Electrophilic aromatic substitution, J. Wiley, Chichester, West Sussex, England, New York, 1990 Search PubMed; (b) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Part B: Reaction and Synthesis, Springer, 5th edn, 2010 Search PubMed.
- C. Friedel and J. Crafts, J. Chem. Soc., 1877, 32, 725–791 RSC.
- (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS; (b) H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550–11571 RSC; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS; (d) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC; (e) X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058–5061 RSC; (f) M. Font, A. R. A. Spencer and I. Larrosa, Chem. Sci., 2018, 9, 7133–7137 RSC.
- (a) C. Verrier, P. Lassalas, L. Théveau, G. Quéguiner, F. Trécourt, F. Marsais and C. Hoarau, Beilstein J. Org. Chem., 2011, 7, 1584–1601 CrossRef CAS; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS; (c) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS.
- (a) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS; (c) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS; (d) L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821–6836 CrossRef CAS.
- (a) D. Whitaker, J. Burés and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 8384–8387 CrossRef CAS; (b) O. René and K. Fagnou, Org. Lett., 2010, 12, 2116–2119 CrossRef; (c) L. Zhou and W. Lu, Organometallics, 2012, 31, 2124–2127 CrossRef CAS; (d) M. Simonetti, G. J. P. Perry, X. C. Cambeiro, F. Juliá-Hernández, J. N. Arokianathar and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 3596–3606 CrossRef CAS; (e) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef CAS.
- (a) P. Wang, M. E. Farmer, X. Huo, P. Jain, P.-X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269–9276 CrossRef CAS; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (c) Y. Aihara and N. Chatani, Chem. Sci., 2013, 4, 664–670 RSC; (d) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS; (e) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS; (f) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS.
- (a) A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–15713 CrossRef CAS; (b) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS.
- R. G. Kalkhambkar and K. K. Laali, Tetrahedron Lett., 2011, 52, 5525–5529 CrossRef CAS.
- Y.-X. Luan, T. Zhang, W.-W. Yao, K. Lu, L.-Y. Kong, Y.-T. Lin and M. Ye, J. Am. Chem. Soc., 2017, 139, 1786–1789 CrossRef CAS.
- (a) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2010, 292, 1–33 CAS; (b) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS; (c) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed., 1997, 36, 119–122 CrossRef CAS.
- (a) L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 13831–13835 CrossRef CAS; (b) L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 14870–14877 CrossRef CAS.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS.
- A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 2629–2638 RSC.
- M. Gómez, J. Granell and M. Martinez, Organometallics, 1997, 16, 2539–2546 CrossRef.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS.
- (a) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400–3407 CrossRef CAS; (b) Y. Tanji, N. Mitsutake, T. Fujihara and Y. Tsuji, Angew. Chem., Int. Ed., 2018, 57, 10314–10317 CrossRef CAS; (c) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (d) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS.
- (a) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS; (b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS; (c) S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS.
- (a) Y.-N. Wang, X.-Q. Guo, X.-H. Zhu, R. Zhong, L.-H. Cai and X.-F. Hou, Chem. Commun., 2012, 48, 10437–10439 RSC; (b) Q. Zhou, Y.-N. Wang, X.-Q. Guo, X.-H. Zhu, Z.-M. Li and X.-F. Hou, Organometallics, 2015, 34, 1021–1028 CrossRef CAS.
- (a) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef; (b) D. García-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880–6886 CrossRef.
- Y. Tan, F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 3683–3686 CrossRef CAS.
- S. I. Gorelsky, Organometallics, 2012, 31, 4631–4634 CrossRef CAS.
- J. Kim and S. H. Hong, ACS Catal., 2017, 7, 3336–3343 CrossRef CAS.
- L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020–18021 CrossRef CAS.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308–3311 CrossRef CAS.
- (a) Y. Tanji, R. Hamaguchi, Y. Tsuji and T. Fujihara, Chem. Commun., 2020, 56, 3843–3846 RSC; (b) L. Caron, L.-C. Campeau and K. Fagnou, Org. Lett., 2008, 10, 4533–4536 CrossRef CAS.
- D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS.
- The kinetic order could not be precisely determined due to the solubility issue of KOPiv.
- D. Wang, Y. Izawa and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 9914–9917 CrossRef CAS.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS.
- C. Colletto, S. Islam, F. Juliá-Hernández and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 1677–1683 CrossRef CAS.
- Attempts to synthesize the aryl–Pd complex with acetonitrile or DMA ligands were unsuccessful, presumably due to the limited stability of the resulting Pd complexes.
- S.-C. Sha, S. Tcyrulnikov, M. Li, B. Hu, Y. Fu, M. C. Kozlowski and P. J. Walsh, J. Am. Chem. Soc., 2018, 140, 12415–12423 CrossRef CAS.
- (a) C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2012, 18, 6616–6625 CrossRef CAS; (b) C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS.
- (a) J. M. Brown and N. A. Cooley, Chem. Rev., 1988, 88, 1031–1046 CrossRef CAS; (b) V. P. Ananikov, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 2002, 124, 2839–2852 CrossRef CAS.
- (a) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC; (b) P. Veerakumar, P. Thanasekaran, K.-L. Lu, K.-C. Lin and S. Rajagopal, ACS Sustainable Chem. Eng., 2017, 5, 8475–8490 CrossRef CAS; (c) M. Pérez-Rodríguez, A. A. C. Braga, M. Garcia-Melchor, M. H. Pérez-Temprano, J. A. Casares, G. Ujaque, A. R. de Lera, R. Alvarez, F. Maseras and P. Espinet, J. Am. Chem. Soc., 2009, 131, 3650–3657 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- (a) P. Nilsson, G. Puxty and O. F. Wendt, Organometallics, 2006, 25, 1285–1292 CrossRef CAS; (b) A. L. Casado and P. Espinet, J. Am. Chem. Soc., 1998, 120, 8978–8985 CrossRef CAS; (c) A. L. Casado, P. Espinet and A. M. Gallego, J. Am. Chem. Soc., 2000, 122, 11771–11782 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, computational details, and spectroscopic data for all new compounds. CCDC 2000400. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05414c |
| This journal is © The Royal Society of Chemistry 2021 |