
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective transition metal catalysis directed by chiral cations
Xinyi
Ye
*a and
Choon-Hong
Tan
*b
aCollege of Pharmaceutical Science & Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou 310014, P. R. China. E-mail: xinyiye1020@zjut.edu.cn
bDivision of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: choonhong@ntu.edu.sg
First published on 21st December 2020
Abstract
Enantioselective transition metal catalysis directed by chiral cations is the amalgamation of chiral cation catalysis and organometallic catalysis. Thus far, three strategies have been revealed: ligand scaffolds incorporated on chiral cations, chiral cations paired with transition metal ‘ate’-type complexes, and ligand scaffolds incorporated on achiral anions. Chiral cation ion-pair catalysis has been successfully applied to alkylation, cycloaddition, dihydroxylation, oxohydroxylation, sulfoxidation, epoxidation and C–H borylation. This development represents an effective approach to promote the cooperation between chiral cations and transition metals, increasing the versatility and capability of both these forms of catalysts. In this review, we present current examples of the three strategies and suggest possible inclusions for the future.
1. Introduction
In some chemical reactions, charged intermediates evolve and transform to neutral state target molecules. In order to overcome the energy barrier of such chemical reactions, using a suitable catalyst is the most straightforward and effective strategy. An ion-pair catalyst can be utilised to stabilise charged intermediates through ionic interactions, thereby allowing these reactions to proceed smoothly.1,2 Chiral cations such as quaternary ammonium, guanidinium and quaternary phosphonium have been developed to control anionic intermediates2a while chiral anions such as borate and phosphate are utilised to control various cationic intermediates.2bIn chiral cation catalysis, the catalyst is paired with an anionic intermediate, usually an enolate resulting from proton abstraction of a reactant by an inorganic base. For example, reactions such as alkylation, Michael addition, Aldol reaction and Mannich reaction have enolate intermediates, and can be efficiently promoted using chiral cationic phase transfer catalysts.1b Other anions such as cyanide and fluoride can also be activated for cyanation and fluorination respectively using this approach. However, reactions involving reactants in a neutral electronic state or which are inert to inorganic bases cannot be catalysed using chiral cationic catalysts. Therefore, developing strategies to circumvent this weakness is eagerly anticipated in order to expand the scope of chiral cation catalysis. In order to activate allylic acetates for addition with glycinate Schiff base, Gong3a and Takemoto3b,c added palladium complexes in the presence of chiral quaternary ammonium salts. Similar asymmetric allylations were also undertaken by Takemoto3d,e and Han3f using iridium complexes. While none of these reports have extensive mechanistic investigations, it was proposed that transition metals function to activate the electrophile in an independent catalytic cycle and work cooperatively with the chiral cation catalyst during the reaction pathway. The hypothesis since then is that if it is possible to allow organometallic catalysis to work synergistically through ion-pair interactions with chiral cations, new chemistry can be developed.2c,d Although this new approach is still in its infancy, three main categories have been identified; they are: (i) transition metal associated with a ligand scaffold incorporated on a chiral cation; (ii) chiral cation is paired with transition metal ‘ate’ complexes; and (iii) chiral cation paired with a transition metal, which is associated with an achiral anion containing a ligand scaffold (Fig. 1).
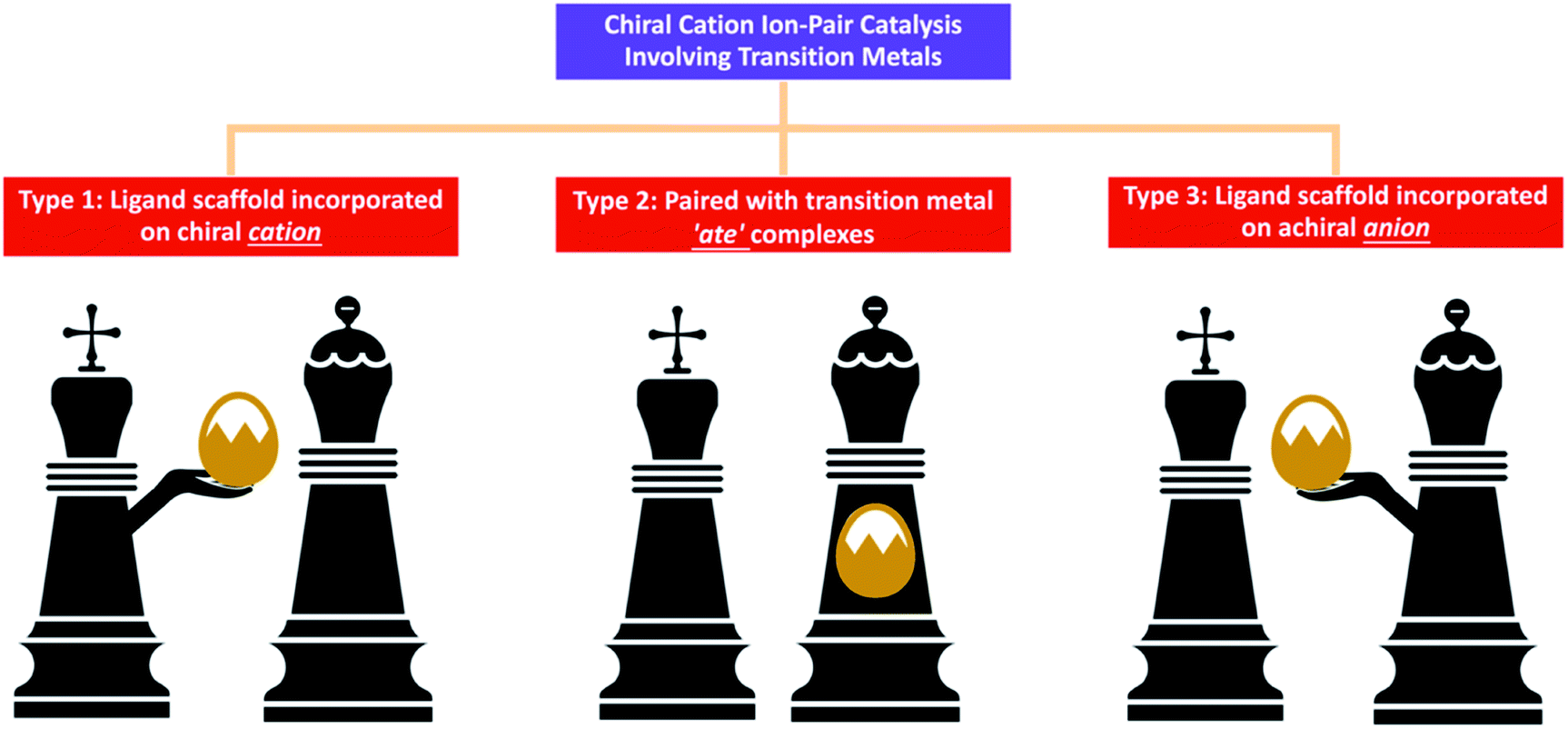 | ||
| Fig. 1 Main strategies for incorporating organometallic complexes in chiral cationic ion-pair catalysis (king chess piece represents the chiral cation, queen chess piece represents the anion, and golden egg represents the metal complex). | ||
2. Strategy 1: ligand scaffold incorporated on a chiral cation
In 2014, Ooi and co-workers placed a phosphine on a side chain of a quaternary ammonium salt, allowing the cation to be an excellent ligand to Pd0 (Scheme 1).4 This novel cationic palladium complex achieved [3 + 2] cycloaddition between 5-vinyloxazolidinones and activated alkenes.4a A similar strategy was utilized in the addition of 5-vinyloxazolidinone to an imine.4b The authors hypothesised that the transition state consisted of a zwitterionic π-allylpalladium intermediate paired with quaternary ammonium (Scheme 1). Incorporation of a quaternary ammonium halide component allows both desirable halide–palladium contact and recognition of the anionic site through facile pairing with the chiral ammonium ion.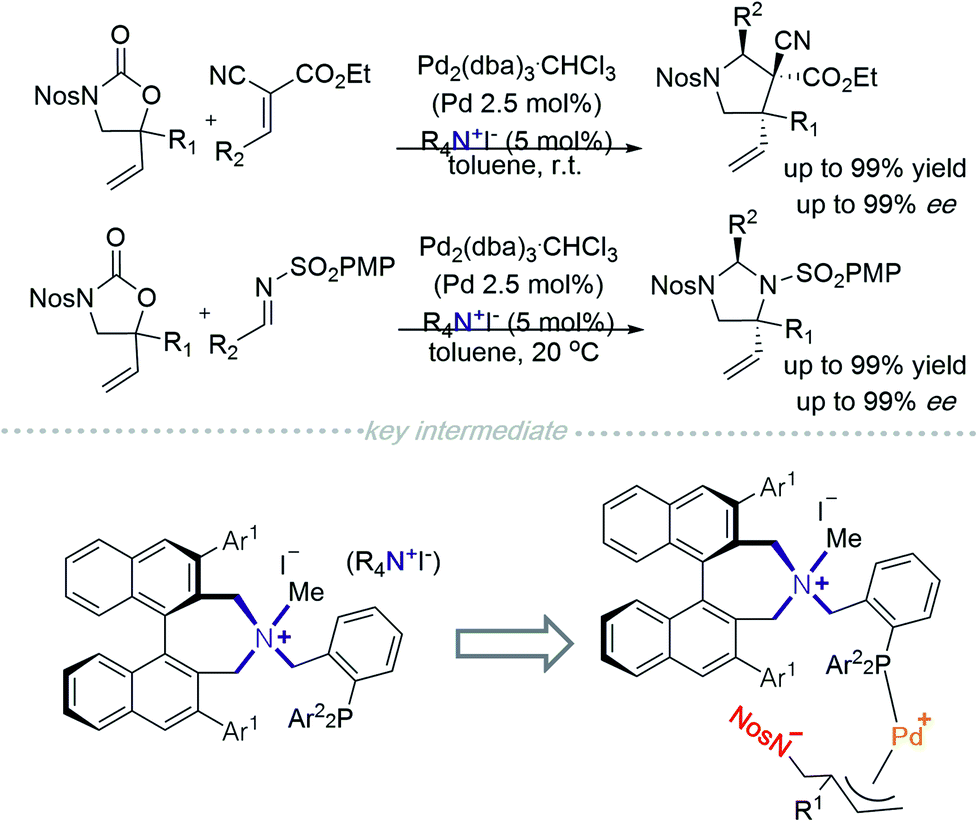 | ||
| Scheme 1 Pd0/Phosphine scaffold incorporated quaternary ammonium-catalysed asymmetric [3 + 2] cycloaddition. | ||
In 2015, the Peters group designed a multi-functional catalyst for diastereodivergent asymmetric 1,4-addition between oxindoles and nitroolefins (Scheme 2).5 The polyfunctional catalyst consisted of a NiII-bis(polyoxyimine) motif with free hydroxyl groups and an axially chiral bis-imidazolium moiety as a chiral linker. Multiple non-covalent interactions were achieved through the bis-imidazolium cation, which provides electrostatic interactions, C–H hydrogen-bond donors and π-interactions. A NiII/Schiff-base complex functioned as a Lewis acid/Brønsted base and free hydroxyl groups allowed hydrogen-bonding. Each of the above factors (including the counterions) was evaluated by a series of control experiments. The authors proposed that the two reactants were controlled separately; one was activated by the chiral cation, and the other by the metallic complex. At this moment, it is still unclear as to which non-covalent interaction is responsible for the stereochemical outcomes.
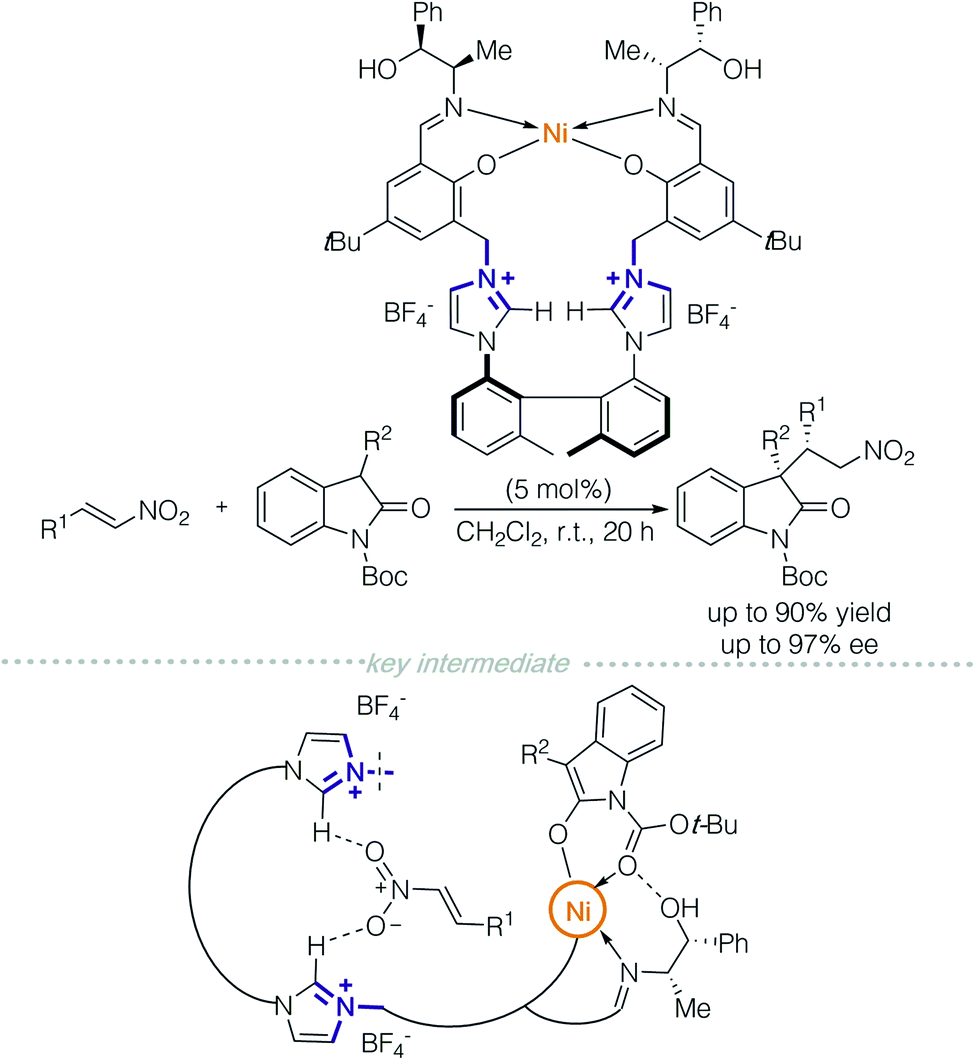 | ||
| Scheme 2 NiII/Schiff-base polyfunctional catalyst for an asymmetric 1,4-addition reaction. | ||
In 2019, Peters and co-workers reported another polyfunctional catalyst with two cobalt centres chelated in a tridentate manner to a Schiff base moiety that is connected to two triazolium cations and in turn linked via a chiral BINOL-based scaffold (Scheme 3).6 A 1,4-addition reaction between 2-oxindoles and maleimides, catalysed by a CoIII/triazolium polyfunctional catalyst can be conducted at room temperature. They discovered that additives have a dramatic impact on reaction outcomes. For instance, AcOH provided the best results while NaOAc decreased the diastereo- and enantioselectivity drastically. Although they were unable to elucidate the mechanism via single-crystal analysis of the catalytically active species, they observed the existence of one or two AcOH ligands on CoIII using ESI-MS. Probing further using DFT calculations suggests that equilibrium exists between monoacid coordinated CoIII and diacid coordinated CoIII. Evidence showed that a triplet spin state complex monoacid coordinated complex is more stable than a singlet diacid coordinated complex. Furthermore, the free coordination site on the paramagnetic monoacid coordinated CoIII allows binding of the pronucleophile suggesting that it is the catalytically active species. Recently, this group has also developed a new strategy for multi-functional cooperative Lewis acid/betaine (zwitterion) catalysis.7a,b
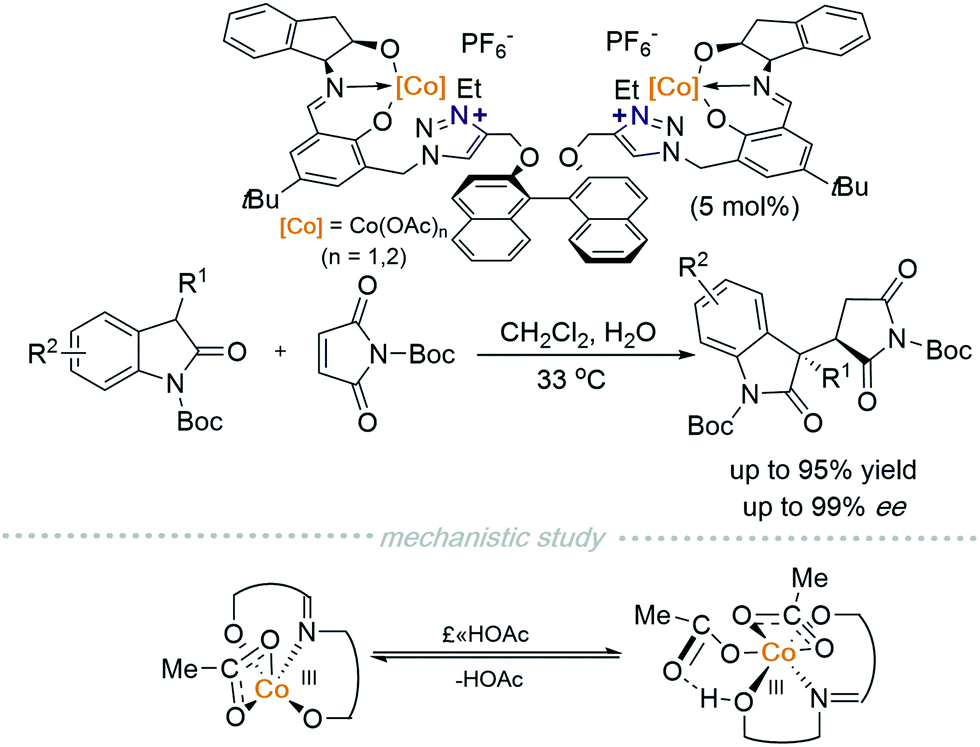 | ||
| Scheme 3 CoIII/Schiff-base polyfunctional catalyst for an asymmetric 1,4-addition reaction. | ||
3. Strategy 2: chiral cations paired with transition metal ‘ate’ complexes
The rudiment of strategy 2 can be tracked back to the first report of alkaloid-catalysed oxidations,8,9 in which stoichiometric permanganate worked as an oxidant but directed by a chiral cation to achieve enantioselectivity. In 2001, Brown and co-workers used cinchonidine-derived salts to promote oxidative cyclisation of 1,5-dienes using potassium permanganate, buffered with acetic acid (Scheme 4).8 The reaction was proposed to proceed in a step-wise fashion consisting of dihydroxylation of dienes, oxidation by permanganate and condensation of diols. Through their study, they found that slightly acidic conditions led to an oxidative cyclisation reaction of 1,5-dienes and the formation of α-ketols while slightly basic conditions promoted dihydroxylation.9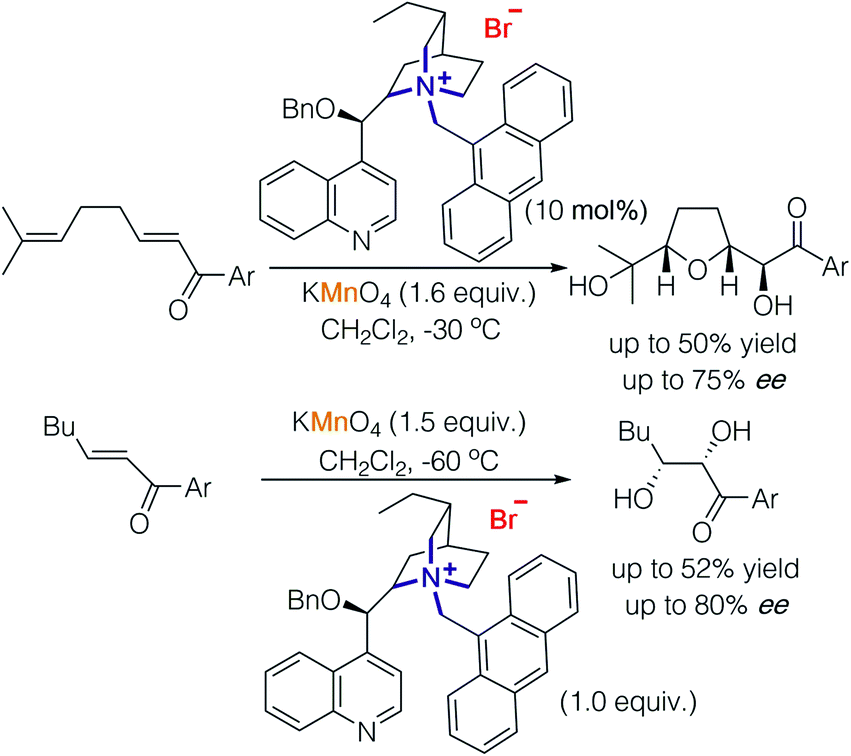 | ||
| Scheme 4 Cinchonidine-derived chiral cation paired with permanganate in asymmetric oxidative cyclisation and dihydroxylation. | ||
Basic conditions result in the decomposition of cinchonidine-derived chiral cations; hence, enantioselective dihydroxylation of enones was carried out with a stoichiometric amount of cinchonidine-derived chiral cations and permanganate furnishing diols with good enantioselectivity (up to 80% ee).
In 2015, the Tan group demonstrated the use of permanganate for asymmetric oxidation in the presence of bisguanidinium (BG). Asymmetric dihydroxylation of α-aryl acrylates and oxohydroxylation of trisubstituted enoates were achieved (Scheme 5).10 The key intermediate was proposed to be a metallo-enolate formed as a result of permanganate insertion into an α,β-unsaturated alkene. This mechanistic insight was deduced through observation of reaction by-products.
 | ||
| Scheme 5 Bisguanidinium permanganate in asymmetric dihydroxylation and oxohydroxylation. | ||
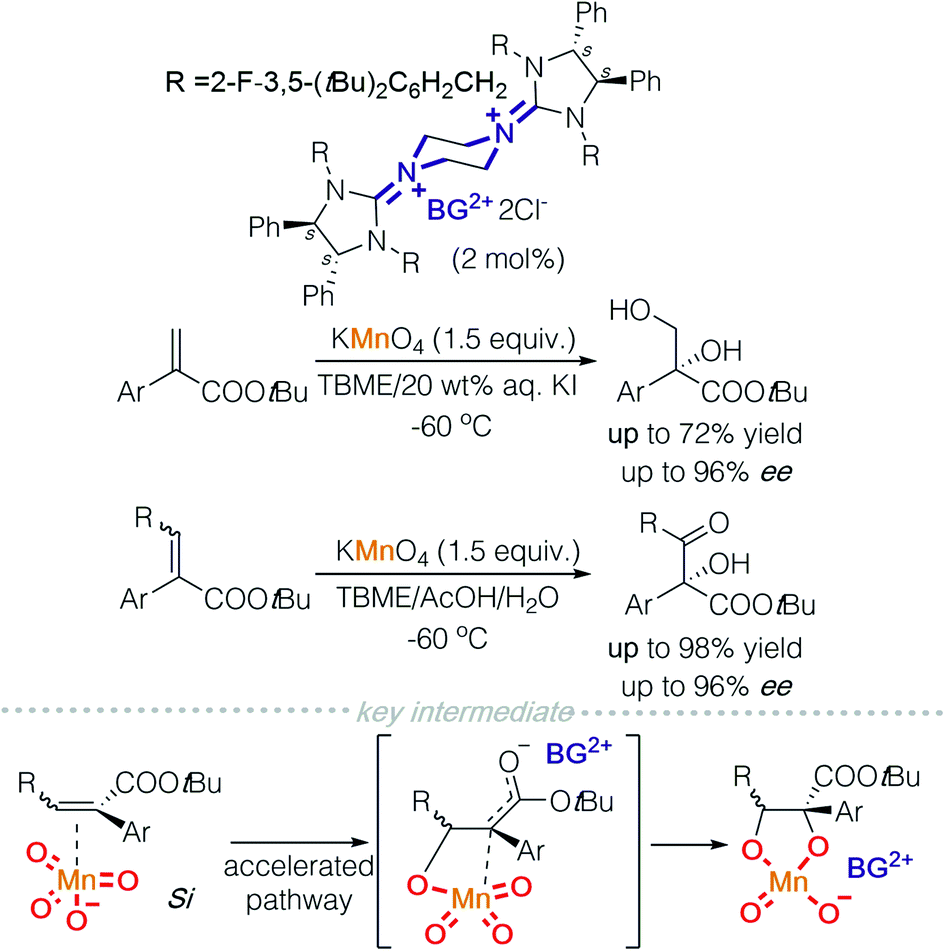
If a readily available external oxidant, such as hydrogen peroxide is present, it is then possible to use a catalytic amount of metal oxides. On this basis, Tan and co-workers took advantage of this possibility and used tungstate to catalyse asymmetric sulfoxidation of heterocyclic substituted thioethers and asymmetric epoxidation of allylic and homoallylic amides (Scheme 6).11a,b In a mechanistic study of the asymmetric sulfoxidation, the active counter ion was characterized using Raman spectroscopy and DFT calculations. A dihydrogen phosphate additive acts as a ligand coordinated to tungsten and DFT calculations indicate that a hydrogen bond exists between the two phosphate ligands. This strategy was successfully employed to synthesise (S)-lansoprazole.11a Homoallylic amides have been a difficult target for epoxidation. Tan and colleagues achieved a highly enantioselective asymmetric epoxidation on homoallylic amides using bisguanidinium tungstate. The active catalyst was isolated, characterised using single crystal X-ray diffraction and identified to be bisguanidinium tetraperoxyditungstate, [BG]2+ [W2O2(μ-O)(O2)4]2−. This strategy was successfully employed to synthesise (−)-venlafaxine.11b
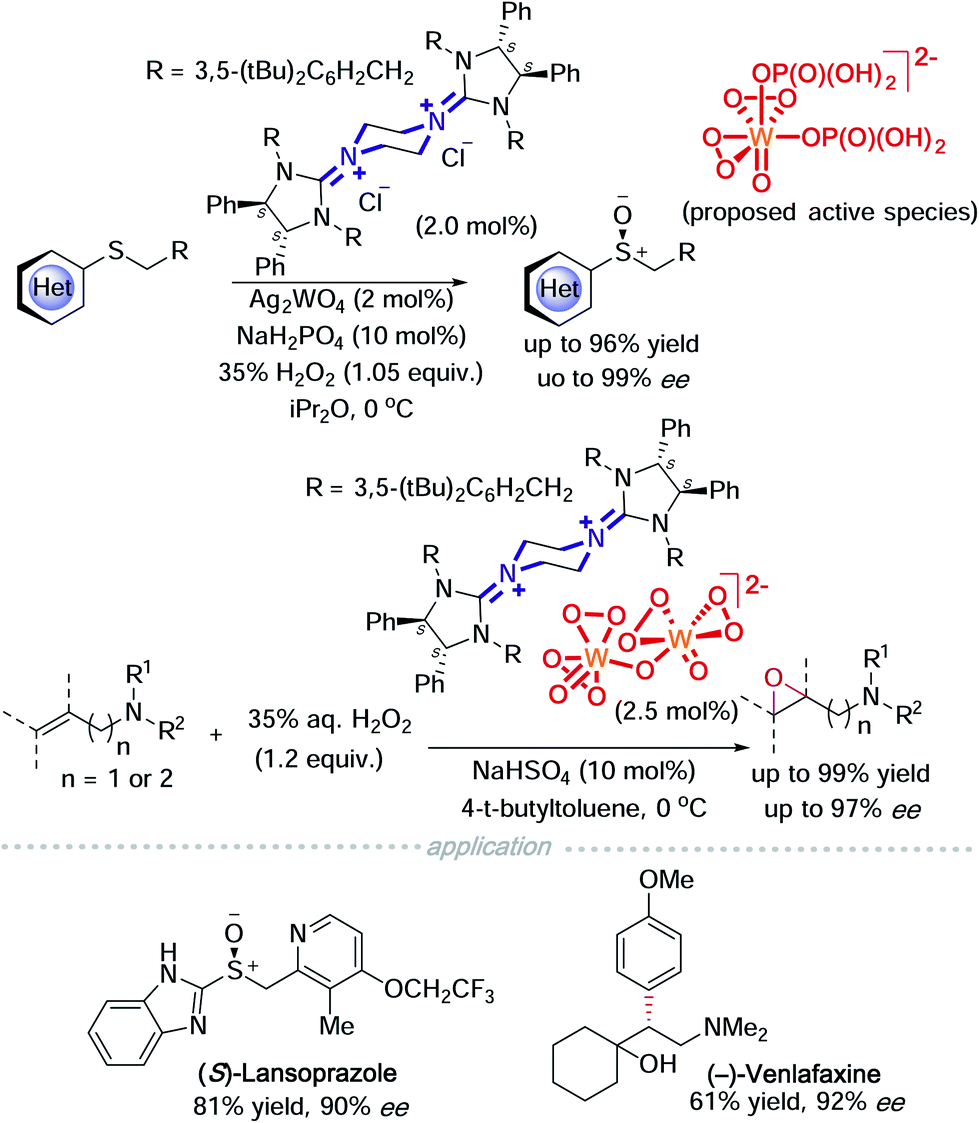 | ||
| Scheme 6 Bisguanidinium heteropolytungstate in asymmetric sulfoxidation and epoxidation. | ||
Another successful application of a transition metal ‘ate’-type counterion is heteropolymolybdate. The active catalyst, bisguanidinium dinuclear oxodiperoxomolybdosulfate [(μ-SO4)Mo2O2(μ-O2)2(O2)2]2−, was isolated and determined using single X-ray crystallography (Scheme 7). It proved effective in asymmetric sulfoxidation of electron-rich thioethers and successfully realised the synthesis of (R)-armodafinil on a gram-scale.12
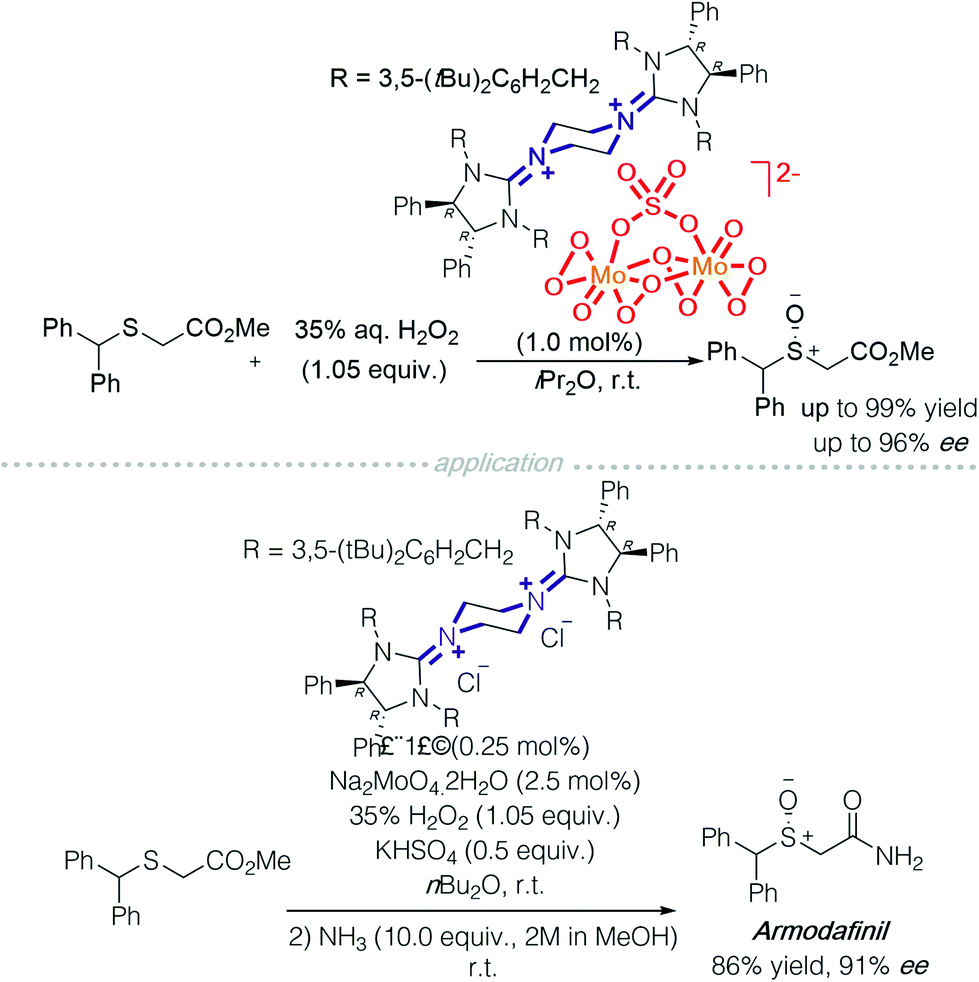 | ||
| Scheme 7 Bisguanidinium heteropolymolybdate in asymmetric sulfoxidation. | ||
In 2019, the Maruoka group reported an alkynylation of isatin derivatives catalysed using a chiral quaternary ammonium salt in the presence of silver acetate (Scheme 8).13 A plausible mechanism of the reaction is described as follows: (1) the chiral quaternary ammonium salt interacts with silver acetate to generate Q+[Ag(OAc)Br]−; (2) the terminal alkyne deprotonated by K2CO3 then associates with Ag, producing an alkynyl Ag-complex; (3) a substitution reaction between the alkynyl Ag-complex and isatin generates the key intermediate with high stereoselectivity; (4) finally, protonation by another alkyne molecule forms the final product and concomitantly regenerates the alkynyl Ag-ammonium ion pair.
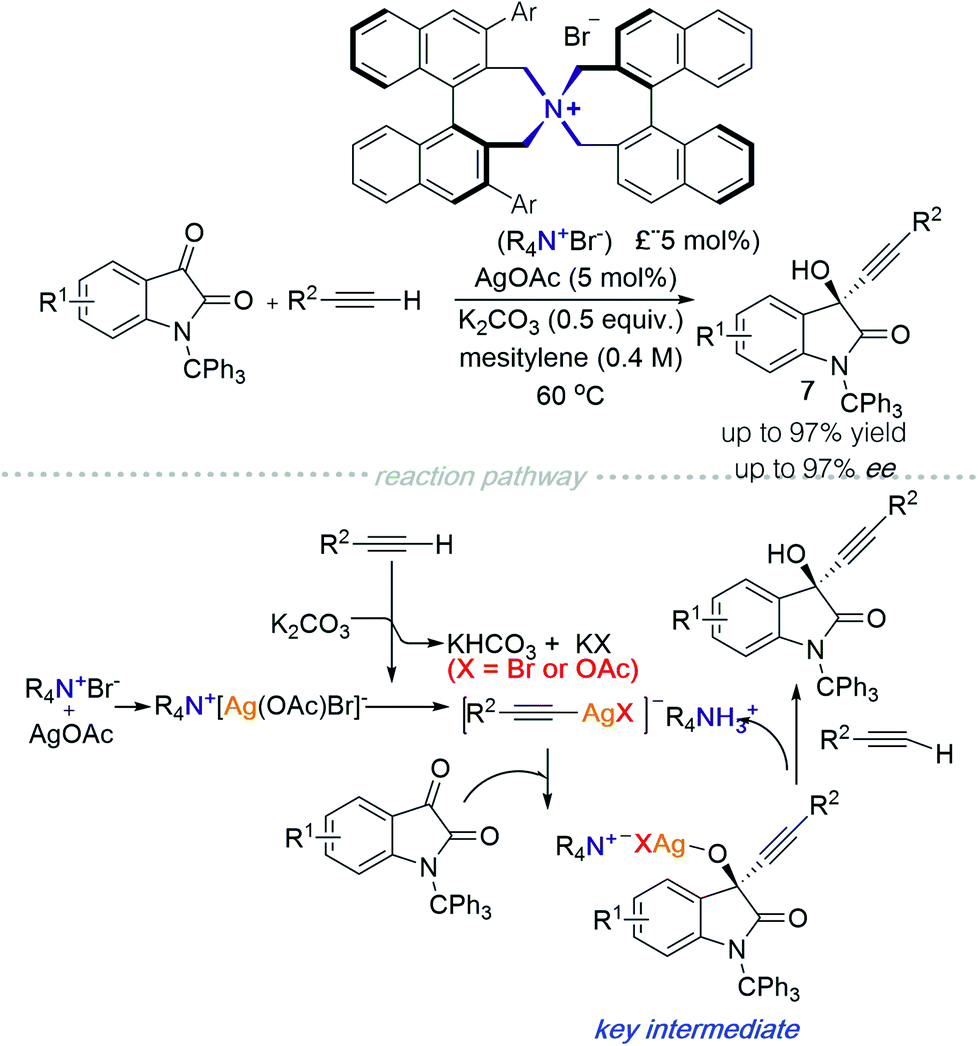 | ||
| Scheme 8 C 2-symmetric quaternary ammonium argentinate in asymmetric alkynylation. | ||
4. Strategy 3: ligand scaffold incorporated on an achiral anion
In 2016, Phipps and co-workers developed a strategy for C–H functionalisation through the use of an anionic Ir-complex to control both cationic and neutral substrates (Scheme 9).14 They demonstrated two modes of non-covalent interactions between substrates and the anionic Ir-complex. The first is quaternary ammonium containing benzylamine that is paired with a ligand possessing anionic sulfonate (model I).14a,c In the second approach, an amide protected benzylamine underwent hydrogen bonding with the sulfonate group on the ligand (model II).14b It should be noted that only model II falls under the scope of Strategy 3.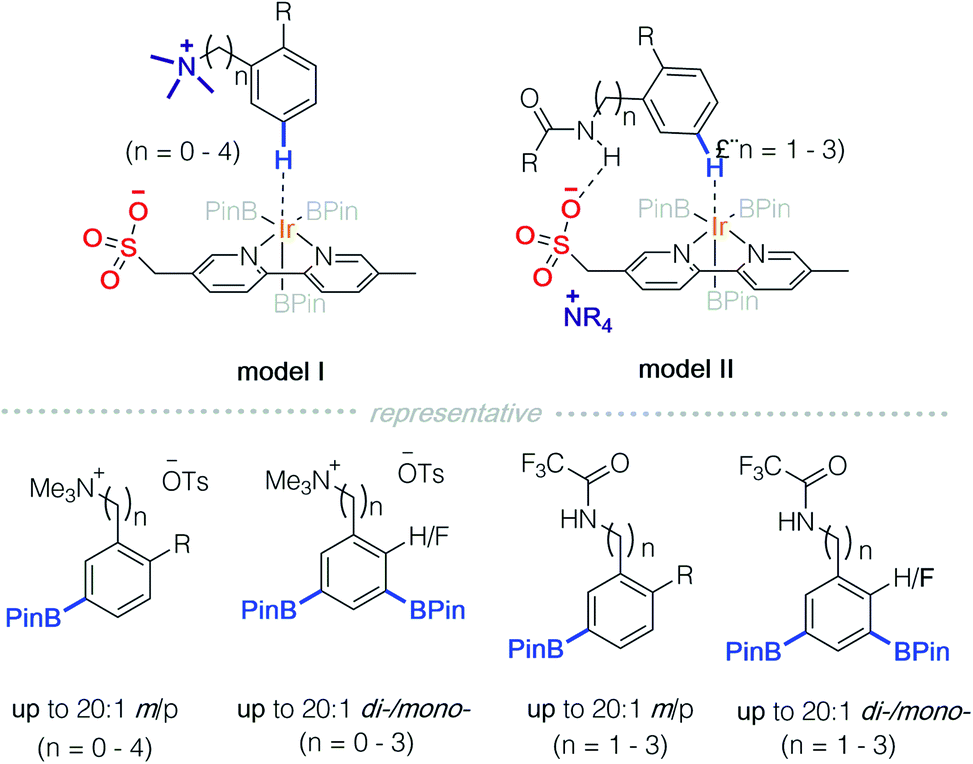 | ||
| Scheme 9 Substrate interaction with a ligand scaffold incorporated on an achiral anion. | ||
The essential characteristic of ion-pair catalysis is its catalytic efficiency due to effective interactions between its charged intermediates. Recently, Phipps and colleagues reported an enantioselective desymmetrisation through the use of an anionic Ir-complex, in conjunction with a chiral cationic catalyst (Scheme 10).15 A quinine-derived chiral cation and sulfonyl modified bipyridine counterion were prepared as an ion pair catalyst. In the presence of iridium, borylation of an arene on benzhydrylamides and diaryl phosphinamides was achieved successfully. It was deduced that the counterion is an Ir-complex with a sulfonyl bipyridine ligand. The sulfonyl bipyridine ligand is both an ion pair to the chiral cation and site for hydrogen bonding with the acylamino group on the substrate. Consequently, remote C–H bond activation is achieved via the charge-inverted Ir-complex and enantioselectivity is induced by the quaternized alkaloid derivatives.
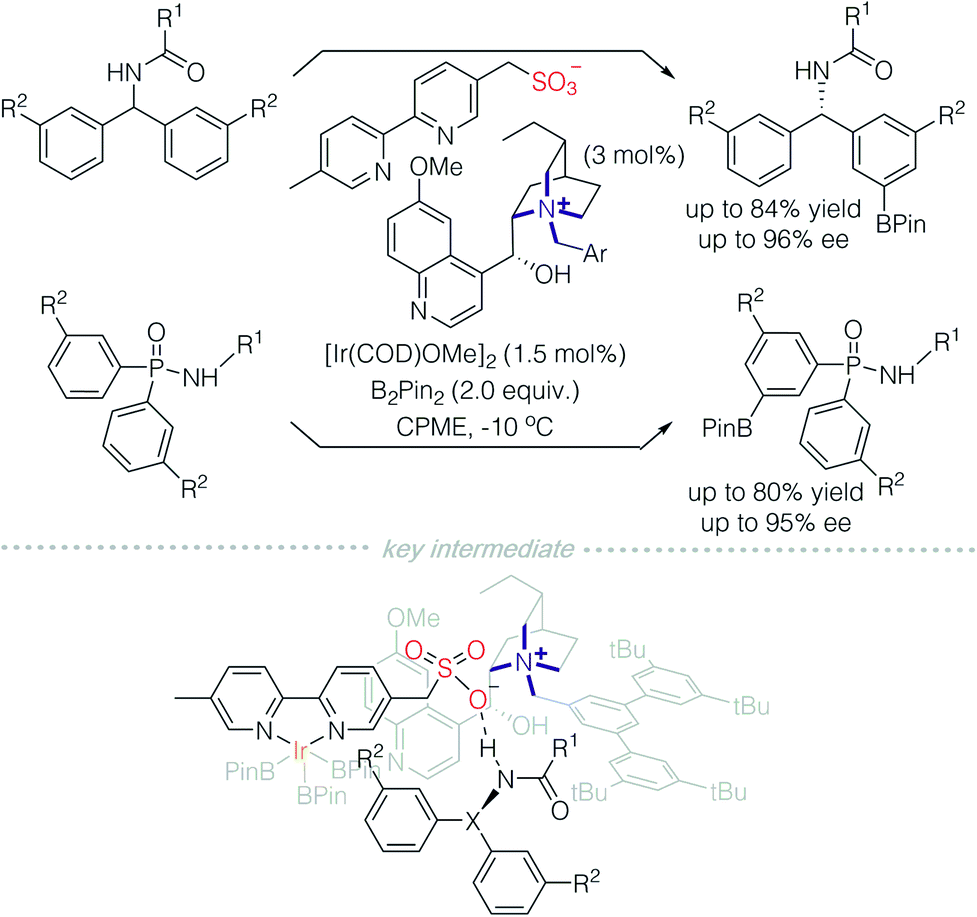 | ||
| Scheme 10 Remote C–H activation catalysed by a quinine-derived chiral cation paired with an anionic Ir-complex. | ||
This methodology demonstrated excellent regioselectivity on aromatic rings. Meta-substitution is dominant even when R2 is on the ortho-position, despite a slight decrease in enantioselectivity. In comparison, when an uncharged bipyridine (dtbpy) was used, the regioselectivity was poor. This powerful methodology is also tolerant of a wide range of substituents on aromatic rings, in particular, iodo-substituted arenes, which otherwise are incompatible with palladium catalysis.16
5. Perspective
In accordance with the aforementioned strategies, several anionic transition metal complexes can be further developed for ion-pair catalysis. For instance, Lavigne and colleagues prepared an N-heterocyclic carbene decorated with a malonate backbone (maloNHC) (Scheme 11).17a It functions as an anionic ligand scaffold and can coordinate to metals such as rhodium,17b silver,17c iron,17a,d and copper. Metal-maloNHC was paired with Li(THF)2+ or quaternary ammonium and successfully used in skeletal rearrangement of enynes, hydrosilylation, hydroboration, and cyclopropanation. These reactions can potentially be carried out in an enantioselective manner with chiral quaternary ammonium salts.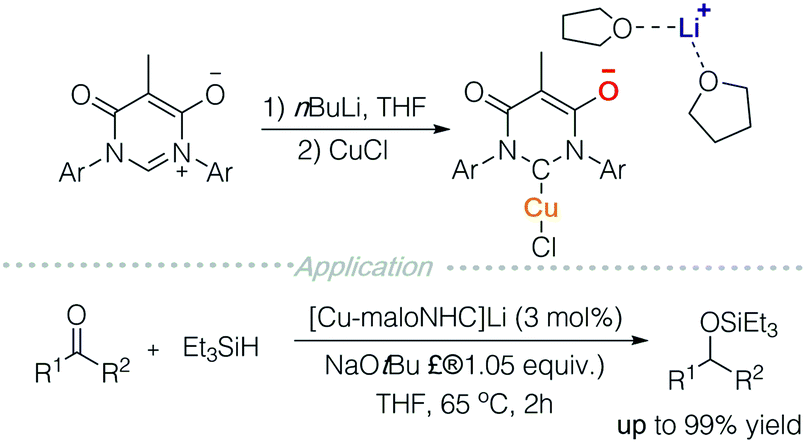 | ||
| Scheme 11 Anionic NHC-complexed metal catalysis. | ||
Ion-pair transition metal complexes can be useful in electrochemistry and photochemistry. Lugan and co-workers prepared a μ-vinylbis(carbene)-dimanganese complex (Scheme 12A), which is reduced to generate a radical anion and dianion via cyclic voltammetry (CV).18 The radical anion was characterized using EPR, showing a mixed-valence pattern of Mn0/MnI, implying that the unpaired electron delocalized over two metal centres. The ability of the above-mentioned complexes to emit and absorb electrons may lead to potential application in electron transfer reactions.
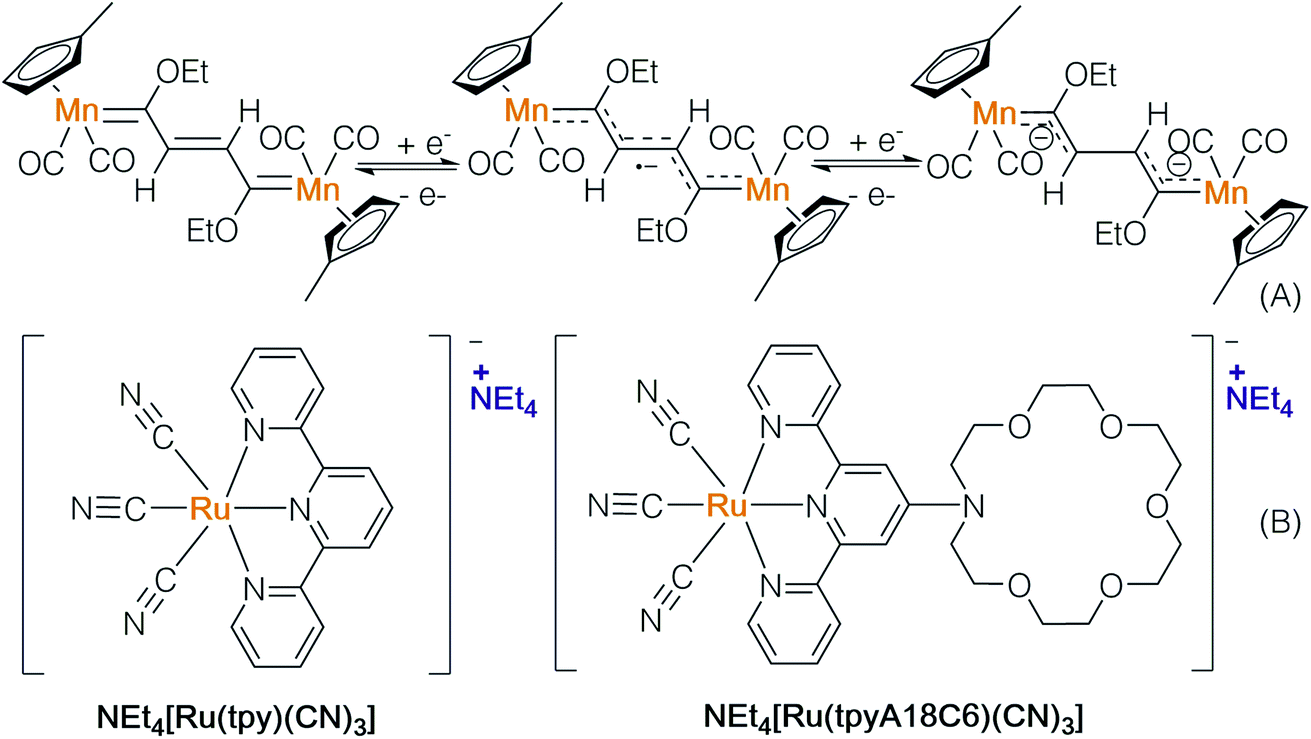 | ||
| Scheme 12 Electron transfer processes of anionic metal complexes. | ||
The Yam group prepared a novel ruthenium anion complex NEt4[Ru(tpyA18C6)(CN)3] and used it as a mobile-phase additive to separate metal-ions in high-performance liquid chromatography (Scheme 12, B).19 The solvatochromic behavior and photoluminescence of [Ru(tpyA18C6)(CN)3]− indicate its UV absorption properties. The solvent dependence of metal-to-ligand charge transfer (MLCT) absorption bands shows that active wavelengths are in the visible region. Through binding with a photosensitizer, the ion-pair ruthenium complex may serve as a catalyst in single electron transfer processes.
In 1979, Griffith and colleagues systematically studied ruthenate entities and their varied oxidative capacities (Scheme 13).20a In their initial report, a catalytic amount of ruthenate together with stoichiometric alkaline persulfate led to over-oxidation of an alcohol and amine to their corresponding acid and nitrile. The second oxidation step is inhibited by tuning the ruthenate and external oxidant. When per-ruthenate and N-methyl morpholine N-oxide (NMO) were used, primary and secondary alcohols were oxidized to aldehydes and ketones respectively.20b Subsequently, a new species [LPh4][RuO2Cl3] (L = P, As) was found to be able to oxidise alcohols in the presence of alkenes.20c The tunable nature of Ru-complexes makes it possible to perform chemoselective oxidation.
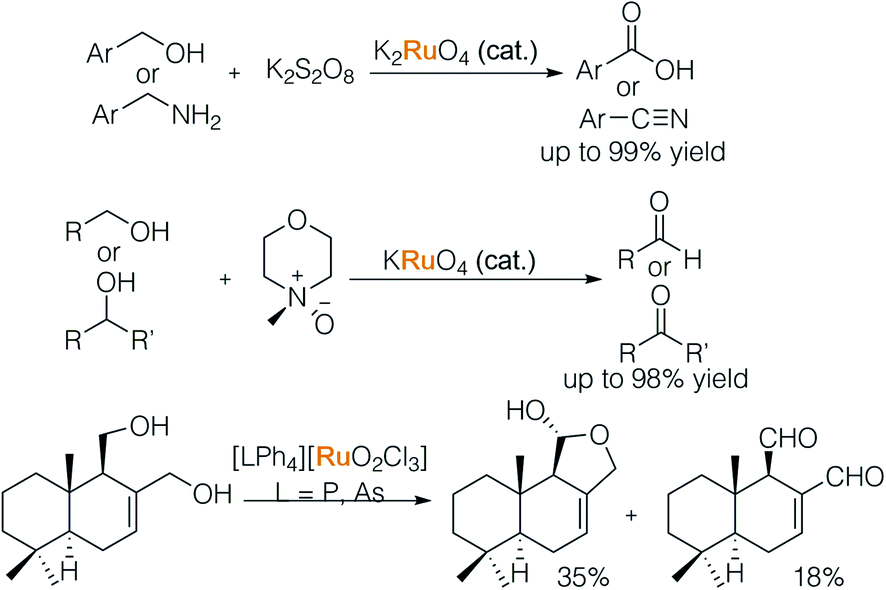 | ||
| Scheme 13 Tunable ruthenates in oxidation of alcohols and amines. | ||
Potassium ferrate is a common water purifying agent,21 and can be used as an oxidant in organic synthesis. In 2017, Ray and co-workers used acid-activated potassium ferrate to oxidize caffeine (Scheme 14A).22 Soon after, in 2018, Jiang and co-workers reported a photooxygenation of benzylic sp3 C–H using iron chloride and lithium bromide under visible light irradiation (Scheme 14B).23 In order to study the active species in the reaction, they prepared TBA[FeCl3Br] and characterized it via single crystal X-ray diffraction (Scheme 14, mechanistic study).
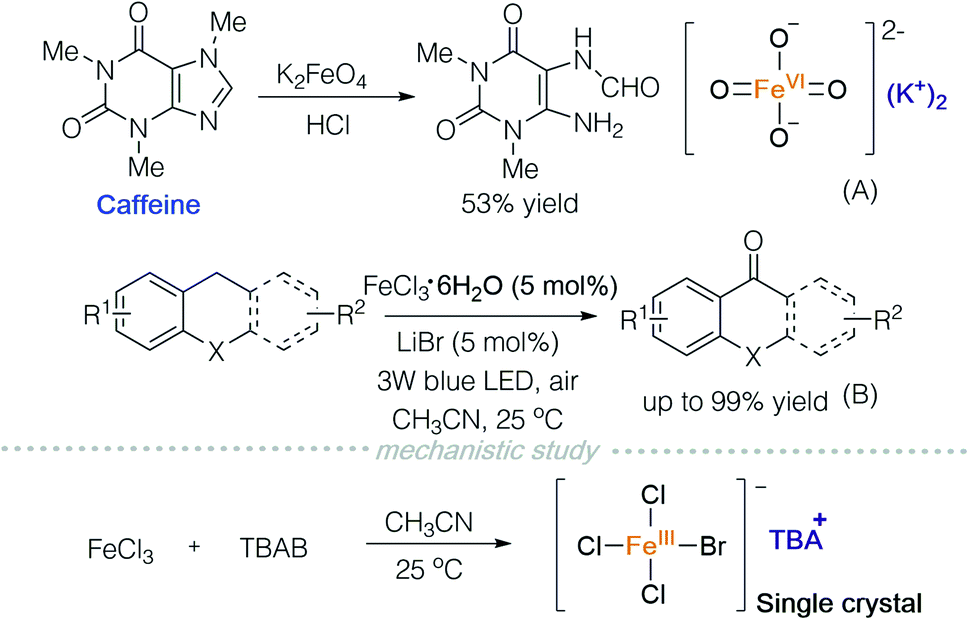 | ||
| Scheme 14 Ferrate and iron ‘ate’-type complexes in oxidations. | ||
6. Conclusion
The epitome of a chiral catalyst is one that it can be used for all enantioselective chemical reactions. This seems to be an impossible dream as each reaction will have a different mode of activation. There lies a possibility of a universal chiral cation that can be paired with different anionic co-catalysts. The anion co-catalyst is responsible for promoting the reaction while the chiral cation provides the ‘chiral space’. Inherently, optimisation of reactions will mainly involve the design of the non-chiral anionic co-catalyst instead of preparing a large number of chiral ligands as per current state-of-the-art approaches.We have provided a synopsis of all major studies in chiral cation catalysis incorporating organometallic catalysis. We found that these new chemistries assimilated the advantages of both phase transfer and organometallic catalysis, leading to reduction of catalyst loading and expansion of scope of synthetic chemistry. We are confident that more developments in this area are forthcoming.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the Zhejiang University of Technology (2020414801729), Nanyang Technological University (RG1/19 and RG2/20) and Ministry of Education Singapore (MOE2019-T2-1-091) for financial support.Notes and references
- For selected reviews on asymmetric phase-transfer reactions see: (a) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS; (b) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS.
- For selected reviews on asymmetric ion-pair catalysis see: (a) K. Bark and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS; (c) L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842–856 CrossRef CAS; (d) K. Ohmatsu and T. Ooi, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS.
- (a) G. Chen, Y. Deng, L. Gong, A. Mi, X. Cui, Y. Jiang, M. C. Chio and A. S. Chan, Tetrahedron: Asymmetry, 2001, 12, 1567–1571 CrossRef CAS; (b) M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Org. Lett., 2001, 3, 3329–3331 CrossRef CAS PubMed; (c) M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, J. Org. Chem., 2002, 67, 7418–7423 CrossRef CAS PubMed; (d) T. Kanayama, K. Yoshida, H. Miyabe, T. Kimachi and Y. Takemoto, J. Org. Chem., 2003, 68, 6197–6201 CrossRef CAS PubMed; (e) T. Kanayama, K. Yoshida, H. Miyabe and Y. Takemoto, Angew. Chem., Int. Ed., 2003, 42, 2054–2056 CrossRef CAS PubMed; (f) Y. Su, Y. Li, Y. Chen and Z. Han, Chem. Commun., 2017, 53, 1985–1988 RSC.
- (a) K. Ohmatsu, N. Imagawa and T. Ooi, Nat. Chem., 2014, 6, 47–51 CrossRef CAS; (b) K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304–4306 CrossRef CAS.
- M. Mechler and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 10303–10307 CrossRef CAS.
- J. Schmid, T. Junge, J. Lang and R. Peters, Angew. Chem., Int. Ed., 2019, 58, 5447–5451 CrossRef CAS.
- (a) F. Willig, J. Lang, A. C. Hans, M. R. Ringenberg, D. Pfeffer, W. Frey and R. Peters, J. Am. Chem. Soc., 2019, 141, 12029–12043 CrossRef CAS PubMed; (b) V. Miskov-Pajic, F. Willig, D. M. Wanner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2020, 59, 19873–19877 CrossRef CAS.
- R. C. D. Brown and J. F. Keily, Angew. Chem., Int. Ed., 2001, 40, 4496–4498 CrossRef CAS.
- R. A. Bhunnoo, Y. Hu, D. I. Laine and R. Brown, Angew. Chem., Int. Ed., 2002, 41, 3479–3480 CrossRef CAS.
- C. Wang, L. Zong and C.-H. Tan, J. Am. Chem. Soc., 2015, 137, 10677–10682 CrossRef CAS.
- (a) X. Ye, A. M. P. Moeljadi, K. F. Chin, H. Hirao, L. Zong and C.-H. Tan, Angew. Chem., Int. Ed., 2016, 55, 7101–7105 CrossRef CAS; (b) K. F. Chin, X. Ye, Y. Li, R. Lee, A. M. Kabylda, D. Leow, X. Zhang, C. X. E. Ang and C.-H. Tan, ACS Catal., 2020, 10, 2684–2691 CrossRef CAS.
- L. Zong, C. Wang, A. M. P. Moeljadi, X. Ye, H. Hirao and C.-H. Tan, Nat. Commun., 2016, 7, 13455 CrossRef PubMed.
- S. Paria, H. Lee and K. Maruoka, ACS Catal., 2019, 9, 2395–2399 CrossRef CAS.
- (a) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed; (b) H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef CAS; (c) H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764–3769 CrossRef.
- G. R. Genov, J. L. Douthwaite, A. S. Lahdenpera, D. Gibson and R. J. Phipps, Science, 2020, 367, 1246–1251 CrossRef CAS PubMed.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J. Yu, Nature, 2018, 558, 581–585 CrossRef CAS PubMed.
- (a) V. Cesar, N. Lugan and G. Lavigne, J. Am. Chem. Soc., 2008, 130, 11286–11287 CrossRef CAS; (b) V. Cesar, N. Lugan and G. Lavigne, Chem.–Eur. J., 2010, 16, 11432–11442 CrossRef CAS; (c) S. Kronig, E. Theuergarten, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 3240–3244 CrossRef CAS; (d) V. Cesar, C. Barthes, Y. Farre, S. Cuisiat, B. Y. Vacher, R. Brousses, N. Lugan and G. Lavigne, Dalton Trans., 2013, 42, 7373–7385 RSC.
- A. Rabier, N. Lugan, R. Mathieu and G. L. Geoffroy, Organometallics, 1994, 13, 4676–4678 CrossRef CAS.
- M.-J. Li, B. W.-K. Chu and V. W.-W. Yam, Chem.–Eur. J., 2006, 12, 3528–3537 CrossRef CAS PubMed.
- (a) M. Schroder, W. P. Griffith and J. C. S. Chem, Comm, 1979, 58–59 Search PubMed; (b) G. Green, W. P. Griffith, D. M. Hollinshead, S. V. Ley and M. Schroder, J. Chem. Soc., Perkin Trans. 1, 1984, 15, 681–686 RSC; (c) W. P. Griffith, S. V. Ley, G. P. Whitcombe, A. D. White and J. C. S. Chem, Comm, 1987, 1625–1627 CAS.
- For selected reports of ferrate in oxidation see: (a) V. K. Sharma, J. T. Bloom and V. N. Joshi, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 1998, 33, 635–650 CrossRef; (b) V. K. Sharma, W. Rivera, V. N. Joshi, F. J. Millero and D. O'Connor, Environ. Sci. Technol., 1999, 33, 2545–2650 CrossRef; (c) M. Feng and V. K. Sharma, Chem. Eng. J., 2018, 341, 137–145 CrossRef CAS; (d) C. Luo, M. Feng, V. K. Sharma and C.-H. Huang, Environ. Sci. Technol., 2019, 53, 5272–5281 CrossRef CAS PubMed.
- K. Manoli, G. Nakhla and A. K. Ray, AIChE J., 2017, 63, 4998–5006 CrossRef CAS.
- S. Li, B. Zhu, R. Lee, B. Qiao and Z. Jiang, Org. Chem. Front., 2018, 5, 380–385 RSC.
| This journal is © The Royal Society of Chemistry 2021 |