
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficacy analysis of compartmentalization for ambient CH4 activation mediated by a RhII metalloradical in a nanowire array electrode†
Benjamin S.
Natinsky‡
 a,
Brandon J.
Jolly‡
a,
David M.
Dumas
a and
Chong
Liu
*ab
a,
Brandon J.
Jolly‡
a,
David M.
Dumas
a and
Chong
Liu
*ab
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: chongliu@chem.ucla.edu
bCalifornia NanoSystems Institute (CNSI), University of California, Los Angeles, CA 90095, USA
First published on 8th December 2020
Abstract
Compartmentalization is a viable approach for ensuring the turnover of a solution cascade reaction with ephemeral intermediates, which may otherwise deactivate in the bulk solution. In biochemistry or enzyme-relevant cascade reactions, extensive models have been constructed to quantitatively analyze the efficacy of compartmentalization. Nonetheless, the application of compartmentalization and its quantitative analysis in non-biochemical reactions is seldom performed, leaving much uncertainty about whether compartmentalization remains effective for non-biochemical reactions, such as organometallic, cascade reactions. Here, we report our exemplary efficacy analysis of compartmentalization in our previously reported cascade reaction for ambient CH4-to-CH3OH conversion, mediated by an O2-deactivated RhII metalloradical with O2 as the terminal oxidant in a Si nanowire array electrode. We experimentally identified and quantified the key reaction intermediates, including the RhII metalloradical and reactive oxygen species (ROS) from O2. Based on such findings, we experimentally determined that the nanowire array enables about 81% of the generated ephemeral intermediate RhII metalloradical in air, to be utilized towards CH3OH formation, which is 0% in a homogeneous solution. Such an experimentally determined value was satisfactorily consistent with the results from our semi-quantitative kinetic model. The consistency suggests that the reported CH4-to-CH3OH conversion surprisingly possesses minimal unforeseen side reactions, and is favorably efficient as a compartmentalized cascade reaction. Our quantitative evaluation of the reaction efficacy offers design insights and caveats into application of nanomaterials to achieve spatially controlled organometallic cascade reactions.
Introduction
Compartmentalization, ubiquitous in biology, allows efficient transfer of reaction intermediates or ephemeral species within a multienzyme cascade reaction in an intracellular medium.1–3 By segregating subsequent catalytic sites from the bulk environment at the microscopic or even nanoscopic level, spatial control of catalytic reactions ensures the functionality of biological metabolism in a factory-like manner with high efficiency.4–6 Here the key to a successful compartmentalized cascade reaction is the capability of confining a transient intermediate within the compartment and preventing its outflux that leads to either intermediate loss or deactivation (Fig. 1A). A number of theoretical models have been established to evaluate the capability of compartmentalized cascade reactions in fulfilling this task,7,8 predominantly in the context of enzymatic catalytic reactions.9–13 Reaction efficiency (γ), defined as the ratio between the product outflux and the substrate influx to the compartment for a one-to-one stoichiometric reaction (Fig. 1A), quantitatively measures the efficacy of a cascade system in retaining and utilizing the intermediate species generated within the compartment.6,9,14,15 With the value of γ commonly approaching unity for cascade reactions in biology,4–6,14 nature masters the design strategy of compartmentalization for enzymatic reactions.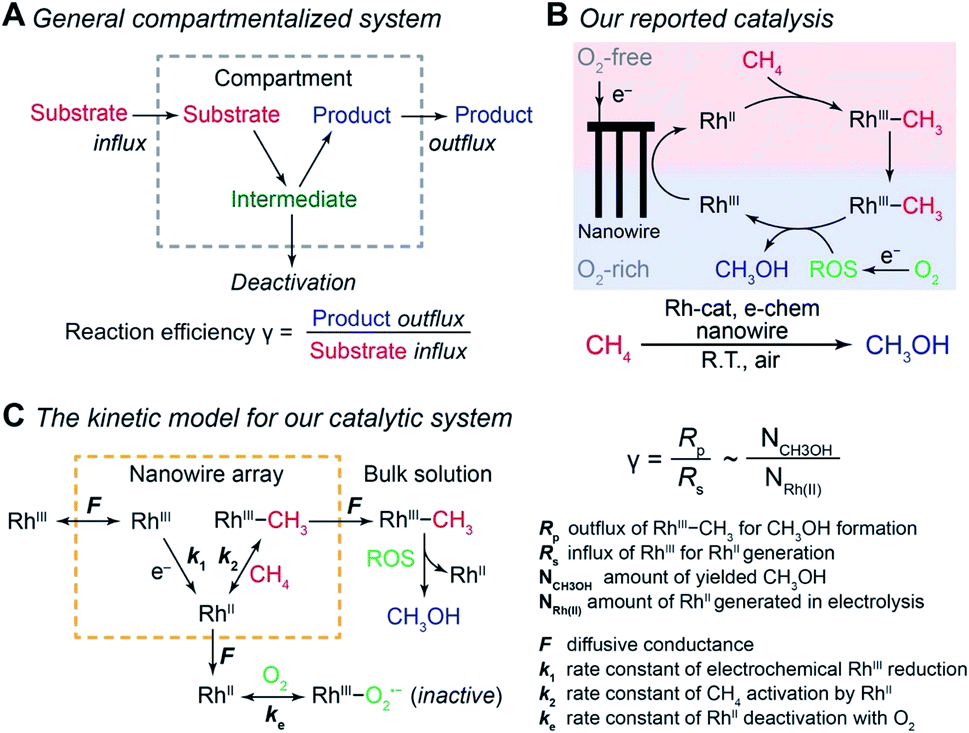 | ||
| Fig. 1 Reaction efficiency γ in compartmentalized cascade reactions. (A) A generalized schematic for the compartmentalization cascade. (B) The proposed mechanism of our previously reported system for ambient CH4-to-CH3OH conversion.20 RhIII, rhodium(III) tetramesityl porphyrin iodide; RhII, rhodium(II) tetramesityl porphyrin metalloradical; RhIII–CH3, methylated rhodium(III) tetramesityl porphyrin; ROS, reactive oxygen species; Rh-cat, rhodium(III) tetramesityl porphyrin iodide; e-chem, electrochemistry; R.T., room temperature. (C) A theoretical framework of a kinetic model for the CH4-to-CH3OH system in the context of a compartmentalized cascade. A detailed mathematical derivation of γ is available in Section 1B of the ESI.† | ||
The benefits of confining catalytic reactions spatially at microscopic and nanoscopic levels in nature inspire the development of other non-enzymatic cascades, mostly with surface-attached molecular, nanoparticle, or enzyme catalysts in porous media such as metal–organic frameworks16–18 or superposed on scaffolds.10–12 Nonetheless, analyzing and determining the value of γ, a quantitative figure-of-merit for the efficacy of compartmentalization, remains infrequent, to say the least, for non-enzymatic scenarios. As reported in recent publications,9,16,19 the scarcity of experimentally determined γ values casts uncertainty about the effectiveness of a certain design strategy of compartmentalization. A synergistic study comparing experimentally determined γ values with the one from a theoretical model will offer valuable insights whether the designed cascade reaction is effective without undesirable side effects, ascertain the merits of designed compartmentalization, and further justify the design strategy of non-enzymatic compartmentalized systems. This research report aims to offer an exemplary case of such a study for organometallic catalysis with compartmentalized systems.
We are interested in employing microscopic concentration gradients within nanomaterials to design organometallic catalytic cycles of seemingly incompatible steps, in which key reaction intermediates will be quickly deactivated once they diffuse out of the nanomaterials. In our previous report,20 a cascade catalysis with O2-sensitive reaction intermediates was established to achieve ambient CH4-to-CH3OH conversion with O2 as the terminal oxidant assisted by electricity. As shown in Fig. 1B, the nanowire array electrode electrochemically reduces rhodium(III) tetramesityl porphyrin iodide (RhIII) into rhodium(II) tetramesityl porphyrin metalloradical (RhII) in aprotic solvent 1,2-difluorobenzene (1,2-DFB). At the same time, the nanowire array reduces O2 into reactive oxygen species (ROS) and creates a sharp O2 concentration gradient in a local O2-free environment near the bottom of the nanowire array under aerobic conditions. In the O2-free microenvironment, two equivalents of the generated RhII species, while highly reactive with O221, activate one equivalent of CH4 ambiently to yield methylated rhodium(III) tetramesityl porphyrin (RhIII–CH3) and rhodium(III) tetramesityl porphyrin hydride (RhIII–H). Both species are proposed to react with the generated ROS that remain to be further identified (vide infra), leading to the formation of CH3OH. While no barrier is present to physically separate the liquid phase within the nanowire array from the bulk solution, the whole system can be considered to be a compartmentalized cascade with the nanowire array as the compartment with its unique microenvironment (Fig. 1C), analogous to the previous argument in the case of two-enzyme cascades in which enzymes are placed in close proximity at the nano-scale.12,22 The overall process was reported to be catalytic, achieving a turnover number up to 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 within 24 hours.20 The synergy between nanomaterials and organometallic chemistry warrants a new catalytic route of CH4 functionalization, while additional studies are needed to understand the underlying mechanism and quantitatively evaluate the efficacy of the strategy that interfaces nanowires with organometallics.23
000 within 24 hours.20 The synergy between nanomaterials and organometallic chemistry warrants a new catalytic route of CH4 functionalization, while additional studies are needed to understand the underlying mechanism and quantitatively evaluate the efficacy of the strategy that interfaces nanowires with organometallics.23
Here we report our analysis of the above-mentioned CH4-to-CH3OH catalysis in the context of a compartmentalized cascade. We translated the reported catalytic system into a theoretical model that estimates the numerical value of γ. Electron paramagnetic resonance (EPR) spectroscopy with a spin-trap agent, along with other characterization experiments, unraveled that the predominant ROS present in the system is a superoxide and illustrated its role in CH3OH formation. This piece of mechanistic insight allowed us to subsequently determine the value of γ in the catalysis, which surprisingly amounts to more than 80%. The high value of measured γ is consistent with our theoretical framework and illustrates the efficacy of the created nanoscopic concentration gradient with minimal side reactions. Our results demonstrate that carefully designed compartmentalization, spatially controlling the occurrence of organometallic reactions in solution at the microscopic length scale, can circumvent undesirable reactions efficiently and create organometallic catalytic cycles impossible in homogeneous solutions.
Results and discussion
A theoretical framework of cascade reactions in a nanowire array electrode
Generally applicable for non-enzymatic cascade design as noted above, one question that we ask is whether our previously reported design employing the nanowire array and a concentration gradient is effective in utilizing the ephemeral O2-sensitive intermediate, RhII, whose activation of CH4 was considered turnover-limiting.20,24 Our approach to this question is to construct a numerical model in the context of compartmentalization and analyze the reaction efficiency γ that will be valuable for future design consideration. Fig. 1C shows the reported reaction in the context of compartmentalized cascade reactions. Here the compartment, schematically shown as a yellow-colored dashed box, is defined as the O2-free liquid phase within the nanowire array where RhII-initiated CH4 activation is proposed to take place. Following its definition,9,15γ is expressed as the ratio between the outward flux of RhIII–CH3 for CH3OH formation (Rp) and the rate of RhII generation (Rs) during the electrolysis. Based on the proposed reaction mechanism (Fig. 1B and C), Rp and Rs are dictated by the kinetic rate constant of electrochemical reduction of RhIII into RhII (k1) as well as the RhII-initiated CH4 activation (k2), respectively. We note that γ could also be interpreted as the competition between the rate of CH4 activation (Rp) in the compartment and the deactivation of RhII with O2 in the bulk, whose rate constant is denoted as ke. Because such a competition depends on the diffusive mass transport at a steady state, another important factor is the diffusive conductance F, as used in the design of enzymatic cascades,6,9,13 which describes the rate of mass transport for chemical species crossing into and out of the compartment boundary. However, in order to obtain the flux of a particular species in and out of the compartment, F must be normalized to the volume of the compartment V and Avogadro's number NA.9 We anticipate that F and V will be co-dependent, therefore we derive an expression for F/(VNA), denoted FV, in terms of the nanowire geometry (Section 1A of the ESI†). Since the RhII, RhIII, and RhIII–CH3 species share the bulky metalloporphyrin structure, as a first-order approximation FV is assumed to be the same among RhII, RhIII, and RhIII–CH3, and only dependent on the morphology of the nanowire array. Assuming ke → ∞ due to the reported rapid deactivation,21 we incorporated the above-mentioned components and derived a numerical model (Sections 1B of the ESI†), which describes γ in a compartmentalized cascade as shown below: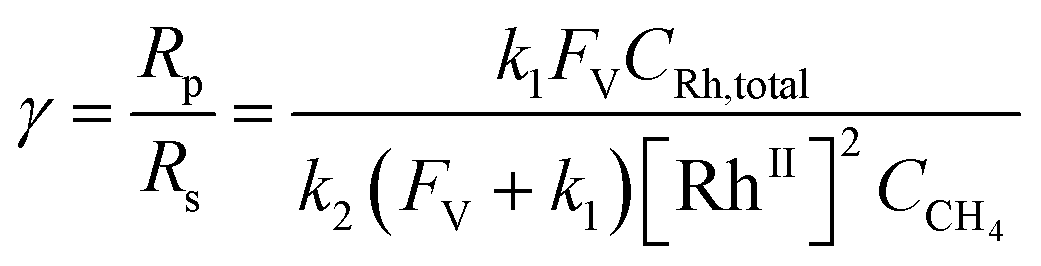 | (1) |
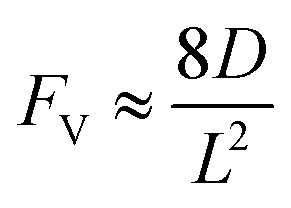 | (2) |
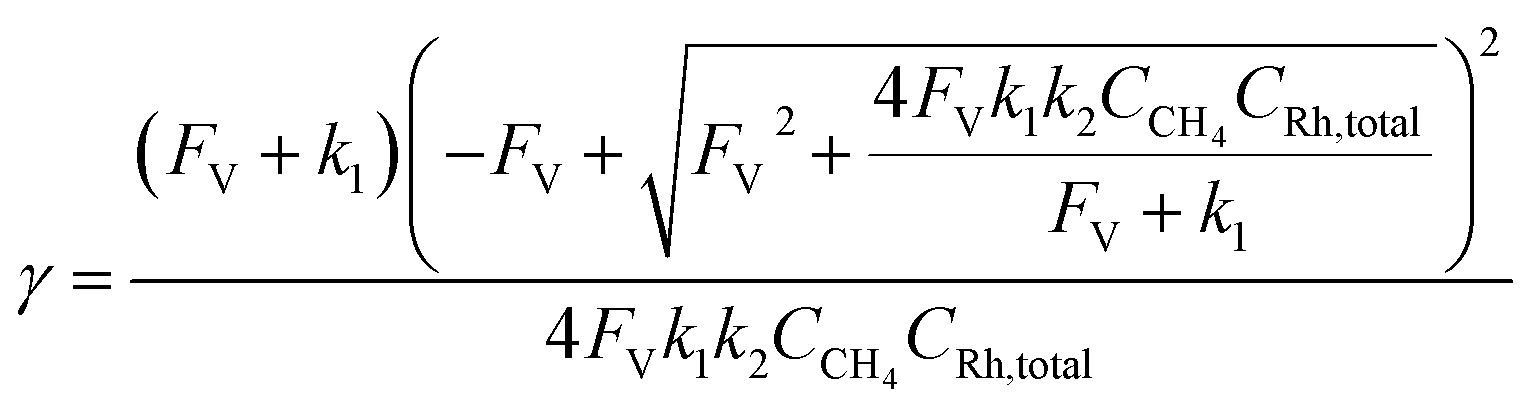 | (3) |
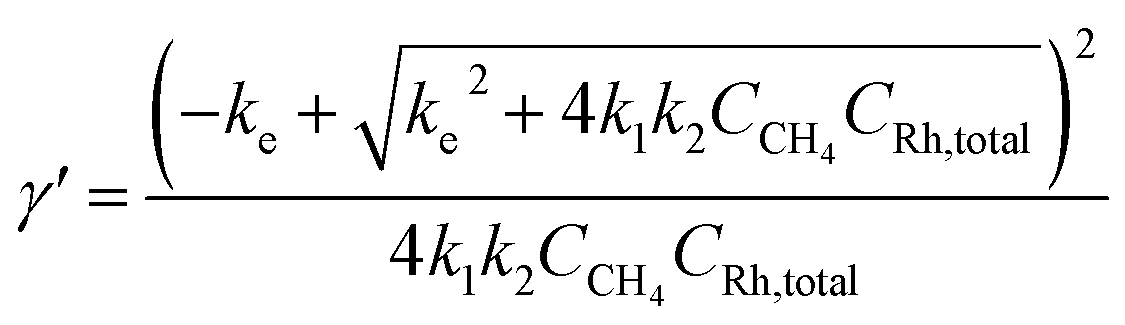 | (4) |
The established theoretical model demands input from experimental results. Because of the high reactivity between RhII and O2 outside of the O2-free microenvironment in the nanowire array,21i.e. ke → ∞, γ can be approximated as the ratio between the amount of generated CH3OH (NCH3OH) and RhII from electrochemical reduction of RhIII (NRh(II)) (Fig. 1C), if we presume that the conversion from RhIII–CH3 to CH3OH is stoichiometric as corroborated by using our prior experimental evidence.20 While NCH3OH is experimentally readily measurable as we have done before,20 the value of NRh(II) is less accessible and requires the quantification of ROS because the electrochemical reduction of RhIII into RhII is concurrent with the electrochemical reduction of O2 into the ROS (Fig. 1B). Therefore, identification and quantification of the ROS in this catalytic system is needed, not only for deeper insights about the chemical steps involved in the catalysis but also for a quantitative determination of γ.
Identification and quantification of reactive oxygen species in the catalysis
The identity of the predominant ROS was probed by EPR spectroscopy with the addition of spin trap agent 5,5-dimethyl-1-pyrroline N-oxide (DMPO) during electrolysis.25,26 Superoxide (O2˙−), hydrogen peroxide (H2O2), and hydroxyl radical (˙OH) were presumed to be the possible ROS from O2 reduction in aprotic solvent systems.27,28 Among these possible ROS, H2O2, along with other hydroperoxide species, react with DMPO to yield the adduct DMPO–OH˙, which can be detected by EPR spectroscopy.29 While O2˙− and ˙OH are short-lived,27,30 their reactions with DMPO yield more stable adducts DMPO–O2˙− and DMPO–OH˙, whose prolonged lifetimes are roughly 1 and 20 minutes, respectively, at room temperature and much longer under liquid N2 conditions.26 By trapping the generated ROS with DMPO during the electrolysis and discerning the trapped radicals in EPR spectroscopy26,31,32 the possible presence of O2˙− and H2O2/˙OH could be unveiled.EPR spectra indicate that O2˙− is the predominant ROS during the CH4-to-CH3OH conversion. As reported in our previous work (Section 3A in the ESI†),20 CH4-to-CH3OH catalysis was conducted in a customized single-chamber electrochemical reactor (Fig. S1†), which was fed with a mixture of CH4 and air (PCH4/Pair = 35) at a constant flow rate at ambient pressure and room temperature. Chronoamperometry of −1.4 V vs. the Saturated Calomel Electrode (SCE) was performed in 1,2-DFB with 1 mM of RhIII as the pre-catalyst and 0.1 M tetrabutylammonium perchlorate (TBAClO4) as the supporting electrolyte. A Si nanowire array with an average wire length of 15 μm and diameter of 50 nm (Fig. S2†), prepared based on the literature,20,33 was used as the working electrode with a Pt wire counter electrode and a Ag+/Ag pseudo-reference electrode equipped with a glass frit. 50 mM DMPO was added during the electrolysis to trap the generated ROS, and aliquots were taken for EPR experiments 5, 15, and 60 minutes after the addition of DMPO. Unless stated specifically, the aliquots were stored in liquid N2 before the EPR measurements, although the DMPO adduct has been observed to be stable at room temperature for at least 90 minutes (Fig. S3†). We note that the addition of DMPO per se does not significantly alter the electrochemistry in the system, if any, because previous studies have shown that DMPO is cathodically stable up to −2.35 V vs. SCE.34 The EPR spectrum from the aliquot 15 minutes after DMPO addition is shown in Fig. 2A, which is similar to the ones from the aliquot taken at 5 and 60 minutes after DMPO addition (Fig. S4†). This similarity suggests that the radical species observed in EPR spectroscopy is the predominant species in the steady state during the electrolysis and not all of the transient ROS involved in the catalytic cycle may be captured in our experiments. Control experiments include the DMPO-added electrolysis without the RhIII pre-catalyst (Fig. 2B), the reaction between DMPO and 0.5 mM KO2 as a surrogate of O2˙− (Fig. 2C and S5†), as well as the reaction between DMPO and 0.5 mM 2-hydroperoxypropan-2-ylbenzene (PhC(CH3)2OOH, cumene hydroperoxide) as a surrogate of H2O2/˙OH (Fig. 2D and S5†). The captured ROS during electrolysis is predominantly O2˙−, based on the similar spectra shown in Fig. 2A and C. Nonetheless, we note that the spectral pattern observed for the DMPO–O2˙− in 0.1 M TBAClO4 in 1,2-DFB solution is noticeably distinct from the DMPO–OOH observed in the aqueous solvent.25,26 This difference can be rationalized by the employment of 1,2-DFB in place of H2O and the resulting solvation sphere surrounding the adduct, the reduced proton concentration, and the possibility of TBA+ cations to coordinate the anionic complex, which could explain the increased stability of the observed adduct upwards of 90 min. The similarity between Fig. 2A and B suggests that it is likely that the electrochemistry of Si nanowires rather than the Rh species is responsible for the generation of O2˙−.
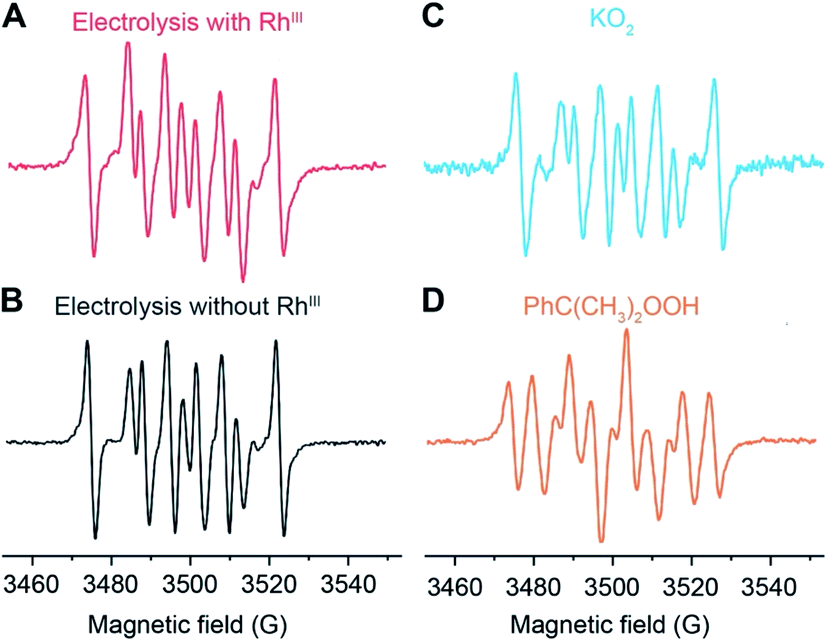 | ||
| Fig. 2 Electron paramagnetic resonance (EPR) spectra depicting the adducts formed in the reaction of 50 mM DMPO (5,5-dimethyl-1-pyrroline N-oxide): (A) the electrolysis with the Rh catalyst during CH4-to-CH3OH conversion; (B) control electrolysis in the absence of the Rh catalyst; (C) 0.5 mM potassium superoxide (KO2); (D) 0.5 mM 2-hydroperoxypropan-2-ylbenzene (PhC(CH3)2OOH, cumene hydroperoxide). Unless otherwise noted, the electrolyte solution is 0.1 M tetrabutylammonium perchlorate (TBAClO4) in 1,2-difluorobenzene (1,2-DFB). Following previously reported conditions,20 chronoamperometry was conducted at −1.4 V vs. SCE with the Si nanowire working electrode under a constant flow of a CH4/air mixture PCH4/Pair = 35 in a customized electrochemical reactor (Fig. S1†) under ambient pressure. | ||
We propose to quantify the generated ROS, predominantly O2˙− at the steady state, by colorimetric assay with the use of nitroblue tetrazolium (NBT) chloride as a O2˙−-selective chromogen. NBT is reported to selectively react with the O2˙− over H2O2 and other ROS,35 leading to the emergence of a purple color in monoformazan, with a maximum absorption peak at ∼530 nm,35,36 from a pale yellow background. The stoichiometric reactivity between NBT and O2˙− (1:2), ensured by utilizing a concentration of NBT/O2˙− < 2, enables the use of UV-Vis absorption spectroscopy for the quantification of O2˙− generation.35,37–39 Additionally, NBT exhibits minimal reductive activity in organic solvent within its electrochemical window, which was not only reported in the previous literature36 but also shown in our cyclic voltammograms (Fig. S6†) and chronoamperometry (Fig. 3A) on a glassy carbon working electrode in 0.1 M TBAClO4 in 1,2-DFB solution. Thus, when added during the electrolysis under CH4-to-CH3OH conditions, NBT will have minimal interference with the O2-reducing electrode and act as an O2˙− scavenger for a colorimetric quantification of the accumulated O2˙−. While multiple methods including fluorescence measurements are viable for the O2˙− quantification,38 the absorbance at 600 nm from monoformazan after the reaction between NBT and O2˙− (ref. 36) was chosen in order to mitigate interference from the optical absorbance and phosphorescence of Rh species (630–750 nm).40 Furthermore, due to the ephemeral nature of O2˙− that hinders the preparation of a standard solution of known O2˙− concentration,35,41 we specifically designed experiments that establish a calibration curve of the absorbance at 600 nm which accounts for the stoichiometric reaction between NBT and KO2, the O2˙− surrogate.35 When an increasing concentration of NBT was mixed with a fixed concentration of KO2 in 1,2-DFB, the absorbance at 600 nm follows a linear correlation before plateauing (Fig. 3B), illustrating a stoichiometric reaction between NBT and O2˙− without other chromogenic side reactions. This led us to determine the absorption coefficient at 600 nm of the yielded monoformazan, ε600 nm = 4327 M−1 cm−1 (R2 = 0.97) (Section 3B of the ESI†). A similar linear response between NBT and O2˙− was also observed in the presence of Rh catalysts in 1,2-DFB (Fig. S7†), which suggests that the presence of RhIII in the bulk solution does not interfere with the colorimetric assay.
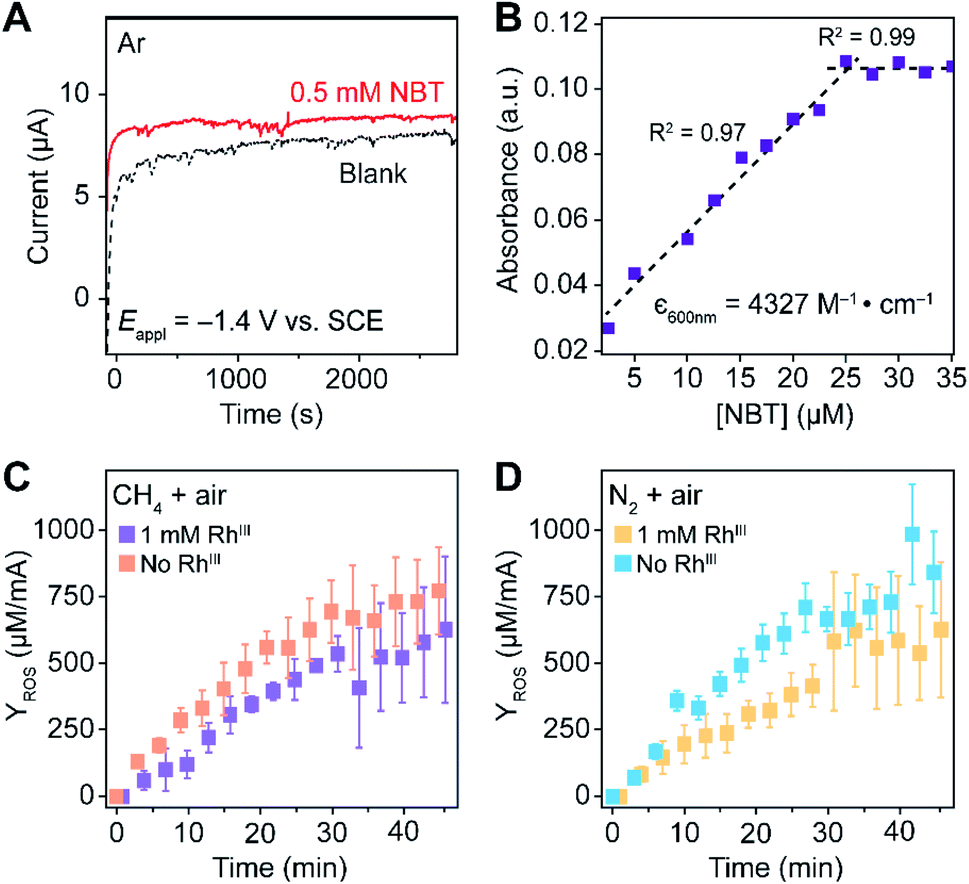 | ||
| Fig. 3 Quantification of superoxide (O2˙−) with O2˙−-selective chromogen nitroblue tetrazolium (NBT). (A) Chronoamperometry with a glassy carbon working electrode in Ar. (B) The absorbance at 600 nm in 1,2-DFB of a fixed concentration of KO2, a surrogate of ephemeral O2˙−, for varying concentrations of NBT. ε600nm, the established absorption coefficient at 600 nm of the yielded monoformazan from a stoichiometric reaction between NBT and O2˙−.35 (C and D) The yield of ROS (YROS), determined as the detected O2˙− normalized by the average electric current, is displayed against electrolysis duration with a Si nanowire array of 15 μm length as the working electrode. (C) A CH4/air atmosphere (PCH4/Pair = 35) with (purple, n = 4) and without (orange, n = 3) 1 mM RhIII. (D) N2/air atmosphere (PN2/Pair = 35) with (yellow, n = 5) and without (blue, n = 5) 1 mM RhIII. −1.4 V vs. SCE 0.1 M TBAClO4 in 1,2-DFB. Error bars denote standard deviations. | ||
The rate of O2˙− generation was quantified with the use of NBT in the nanowire-based cascade system for CH4-to-CH3OH conversion. Under the same electrochemical conditions with the use of the Si nanowire array electrode of 15 μm length at −1.4 V vs. SCE, aliquots of the electrolyte solution were sequentially sampled, measured for optical absorbance at 600 nm, and applied to calculate the amount of accumulated O2˙− (Section 3C of the ESI†). The yields of the net accumulated O2˙− (YROS), normalized by the average electric current during electrolysis, were shown as a function of electrolysis duration in CH4/air and N2/air atmospheres (PCH4/Pair and PN2/Pair = 35; Fig. 3C and D, respectively). The initial slopes of the YROS before plateauing (∂YROS/∂t) were determined as the rate of electrochemical ROS generation in the compartmentalized cascade reaction. ∂YROS/∂t = 18 ± 4 and 22 ± 4 μM mA−1˙min−1 in the CH4/O2 atmosphere with the presence and absence of 1 mM RhIII (n = 4 and n = 3; purple and orange traces in Fig. 3C, respectively); ∂YROS/∂t = 16 ± 4 and 23 ± 3 μM mA−1˙min−1 in the N2/air with the presence and absence of 1 mM RhIII (n = 5; yellow and blue traces in Fig. 3D, respectively). The similar values of ∂YROS/∂t between the CH4 and N2 atmosphere suggests that the ROS formation is independent of the gaseous environment. The similar values of ∂YROS/∂t with and without RhIII suggests that the electrode surface of Si nanowires is primarily responsible for ROS generation, albeit the presence of RhIII does seem to lower the ROS yield. Here we note that ∂YROS/∂t could be underestimated, because NBT is less reactive towards other ROS that may be concurrently generated during the electrochemical process.
Experimental determination and analysis of reaction efficiency γ
Our successful quantification of the electrochemical ROS generation rate (∂YROS/∂t) led to an experimentally determined value of reaction efficiency γ. As the reductive current is responsible for the generation of both ROS and CH4-activating RhII, the equation for γ in Fig. 1C can be written as: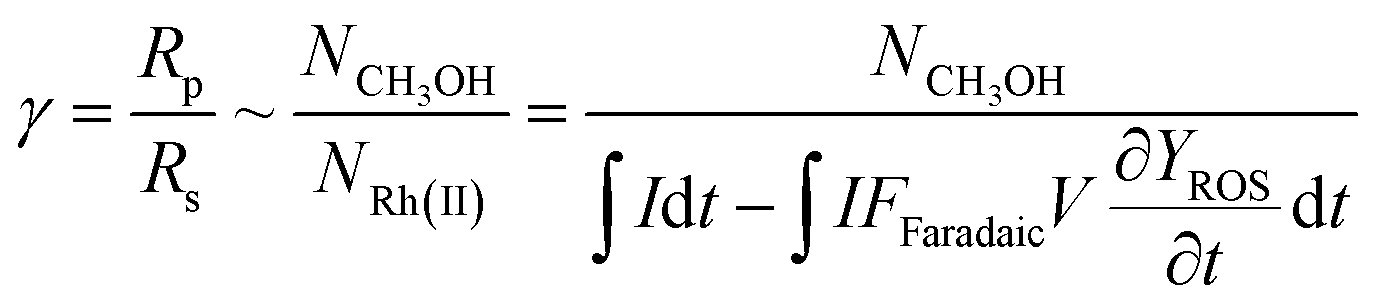 | (5) |
In an effort to compare our experimentally determined γ values with the theoretical maximum, we numerically calculated the values of γ based on the established theoretical framework for a nanowire-based compartmentalization. Following the model displayed in Fig. 1C, the calculated values of Rs (Fig. 4A and D), Rp (Fig. 4B and E), and the RhII flux diffusing out of the compartment Ri (Fig. 4C and F) were displayed as a function of k1 and FV (Section 1B of the ESI†). These calculations use ke → ∞, CCH4 = 9.5 mM,20CRh,total = 1.0 mM, and k2 = 2.9 × 104 M−2 s−1 for RhII-initiated CH4 activation within the nanowire array as determined in our previous report.20 Because the rate of electrochemical reduction of RhIII (k1) was not readily determinable in our system, k1 was assigned as a range of values spanning four orders of magnitude (106 to 1010 s−1) based on a typical range reported for electron transfer of metalloporphyrin complexes in the literature.42 Nonetheless, Fig. 4 shows that the effect of compartmentalization is not sensitive to the value of k1, evident as the k1 term cancels out in eqn (S14), (S18), and (S19)† during the derivation as long as k1 is sufficiently larger (≥106 s−1) than the value of FV, with an increased FV value leading to increased fluxes both outward and inward of the compartment. Because the value of FV is dependent on the nanowire array's morphology, such a trend suggests that we can control and possibly optimize the mass transport across the compartment by controlling the length L of the nanowire array based on eqn (2).
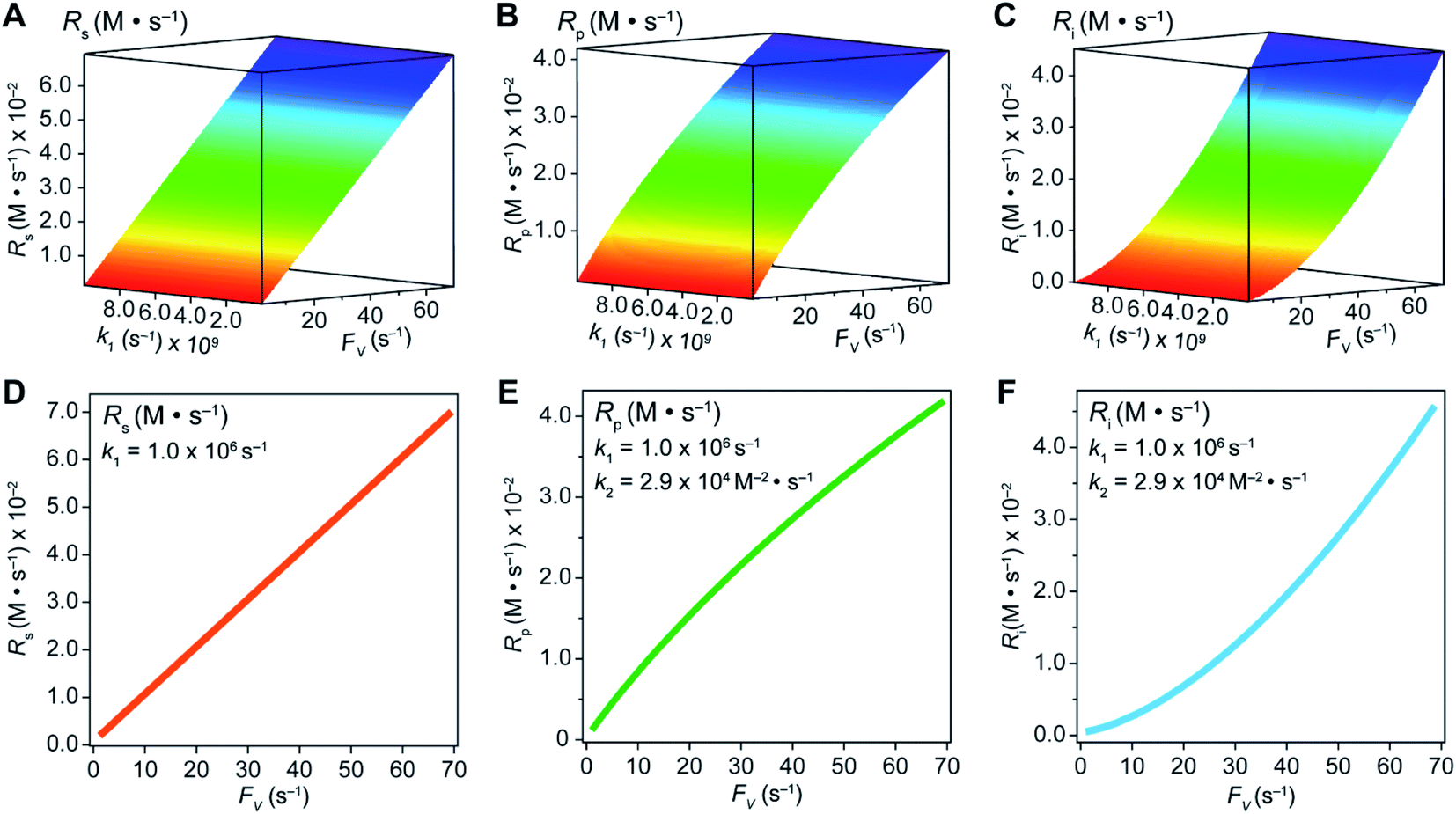 | ||
| Fig. 4 Graphical representations of substrate conversion (Rs), product formation (Rp), and intermediate outflux (Ri) in the compartmentalized system. Panels (A), (B), and (C) represent Rs, Rp, and Ri, respectively, as a function of both FV and k1. Panels (D), (E), and (F) represent Rs, Rp, and Ri, respectively, as a function of FV for a constant value of k1 (1.0 ×106 s−1). k2 was experimentally determined to be 2.9 × 104 M−2 s−1 within the nanowire array in our previous report.20 | ||
A comparison between the experimental value of γ with simulation results suggests that the Rh-based organometallic catalysis enabled by the nanowire array is indeed functioning near its theoretical limit. Given the strong dependence on the value of FV for Rs, Rp, and Ri, we envisioned that γ is mostly a function of FV, whose relationship is displayed as the red trace in Fig. 5. While Rs, Rp, and Ri all increase with larger values of FV (vide supra), overall a larger FV value tends to decrease the value of γ (Fig. 5). Such a trend is corroborated with our previous observation that a nanowire array of longer length L, hence smaller FV value based on eqn (2), corresponds to a larger yield of CH3OH before mass transport becomes rate-limiting.20 One thing to note is that our developed theoretical framework does not account for those RhIII molecules that enter and leave the nanowire compartment without undergoing the initial electrochemical reduction reaction. However, the alignment of the experimental and theoretical values indicates that this phenomenon, while it cannot be excluded completely, remains minimal in our system. In comparison, the value of γ for a non-compartmentalized scenario, i.e. homogeneous solution without a nanostructured electrode, is also shown as the black trace in Fig. 5 (Section 1C in the ESI†). A clear difference is observable as the value of γ for the non-compartmentalized scenario is virtually near zero (at most 0.001% indeed), in line with our previous experimental work that utilized a planar electrode surface and resulted in no CH3OH generation.20 We also positioned our experimentally determined γ value in Fig. 5, after we determined FV = 20. s−1 given L = 15 μm and D = 5.6 × 10−10 m2 s−1 in 1,2-DFB as measured before.20 The error bar in Fig. 5 represents the approximation incurred when deriving the expression for FV (eqn (2)) (Section 1A in the ESI†). A good agreement between the experimental and theoretical values of γ for the nanocompartment was observed (Fig. 5), yet the slightly higher value of experimentally derived γ might originate from the underestimation of ∂YROS/∂t (vide supra). The agreement indicates that minimal unforeseen side reactions, if any, are present in the catalyst system and the proposed benefit of a nanowire-generated O2-free microenvironment for RhII-initiated CH4 activation is well demonstrated.
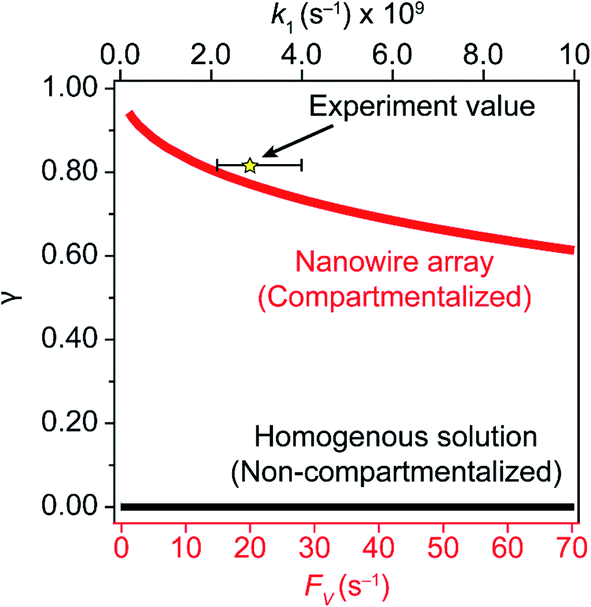 | ||
| Fig. 5 Reaction efficiency γ plotted as a function of diffusive conductance FV for the scenarios with nanowire-enabled compartmentalization (red trace) and γ plotted as a function of k1 without nanowire-enabled compartmentalization (black trace). The experimentally determined data point of γ for nanowire array of 15 μm length is represented by the star. The error bar presents the approximation when deriving the experimental value of FV (Section 1A in the ESI†). | ||
Despite the findings that O2˙− is the dominant ROS at steady state, we propose that O2˙− is not the immediate oxidant that reacts with RhIII–CH3 for CH3OH formation. We found that under strictly dry conditions, no CH3OH was observed in a mixture of KO2 and the as-synthesized RhIII–CH3. Yet a stoichiometric amount of CH3OH, calculated using 1H NMR, was observed in experiments with RhIII–CH3 and KO2 “wet” in 1,2-DFB (5.1 ± 0.3 mM H2O based on Karl Fischer titration). Along the same lines, 1:1 reactivity was observed upon mixing RhIII–CH3 with hydroperoxide species such as cumene hydroperoxide or t-butyl hydroperoxide in dry 1,2-DFB (Section 4 of the ESI†).20 Such observations prompted us to propose that trace hydroperoxide, namely H2O2, is constantly generated during the electrolysis and is the immediate reactant towards RhIII–CH3 to afford CH3OH as the product. We speculate that the electrochemical reduction of O2 to O2˙− is followed by a chemical protonation step to generate other ROS such as a hydroperoxyl radical (HO2˙),43 which is susceptible to heterolytic and homolytic cleavages and eventually converges on H2O2.44–46 Whether the conversion of HO2˙ to H2O2 proceeds electrochemically or chemically should be dependent on the proton donor concentration in solution and the reduction potential applied.46 Once the hydroperoxide species is formed, CH3OH formation proceeds from its reaction with RhIII–CH3. Such mechanistic consideration would be useful in designing other ROS-initiated CH4-activation reactions.
Additional design insights are available from our results. We examined the dependence of γ on different hypothetical values of the CH4-activation rate constant k2. Smaller values of k2 significantly lower γ, i.e. the efficiency of compartmentalization created by the nanowire array (Fig. S8†). Such a trend is reasonable since a lower value of k2 will lead to more prominent outward flux Ri of the unreacted RhII species which will introduce a larger percentage of undesirable RhII deactivation. Therefore, while our Rh-based catalysis demonstrates the benefits of compartmentalization by constructing a catalytic cycle of seemingly incompatible steps for organometallics, we caution that the efficacy of this strategy depends on the specific chemical system under consideration. As the value of γ depends on FV and thus the nanostructure's morphology, such as nanowire length L in our case, there exists an optimal morphology of nanostructures for specific organometallic compounds' reactivities in order to create an effective microenvironment and efficient cascade reactions with minimal detrimental deactivation. A quantitative evaluation of reaction efficiency γ, however estimated, is recommended in order to justify the introduction of nanostructures and a micro-environment. A general numerical design framework of nanostructures for a typical organometallic catalytic cycle that includes oxidative addition and reductive elimination is currently being developed in the authors' laboratory.
Conclusion
In this work, we applied the concept of reaction efficiency γ in biochemical cascades to quantitatively evaluate the efficacy of compartmentalization for organometallic reactions with the use of nanowire array electrodes. A high γ value approaching unity, the theoretical limit, was experimentally observed, suggesting minimal detrimental side reactions. This observation indicates that with suitable design it is possible to employ nanomaterials to spatially control organometallic reactions and achieve efficient cascade with ephemeral intermediates, analogous to the biological counterparts with near-unity γ values. This work quantitatively highlights the transformative power of spatial control at the nanoscale for new chemical reactivity.Author's contribution
C. L. supervised the project. C. L. and B. S. N. designed experiments and wrote the paper. B. S. N. synthesized the compounds and conducted electrochemical characterization experiments with assistance from D. M. D. B. J. J. performed the mathematical derivations on the compartmentalized system. All the authors discussed the results and assisted during the manuscript preparation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to acknowledge Paul Ayala at the California Institute of Technology for the use of the EPR spectrometer. We thank the Molecular Instrumentation Center lab at the University of California, Los Angeles for sample characterization experiments. C. L. acknowledges the NSF Award (CHE-2027330), the startup fund from the University of California, Los Angeles, and the financial support of the Jeffery and Helo Zink Endowed Professional Development Term Chair.References
- J. Shi, Y. Wu, S. Zhang, Y. Tian, D. Yang and Z. Jiang, Bioinspired construction of multi-enzyme catalytic systems, Chem. Soc. Rev., 2018, 47, 4295–4313 RSC.
- M. Vázquez-González, C. Wang and I. Willner, Biocatalytic cascades operating on macromolecular scaffolds and in confined environments, Nat. Catal., 2020, 3, 256–273 CrossRef.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, Engineering Compartmentalized Biomimetic Micro- and Nanocontainers, ACS Nano, 2017, 11, 6549–6565 CrossRef CAS PubMed.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Mimicking the Cell: Bio-Inspired Functions of Supramolecular Assemblies, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS PubMed.
- J.-L. Lin, L. Palomec and I. Wheeldon, Design and Analysis of Enhanced Catalysis in Scaffolded Multienzyme Cascade Reactions, ACS Catal., 2014, 4, 505–511 CrossRef CAS.
- F. Hinzpeter, U. Gerland and F. Tostevin, Optimal Compartmentalization Strategies for Metabolic Microcompartments, Biophys. J., 2017, 112, 767–779 CrossRef CAS.
- P. B. Weisz, Diffusion and Chemical Transformation, Science, 1973, 179, 433–440 CrossRef CAS PubMed.
- P. B. Weisz, Stepwise Reaction via Intermediates on Separate Catalytic Centers, Science, 1956, 123, 887–888 CrossRef CAS PubMed.
- S. Tsitkov and H. Hess, Design Principles for a Compartmentalized Enzyme Cascade Reaction, ACS Catal., 2019, 9, 2432–2439 CrossRef CAS.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Enzyme cascades activated on topologically programmed DNA scaffolds, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS.
- S.-H. Park, A. Zarrinpar and W. A. Lim, Rewiring MAP Kinase Pathways Using Alternative Scaffold Assembly Mechanisms, Science, 2003, 299, 1061–1064 CrossRef CAS PubMed.
- O. Idan and H. Hess, Origins of Activity Enhancement in Enzyme Cascades on Scaffolds, ACS Nano, 2013, 7, 8658–8665 CrossRef CAS PubMed.
- Y. Zhang and H. Hess, Toward Rational Design of High-efficiency Enzyme Cascades, ACS Catal., 2017, 7, 6018–6027 CrossRef CAS.
- M. Castellana, M. Z. Wilson, Y. Xu, P. Joshi, I. M. Cristea, J. D. Rabinowitz, Z. Gitai and N. S. Wingreen, Enzyme clustering accelerates processing of intermediates through metabolic channeling, Nat. Biotechnol., 2014, 32, 1011–1018 CrossRef CAS PubMed.
- K. S. Chavan and S. Calabrese Barton, Simulation of Intermediate Channeling by Nanoscale Confinement, J. Phys. Chem. C, 2018, 122, 14474–14480 CrossRef CAS.
- W.-H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Biocatalytic cascades driven by enzymes encapsulated in metal–organic framework nanoparticles, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- Y. Ikezoe, G. Washino, T. Uemura, S. Kitagawa and H. Matsui, Autonomous motors of a metal–organic framework powered by reorganization of self-assembled peptides at interfaces, Nat. Mater., 2012, 11, 1081–1085 CrossRef CAS.
- R. Riccò, W. Liang, S. Li, J. J. Gassensmith, F. Caruso, C. Doonan and P. Falcaro, Metal–Organic Frameworks for Cell and Virus Biology: A Perspective, ACS Nano, 2018, 12, 13–23 CrossRef PubMed.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Substrate channelling as an approach to cascade reactions, Nat. Chem., 2016, 8, 299–309 CrossRef CAS PubMed.
- B. S. Natinsky, S. Lu, E. D. Copeland, J. C. Quintana and C. Liu, Solution Catalytic Cycle of Incompatible Steps for Ambient Air Oxidation of Methane to Methanol, ACS Cent. Sci., 2019, 5, 1584–1590 CrossRef CAS PubMed.
- W. Cui and B. B. Wayland, Superoxo, Peroxo, and Hydroperoxo Complexes Formed from Reactions of Rhodium Porphyrins with Dioxygen: Thermodynamics and Kinetics, J. Am. Chem. Soc., 2006, 128, 10350–10351 CrossRef CAS.
- O. Idan and H. Hess, Diffusive transport phenomena in artificial enzyme cascades on scaffolds, Nat. Nanotechnol., 2012, 7, 769–770 CrossRef CAS PubMed.
- L. A. Berben and N. D. Loewen, Control of Substrates Beyond the Catalyst Active Site, ACS Cent. Sci., 2019, 5, 1485–1487 CrossRef CAS PubMed.
- B. B. Wayland, S. Ba and A. E. Sherry, Activation of methane and toluene by rhodium(II) porphyrin complexes, J. Am. Chem. Soc., 1991, 113, 5305–5311 CrossRef CAS.
- G. R. Buettner and L. W. Oberley, Considerations in the spin trapping of superoxide and hydroxyl radical in aqueous systems using 5,5-dimethyl-1-pyrroline-1-oxide, Biochem. Biophys. Res. Commun., 1978, 83, 69–74 CrossRef CAS PubMed.
- K. Makino, T. Hagiwara and A. Murakami, A mini review: Fundamental aspects of spin trapping with DMPO, Int. J. Radiat. Appl. Instrum. C Radiat Phys. Chem., 1991, 37, 657–665 CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide Ion: Generation and Chemical Implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Metal-Free Catalysts for Oxygen Reduction Reaction, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- L. Chen, S. Kutsuna, S. Yamane and J. Mizukado, ESR spin trapping determination of the hydroperoxide concentration in polyethylene oxide (PEO) in aqueous solution, Polym. Degrad. Stab., 2017, 139, 89–96 CrossRef CAS.
- F.-J. Schmitt, G. Renger, T. Friedrich, V. D. Kreslavski, S. K. Zharmukhamedov, D. A. Los, V. V. Kuznetsov and S. I. Allakhverdiev, Reactive oxygen species: Re-evaluation of generation, monitoring and role in stress-signaling in phototrophic organisms, Biochim. Biophys. Acta Bioenerg., 2014, 1837, 835–848 CrossRef CAS PubMed.
- G. R. Buettner, The spin trapping of superoxide and hydroxyl free radicals with DMPO (5,5-dimethylpyrroline-N-oxide): more about iron, Free Radic. Res. Commun., 1993, 19, S79–S87 CrossRef CAS PubMed.
- E. Finkelstein, G. M. Rosen and E. J. Rauckman, Spin trapping of superoxide and hydroxyl radical: Practical aspects, Arch. Biochem. Biophys., 1980, 200, 1–16 CrossRef CAS.
- Z. Huang, N. Geyer, P. Werner, J. de Boor and U. Gösele, Metal-Assisted Chemical Etching of Silicon: A Review, Adv. Mater., 2011, 23, 285–308 CrossRef CAS.
- G. L. McIntire, H. N. Blount, H. J. Stronks, R. V. Shetty and E. G. Janzen, Spin trapping in electrochemistry. 2. Aqueous and nonaqueous electrochemical characterizations of spin traps, J. Phys. Chem., 1980, 84, 916–921 CrossRef CAS.
- R.-h. Liu, S.-y. Fu, H.-y. Zhan and L. A. Lucia, General Spectroscopic Protocol to Obtain the Concentration of the Superoxide Anion Radical, Ind. Eng. Chem. Res., 2009, 48, 9331–9334 CrossRef CAS.
- D. Hirshberg, D. Sharon, M. Afri, R. Lavi, A. A. Frimer, N. Metoki, N. Eliaz, W.-j. Kwak, Y.-K. Sun and D. Aurbach, Shedding Light on the Oxygen Reduction Reaction Mechanism in Ether-Based Electrolyte Solutions: A Study Using Operando UV–Vis Spectroscopy, ACS Appl. Mater. Interfaces, 2018, 10, 10860–10869 CrossRef CAS PubMed.
- H. S. Choi, J. W. Kim, Y. N. Cha and C. Kim, A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells, J. Immunoassay Immunochem., 2006, 27, 31–44 CrossRef CAS.
- K. K. Griendling, R. M. Touyz, J. L. Zweier, S. Dikalov, W. Chilian, Y.-R. Chen, D. G. Harrison and A. Bhatnagar, Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association, Circ. Res., 2016, 119, e39–e75 CrossRef CAS PubMed.
- C. Beauchamp and I. Fridovich, Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS.
- S. E. Vitols, D. A. Friesen, D. S. Williams, D. Melamed and T. G. Spiro, Excited State Dynamics of Rh(II) Tetramesityl Porphyrin Monomer from Nanosecond Transient Absorption and Emission Spectroscopy, J. Phys. Chem., 1996, 100, 207–213 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, How super is superoxide?, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Elsevier Inc., 2003, Vol. 16 Search PubMed.
- Y. Che, M. Tsushima, F. Matsumoto, T. Okajima, K. Tokuda and T. Ohsaka, Water-Induced Disproportionation of Superoxide Ion in Aprotic Solvents, J. Phys. Chem., 1996, 100, 20134–20137 CrossRef CAS.
- D. T. Sawyer, M. S. McDowell and K. S. Yamaguchi, Reactivity of perhydroxyl (HOO˙) with 1,4-cyclohexadiene (model for allylic groups in biomembranes), Chem. Res. Toxicol., 1988, 1, 97–100 Search PubMed.
- I. B. Afanas′ev and N. S. Kuprianova, Kinetics and mechanism of the reactions of the superoxide ion in solutions. II. The kinetics of protonation of the superoxide ion by water and ethanol, Int. J. Chem. Kinet., 1983, 15, 1057–1062 CrossRef.
- P. Cofre and D. T. Sawyer, Electrochemical reduction of dioxygen to perhydroxyl (HOO˙) in aprotic solvents that contain Broensted acids, Anal. Chem., 1986, 58, 1057–1062 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, synthetic procedures, electrochemical characterization, mathematical derivations, and additional figures. See DOI: 10.1039/d0sc05700b |
| ‡ Equal contributions of authorship. |
| This journal is © The Royal Society of Chemistry 2021 |