
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric construction of pyrido[1,2-a]-1H-indole derivatives via a gold-catalyzed cycloisomerization†
Feng
Jiang
 ,
Chunling
Fu
and
Shengming
Ma
*
,
Chunling
Fu
and
Shengming
Ma
*
Laboratory of Molecular Recognition and Synthesis, Department of Chemistry, Zhejiang University, Hangzhou 310027, Zhejiang, People's Republic of China. E-mail: masm@sioc.ac.cn
First published on 22nd October 2020
Abstract
Pyrido[1,2-a]-1H-indoles are important scaffolds found in many biologically active compounds. Herein, we first developed an IPrAuCl/AgSbF6-catalyzed cycloisomerization of N-1,3-disubstituted allenyl indoles affording pyrido[1,2-a]-1H-indoles. Then the axial-to-central chirality transfer starting from enantio-enriched N-1,3-disubstituted allenylindoles affording optically active pyrido[1,2-a]-1H-indoles has been realized in excellent yields and enantioselectivities. A mechanism has been proposed based on mechanistic studies. Synthetic applications have also been demonstrated.
Introduction
Polycycles containing indole units are prevalent ring systems distributed in many bioactive alkaloids and pharmaceuticals.1 Gold-catalyzed cycloisomerizations of different types of allenyl indoles have contributed greatly to this topic:2–10 In 2006, Widenhoefer and his group reported the pioneering work on the synthesis of tetrahydrocarbazoles and cyclohepta[b]indoles via a 6- and 7-exo cycloisomerization of 2-allenyl indoles (Scheme 1a).4 As for 3-allenyl indoles, the gold-catalyzed 6-endo,5 6-exo,6 and 5-endo7 annulations as well as [2 + 2] cycloaddition8 successfully furnished the construction of varied indole scaffolds (Scheme 1b); however, annulations of N-allenyl indoles have been less explored:9,10 Toste's group reported the construction of dihydropyrroloindole skeletons via a 5-exo cyclization of N-allenyl indoles (Scheme 1(c1)).9 Shi and co-workers realized the construction of indole-fused tricyclic systems via a [3 + 2] or [2 + 2] cycloaddition of N-allenyl indoles under varied reaction conditions (Scheme 1(c1)).10 | ||
| Scheme 1 Gold-catalyzed cycloisomerization of allenylindoles and representative natural products containing pyrido[1,2-a]-1H-indole scaffolds. | ||
Pyrido[1,2-a]-1H-indoles are core motifs in ubiquitous biologically active alkaloids (Scheme 1d).11 Extensive efforts have been devoted to the development of new methods for their efficient synthesis. General approaches developed are the RCM reaction of diallyl indoles,12 Michael addition of α,β-unsaturated ketones13 and radical cyclization of N-alkylindoles,14 mostly suffering from the substrate scope, selectivity, and efficiency. Recently, González's15 and Muñoz's16 groups reported the elegant Au- or Pt–Au catalyzed cycloisomerization of N-2,3-butadienylindoles affording 6,9-dihydropyrido[1,2-a]-1H-indoles, which further reacted with heteroarenes or the electron-rich m-trimethoxybenzene to furnish racemic 9-aryl-6,7,8,9-tetrahydropyrido[1,2-a]-1H-indoles (Scheme 1(c2)). Enantioselective syntheses have not been developed. We envisaged that the cycloisomerization of optically active N-(2,3-allenyl)indoles would provide an efficient approach to various 9-substituted pyrido[1,2-a]-1H-indoles with high ee provided that the efficiency of chirality transfer may be ensured (Scheme 1(c3)).
Results and discussion
Initially, we conducted the reaction of racemic N-(2,3-allenyl)indole 1a in toluene under the catalysis of [IPrAuCl]/[AgNTf2], and the expected 6-endo cycloisomerization product 2a was afforded in 81% yield, accompanied by 1% of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond migration by-product 3a (entry 1, Table 1). Then we tested different neutral gold catalysts, AuCl, AuCl(PPh3), AuCl(PMe3), Au2Cl2(dppm), and AuCl(LB-phos); they all led to high recoveries of 1a (entries 2–6, Table 1). We reasoned that the cationic gold catalyst may have a much higher philicity towards the CC bond in allenes to increase the overall reactivity. Thus, different silver salts were added:17 when AgSbF6 was applied, the expected product 2a and the CC bond migration product 3a were formed in the yields of 70% and 13%, respectively (entry 10, Table 1). Considering that the high reaction temperature may have accelerated the CC bond migration, the reaction was conducted at room temperature to successfully form 2a in 92% yield, exclusively (entry 11, Table 1). Further screening of solvents (entries 12–14, Table 1) revealed that CHCl3, CH2Cl2, and toluene were the best. Finally, we defined the standard conditions as follows: IPrAuCl and AgSbF6 in CHCl3 at room temperature.
C bond migration by-product 3a (entry 1, Table 1). Then we tested different neutral gold catalysts, AuCl, AuCl(PPh3), AuCl(PMe3), Au2Cl2(dppm), and AuCl(LB-phos); they all led to high recoveries of 1a (entries 2–6, Table 1). We reasoned that the cationic gold catalyst may have a much higher philicity towards the CC bond in allenes to increase the overall reactivity. Thus, different silver salts were added:17 when AgSbF6 was applied, the expected product 2a and the CC bond migration product 3a were formed in the yields of 70% and 13%, respectively (entry 10, Table 1). Considering that the high reaction temperature may have accelerated the CC bond migration, the reaction was conducted at room temperature to successfully form 2a in 92% yield, exclusively (entry 11, Table 1). Further screening of solvents (entries 12–14, Table 1) revealed that CHCl3, CH2Cl2, and toluene were the best. Finally, we defined the standard conditions as follows: IPrAuCl and AgSbF6 in CHCl3 at room temperature.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yield of 2ab (%) | Yield of 3ab (%) | Recoveryb (%) |
| a Reaction conditions (unless otherwise noted): 1a (0.2 mmol) and catalyst (5 mol%) in 5.0 mL of solvent at 65 °C. b Yields were determined by the 1H NMR analysis of the crude products with mesitylene as the internal standard. c 2.5 mol% [Au2Cl2(dppm)] was used. d The reaction was carried out at room temperature for 12 h. | |||||
| 1 | IPrAuCl/AgNTf2 | Toluene | 81 | 1 | — |
| 2 | AuCl/AgNTf2 | Toluene | — | — | 84 |
| 3 | AuCl(PPh3)/AgNTf2 | Toluene | 14 | 3 | 61 |
| 4 | AuCl(PMe3)/AgNTf2 | Toluene | 14 | 4 | 62 |
| 5c | [Au2Cl2(dppm)]/AgNTf2 | Toluene | 15 | 4 | 50 |
| 6 | AuCl(LB-phos)/AgNTf2 | Toluene | 26 | 3 | 50 |
| 7 | IPrAuCl/AgBF4 | Toluene | 66 | 16 | — |
| 8 | IPrAuCl/AgOTs | Toluene | — | — | 91 |
| 9 | IPrAuCl/AgPF6 | Toluene | — | 69 | — |
| 10 | IPrAuCl/AgSbF6 | Toluene | 70 | 13 | — |
| 11d | IPrAuCl/AgSbF6 | Toluene | 92 | — | — |
| 12d | IPrAuCl/AgSbF6 | CH3CN | 9 | — | 87 |
| 13d | IPrAuCl/AgSbF6 | CH2Cl2 | 91 | — | — |
| 14d | IPrAuCl/AgSbF6 | CHCl3 | 95 | — | — |

With the optimal conditions in hand, the substrate scope of this gold-catalyzed cycloisomerization of racemic N-(2,3-allenyl) indoles has been examined (Table 2). Both alkyl- and aryl-substituted allenylindoles could work smoothly. Functional groups such as the CC bond, OMe, Cl, OTBS, Br, F, are all compatible (entries 3–4, 6, 12, and 16–17, Table 2). Substitution on the indoles with electron-withdrawing and -donating substituents at positions 3, 4, 5, or 6 has a very little impact on the yield (entries 4–7 and 11–14, Table 2). Furthermore, it was worth noting that when R3 = Me, Et, Cl, TBSO(CH2)2, the yields of 2 were much higher, as compared with R3 = H (compare entries 10–14 with entries 1–6, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | t/h | Yield of 2 (%) | ||
| R1 | R2 | R3 | |||
| a Reaction conditions: 1 (1 mmol), IPrAuCl (3 mol%) and AgSbF6 (3 mol%) in CHCl3 (5.0 mL) at room temperature unless otherwise noted. b The reaction was performed on a 0.2 mmol scale in 5.0 mL of CHCl3. c The reaction was performed on a 2.8 mmol scale with 1.6 mol% catalyst. | |||||
| 1 | n-C4H9 | H | H (1a) | 12 | 71 (2a) |
| 2 | n-C10H21 | H | H (1b) | 12 | 68 (2b) |
| 3 |
CH
2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH(CH2)8 CH(CH2)8 |
H | H (1c) | 12 | 72 (2c) |
| 4 | n-C4H9 | 5-OMe | H (1d) | 12 | 78 (2d) |
| 5 | n-C4H9 | 4-Me | H (1e) | 12 | 77 (2e) |
| 6 | n-C4H9 | 6-Cl | H (1f) | 12 | 84 (2f) |
| 7 | n-C4H9 | H | Me (1g) | 17 | 96 (2g) |
| 8 | Cl(CH2)5 | H | Me (1h) | 10 | 92 (2h) |
| 9 | BnCH2 | H | Me (1i) | 12 | 94 (2i) |
| 10 | 3-Pentyl | H | Me (1j) | 12 | 98 (2j) |
| 11 | n-C4H9 | H | Ethyl (1k) | 14 | 97 (2k) |
| 12 | n-C4H9 | H | TBSO(CH2)2 (1l) | 12 | 100 (2l) |
| 13 | n-C4H9 | 6-Cl | Me (1m) | 12 | 96 (2m) |
| 14 | n-C4H9 | 5-OMe | Cl (1n) | 13 | 98 (2n) |
| 15b | Ph | H | Me (1o) | 16 | 96 (2o) |
| 16b | 2-BrC6H4 | H | Me (1p) | 12 | 93 (2p) |
| 17 | 4-FC6H4 | H | Me (1q) | 12 | 86 (2q) |
| 18 | 4-BrC6H4 | H | Me (1r) | 12 | 80 (2r) |
| 19c | 4-BrC6H4 | H | TBSO(CH2)2 (1s) | 14 | 83 (2s) |

With these encouraging results, we then began the investigation of the cycloisomerization of the optically active substrates with (Ra)-1a as the model substrate. Such optically active substrates are readily available via the enantioselective allenation of terminal alkynes (EATA) reaction.18 Initially, the combination of IPrAuCl and AgSbF6 was tested in CHCl3 at 0 °C, the product (R)-2a was formed in 93% yield and 91% ee (entry 1, Table 3). Additional screening of solvents showed inferior results (entries 2–8, Table 3). Control experiments showed that essentially no reaction occurred when either IPrAuCl or AgSbF6 was omitted (entries 9 and 10, Table 3). However, when the reaction was conducted on a 1 mmol scale, the ee of (R)-2a dropped again (entry 11, Table 3). Reducing the catalyst loading solved this problem, thus, the optimal conditions have been successfully re-defined by running the reaction with 3 mol% each of IPrAuCl and AgSbF6 in CHCl3 at 0 °C (entry 12, Table 3).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Yield/ee of (R)-2a (%)b (%)c | Recovery/ee of (Ra)-1a (%)b (%)c |
| a Reaction conditions: (Ra)-1a (0.2 mmol), catalyst (5 mol%) in solvent (5.0 mL) at 0°C under a N2 atmosphere unless otherwise noted. b Yields were determined by 1H NMR using mesitylene as the internal standard. c The ee was determined by HPLC analysis using a chiral stationary phase. d Without AgSbF6. e Without IPrAuCl. f Reaction was carried out at 0 °C for 48 h. g The reaction was performed on a 1 mmol scale. h 3 mol% catalyst was used. | |||
| 1 | CHCl3 | 93/91 | — |
| 2 | DCE | 74/90 | 13/84 |
| 3 | CH2Cl2 | 98/90 | — |
| 4 | Toluene | 56/92 | 47/93 |
| 5 | CH3CN | — | 100/98 |
| 6 | THF | 88/84 | — |
| 7 | Dioxane | 52/60 | — |
| 8 | DMF | 54/66 | 31/87 |
| 9d,f | CHCl3 | — | 92/98 |
| 10e,f | CHCl3 | — | 91/98 |
| 11g | CHCl3 | 90/87 | — |
| 12f,g,h | CHCl3 | 94/90 | — |

Having identified the optimal conditions, we aimed to define the scope of both the allene part and the indole part. First, we conducted a brief study of the substrate scope for R3 = H. As shown in Table 4, R1 could be a butyl, decyl, or decenyl group (entries 1–3, Table 4). For the indole part, substrates with substituents at positions of 4 or 5 also reacted smoothly with high yields and ee values (entries 4 and 5, Table 4). However, when the weak electron-withdrawing group Cl was employed, the ee of the product decreased (entry 6, Table 4).
|
|
||||
|---|---|---|---|---|
| Entry | (Ra)-1/(ee%) | t (h) | 2 | |
| R1/R2 | Yield (%) | ee (%) | ||
| a Reaction conditions: (Ra)-1 (1 mmol), IPrAuCl (3 mol%), and AgSbF6 (3 mol%) in CHCl3 (25.0 mL) at 0 °C unless otherwise noted. b 5 mol% catalyst was used. | ||||
| 1 | n-C4H9/H ((Ra)-1a)/97 | 48 | 94 ((R)-2a) | 90 |
| 2 | n-C10H21/H ((Ra)-1b)/98 | 48 | 95 ((R)-2b) | 90 |
| 3 | CH2CH(CH2)8/H ((Ra)-1c)/96 |
51 | 96 ((R)-2c) | 92 |
| 4 | n-C4H9/5-OMe ((Ra)-1d)/98 | 53 | 95 ((R)-2d) | 91 |
| 5b | n-C4H9/4-Me ((Ra)-1e)/98 | 48 | 88 ((R)-2e) | 90 |
| 6 | n-C4H9/6-Cl ((Ra)-1f)/98 | 58 | 93 ((R)-2f) | 78 |
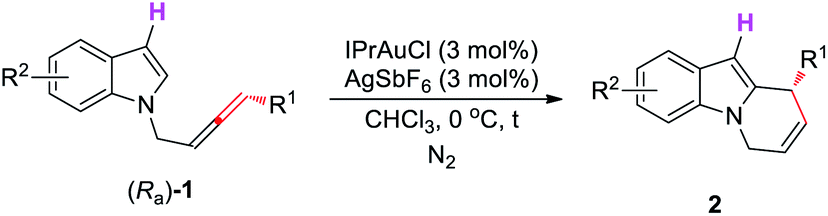
We further studied the scope of substrates for R3 ≠ H and observed higher yields and ee values. As shown in Table 5, both alkyl- (entries 1–8) and aryl-substituted allenes (entries 9–13) could work smoothly. For aryl-substituted allenes, both F and Br may survive (entries 10–13, Table 5). Furthermore, the Br atom installed at the ortho- or para-positions of the phenyl ring had little effect on the yield and ee of (S)-2. For the indole part, electron-donating as well as electron-withdrawing substituents and functional groups, including OTBS, OMe, and Cl, which could further be elaborated, were well tolerated (entries 6–8 and 13, Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | (Ra)-1/(ee%) | t (h) | 2 | |
| R1/R2/R3 | Yield (%) | ee (%) | ||
| a Reaction conditions: (Ra)-1 (1 mmol), IPrAuCl (3 mol%) and AgSbF6 (3 mol%) in CHCl3 (25.0 mL) at 0 °C unless otherwise noted. b 5 mol% catalyst was used. | ||||
| 1b | n-C4H9/H/Me ((Ra)-1g)/98 | 59 | 90 ((R)-2g) | 96 |
| 2 | Cl(CH2)5/H/Me ((Ra)-1h)/98 | 48 | 99 ((R)-2h) | 99 |
| 3 | BnCH2/H/Me ((Ra)-1i)/97 | 48 | 98 ((R)-2i) | 98 |
| 4 | 3-Pentyl/H/Me ((Ra)-1j)/98 | 48 | 89 ((R)-2j) | 90 |
| 5 | n-C4H9/H/ethyl ((Ra)-1k)/99 | 48 | 98 ((R)-2k) | 97 |
| 6 | n-C4H9/H/TBSO(CH2)2 ((Ra)-1l)/98 | 48 | 98 ((R)-2l) | 96 |
| 7 | n-C4H9/6-Cl/Me ((Ra)-1m)/98 | 48 | 97 ((R)-2m) | 95 |
| 8 | n-C4H9/5-OMe/Cl ((Ra)-1n)/98 | 42 | 98 ((R)-2n) | 98 |
| 9 | Ph/H/Me ((Ra)-1o)/99 | 48 | 99 ((S)-2o) | 95 |
| 10 | 2-BrC6H4/H/Me ((Ra)-1p)/96 | 48 | 98 ((S)-2p) | 96 |
| 11 | 4-FC6H4/H/Me ((Ra)-1q)/98 | 48 | 89 ((S)-2q) | 95 |
| 12 | 4-BrC6H4/H/Me ((Ra)-1r)/98 | 48 | 91 ((S)-2r) | 96 |
| 13 | 4-BrC6H4/H/TBSO(CH2)2 ((Ra)-1s)/99 | 48 | 90 ((S)-2s) | 92 |

Notably, this reaction could be easily scaled up to 3.0 mmol and the loading of IPrAuCl/AgSbF6 may further be reduced to 1.5 mol%, providing 1.27 g of (S)-2s (88% yield) in 97% ee (Scheme 2). To illustrate the synthetic potential of this protocol, the transformations of cycloisomerization product (S)-2s were demonstrated: Suzuki coupling of (S)-2s with 3-methoxyphenyl boronic acid gave product (S)-4 in 66% yield and 91% ee.19 Treatment of (S)-2s with HCl and IBX successively afforded aldehyde (S)-5 in 78% yield and 97% ee. The hydrogenation of (S)-2s gave product (S)-6 in 89% yield and 90% ee (Scheme 2). Products (S)-2s and (S)-6 contain the core unit in goniomitine.11b Then, an extra chiral centre was further introduced to the 1-position of the allene side chain by including a methyl group. The reaction of (R,Ra)-1u and (S,Ra)-1u could also work smoothly, providing diastereomers (R,S)-2u (92% yield) and (S,S)-2u (89% yield) in 99% ee and 98% ee respectively. These results provide a possibility to synthesize a library of (−)-goniomitine derivatives (Scheme 2).
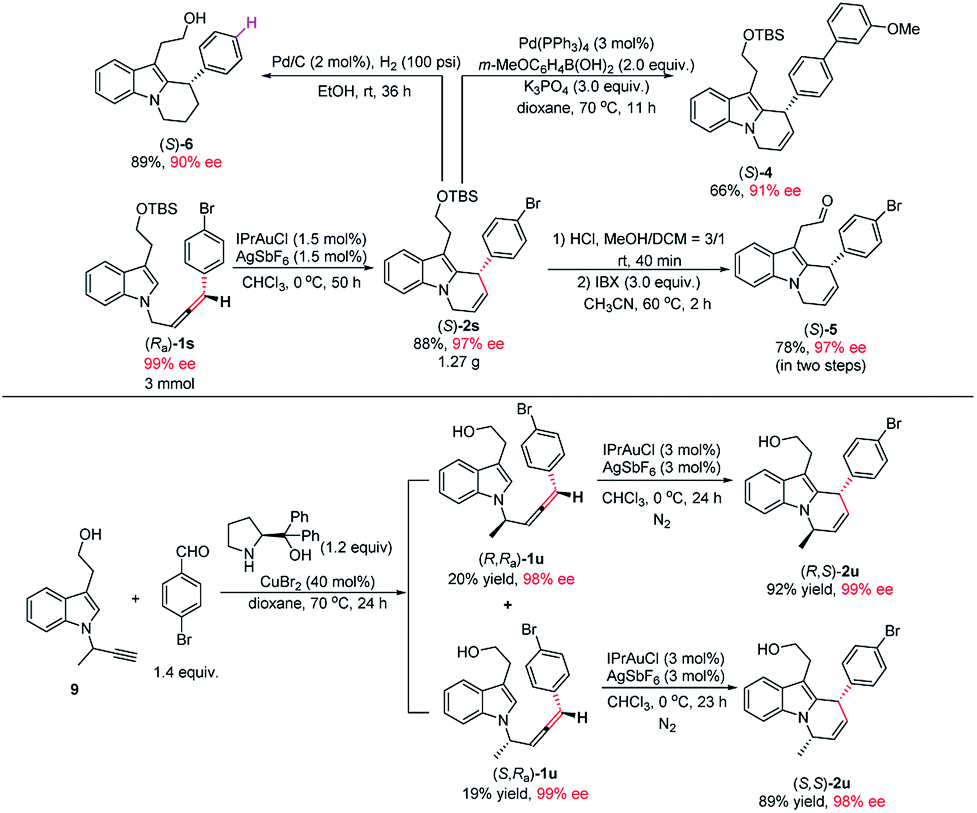 | ||
| Scheme 2 A gram–scale reaction and the syntheses of simplified analogues of (−)-goniomitine. | ||
In order to unveil the nature of the stereoselectivity of the reaction, the absolute configurations of the starting materials (Ra)-1n and (Ra)-1r have been established based on the literature.18c,20 The absolute configurations of their corresponding products were unambiguously established by X-ray single crystal diffraction analysis of (R)-2n and (S)-2r (eqn (1) and (2), Scheme 3). The electronic effect of the 3-position substituent of indole on chirality transfer has been studied by conducting the parallel reactions of (Ra)-1g (R3 = Me) and (Ra)-1t (R3 = CO2Me) under standard conditions (eqn (3) and (4), Scheme 3): the reaction of (Ra)-1g proceeded smoothly to afford the product (R)-2g in 91% yield and 96% ee while the reaction of (Ra)-1t afforded the target product (R)-2t in 21% yield with only 24% ee. Interestingly, we found the ee value of the recovered (Ra)-1t is 0, indicating the potential competitive racemization and cycloisomerization of N-(2,3-allenyl)indoles. This also explains why the loading of the catalyst has an effect on the efficiency of chirality transfer. Furthermore, a control experiment of exposing 1g and CD3OD to the gold catalyst in CHCl3 afforded 2g-D with 50% deuterium incorporation, confirming the process of proto-deauration (eqn (5), Scheme 3).
 | ||
| Scheme 3 Determination of absolute configurations and control experiments. | ||
Based on these experimental data, we proposed a plausible mechanism15,21 of the reaction (Scheme 4): firstly, the catalytically active cationic Au(I) species generated in situ coordinated with the “distal” double bond of the allene unit (Ra)-I. Subsequent anti-attack of the C2 atom at the coordinated CC bond formed (R)-II with a high stereoselectivity. At this stage, when R3 = Me, Et, Cl, and TBSO(CH2)2, because of the higher stability of carbon cation (R)-II, the direct aromatization (path a) was prone to proceed, affording the alkenyl-gold species (R)-IV. Alternatively, highly active cationic species (R)-II could also undergo the 1,2-H shift to afford intermediate (R)-III (path b), which would undergo subsequent aromatization and proto-deauration to afford the optically active final products (R)-2, and released the catalytically active cationic gold catalyst back into the catalytic cycle. The final step was confirmed by the D-labeling experiment in eqn (5) of Scheme 3. While deprotonation of (R)-III followed by a 1,3-H shift of complex V and proto-deauration of complex VI would generate rac-2 (path c, Scheme 4), explaining the lower ee and yield for R3 being H and CO2Me. Also, racemization of the starting allene (Ra)-I and further cycloisomerization may be an alternative pathway for the erosion of the optical purity as confirmed by the parallel reaction in eqn (3) and (4) of Scheme 3.
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have developed a Au+-catalyzed intramolecular cycloisomerization of N-1,3-disubstituted allenylindoles. Various functionalized 6,9-dihydro-pyrido[1,2-a]-1H-indoles can be accessed directly. Furthermore, the optically active 6,9-dihydro-pyrido[1,2-a]-1H-indoles can be prepared with high yields and ee values via an efficient transfer of the axial chirality to central chirality. The core structure of (−)-goniomitine has been prepared in excellent yields and ee values. This protocol features simple operation, mild conditions, and good functional group compatibility. We anticipated that this protocol may provide a new route for the synthesis of natural product analogues and pharmaceuticals containing the core unit of the pyrido[1,2-a]-1H-indole unit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21690063 and 21572202) is greatly appreciated. Shengming Ma is a Qiu Shi Adjunct Professor at Zhejiang University. We thank Mr Weifeng Zheng in our group for reproducing the results: rac-2n, (R)-2e, and (S)-2r.Notes and references
- (a) J. Song, D.-F. Chen and L.-Z. Gong, Natl. Sci. Rev., 2017, 4, 381 CrossRef CAS; (b) J. Hájíček, Collect. Czech. Chem. Commun., 2011, 76, 2023 CrossRef; (c) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC; (d) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278 RSC.
- For recent reviews on gold-catalyzed cyclization reactions, see: (a) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465 CrossRef; (b) Y. Wei and M. Shi, ACS Catal., 2016, 6, 2515 CrossRef CAS; (c) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC; (d) B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939 CrossRef CAS; (e) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884 RSC; (f) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS; (g) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS; (h) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC.
- For selected recent examples of gold-catalyzed cyclization reactions, see: (a) F. M. Miloserdov, M. S. Kirillova, M. E. Muratore and A. M. Echavarren, J. Am. Chem. Soc., 2018, 140, 5393 CrossRef CAS; (b) Z. L. Niemeyer, S. Pindi, D. A. Khrakovsky, C. N. Kuzniewski, C. M. Hong, L. A. Joyce, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 12943 CrossRef CAS; (c) W. Zi, H. Wu and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3225 CrossRef CAS; (d) L. Huang, H.-B. Yang, D.-H. Zhang, Z. Zhang, X.-Y. Tang, Q. Xu and M. Shi, Angew. Chem., Int. Ed., 2013, 52, 6767 CrossRef CAS; (e) A. S. K. Hashmi, W. Yang and F. Rominger, Adv. Synth. Catal., 2012, 354, 1273 CrossRef CAS; (f) C. Ferrer, C. H. M. Amijs and A. M. Echavarren, Chem.–Eur. J., 2007, 13, 1358 CrossRef CAS; (g) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105 CrossRef CAS; (h) P.-L. Zhu, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 7245 RSC; (i) B. Alcaide, P. Almendros, I. Fernández, F. Herrera and A. Luna, Chem.–Eur. J., 2018, 24, 1448 CrossRef CAS; (j) For the seminal report on gold-catalyzed cyclization of allenes, see: A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem. Int. Ed., 2000, 39, 2285 CrossRef CAS.
- (a) Z. Zhang, C. Liu, R. E. Kinder, X. Han, H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2006, 128, 9066 CrossRef CAS; (b) C. Liu and R. A. Widenhoefer, Org. Lett., 2007, 9, 1935 CrossRef CAS.
- E. Álvarez, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, J. Org. Chem., 2013, 78, 9758 CrossRef.
- Y. Wang, P. Zhang, X. Di, Q. Dai, Z.-M. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 15905 CrossRef CAS.
- B. Chen, W. Fan, G. Chai and S. Ma, Org. Lett., 2012, 14, 3616 CrossRef CAS.
- (a) V. Magné, Y. Sanogo, C. S. Demmer, P. Retailleau, A. Marinetti, X. Guinchard and A. Voituriez, ACS Catal., 2020, 10, 8141 CrossRef; (b) Y.-Y. Zhang, Y. Wei and M. Shi, Chem. Commun., 2019, 55, 4210 RSC.
- R. M. Zeldin and F. D. Toste, Chem. Sci., 2011, 2, 1706 RSC.
- L.-Y. Mei, Y. Wei, X.-Y. Tang and M. Shi, J. Am. Chem. Soc., 2015, 137, 8131 CrossRef CAS.
- (a) X. Liang, S.-Z. Jiang, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2016, 138, 2560 CrossRef CAS; (b) M. Mizutani, F. Inagaki, T. Nakanishi, C. Yanagihara, I. Tamai and C. Mukai, Org. Lett., 2011, 13, 1796 CrossRef CAS; (c) M. Mori, Heterocycles, 2010, 81, 259 CrossRef CAS; (d) T.-S. Kam and K.-M. Sim, Heterocycles, 1999, 51, 345 CrossRef CAS.
- For selected examples of RCM reactions of indoles, see: (a) T. Mandal, G. Chakraborti, S. Karmakar and J. Dash, Org. Lett., 2018, 20, 4759 CrossRef CAS; (b) M. A. Abozeid, S. Sairenji, S. Takizawa, M. Fujita and H. Sasai, Chem. Commun., 2017, 53, 6887 RSC; (c) P. González-Pérez, L. Pérez-Serrano, L. Casarrubios, G. Domínguez and J. Pérez-Castells, Tetrahedron Lett., 2002, 43, 4765 CrossRef.
- For selected examples of Michael addition reactions of indoles, see: (a) C. Zhao, F. D. Toste and R. G. Bergman, J. Am. Chem. Soc., 2011, 133, 10787 CrossRef CAS; (b) Q. Cai, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 8666 CrossRef CAS.
- For selected examples of radical cyclization of N-alkylindoles, see: (a) A. R. O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731 CrossRef CAS; (b) J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368 CrossRef CAS; (c) S.-C. Lu, X.-Y. Duan, Z.-J. Shi, B. Li, Y.-W. Ren, W. Zhang, Y.-H. Zhang and Z.-F. Tu, Org. Lett., 2009, 11, 3902 CrossRef CAS; (d) J. Magolan and M. A. Kerr, Org. Lett., 2006, 8, 4561 CrossRef CAS; (e) C. J. Moody and C. L. Norton, J. Chem. Soc. Perkin Trans. I, 1997, 1, 2639 RSC; (f) S. Caddick, K. Aboutayab and R. West, Synlett, 1993, 231 CrossRef CAS.
- J. Barluenga, M. Piedrafita, A. Ballesteros, Á. L. Suárez-Sobrino and J. M. González, Chem.–Eur. J., 2010, 16, 11827 CrossRef CAS.
- (a) J. M. Alonso and M. P. Muñoz, Angew. Chem., Int. Ed., 2018, 57, 4742 CrossRef CAS; (b) L. Cooper, J. M. Alonso, L. Eagling, H. Newson, S. Herath, C. Thomson, A. Lister, C. Howsham, B. Cox and M. P. Muñoz, Chem.–Eur. J., 2018, 24, 6105 CrossRef CAS.
- For the significance of counter anions in the optimization of gold catalysts, see: (a) J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 3949 CrossRef; (b) J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2493 CrossRef; (c) M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638 CrossRef CAS; (d) Z. Lu, J. Han, H. B. Hammond and B. Xu, Org. Lett., 2015, 17, 4534 CrossRef CAS.
- (a) X. Huang and S. Ma, Acc. Chem. Res., 2019, 52, 1301 CAS; (b) D. Ma, X. Duan, C. Fu, X. Huang and S. Ma, Synthesis, 2018, 50, 2533 CrossRef CAS; (c) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956 RSC.
- For other sequences using gold catalysis of halogenated substrates and subsequent palladium-catalyzed steps, see: (a) P. García-Domínguez and C. Nevado, J. Am. Chem. Soc., 2016, 138, 3266 CrossRef; (b) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. D. B. Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133 CrossRef CAS; (c) Y. Shi, K. E. Roth, S. D. Ramgren and S. A. Blum, J. Am. Chem. Soc., 2009, 131, 18022 CrossRef CAS; (d) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243 CrossRef CAS.
- (a) Y. Qiu, J. Zhou, J. Li, C. Fu, Y. Guo, H. Wang and S. Ma, Chem.–Eur. J., 2015, 21, 15939 CrossRef CAS; (b) J. Ye, S. Li, B. Chen, W. Fan, J. Kuang, J. Liu, Y. Liu, B. Miao, B. Wan, Y. Wang, X. Xie, Q. Yu, W. Yuan and S. Ma, Org. Lett., 2012, 14, 1346 CrossRef CAS.
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS; (b) H. Li, R. J. Harris, K. Nakafuku and R. A. Widenhoefer, Organometallics, 2016, 35, 2242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1956055 and 1956072. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05619g |
| This journal is © The Royal Society of Chemistry 2021 |