
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA β-hairpin epitope as novel structural requirement for protein arginine rhamnosylation†
Liubov
Yakovlieva‡
 a,
Thomas M.
Wood‡
bc,
Johan
Kemmink
a,
Ioli
Kotsogianni
b,
Franziska
Koller
d,
Jürgen
Lassak
d,
Nathaniel I.
Martin
*b and
Marthe T. C.
Walvoort
*a
a,
Thomas M.
Wood‡
bc,
Johan
Kemmink
a,
Ioli
Kotsogianni
b,
Franziska
Koller
d,
Jürgen
Lassak
d,
Nathaniel I.
Martin
*b and
Marthe T. C.
Walvoort
*a
aChemical Biology Group, Stratingh Institute for Chemistry, University of Groningen, Groningen, The Netherlands. E-mail: m.t.c.walvoort@rug.nl
bBiological Chemistry Group, Institute of Biology Leiden, Leiden University, Leiden, The Netherlands. E-mail: n.i.martin@biology.leidenuniv.nl
cDepartment of Chemical Biology & Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Utrecht, The Netherlands
dDepartment of Biology I, Microbiology, Ludwig-Maximilians-Universität München Planegg/Martinsried, Germany
First published on 7th December 2020
Abstract
For canonical asparagine glycosylation, the primary amino acid sequence that directs glycosylation at specific asparagine residues is well-established. Here we reveal that a recently discovered bacterial enzyme EarP, that transfers rhamnose to a specific arginine residue in its acceptor protein EF-P, specifically recognizes a β-hairpin loop. Notably, while the in vitro rhamnosyltransferase activity of EarP is abolished when presented with linear substrate peptide sequences derived from EF-P, the enzyme readily glycosylates the same sequence in a cyclized β-hairpin mimic. Additional studies with other substrate-mimicking cyclic peptides revealed that EarP activity is sensitive to the method used to induce cyclization and in some cases is tolerant to amino acid sequence variation. Using detailed NMR approaches, we established that the active peptide substrates all share some degree of β-hairpin formation, and therefore conclude that the β-hairpin epitope is the major determinant of arginine-rhamnosylation by EarP. Our findings add a novel recognition motif to the existing knowledge on substrate specificity of protein glycosylation, and are expected to guide future identifications of rhamnosylation sites in other protein substrates.
Introduction
Protein glycosylation, an enzymatic process in which a carbohydrate or glycan is covalently added to a specific amino acid residue, is one of the most ubiquitous post-translational modifications in nature.1 Glycosylation confers specific properties on the acceptor protein, such as increased solubility, protection from degradation, tagging for transport or destruction, interaction with receptors, or functional activation. As a result, protein glycosylation influences a myriad of biological processes in all kingdoms of life.Protein asparagine N-glycosylation is universally present and fairly conserved across species, and it involves the en bloc transfer of an oligosaccharide from a lipid-linked carrier to an acceptor protein, catalyzed by a membrane-associated glycosyltransferase, such as eukaryotic OST and prokaryotic PglB.2,3 The requirements for the primary sequence and structural folds are well-established for canonical N-linked glycosylation. In general, asparagine residues are modified in so-called “sequons” – recognition sequences of N-X-S/T, with X being any amino acid except proline.4 In addition, PglB of C. jejuni recognizes an extended sequon of D/E-Z-N-X-S/T (Z and X ≠ Pro).3 The X residue (+1) and Ser/Thr residue (+2) have been shown to play an important role in acceptor recognition by glycosyltransferases.5 In the co-crystal structure of the bacterial oligosaccharyltransferase PglB, the acceptor peptide was shown to adopt a distinct bound conformation, featuring the recognition sequon in a 180 degree loop.6 This structure would be impossible to adopt with proline in the +1 position, explaining the negative selection for Pro in the glycosylation sequon.5 The +2 hydroxy amino acid has been shown to contribute to recognition by interacting with the conserved WWD motif in PglB. Interestingly, when bound in the active site, Thr in position +2 has been shown to be engaged in more stabilizing interactions than Ser in position +2 which is reflected in faster glycosylation rates of Thr-containing sequons.6,7 Another important aspect of N-linked glycosylation is the mechanism of asparagine activation for nucleophilic attack. One of the early explanations implied the importance of the local secondary structure, namely the Asx turn (Fig. 1A).8 Within this structure, protonation of the Asn-amide carbonyl by the hydroxyl of the +2 Ser/Thr residue, in combination with deprotonation of nitrogen by an enzymatic base would result in the formation of a reactive imidol species capable of carrying out the nucleophilic attack (Fig. 1B). An alternative explanation of carboxamide activation in protein glycosylation is the so-called “twisted amide” hypothesis wherein activation of Asn occurs as a result of twisting the nitrogen lone pair out of conjugation with the carbonyl group (Fig. 1C).6,9 It is postulated that Asp and Glu residues in the OST active site form H-bonds with amide hydrogens of the acceptor Asn. As a result of this H-bond stabilization, de-conjugation of the nitrogen lone pair electrons occurs, resulting in a more nucleophilic nitrogen primed for attack.
 | ||
| Fig. 1 (A) Asx-turn: proposed secondary structure formed by the sequon amino acids. The amide group of Asn is forming H-bonds with the side chain hydroxyl of the +2 amino acid. (B) Deprotonation by the enzymatic base leads to the formation of the activated imidol species. (C) The twisted amide as alternative hypothesis for Asn activation. (D) β-turn secondary structure, formed by reversing the direction of the chain over four residues, stabilized by interstrand H-bonds. (H-bonds are showed as dashed lines). (E) Rhamnosylation of EF-P by EarP. Domain I of EF-P (amino acids 1–65) is shown in orange. | ||
Studies have demonstrated that peptides forced into the Asx turn exhibited increased affinity and faster glycosylation rates in comparison to linear peptides or β-turn peptides (Fig. 1D).10
Despite the clear three amino acid sequon in the substrate protein, not all predicted N-X-S/T sites are found to be glycosylated which indicates the role of additional recognition elements. A statistical analysis of eukaryotic N-glycosylation sites revealed that there is a preference for β-turns and -bends around the glycosylation site, whereas α-helices are disfavored.11 Additionally, it has been demonstrated that similarly to eukaryotic protein glycosylation, which occurs co-translationally on unfolded polypeptides, bacterial protein glycosylation preferentially takes place on glycosylation sites located in exposed loops and benefits from moderate structure disorder of the acceptor protein.12 Interestingly, while O-glycosyltransferases do not generally require a specific recognition sequence in their acceptor substrates, several recent examples indicate the preference for properly folded substrate domains, implying a fold recognition mechanism instead.13,14
In addition to the well-established forms of protein glycosylation, novel glycosylation systems have been discovered that are unique to bacteria.15 Recently, arginine rhamnosylation was identified as a novel type of N-glycosylation.16,17 Here the enzyme EarP transfers a rhamnose moiety from dTDP-β-L-rhamnose (TDP-Rha) to a specific arginine residue in the acceptor translation elongation factor P (EF-P) (Fig. 1E).16,18 Arginine glycosylation is a rare modification with only two other examples reported to date, i.e. autocatalytic modification of Arg with glucose (Glc) of sweet corn amygdalin,19 and with N-acetylglucosamine (GlcNAc) of human death receptor domains by the bacterial effector protein NleB.20
Genes associated with the newly discovered EF-P rhamnosylation (earP, efp, and rmlABCD genes for dTDP-β-L-rhamnose (TDP-Rha) donor synthesis) have been identified predominantly in beta- and gamma-proteobacteria, including multiple clinically relevant pathogens, e.g. Pseudomonas aeruginosa, Neisseria meningitidis, and Bordetella pertussis.16 The rhamnose modification has been shown to activate EF-P which alleviates ribosomal stalling during the synthesis of poly-proline stretches in nascent polypeptides.21–23 Abolishing rhamnosylation of EF-P in P. aeruginosa and N. meningitidis led to cellular effects that were detrimental to bacterial fitness and increased susceptibility to antibiotics.18,24 It is hypothesized that these severe effects are associated with importance of polyPro-containing proteins and virulence factors of investigated bacterial pathogens for their survival.18,24
Since the discovery of EF-P rhamnosylation in 2015, a number of studies have contributed to an increased understanding of this unique system. The stereochemistry of the α-glycosidic linkage between Arg and Rha was shown by two research groups independently,25,26 proving that EarP is an inverting glycosyltransferase. An anti-ArgRha antibody has also been developed, allowing for facile (in vitro) monitoring of arginine rhamnosylation.25,27 Several (co)-crystal structures have been reported for EarP (from P. putida,27N. meningitidis28 and P. aeruginosa29), also in complex with its EF-P substrate, providing insight into the specific amino acid interactions and the catalytic mechanism of rhamnosylation.
As only a single Arg residue in EF-P is modified, an important yet unexplored aspect of this novel glycosylation system is the basis for the observed specificity in recognizing this arginine residue. Previous reports indicate that domain I of EF-P (aa 1–65, Fig. 1E) is sufficient for recognition and rhamnosylation by EarP.27 Domain I, commonly referred to as a “KOW-domain”,27 is a conserved domain in various ribosome-associated proteins involved in transcription and translation,30 and it appears to contain all recognition elements necessary to promote Arg rhamnosylation.27–29 A recent study indicates that structural elements are more important than a specific sequon to promote EarP-mediated rhamnosylation.31 However, the precise determinants for recognition by EarP are currently not known. Elucidating the necessary substrate recognition elements will allow us to more fully understand this unique bacterial system, an important step towards exploiting bacterial glycosylation systems for the development of novel anti-virulence strategies.32
Here we report the discovery of a novel β-hairpin recognition element in arginine rhamnosylation of EF-P from P. aeruginosa. Using in vitro rhamnosylation assays and in-depth NMR studies we demonstrate the importance of this key structural motif in acceptor substrate recognition by EarP. Moreover, we report the shortest peptide fragment known to date to be rhamnosylated by EarP. Next to expanding the current knowledge on structural requirements of protein glycosyltransferases, our results have the potential to inform the development of inhibitors and activity assays to screen for inhibitors for EarP based upon the β-hairpin motif of the EF-P KOW domain.
Results
EarP does not rhamnosylate Arg in linear peptide fragments in vitro
A common strategy for studying the activity of N-glycosyltransferases in vitro is using linear peptide fragments corresponding to their protein substrate. We heterologously expressed the rhamnosyltransferase EarP from P. aeruginosa using a previously described procedure,29 and focused our studies on the natural substrate EF-P_Pa. As a first step in deciphering the determinants of substrate recognition in arginine rhamnosylation, we investigated a linear peptide fragment comprised of eight amino acids (8mer, Table S1†). As can be seen from the Fig. S1,† this fragment corresponds to the Arg32-containing loop of EF-P. Unexpectedly, this linear peptide did not prove to be a suitable substrate for EarP as no conversion was observed under in vitro rhamnosylation conditions (analysis with RP-LCMS). These results suggest that EarP does not rely exclusively on a specific amino acid sequence (primary structure) in the protein substrate for recognition, suggesting that there may be secondary structure requirements involved.Arginine in an L-Pro–D-Pro-cyclized peptide is rhamnosylated by EarP
As revealed in various structural studies,27–29 the acceptor binding site of EarP is unusually large and multiple contacts between active site residues of EarP and amino acids of domain I of EF-P are necessary for protein substrate recognition. Upon examining the co-crystal structure of EarP and domain I of EF-P from P. aeruginosa29 (PDB 6J7M) it is evident that the majority of EF-P residues involved in binding to EarP are located in the β-hairpin with Arg32 at its tip (Fig. 2A). Multiple EarP active site residues are involved in acceptor protein recognition and form both main-chain and side-chain promoted H-bonds, salt bridges, and hydrophobic interactions (Table S2†). As can be seen from Fig. 2A, a selected number of EF-P residues (Arg32, Lys29, Ser30, Asn28, Val36, Phe54, Val53, Lys55) are involved in binding to EarP suggesting that both sequence and shape of the bound motif are recognized, rather than just the target arginine. The β-hairpin secondary structure appears to optimally position Arg32 for binding in the EarP active site. Based on this interaction profile, we decided to explore the importance of secondary structure in Arg-rhamnosylation by preparing peptide mimics of the β-hairpin containing Arg32. Mimics of the β-hairpin secondary structure have been extensively studied over the years, and many structure-inducing templates have been developed.33 One of the most widely used methods to nucleate a β-hairpin structure is the introduction of an L-proline/D-proline motif.33 This motif leads to a “kink” in the sequence and brings the strands in close proximity to allow the formation of secondary structure-stabilizing H-bonds between the antiparallel strands.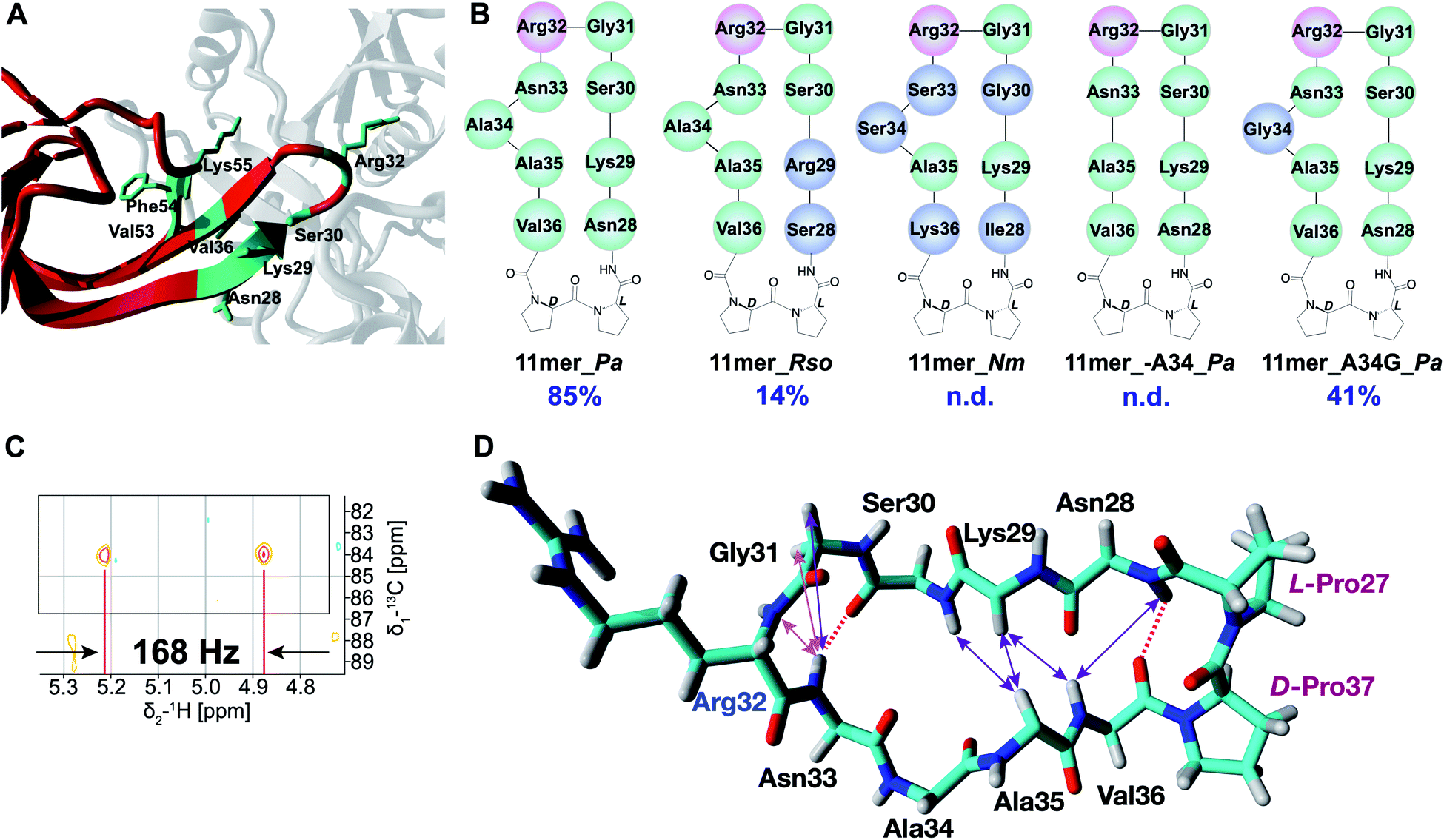 | ||
| Fig. 2 (A) EarP–EF-P complex from P. aeruginosa (generated with YASARA, PDB 6J7M). EarP is depicted in grey, domain I of EF-P is colored red, EF-P residues involved in binding with EarP are in green. (B) Cyclic peptide mimics of the EF-P β-hairpin. Arg32 is shown in pink, altered residues (with respect to 11mer_Pa) are shown in blue. (C) Zoom-in of the coupled HSQC spectrum of crude Rha-11mer_Pa reaction mixture. JCH = 168 Hz (α-glycosidic bond). (D) Experimentally determined NOE signals that are indicative of a β-hairpin structure are mapped on the 11mer L-Pro–D-Pro_Pa fragment (from 3OYY crystal structure). Hydrogen bonds inferred from the NH temperature studies are shown as red dashed lines. NOE signals are shown as double-ended arrows (magenta: medium NOE; purple: weak NOE). | ||
Based on the sequences of EF-P proteins from P. aeruginosa, Ralstonia solanacearum (a Gram-negative plant pathogen) and N. meningitidis, the corresponding cyclic 11mer peptides depicted in Fig. 2B were prepared using solid-phase peptide synthesis (SPPS) starting from Gly31 loaded onto 2-chlorotrityl resin with the peptides assembled terminating with Arg32 (see ESI† for details). Following mild acid treatment, the side-chain protected peptides where then cyclized by amide bond formation between Gly31 and Arg32 by activation of the C-terminal Gly to avoid any possible racemization. Solution-phase cyclization of these peptides proceeded cleanly after which side chain deprotection and HPLC purification provided the desired cyclic peptides (Table S1†). The peptides were tested in the in vitro rhamnosylation reaction with EarP_Pa and TDP-Rha, and the rhamnosylated products were identified by an increase in the mass of +146 Da with RP-LCMS. Gratifyingly, the prepared cyclic peptides revealed successful modification by EarP, albeit to varying extents. The best substrate identified was the L-Pro–D-Pro-cyclized 11mer fragment of EF-P from P. aeruginosa (11mer_Pa, 85% conversion overnight). The extent of rhamnosylation was calculated from ion intensities in the MS spectra and corrected for the relative ionization factor (RIF) values (Table S3†),34 as described in the ESI.† A detailed kinetic analysis of the rhamnosylation of 11mer_Pa and native protein substrate EF-P was obtained through a time course study. This analysis revealed that the cyclic 11-mer is indeed rhamnosylated in a time-dependent manner albeit with a lower rate of conversion relative to EF-P (Fig. S2†).
Interestingly, follow-up experiments with both shorter and longer cyclic peptides inspired by the successful 11mer_Pa peptide, revealed that the 11mer peptide (nine native amino acids, plus the L/D-Pro motif) was favored as a substrate, as EarP exhibited no activity towards the shorter (7mer_Pa) and only low levels of conversion (14%) were achieved with the longer (15mer_Pa) fragment. Detailed NMR analysis of the rhamnosylated 11mer_Pa peptide (Rha-11mer_Pa, prepared enzymatically) revealed that the rhamnose-arginine glycosidic linkage was formed in an α-stereoselective fashion (Fig. 2C and S3†), identical to the linkage described for Arg-rhamnosylation of EF-P.25,26 Interestingly, our initial attempts at purifying Rha-11mer_Pa using anion-exchange under basic conditions, led to full epimerization to the β-linked Arg-Rha species (Fig. S4†), in accordance with previous observations by Payne and co-workers.26
EarP allows for sequence promiscuity, but it is sensitive to the cyclization strategy
Encouraged by the successful rhamnosylation of 11mer_Pa, we set out to map the promiscuity of EarP for the amino acid sequence surrounding Arg32. To this end, the 11mer fragments of EF-P sequences from R. solanacearum and N. meningitidis were tested in the rhamnosylation reaction with EarP from P. aeruginosa (Fig. 2B). Interestingly, the 11mer_Rso peptide with two amino acid mutations compared to 11mer_Pa showed low levels of conversion (14% overnight), whereas the 11mer_Nm peptide bearing five mutations was not accepted as a substrate. While these cyclic 11mer peptides do not show high conversion with EarP_Pa, this does not exclude the possibility that they may be substrates for their associated native enzymes EarP_Rso and EarP_Nm, respectively.The co-crystal structure of EF-P bound to EarP revealed that Ala34 in EF-P undergoes a significant conformational change.29 It appears that upon binding to EarP, the “bulge” formed by Ala34 is significantly reduced, leading to a more narrowly shaped and structured loop than that in free EF-P. To investigate whether Ala34 and the concomitant conformational movement are important for binding, the residue was completely removed in peptide 11mer_A34_Pa. Interestingly, this substrate was not rhamnosylated by EarP, suggesting that the Ala34 bulge is important for recognition. Whereas the co-crystal structure suggests that Ala34 is not directly involved in binding to EarP,29 it is reasonable to assume that this residue contributes to shaping the β-hairpin, which in turn positions Arg32 in the active site. This was further corroborated by substituting Ala34 with glycine (11mer_A34G_Pa), a smaller and more flexible amino acid. This mutant retained its role as a substrate for EarP, albeit with reduced efficiency (41% conversion).
Next, alternate cyclization strategies were compared to assess their impact on recognition by EarP (Table S1, Fig. S5†). Specifically, peptides of varying lengths, based upon the same key EF-P amino acid substrate sequence, were synthesized and cyclized using CLIPS-35 (Chemical Linkage of Peptides onto Scaffolds) and disulfide strategies36 to introduce conformational rigidity and flexibility, respectively. In addition, we also prepared linear peptides containing the same EF-P sequence with Trp and Phe residues to introduce a so-called “tryptophan zipper” motif known to induce interstrand H-bonding as another means of generating a β-turn mimic.37 Notably, the majority of the trp-zip designs (especially longer sequences of 13mers, 17mers and 18mers) led to almost immediate protein precipitation when incubated with EarP, indicating that the hydrophobicity of these substrates mimics is not compatible with forming and maintaining a soluble complex with the enzyme.
NMR studies reveal β-hairpin formation in the active peptide substrates
Suitable EarP substrates were further investigated with several NMR techniques to gain understanding of the secondary structure, with the most pronounced effects depicted in the Fig. 2D. By performing temperature studies of the chemical shift of amide-hydrogens, two NH groups (Asn28 and Asn33) with low temperature coefficients were identified (Table S4†), indicative of the β-hairpin-forming interstrand hydrogen bonds (dashed lines). Additionally, a number of characteristic Nuclear Overhauser Effect (NOE) signals (medium and weak) were observed, indicative of the β-hairpin secondary structure (Table S5†). Interestingly, the majority of the observed NOE signals that are characteristic of a β-hairpin structure (Asn28NH-Val36NH, Lys29HA-Val36NH, Lys29HA-Ala35HA, Ser30NH-Ala35HA) are clustered in close proximity to the L-Pro–D-Pro motif, which supports the ability of this template to induce the twist of the peptide structure and consequently bring the strands together for H-bonding. As can be seen from the Fig. 2D this structure-inducing effect of the L-Pro–D-Pro template tapers off towards the Ala35 residue, as the bulge most likely does not allow strands to come close together. Finally, the NOE signals and another H-bond (Asn33) re-appear around Arg32, presumably induced by the Gly31-Arg32 β-turn.Compared to the original 11mer_Pa peptide, the 11mer_A34G_Pa variant features only one of the two H-bonds (NH of Asn28, Table S4†) and several NOE signals are missing (K29HA-V36NH, S30NH-A35HA, Table S5†). It was also shown to exhibit more flexibility, presumably due to the inclusion of the inherently more flexible glycine in the loop. In the case of 11mer_A34_Pa, that showed no conversion to product, only very few structural elements were preserved (sequential NOE signals of P27HA-N28NH and R32NH-N33NH, Table S5†), and the structure was found to lack one of the two H-bonds (NH of Asn33, Table S4†) present in the 11mer_Pa that enable the formation of the β-hairpin structure. The 11mer_Rso variant structure has both H-bonds found in 11mer_Pa (NH of Ser28 and Asn33, Table S4†) and more of β-hairpin characteristic NOEs, although S28NH-V36NH was absent (Table S5†). On the other hand, it was not possible to determine the structure of the 11mer_Nm peptide, as it appears to be a mixture of several structures due to cis/trans isomerization of Pro residues. It is therefore difficult to conclude whether absence of conversion for 11mer_Nm peptide stems from the sequence variation or lack of structure. NMR analysis of the structures of the linear peptide, CLIPS-, disulfide- and Trp-rich peptides showed that these peptides tend to form disordered structures and show close to none of the β-sheet structure (see ESI† NMR spectra of these peptides).
EarP has a low binding affinity for 11mer_Pa
In order to study the binding between EarP and 11mer_Pa peptide different experimental methods were employed. Saturation-Transfer Difference (STD) NMR is a widely employed strategy to observe binding between proteins and ligands, even at low binding affinities.38 However, several attempts at measuring STD-NMR for the EarP-11mer_Pa peptide complex, in the presence and absence of TDP, provided no definitive proof of binding affinity (data not shown). Similarly, attempts to identify the amino acid residues of 11mer_Pa involved in binding to EarP with (TR)-NOE measurements proved unsuccessful (data not shown), presumably due to the low affinity between the protein and the peptide substrate. In a final attempt to quantify the binding affinity of EarP for the best substrate 11mer_Pa, Isothermal Titration Calorimetry (ITC) studies were performed. Tight binding of the native protein substrate EF-P was measured with a binding constant Kd of 473 ± 94 nM for EarP (Fig. 3A), in agreement with reported values.29 Conversely, binding of 11mer_Pa to EarP could not be detected by ITC using a range of increasing concentrations (Fig. 3B). Even a displacement experiment, in which EF-P was titrated into a solution of pre-formed 11mer_Pa–EarP–TDP complex, did not reveal binding of 11mer-Pa to EarP (data not shown). The absence of clear results from the STD-NMR and ITC suggest that the binding affinity of EarP for 11mer_Pa is too low (mM range or higher) to clearly visualize and quantify using such techniques. | ||
| Fig. 3 (A) ITC studies reveal strong binding between EF-P (233 μM) and EarP (20 μM) in the presence of 60 μM TDP. (B) No apparent binding was observed between 11mer_Pa (10 mM) and EarP (20 μM) with ITC in the presence of 60 μM TDP. (C) Dot blot affinity assay of a two-fold serial dilution of 35 μM Rha-EFP (C1) and 60 μM Rha-11mer_Pa (C2) binding to the anti-ArgRha antibody, and visualized using anti-rabbit Alexa488 secondary antibody. Non-rhamnosylated substrates are not recognized by the anti-ArgRha antibody (C3 and C4). | ||
The rhamnosylated β-hairpin peptide is recognized by anti-ArgRha antibodies
Finally, we investigated the structural similarity between rhamnosylated EF-P (Rha-EF-P), and the in vitro rhamnosylated cyclic peptide (Rha-11mer_Pa). In previous studies, antibodies against the Rha-Arg modification were raised using synthesized linear Rha-peptide coupled to BSA.27 To determine binding of the anti-ArgRha antibodies to the rhamnosylated cyclic peptide, we performed a dot blot affinity assay using freshly rhamnosylated 11mer_Pa (Rha-11mer_Pa) and Rha-EF-P as a control. As can be seen in Fig. 3C, a clear fluorescent signal belonging to the formation of Rha-11mer_Pa (C2) was observed well into low micromolar concentration (up to 4 μM). As can be seen from the Fig. 3C, a similar, albeit more intense signal was obtained for the native substrate (Rha-EFP, C1). At the same time, identical concentrations of non-modified substrates were not detected in this assay (Fig. 3, C3 and C4). Interestingly, a similar dot blot affinity study with the β-linked Rha-11mer_Pa product, serendipitously obtained after complete anomerization during anion exchange chromatography under basic conditions (vide supra), revealed very little binding to anti-ArgRha antibodies (Fig. S6†). This establishes the selectivity of the antibodies for the α-anomeric linkage in the Arg-Rha glycosidic bond.Discussion
As protein glycosylation is a non-templated process, it is generally governed by co-localization of the necessary enzymes and substrates, and specific motifs in the protein substrate that dictate glycosylation. A thorough understanding of the molecular basis underlying site-specific glycosylation is paramount to predicting protein glycosylation, and to combining that knowledge with functional effects to fully understand the impact of protein glycosylation on health and disease. As novel bacterial protein glycosylation systems are identified at a steady pace, knowledge of the chemical and structural requirements at play is also increasing. Bacterial glycoproteins are often involved in cell homeostasis and the initiation of infection, therefore the enzymes involved in production of bacterial glycoproteins are interesting targets for the development of novel antimicrobial strategies.Here we report the successful rhamnosylation of a cyclic peptide fragment, 11mer_Pa, designed to mimic the native EF-P substrate of the EarP rhamnosyltransferase. This 11mer L-Pro–D-Pro_Pa peptide is the smallest fragment of EF-P reported to date to be successfully rhamnosylated by EarP. The combination of enzyme activity assays and NMR structural analysis reveals that activity is directly linked to the propensity of the cyclic peptide to form a structured β-hairpin motif. Moreover, the glycosidic linkage is formed in an α-stereoselective fashion, analogous to the native Rha-EF-P and the resulting α-Rha-11mer epitope is successfully recognized by anti-ArgRha antibody. The results of the various 11mer peptide mutants and different cyclization strategies indicate that substrate recognition by the EarP rhamnosyltransferase is highly dependent on the conformation of the substrate and that some sequence variation is tolerated. Developing a successful structural mimic of EF-P beta-hairpin has proved to be challenging, as the majority of the strategies for secondary structure stabilization investigated led to inactive substrates. It would appear that many of the peptide mimics generated in this project are not able to recapitulate important enzyme–substrate contacts in the enzyme active site. In this regard, our study reveals that both specific amino acid residues and an optimal secondary structure are important for recognition by EarP.
The success of the substrate mimics bearing the L-Pro–D-Pro motif may be attributed to the right-handed twist that brings the strands together to allow the formation of H-bonds that stabilize the secondary structure.39 In contrast, CLIPS-bearing peptides may be too bulky to fit in the narrow active site of EarP, whereas disulfide cyclization may induce too much rotational freedom and a less defined β-hairpin. Trp zippers, although widely reported in literature to form β-sheet structures, did not induce measurable secondary structures in the peptides in our investigation, and proved to be too hydrophobic to be suitable EarP substrates. Alternative methods of secondary structure stabilization, such as N-methylation to reduce flexibility of the structure may be explored in the future.40
As apparent from the co-crystal structure, there are many points of contact between EarP and EF-P that are likely to contribute to substrate recognition (Fig. 2A). Interestingly, while the majority of the EarP–EF-P contacts resides around the Arg32-containing β-hairpin that we chose to mimic, several residues of the neighboring β-strand have also been shown to be involved in binding to EarP.29 Consequently, the low affinity of 11mer_Pa for EarP may be increased by extending the current scaffold to include more contact points. Notably, residue Lys55 is a promising residue to consider, particularly as replacing it by alanine resulted in a 200-fold decrease in affinity for EarP.29 One strategy for improving the affinity for EarP and the rates of conversion of future peptidomimetic substrates might include incorporation of either a fragment of the third strand, or a single Lys residue into the peptide. While it can be expected that expanding the structure to include the β-sheet increases the substrate affinity, β-sheets and other secondary structures are difficult to design as isolated motifs. They are stabilized by a larger protein structure they are found in, and without it, they tend to misfold and aggregate.
Conclusion
Whereas the structural determinants for asparagine-linked protein glycosylation are largely based on the primary sequence (consensus sequence), our results suggest that for bacterial arginine-rhamnosylation a specific secondary structural motif is required. The clear importance of secondary structure, and more specifically a β-hairpin motif, as the minimal structural epitope for protein glycosylation may be a unique characteristic of this class of enzymes. Taken together these findings show that the propensity of the (cyclic) peptides to form a β-hairpin structure is an important substrate prerequisite for EarP rhamnosyltransferase and can be directly correlated to activity of the enzyme towards various peptide substrates. This work provides important insights into the recognition motifs for bacterial arginine rhamnosylation which will be useful for the development of future substrate mimics or structure-guided design of peptide inhibitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ralph Krafczyk for help with optimization of the antibody-based assay. This work was financially supported by the Dutch Organization for Scientific Research (VENI 722.016.006) and the European Union through the Rosalind Franklin Fellowship COFUND project 60021 (both to M. T. C. W.), and the European Research Council (ERC consolidator grant to N. I. M., grant agreement no. 725523) and J. L. is grateful for funding from the Deutsche Forschungsgemeinschaft (LA 3658/1-1).References
- G. A. Khoury, R. C. Baliban and C. A. Floudas, Sci. Rep., 2011, 1, 90 CrossRef CAS PubMed.
- D. J. Kelleher and R. Gilmore, Glycobiology, 2006, 16, 47–62 CrossRef PubMed.
- C. M. Szymanski, R. Yao, C. P. Ewing, T. J. Trust and P. Guerry, Mol. Microbiol., 1999, 32, 1022–1030 CrossRef CAS PubMed.
- A. Dell, A. Galadari, F. Sastre and P. Hitchen, Int. J. Microbiol., 2010, 148178 Search PubMed.
- A. Yan and W. J. Lennarz, J. Biol. Chem., 2005, 280, 3121–3124 CrossRef CAS PubMed.
- C. Lizak, S. Gerber, S. Numao, M. Aebi and K. P. Locher, Nature, 2011, 474, 350–355 CrossRef CAS PubMed.
- E. Bause, Biochem. Soc. Trans., 1984, 12, 514–517 CrossRef CAS PubMed.
- B. Imperiali, K. L. Shannon, M. Unno and K. W. Rickert, J. Am. Chem. Soc., 1992, 114, 7944–7945 CrossRef CAS.
- C. Lizak, S. Gerber, G. Michaud, M. Schubert, Y.-Y. Fan, M. Bucher, T. Darbre, M. Aebi, J.-L. Reymond and K. P. Locher, Nat. Commun., 2013, 4, 2627 CrossRef PubMed.
- B. Imperiali, Acc. Chem. Res., 1997, 30, 452–459 CrossRef CAS.
- A. J. Petrescu, A. L. Milac, S. M. Petrescu, R. A. Dwek and M. R. Wormland, Glycobiology, 2004, 14, 103–114 CrossRef CAS PubMed.
- J. M. Silverman and B. Imperiali, J. Biol. Chem., 2016, 291, 22001–22010 CrossRef CAS PubMed.
- Z. Li, M. Fischer, M. Satkunarajah, D. Zhou, S. G. Withers and J. M. Rini, Nat. Commun., 2017, 8, 185 CrossRef CAS PubMed.
- Z. Li, K. Han, J. E. Pak, M. Satkunarajah, D. Zhou and J. M. Rini, Nat. Chem. Biol., 2017, 13, 757–763 CrossRef CAS PubMed.
- H. Nothaft and C. M. Szymanski, Curr. Opin. Chem. Biol., 2019, 53, 16–24 CrossRef CAS PubMed.
- J. Lassak, E. C. Keilhauer, M. Fürst, K. Wuichet, J. Gödeke, A. L. Starosta, J.-M. Chen, L. Søgaard-Andersen, J. Rohr, D. N. Wilson, S. Häussler, M. Mann and K. Jung, Nat. Chem. Biol., 2015, 11, 266–270 CrossRef CAS PubMed.
- D. Gast, F. Koller, R. Krafczyk, L. Bauer, S. Wunder, J. Lassak and A. Hoffmann-Röder, Org. Biomol. Chem., 2020, 18, 6823–6828 RSC.
- A. Rajkovic, S. Erickson, A. Witzky, O. E. Branson, J. Seo, P. R. Gafken, M. A. Frietas, J. P. Whitelegge, K. F. Faull, W. Navarre, A. J. Darwin and M. Ibba, mBio, 2015, 6, e00823 CrossRef CAS PubMed.
- D. G. Singh, J. Lomako, W. M. Lomako, W. J. Whelan, H. E. Meyer, M. Serwe and J. W. Metzger, FEBS Lett., 1995, 376, 61–64 CrossRef CAS PubMed.
- S. Li, L. Zhang, Q. Yao, L. Li, N. Dong, J. Rong, W. Gao, X. Ding, L. Sun, X. Chen, S. Chen and F. Shao, Nature, 2013, 501, 242–246 CrossRef CAS PubMed.
- S. Ude, J. Lassak, A. L. Starosta, T. Kraxenberger, D. N. Wilson and K. Jung, Science, 2013, 339, 82–85 CrossRef CAS PubMed.
- L. Peil, A. L. Starosta, J. Lassak, G. C. Atkinson, K. Virumäe, M. Spitzer, T. Tenson, K. Jung, J. Remme and D. N. Wilson, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 15265–15270 CrossRef CAS PubMed.
- J. Lassak, D. N. Wilson and K. Jung, Mol. Microbiol., 2016, 99, 219–235 CrossRef CAS PubMed.
- T. Yanagisawa, H. Takahashi, T. Suzuki, A. Masuda, N. Dohmae and S. Yokoyama, PLoS One, 2015, 11, e0147907 CrossRef PubMed.
- X. Li, R. Krafczyk, J. Macošek, Y.-L. Li, Y. Zou, B. Simon, X. Pan, Q.-Y. Wu, F. Yan, S. Li, J. Hennig, K. Jung, J. Lassak and H.-G. Hu, Chem. Sci., 2016, 7, 6995–7001 RSC.
- S. Wang, L. Corcilius, P. P. Sharp, A. Rajkovic, M. Ibba, B. L. Parker and R. J. Payne, Chem. Sci., 2017, 8, 2296–2302 RSC.
- R. Krafczyk, J. Macošek, P. K. A. Jagtap, D. Gast, S. Wunder, P. Mitra, A. K. Jha, J. Rohr, A. Hoffman-Röder, K. Jung, J. Hennig and J. Lassak, mBio, 2017, 8, e014112 CrossRef PubMed.
- T. Sengoku, T. Suzuki, N. Dohmae, C. Watanabe, T. Honma, Y. Hikida, Y. Yamaguchi, H. Takahashi, S. Yokoyama and T. Yanagisawa, Nat. Chem. Biol., 2018, 14, 368–374 CrossRef CAS PubMed.
- C. He, N. Liu, F. Li, X. Jia, H. Peng, Y. Liu and Y. Xiao, J. Bacteriol., 2019, 201, e00128 CrossRef CAS PubMed.
- N. C. Kyrpides, C. R. Woese and C. A. Ouzounis, Trends Biochem. Sci., 1996, 21, 425–426 CrossRef CAS PubMed.
- W. Volkwein, R. Krafczyk, P. K. A. Jagtap, M. Parr, E. Mankina, J. Macošek, Z. Guo, M. J. L. J. Fürst, M. Pfab, D. Frishman, J. Hennig, K. Jung and J. Lassak, Front. Microbiol., 2019, 10, 1148 CrossRef PubMed.
- Q. Lu, S. Li and F. Shao, Trends Microbiol., 2015, 23, 630–641 CrossRef CAS PubMed.
- J. A. Robinson, Acc. Chem. Res., 2007, 41, 1278–1288 CrossRef PubMed.
- W. Kightlinger, L. Lin, M. Rosztoczy, W. Li, M. P. DeLisa, M. Mrksich and M. C. Jewett, Nat. Chem. Biol., 2018, 14, 627–635 CrossRef CAS PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS PubMed.
- C. M. Santiveri, E. León, M. Rico and M. A. Jiménez, Chem.–Eur J., 2008, 14, 488–499 CrossRef CAS PubMed.
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U.S.A., 2001, 98, 5578–5583 CrossRef CAS PubMed.
- A. Viegas, J. Manso, F. L. Nobrega and E. J. Cabrita, J. Chem. Educ., 2011, 88, 990–994 CrossRef CAS.
- J. A. Robinson, Chimia, 2013, 67, 885–890 CrossRef CAS PubMed.
- M. P. Gimeno, A. Glas, O. Koch and T. N. Grossman, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05823h |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |