
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing fluorene to lock electronically active moieties in thermally activated delayed fluorescence emitters for high-performance non-doped organic light-emitting diodes with suppressed roll-off†
Lin
Wu
a,
Kai
Wang
 *a,
Cheng
Wang
a,
Xiao-Chun
Fan
a,
Yi-Zhong
Shi
a,
Xiang
Zhang
a,
Shao-Li
Zhang
a,
Jun
Ye
*b,
Cai-Jun
Zheng
c,
Yan-Qing
Li
a,
Jia
Yu
a,
Xue-Mei
Ou
a and
Xiao-Hong
Zhang
*a
*a,
Cheng
Wang
a,
Xiao-Chun
Fan
a,
Yi-Zhong
Shi
a,
Xiang
Zhang
a,
Shao-Li
Zhang
a,
Jun
Ye
*b,
Cai-Jun
Zheng
c,
Yan-Qing
Li
a,
Jia
Yu
a,
Xue-Mei
Ou
a and
Xiao-Hong
Zhang
*a
aInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou, Jiangsu 215123, P.R. China. E-mail: xiaohong_zhang@suda.edu.cn; wkai@suda.edu.cn
bSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang 315211, P. R. China. E-mail: yejun@nbu.edu.cn
cSchool of Optoelectronic Science and Engineering, University of Electronic Science and Technology of China, Chengdu, Sichuan 610054, P. R. China
First published on 26th November 2020
Abstract
Thermally activated delayed fluorescence (TADF) emitters with aggregation-induced emission (AIE) features are hot candidates for non-doped organic light-emitting diodes (OLEDs), as they are highly emissive in solid states upon photoexcitation. Nevertheless, not every AIE-TADF emitter in the past had guaranteed decent efficiencies in non-doped devices, indicating that the AIE character alone does not necessarily afford ideal non-doped TADF emitters. As intermolecular electron-exchange interaction that involves long-lived triplet excitons plays a dominant role in the whole quenching process of TADF, we anticipate that it is the main reason for the different electroluminescence performances of AIE-TADF emitters. Therefore, in this work, we designed two TADF emitters SPBP-DPAC and SPBP-SPAC by modifying a reported less successful emitter BP-DPAC with extra fluorenes to increase intermolecular distances and attenuate this electron-exchange interaction. With the fluorene lock as steric hindrance, SPBP-DPAC and SPBP-SPAC exhibit significantly higher exciton utilization in non-doped films due to the suppressed concentration quenching. The non-doped OLEDs based on SPBP-DPAC and SPBP-SPAC show an excellent maximum external quantum efficiency (EQE) of 22.8% and 21.3% respectively, and what's even more promising is that ignorable roll-offs at practical brightness (e.g., 1000 and 5000 cd m−2) were realized. These results reveal that locking the phenyl rings as steric hindrance can not only enhance the molecular rigidity, but also cause immediate relief of concentration quenching, and result in significant performance improvement under non-doped conditions. Our approach proposes a feasible molecular modification strategy for AIE-TADF emitters, potentially increasing their applicability in OLEDs.
Introduction
Organic light-emitting diodes (OLEDs) are regarded as promising candidates for flat panel displays as well as lighting owing to their great advantages, such as high-quality colour reproduction, low energy cost, light weight, and flexibility. To take the initiative in a highly competitive market, it is generally required to improve device performances and stabilities meanwhile reducing production costs.1–3 Thermally activated delayed fluorescence (TADF) emitters4–23 are thus the current hotspots in OLED technology as they are pure organic compounds as cheap as conventional host matrixes and can harvest not only singlet excitons, but also triplet excitons via a reverse intersystem crossing (RISC) process. To acquire high device performances, the doping strategy, namely dispersing emitters in host matrixes, has been widely accepted for TADF emitters.24–29 It indeed has proven effective over time in unleashing the full potential of exciton harvesting, while on the other hand, the doping process apparently brings about some cost- and stability-associated disadvantages as well. In order to achieve stable device performance in mass production, precise doping concentration control with complicated device structures is necessary. It brings about high device fabrication costs. By contrast, non-doped OLEDs30–40 are more promising architectures for commercial applications due to the merits of structural or process simplicity which leads to reduced fabrication cost and stable device performance in mass production. Moreover, non-doped systems can well avoid fatal phase separation which may happen in host–guest systems. Unfortunately, most TADF emitters by far are actually excluded as suitable candidates for non-doped devices because of severe concentration quenching. The progress of high-performance non-doped TADF OLED devices is still very limited despite the fast and fruitful development of TADF materials themselves. Among them, emitters exhibiting aggregation-induced emission (AIE) are considered as hot candidates since they are highly emissive in solid states upon photoexcitation. Nevertheless, not every AIE-TADF emitter37,39,41 in the past showed decent efficiency in non-doped devices, indicating that merely possessing the AIE feature is still insufficient to afford ideal non-doped TADF emitters.Previous studies have indicated that long-lived triplet exciton-involving intermolecular electron-exchange interaction, as described by the Dexter energy-transfer model,42,43 plays a dominant role in the whole quenching process of TADF in films. Besides, triplet-related quenching also accounts for the efficiency roll-off at practical brightness with high current densities, which is a widespread issue for electroluminescence (EL). Owing to the short-range nature of intermolecular Dexter interaction and exciton quenching, we anticipate that those AIE-TADF emitters that failed in achieving decent non-doped EL performance still have a great chance to ameliorate behaviours, once they undergo further modification to enlarge the molecular distance, that is, to restrict these adverse intermolecular interactions.
Herein, by remodelling a reported less successful donor (D)–acceptor (A) type compound (4-(9,9-diphenylacridin-10(9H)-yl)phenyl)(phenyl)methanone (BP-DPAC),44 we obtained two novel TADF emitters 2-(9,9-diphenylacridin-10(9H)-yl)-10H-spiro[anthracene-9,9′-fluoren]-10-one (SPBP-DPAC) and 2-(10H-spiro[acridine-9,9′-fluoren]-10-yl)-10H-spiro[anthracene-9,9′-fluoren]-10-one (SPBP-SPAC) (shown in Fig. 1). The two new emitters both retain the electron-donating and electron-withdrawing cores (respectively marked in red and blue in Fig. 1) as in the reference compound. However, in marked contrast to those of BP-DPAC, the phenyl rings of benzophenone (BP) groups are locked via a rigid fluorene group, forming a new designed A moiety named 10H-spiro[anthracene-9,9′-fluoren]-10-one (SPBP), in order to enhance molecular rigidity and reduce the intermolecular interaction. Likewise, in SPBP-SPAC, the diphenyl-9,10-dihydroacridine (DPAC) group is also reinforced into the 10H-spiro[acridine-9,9′-fluoren] (SPAC) moiety by fusing the two peripheral phenyl groups to form one fluorene unit. Detailed physical measurements prove that these three compounds have similar energy levels as well as characteristics of AIE and TADF, while in neat films, SPBP-DPAC and SPBP-SPAC exhibit significantly higher exciton utilization in photoluminescence (PL) and EL processes. This is because the triplet spin densities of SPBP-DPAC and SPBP-SPAC were barely distributed over the fluorene groups, and these rigid fluorenes can act as rigid steric hindrances to hinder intermolecular triplet–triplet interactions. Notably, the non-doped OLEDs based on SPBP-DPAC and SPBP-SPAC respectively displayed an excellent maximum external quantum efficiency (EQE) of 22.8% and 21.3%, which are evidently higher than that of the BP-DPAC-based one (12.9%). Moreover, they both exhibited a well-suppressed efficiency roll-off. In particular, the SPBP-DPAC based non-doped OLED maintained a record-high EQE of 22.4% at a practical brightness of 1000 cd m−2. The behaviour of the SPBP-SPAC based device is more noteworthy, with its EQE climbing up to the maxima at a very high brightness of 2186 cd m−2, suggesting that the exciton quenching is largely suppressed at relatively high exciton densities. These outstanding performances highlight the evident role of using fluorene locks to enhance molecular rigidity and reduce the intermolecular interaction, which not only works on electron-donating cores, but also on electron-withdrawing ones. Our results demonstrate a feasible strategy of molecular modification to improve the non-doped EL performance of practical AIE-TADF molecule candidates.
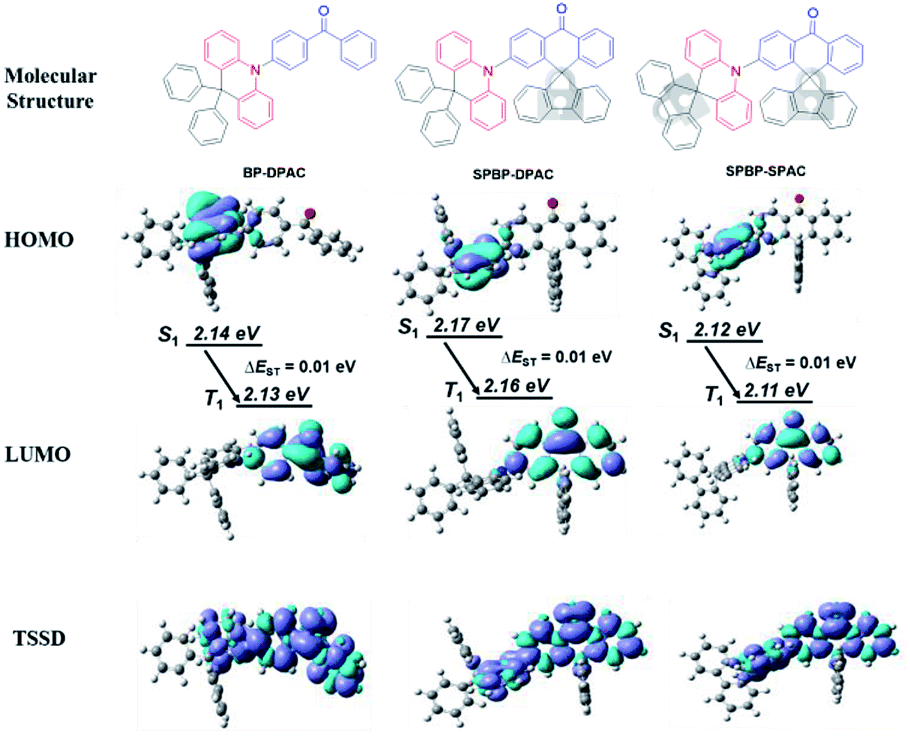 | ||
| Fig. 1 Molecular structures of BP-DPAC, SPBP-DPAC and SPBP-SPAC, in which their electronically active cores are marked in red for D and blue for A segments respectively; and their frontier molecular orbital distributions at optimized ground states and spin-density distributions of T1 states. | ||
Results and discussion
Synthetic procedures
The reference compound BP-DPAC was prepared according to the literature.44 The detailed synthesis of SPBP-DPAC and SPBP-SPAC is demonstrated in Scheme 1 and the ESI.† The brominated A segment SPBP-Br was prepared via the following two steps: (i) a carbonyl addition reaction involving n-butyl lithium with 2-biphenyl bromide and 2-bromoanthraquinone; (ii) a dehydration ring-closure reaction with hydrochloric acid and acetic acid. Then the target compounds SPBP-DPAC and SPBP-SPAC were obtained via a Buchwald–Hartwig coupling reaction with the corresponding D segments. We then used nuclear magnetic resonance (NMR) and mass spectrometry to further confirm their chemical structures. The compounds were further purified by sublimation before investigations.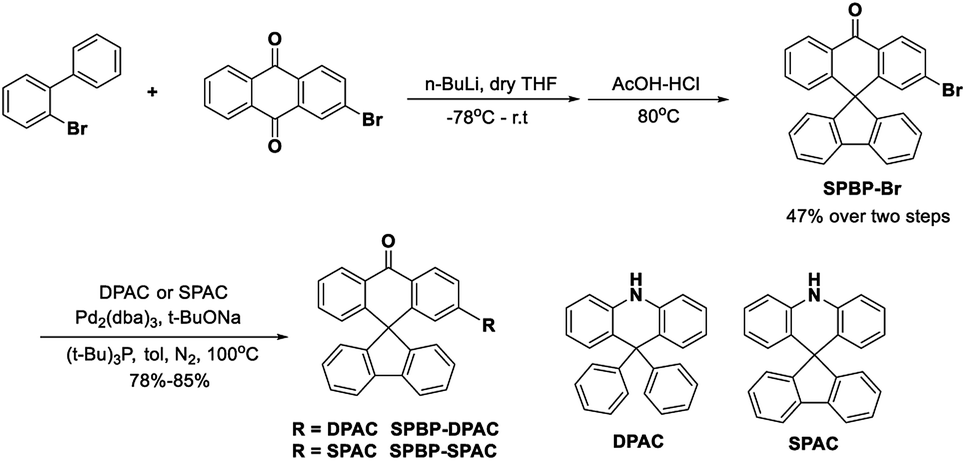 | ||
| Scheme 1 Synthetic route to SPBP-DPAC and SPBP-SPAC. | ||
Theoretical calculation
Theoretical calculations were first performed to investigate the electronic delocalization of the compounds (Fig. 1). Ground state (S0) geometries were optimized using density functional theory (DFT) on the basis of the B3LYP/6-31G(d) level. Due to similar evident steric hindrances, similar large dihedrals between D and A segments are estimated to be 82.8° for BP-DPAC, 84.7° for SPBP-DPAC and 88.3° for SPBP-SPAC, respectively. Meanwhile for both new SPBP derivatives, locking the two phenyl rings of the BP group endows them with coplanar rigidity instead of free rotation in BP-DPAC. Similarly, the extra lock in the SPAC moiety also restricts the rotation of the phenyl rings in the DPAC moiety and results in an enhanced rigidity. Based on the optimized geometries, their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions were calculated. As shown in Fig. 1, the delocalization of the HOMO and LUMO of both SPBP derivatives is very similar to that of BP-DPAC with evident separations, which is essential for facilitating TADF features. Notably the delocalization can barely cross over the sp3 carbon and extend to the fluorene groups, suggesting that the electronically active cores are similar to those present in BP-DPAC. Therefore, the HOMO and LUMO energy levels are predicted to be very similar for all three compounds (Table S1†). We further simulated their natural transition orbitals (NTOs) by a time-dependent DFT (TD-DFT) method. As shown in Fig. S1,† like those of ground states, the “hole” and “particle” of their lowest singlet (S1) and triplet (T1) excited states only distribute in the electronically active cores, respectively, suggesting their intramolecular charge-transfer (ICT) characteristics. Accordingly, the calculated S1–T1 energy offsets (ΔEST) are all estimated to be extremely small values of ∼0.01 eV, further indicating that they are TADF emitters. To further clarify the roles of the moieties beyond the sp3 carbon that barely show orbital distributions, we also calculated the triplet spin-density distributions (TSSDs) (Fig. 1). It is apparently shown that the triplet excitons of the three compounds almost exclusively localize on the electronically active cores (coloured in molecular structures in Fig. 1), while the fluorenes and phenyl rings only act as shielding protectors for these active cores, contributing no TSSDs. As a result, it is expected that the intermolecular interaction-associated concentration quenching in the solid state should be effectively suppressed, when involving the protectors on both D and A moieties in SPBP-DPAC and SPBP-SPAC, especially these rigid bulky fluorene protectors. In comparison, the reference compound BP-DPAC, which only has two rotation-free phenyl rings at the acridine side, could most likely expose its electronically active cores to each other in pure thin films, fatally confronting strong intermolecular interactions.Crystal structures
Single crystal analysis was then carried out for SPBP-DPAC and SPBP-SPAC to determine their geometries and packing patterns. As shown in Fig. S2,† both emitters adopt highly twisted molecular geometries, which are well consistent with our previous estimation. Moreover, with the rigid fluorene groups as steric hindrance, direct face-to-face close packing of both the D and A cores is unavailable in both emitters, which would effectively suppress the intermolecular electron-exchange interactions. Close intermolecular distances are observed between D and A moieties and estimated to be 3.279 Å for SPBP-DPAC and 3.597 Å for SPBP-SPAC respectively (shown in Fig. S2d and h†). Concentration-caused emission quenching and exciton annihilation can thus be expected to be largely suppressed.Electrochemical and thermal properties
We then carried out cyclic voltammetry (CV) measurement to characterize the energy levels of the studied compounds. As shown in Fig. S3,† all compounds exhibited similar curve shapes due to their similar D and A moieties. From the onsets of oxidation and reduction curves, we estimated the HOMO and LUMO levels as summarized in Table 1. In particular, the HOMO energy level of SPAC is only slightly shallower than those of DPAC derivatives, confirming the similar electron-donating abilities of DPAC and SPAC, while the LUMO energy levels of SPBP derivatives are evidently lower than that of BP-DPAC. This could be ascribed to the enhanced conjugation of BP cores in SPBP moieties because of the lock.| Compound | λ abs [nm] | λ em [nm] | S1c [eV] | T1c [eV] | ΔESTd [eV] | HOMOe [eV] | LUMOe [eV] | Φ PL [%] |
|---|---|---|---|---|---|---|---|---|
| a Measured in toluene at room temperature. b Measured in toluene, THF and neat films at room temperature (298 K). c Measured in non-doped films and 2-MeTHF at 77 K. d ΔEST = S1 − T1. e Determined using cyclic voltammetry curves in DMF at 298 K. f Absolute PLQY of 30 wt% emitters doped in mCP films measured using an integrating sphere under a nitrogen atmosphere and coefficient of error within ±1.0%. | ||||||||
| BP-DPAC | 305, 360 | 491/520/486 | 2.77/2.97 | 2.71/2.93 | 0.06/0.04 | 5.70 | 3.06 | 88 |
| SPBP-DPAC | 307, 380 | 500/535/495 | 2.67/2.95 | 2.64/2.88 | 0.03/0.07 | 5.70 | 3.24 | 98 |
| SPBP-SPAC | 308, 410 | 497/542/504 | 2.52/2.92 | 2.50/2.84 | 0.02/0.08 | 5.65 | 3.23 | 99 |
To confirm that their thermal stabilities are suitable for thermal evaporation, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were carried out for the new compounds. As shown in Fig. S4,† the decomposition temperatures (corresponding to 5% weight loss) were estimated to be 387 °C for SPBP-DPAC and 412 °C for SPBP-SPAC respectively. Such high decomposition temperatures suggest they are suitable for the evaporation process. Moreover, both compounds did not show obvious glass transition or melting during the whole range from room temperature to 200 °C, guaranteeing their application in OLED devices.
Photophysical properties
The photophysical properties of these compounds were then investigated under different conditions. Fig. 2a shows the absorption and PL spectra in dilute toluene. Due to their similar electron-donating and electron-withdrawing cores, all these emitters exhibit similar locally excited absorption bands below ∼340 nm. Meanwhile due to their similar D–A interactions, weak bands assigned to ICT transitions can be noticed to be centred at 360 nm for BP-DPAC, 380 nm for SPBP-DPAC and 410 nm for SPBP-SPAC respectively. In the PL spectra, clearly broad and structureless emission can be observed similarly at 500 nm for SPBP-DPAC and 497 nm for SPBP-SPAC, respectively, which has a slight bathochromic shift compared with that of BP-DPAC (491 nm), well consistent with their different HOMO–LUMO gaps. In particular, the Stokes shift of SPBP-SPAC is evidently smaller than that of SPBP-DPAC and BP-DPAC, indicating that locking these rotation-free phenyl rings can instantly suppress the molecular excited-state relaxation. We then changed the solution to polar tetrahydrofuran (THF) (Fig. S5b†), in which all these ICT-type emissions showed redshifted PL spectra compared with the results in toluene. As shown in Fig. 3, the three emitters all display weak yellow emissions in dilute THF solutions, while the PL intensities were greatly enhanced along with the aggregates formed by adding a large quantity of water into THF solutions. Correspondingly, noticeably increased delayed fluorescence lifetimes are observed which clearly reveal the AIE features of these three compounds (Fig. S6†).37,45 Apparently, although using spiro fluorene gives rise to evidently increased rigidity of D and A moieties in SPBP-DPAC and SPBP-SPAC, there are still important single bonds remaining, which are the bridges between D and A segments and would suffer evident geometrical changes during CT excitation, radiation and relaxations. Therefore, there are still serious molecular rotations which will induce serious energy loss and weak emission in highly polar THF, while upon formation of the aggregates, such molecular structural relaxation processes can thus be well restrained, inducing enhanced emission (i.e. AIE). Moreover, the spiro fluorene moieties also help prevent the formation of close π–π stacking in the aggregate state and the unfavourable ACQ can thus be effectively relieved. Therefore, both emitters could depict an AIE phenomenon. It should be noted that with different aggregation degrees (i.e. water fraction), the emission peaks are observed to red or blue shift. This could be attributed to the combined results of several factors such as increased environmental polarity intensity and molecular packing (redshift the emission), restricted molecular free rotations upon aggregation (blueshift the emission), etc.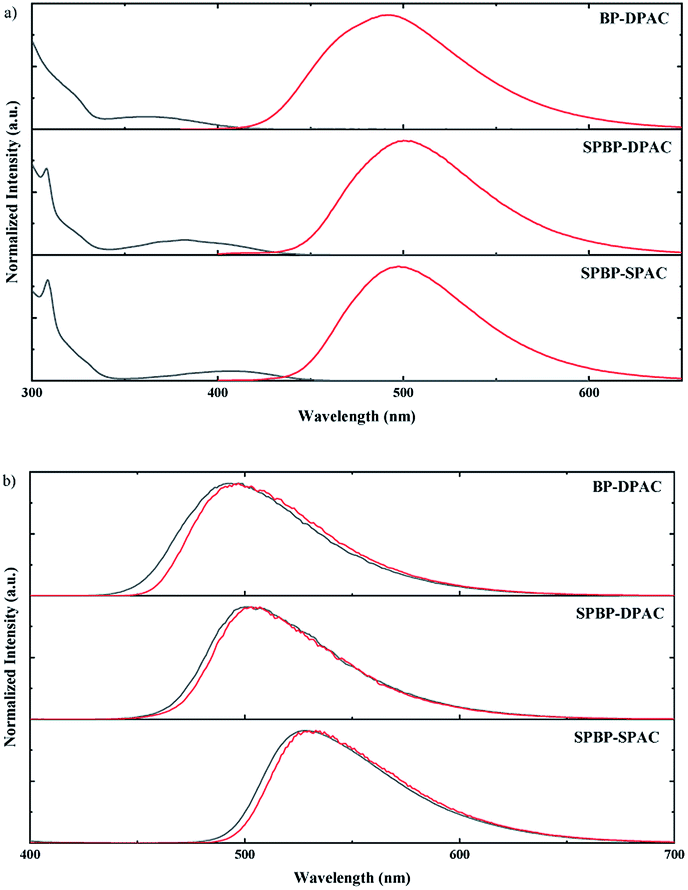 | ||
| Fig. 2 (a) Normalized UV/Vis absorption (black) and photoluminescence (red) spectra of the studied compounds in toluene solution at room temperature and (b) their fluorescence (black) and phosphorescence (red) spectra in amorphous neat films at 77 K. | ||
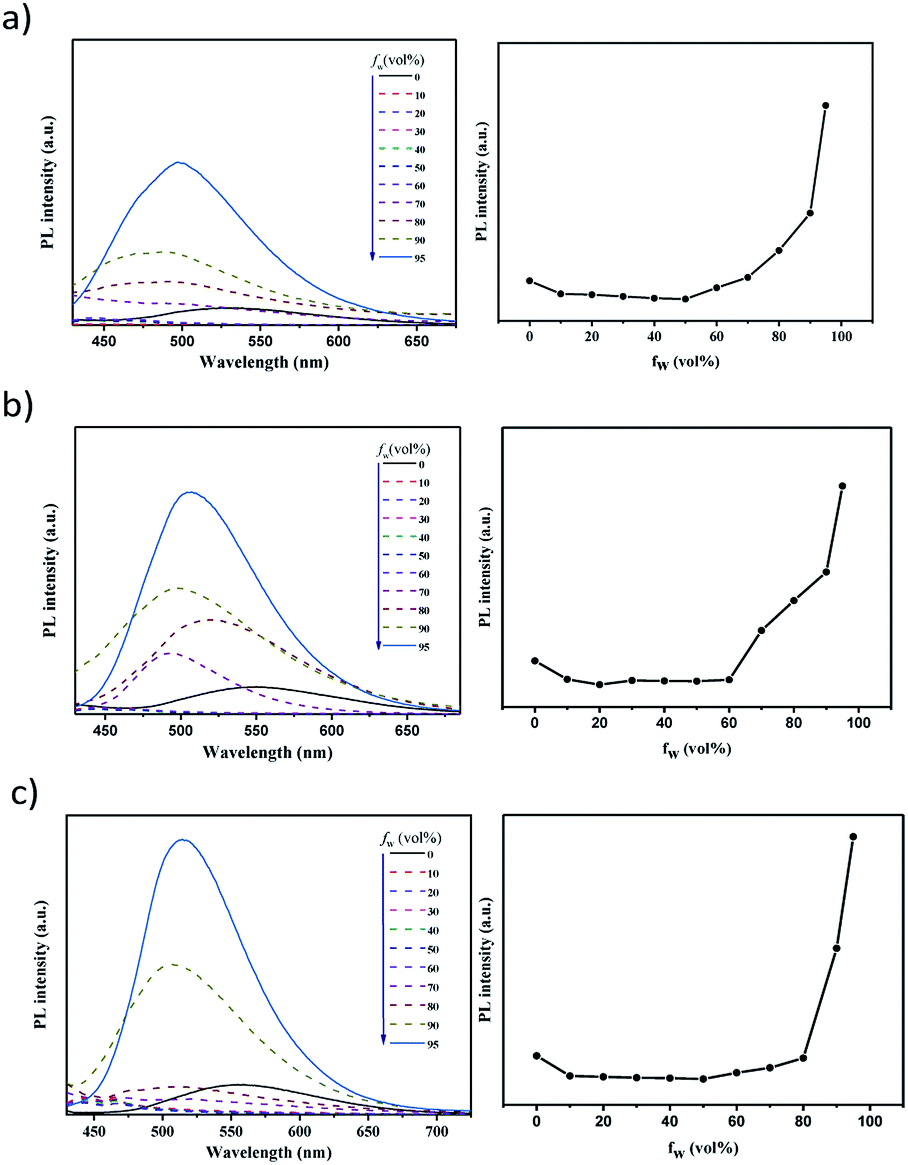 | ||
| Fig. 3 PL spectra of (a) BP-DPAC, (b) SPBP-DPAC and (c) SPBP-SPAC in THF/water mixtures with different water fractions (fw) (left) and the corresponding PL intensity versus fw (right). | ||
We then characterized the PL properties of these emitters in amorphous neat films, in which the molecular geometry relaxation is effectively suppressed while molecular energy levels and packing modes are the determining factors to their emission. As shown in Fig. S5c,† the PL spectral peak wavelength of SPBP-SPAC became slightly longer than that of SPBP-DPAC, consistent with the narrower HOMO–LUMO bandgap of SPBP-SPAC. To further characterize the energy levels of these emitters, we measured their fluorescence and phosphorescence spectra in neat films and 2-methyltetrahydrofuran (2-MeTHF) at 77 K. As shown in Fig. 2b and S7,† similar fluorescence and phosphorescence spectra with structureless shapes were observed, indicative of their notable ICT transition characteristics. Table 1 summarizes the results by calculations from their corresponding spectral onsets. The S1/T1 energy levels of BP-DPAC, SPBP-SPAC and SPBP-DPAC showed regular declines, and coincide well with their HOMO and LUMO energy levels. Based on these energy differences, their ΔESTs are experimentally calculated to be less than 0.1 eV in both the amorphous neat film and 2-MeTHF solution, and are small enough to promote the transition of triplet excitons to singlet excitons via the thermally activated RISC process.
The TADF nature of BP-SPAC has been proved by previous literature.44 In order to verify our modified compounds retaining TADF properties, transient PL decay measurements were proposed, as shown in Fig. S8.† In dilute toluene and THF solution, both emitters showed obvious delayed fluorescence lifetime, indicating that they have TADF characteristics in the dilute solution (Fig. S8a and b†). Likewise, we measured their transient PL profiles in neat films. As shown in Fig. S8c,† both SPBP-DPAC and SPBP-SPAC exhibited prompt components with lifetimes estimated to be 24.4 and 24.9 ns, and delayed components with very short delayed lifetimes of 2.5 and 1.3 μs, respectively. Such short delayed lifetimes are beneficial to suppress the device efficiency roll-off by accelerating triplet conversion to delayed fluorescence. With obvious TADF contribution, the photoluminescence quantum yields (PLQYs) of SPBP-DPAC and SPBP-SPAC neat films are estimated to be as high as 93% and 98%, respectively, almost double that of BP-DPAC (47%). When diluting these emitters in 1,3-bis(N-carbazolyl)benzene (mCP) films, their PLQY difference is not so obvious (BP-DPAC: 88%; SPBP-DPAC: 98%; SPBP-SPAC: 99%). The enhanced molecular rigidities due to the fluorene locks apparently contribute to the PLQY enhancement of the modified molecules; more importantly, concentration quenching is considerably suppressed in non-doped thin films of the modified compounds resulting in great progress in exciton utilization relative to the reference molecule under non-doped conditions.
To further analyse the mechanism behind, we calculated the photophysical kinetic parameters. As summarized in Table 2, the rate constants of the RISC process (kRISC) of SPBP-DPAC and SPBP-SPAC are estimated to be 1.2 × 106 and 1.4 × 106 s−1, receptively. These values are evidently higher than those for most TADF competitors, suggesting that the triplet energy loss routes could be effectively blocked for SPBP-DPAC and SPBP-SPAC. These improvements then clearly highlight the effect of introducing steric hindrance on relieving concentration quenching. Notably, with the extra lock of SPAC, the SPBP-SPAC molecule exhibited a further suppressed non-radiative process of triplets (kTnr) compared with SPBP-DPAC, revealing the effect of locking via fluorene on increasing segmental rigidity and thus suppressing non-radiative decays.
| Compound | Φ PL [%] | Φ p/Φdb [%] | τ p [ns] | τ d [μs] | k p [107 s−1] | k d [105 s−1] | k r [107 s−1] | k ISC [107 s−1] | k RISC [106 s−1] | k Tnr [105 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|
a Absolute PLQY measured using an integrating sphere under a nitrogen atmosphere and coefficient of error within ±1.0%.
b Prompt and delayed components of PLQY obtained by fitting the transient curve at 298 K.
c
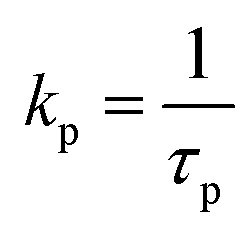 .
d .
d
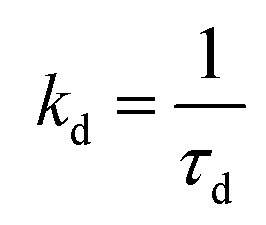 .
e Rate constant of radiative decay, kr = Φpkp.
f Rate constant of the intersystem crossing (ISC) process (S1 → T1), kISC = kp − kr.
g Rate constant of the RISC process (T1 → S1), .
e Rate constant of radiative decay, kr = Φpkp.
f Rate constant of the intersystem crossing (ISC) process (S1 → T1), kISC = kp − kr.
g Rate constant of the RISC process (T1 → S1),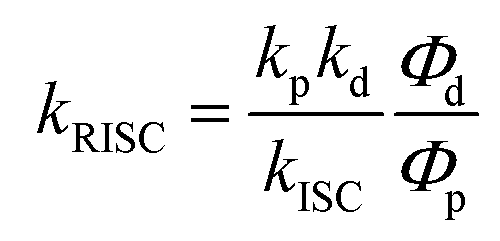 .
h Rate constant of nonradiative transitions of triplet excitons, kTnr = kd − ΦpkRISC. .
h Rate constant of nonradiative transitions of triplet excitons, kTnr = kd − ΦpkRISC.
|
||||||||||
| BP-DPAC | 47 | 14/33 | 17.9 | 5.3 | 5.6 | 1.9 | 0.8 | 4.8 | 0.5 | 1.2 |
| SPBP-DPAC | 93 | 31/62 | 24.4 | 2.5 | 4.1 | 4.0 | 1.3 | 2.8 | 1.2 | 0.4 |
| SPBP-SPAC | 98 | 53/45 | 24.9 | 1.3 | 4.0 | 7.7 | 2.1 | 1.9 | 1.4 | 0.3 |
EL performance
Inspired by all these above properties, we investigated the potential of the two emitters in non-doped OLEDs. The detailed device structures and their corresponding energy level diagram are illustrated in Fig. 4. In the fabrication of devices with structures of ITO/TAPC (40 nm)/TCTA (10 nm)/mCP (10 nm, except for SPBP-DPAC)/emitter (20 nm)/TmPyPb (40 nm)/LiF (1 nm)/Al (120 nm), indium tin oxide (ITO) and LiF/Al were used as the anode and the cathode, respectively; 1,1-bis[4-[N,N-di(p-tolyl)amino]phenyl]cyclohexane (TAPC) and 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPb) respectively acted as the hole-transporting and electron-transporting layer; 4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TCTA) was used as electron-blocking layers; and in particular, mCP was inserted as the exciton-blocking layer except for SPBP-DPAC to optimize the device performance.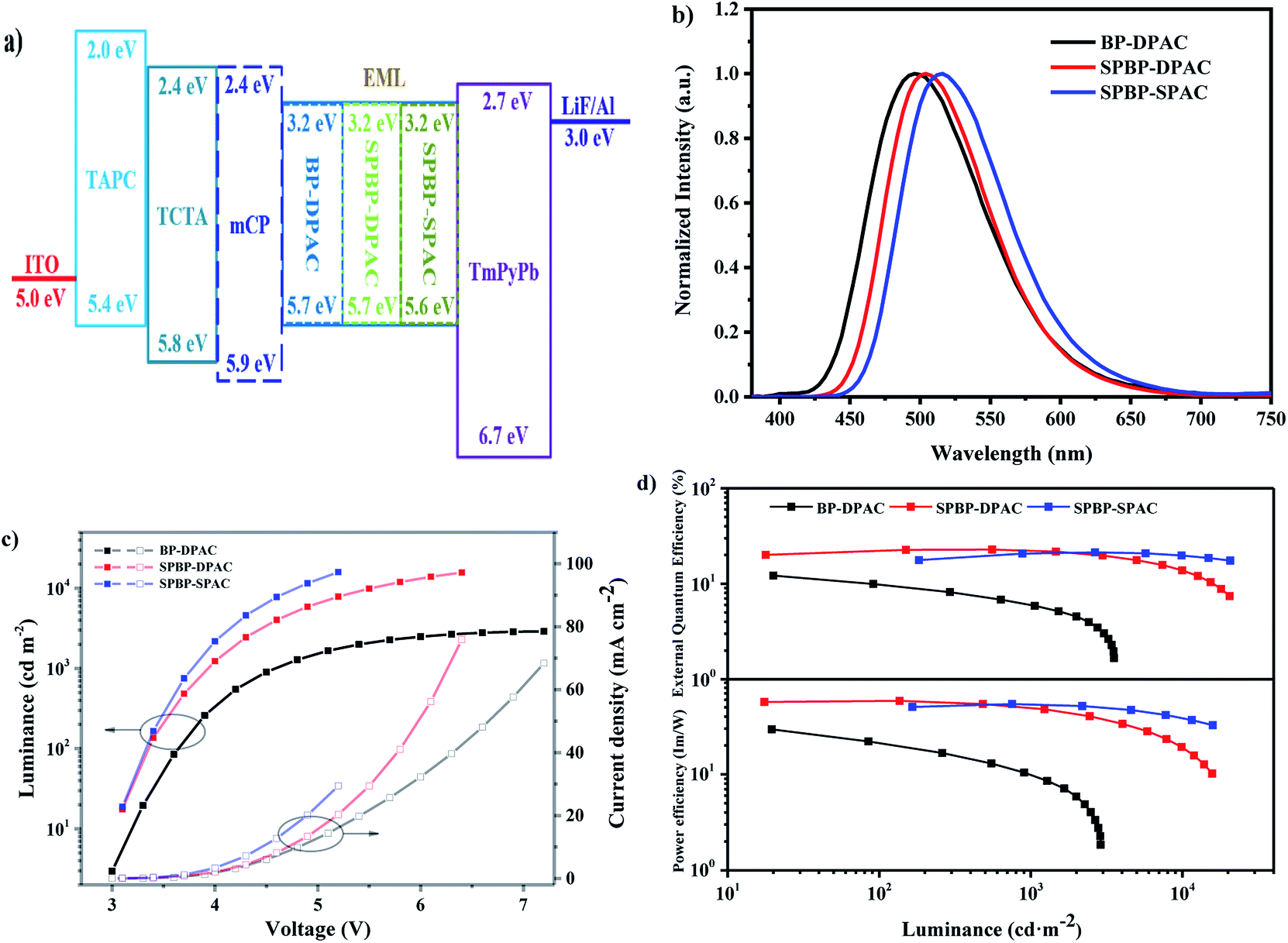 | ||
| Fig. 4 (a) Device structures and energy level diagrams of non-doped OLEDs and the corresponding (b) electroluminescence (EL) spectra at 1000 cd m−2, (c) current density and luminance versus voltage curves, and (d) power efficiency and EQE versus luminance curves. | ||
Table 3 summarizes the performance of the non-doped OLEDs with optimized device structures. As shown in Fig. 4, the AIE feature of BP-DPAC does not lead to a high exciton utilization in EL. Its non-doped OLED can only achieve a maximum EQE of 12.9%. Compared with BP-DPAC, the remodelled emitters SPBP-DPAC and SPBP-SPAC only slightly redshifted the EL spectra of their non-doped devices, which are well consistent with their non-doped PL spectra. The optimized OLEDs respectively displayed stable green EL with a peak at 504 nm and a CIE coordinate of (0.23, 0.50) for SPBP-DPAC, and at 516 nm and (0.28, 0.56) for SPBP-SPAC. This tendency is well consistent with their photophysical properties. On the other hand, these devices showed much improved performance. The SPBP-DPAC and SPBP-SPAC based devices exhibited maximum EQEs of 22.8% and 21.3% respectively, markedly improved compared with those of the reference compound BP-DPAC. This should be primarily attributed to the introduction of the fluorene locks onto the electronically active moieties, which not only enhance the molecular rigidity, but also successfully distance the excitons and reduce the possibility of exciton annihilation during electro-excitations. Meanwhile, it should be noted that although SPBP-SPAC shows a higher PLQY and superior dynamic parameters, its non-doped device displays a slightly inferior EQE maximum to that of SPBP-DPAC. To figure out the reason, we further prepared hole- and electron-only devices based on these compounds. As shown in Fig. S9,† the hole and electron mobilities are evidently more balanced in the non-doped SPBP-DPAC film than in SPBP-SPAC and BP-DPAC, giving the SPBP-DPAC device a higher level of carrier balance. This should be attributed to the reasonably closer packing mode of SPBP-DPAC under non-doped conditions. On the other hand, the looser molecular packing of SPBP-SPAC helps to reduce the concentration quenching especially at high current density. Therefore the SPBP-SPAC based device reversely showed better performance at a practical brightness of over ∼1500 cd m−2.
| Compound | V on [V] | λ EL [nm] | CIEb (x,y) | EQEc/PEd/CEe [%/lm W−1/cd A−1] | ||
|---|---|---|---|---|---|---|
| Maximum | at 1000 cd m−2 | at 5000 cd m−2 | ||||
| a Turn-on voltage. b Commission Internationale de l'Eclairage (CIE) coordinates. c External quantum efficiency. d Power efficiency. e Current efficiency. | ||||||
| BP-DPAC | 3.0 | 496 | (0.21, 0.42) | 12.9/34.3/32.7 | 4.3/10.0/13.8 | n/a |
| SPBP-DPAC | 3.1 | 504 | (0.23, 0.50) | 22.8/54.1/63.7 | 22.4/49.9/61.4 | 17.1/31.1/46.5 |
| SPBP-SPAC | 3.1 | 516 | (0.27, 0.56) | 21.3/51.6/65.7 | 20.8/53.4/64 | 20.9/46.1/63.8 |
In fact, the performances of the optimized doped devices of SPBP-DPAC and SPBP-SPAC are only slightly higher than those under non-doped conditions (seen Fig. S10 and Table S2†). Such weak concentration sensitivity thus indicates that for SPBP-DPAC and SPBP-SPAC the role of the host matrix is not as important as it is for most common TADF emitters. Moreover, as the delayed lifetimes of both emitters are as short as ∼2 μs, the suppression of their device efficiency roll-offs has also been significantly ameliorated. The SPBP-DPAC based non-doped device maintained an excellent EQE of 22.4% at 1000 cd m−2, corresponding to an extremely small roll-off of 1.8%. To the best of our knowledge, this is the highest value among reported non-doped TADF OLEDs (Table S3†). Moreover the device still maintained an impressive EQE of 17.1% at 5000 cd m−2 with a relative efficiency roll-off of only 25.0%. Meanwhile the device based on SPBP-SPAC with two rigid fluorene locks showed more inspiring results. With increasing current densities, the device EQE climbed up to its maximum at a very high brightness of 2186 cd m−2, and maintained as high as 20.9% at 5000 cd m−2, corresponding to a negligible roll-off of ∼2%. All these results thus prove that with appropriate intermolecular steric hindrance to isolate excitons and an extremely short exciton lifetime to accelerate exciton-to-light conversion, the exciton quenching problem could be perfectly solved in non-doped emitting layers.
Conclusion
In summary, we designed two TADF emitters SPBP-DPAC and SPBP-SPAC by modifying a reported less successful emitter BP-DPAC with extra fluorene moieties to enhance the molecular rigidity and increase the intermolecular distance. Though all three compounds have similar energy levels and prominent characteristics of AIE and TADF, SPBP-DPAC and SPBP-SPAC exhibit significantly higher exciton utilization in non-doped films because of the suppressed concentration quenching. Notably, the non-doped OLEDs based on SPBP-DPAC and SPBP-SPAC respectively displayed an excellent maximum external quantum efficiency (EQE) of 22.8% and 21.3%, which are evidently higher than that of the BP-DPAC based one (12.9%). Moreover, the performances of SPBP-DPAC and SPBP-SPAC are even more promising with ignorable roll-offs at practical brightness (i.e. 1000 and 5000 cd m−2). In particular the EQE of the SPBP-DPAC based non-doped device at 1000 cd m−2 is record-high compared to those of reported TADF-based non-doped OLEDs. Our work not only provided two highly efficient non-doped TADF emitters by simply using fluorene to lock the electron-donating and electron-withdrawing moieties, but more importantly, by comparing three AIE-active materials with different behaviours in nondoped devices, we figured out the key for AIE-active materials to obtain decent performance in non-doped OLEDs, which was not fully clarified in previous literature. Accordingly we proposed a feasible molecular modification strategy for AIE-TADF molecules with low performance in nondoped OLEDs to increase their applicability in OLEDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research & Development Program of China (Grant No. 2016YFB0401002), the National Natural Science Foundation of China (Grant No. 51533005, 51821002, and 51773029), the China Postdoctoral Science Foundation (Grant No. 2019M661924 and 2018M642307), Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 111 Project, and Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.References
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS.
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- X. Cai, X. Li, G. Xie, Z. He, K. Gao, K. Liu, D. Chen, Y. Cao and S. J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- X. D. Cao, D. Zhang, S. M. Zhang, Y. T. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- S. Gan, S. Hu, X. L. Li, J. Zeng, D. Zhang, T. Huang, W. Luo, Z. Zhao, L. Duan, S. J. Su and B. Z. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 17327–17334 CrossRef CAS.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS.
- M. Kim, S. K. Jeon, S. H. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515–2520 CrossRef CAS.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS.
- X. K. Liu, Z. Chen, C. J. Zheng, C. L. Liu, C. S. Lee, F. Li, X. M. Ou and X. H. Zhang, Adv. Mater., 2015, 27, 2378–2383 CrossRef CAS.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, P. Y. Huang, M. J. Huang, C. Z. Ren-Wu, C. Y. Yang, M. J. Chiu, L. K. Chu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2016, 138, 628–634 CrossRef CAS.
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS.
- Y. Seino, S. Inomata, H. Sasabe, Y. J. Pu and J. Kido, Adv. Mater., 2016, 28, 2638–2643 CrossRef CAS.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef.
- T. L. Wu, M. J. Huang, C. C. Lin, P. Y. Huang, T. Y. Chou, R. W. Chen-Cheng, H. W. Lin, R. S. Liu and C. H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- D. Zhang, X. Song, M. Cai, H. Kaji and L. Duan, Adv. Mater., 2018, 30, 1705406 CrossRef.
- D. D. Zhang, L. A. Duan, Y. L. Li, D. Q. Zhang and Y. Qiu, J. Mater. Chem. C, 2014, 2, 8191–8197 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS.
- K. Albrecht, K. Matsuoka, K. Fujita and K. Yamamoto, Mater. Chem. Front., 2018, 2, 1097–1103 RSC.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef.
- P. L. Dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS.
- J. A. Seo, Y. Im, S. H. Han, C. W. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 37864–37872 CrossRef CAS.
- Y. Yuan, Y. Hu, Y. X. Zhang, J. D. Lin, Y. K. Wang, Z. Q. Jiang, L. S. Liao and S. T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef.
- X. L. Chen, J. H. Jia, R. Yu, J. Z. Liao, M. X. Yang and C. Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS.
- J. J. Guo, X. L. Li, H. Nie, W. W. Luo, S. F. Gan, S. M. Hu, R. R. Hu, A. J. Qin, Z. J. Zhao, S. J. Su and B. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S. J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef CAS.
- Z. Huang, Z. Bin, R. Su, F. Yang, J. Lan and J. You, Angew. Chem., Int. Ed., 2020, 59, 9992–9996 CrossRef CAS.
- H. Liu, J. Zeng, J. Guo, H. Nie, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 9290–9294 CrossRef CAS.
- W. Liu, S. Ying, R. D. Guo, X. F. Qiao, P. P. Leng, Q. Zhang, Y. X. Wang, D. G. Ma and L. Wang, J. Mater. Chem. C, 2019, 7, 1014–1021 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- Y. Z. Shi, K. Wang, X. Li, G. L. Dai, W. Liu, K. Ke, M. Zhang, S. L. Tao, C. J. Zheng, X. M. Ou and X. H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9480–9484 CrossRef CAS.
- S. P. Xiang, Z. Huang, S. Q. Sun, X. L. Lv, L. W. Fan, S. F. Ye, H. T. Chen, R. D. Guo and L. Wang, J. Mater. Chem. C, 2018, 6, 11436–11443 RSC.
- Z. Yang, Z. Mao, C. Xu, X. Chen, J. Zhao, Z. Yang, Y. Zhang, W. Wu, S. Jiao, Y. Liu, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 8129–8134 RSC.
- J. J. Guo, Z. J. Zhao and B. Z. Tang, Adv. Opt. Mater., 2018, 6, 1800264 CrossRef.
- J. Lee, N. Aizawa, M. Numata, C. Adachi and T. Yasuda, Adv. Mater., 2017, 29, 1604856 CrossRef.
- D. Zhang, P. Wei, D. Zhang and L. Duan, ACS Appl. Mater. Interfaces, 2017, 9, 19040–19047 CrossRef CAS.
- R. J. Yang, Q. Guan, Z. Y. Liu, W. Song, L. Hong, T. Lei, Q. Wei, R. X. Peng, X. Fan and Z. Y. Ge, Chem.–Asian J., 2018, 13, 1187–1191 CrossRef CAS.
- H. Liu, H. Liu, J. Fan, J. Guo, J. Zeng, F. Qiu, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 2001027 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details, NMR spectra of compounds, cyclic voltammetry, TGA and DSC thermograms, photophysical properties and EL performances. CCDC 2032631 and 2032633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05631f |
| This journal is © The Royal Society of Chemistry 2021 |