
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn engineered biosynthetic–synthetic platform for production of halogenated indolmycin antibiotics†
Elesha R.
Hoffarth
 ,
Sunnie
Kong
,
Hai-Yan
He‡
and
Katherine S.
Ryan
*
,
Sunnie
Kong
,
Hai-Yan
He‡
and
Katherine S.
Ryan
*
Department of Chemistry, The University of British Columbia, Vancouver, Canada. E-mail: ksryan@chem.ubc.ca
First published on 1st June 2021
Abstract
Indolmycin is an antibiotic from Streptomyces griseus ATCC 12648 with activity against Helicobacter pylori, Plasmodium falciparum, and methicillin-resistant Staphylococcus aureus. Here we describe the use of the indolmycin biosynthetic genes in E. coli to make indolmycenic acid, a chiral intermediate in indolmycin biosynthesis, which can then be converted to indolmycin through a three-step synthesis. To expand indolmycin structural diversity, we introduce a promiscuous tryptophanyl-tRNA synthetase gene (trpS) into our E. coli production system and feed halogenated indoles to generate the corresponding indolmycenic acids, ultimately allowing us to access indolmycin derivatives through synthesis. Bioactivity testing against methicillin-resistant Staphylococcus aureus showed modest antibiotic activity for 5-, 6-, and 7-fluoro-indolmycin.
Antibiotic-resistant bacteria pose a great threat to human health,1–4 and the rates of new antibiotic discoveries and clinical approvals have been in a steep decline since the 1980s.1 Without the discovery and development of new antibiotics, drug-resistant pathogens, such as methicillin-resistant Staphylococcus aureus (MRSA), will become increasingly prevalent.2–4 One strategy to increase antibiotic development has been to “rediscover” known, but underdeveloped, antibiotics.1 One such example is indolmycin, which was originally discovered in 1960 from Streptomyces griseus ATCC 126485 but was not originally developed for clinical use because of its narrow spectrum of activity6–10 and its interference with tryptophan catabolism in the liver.9,10 However, reignited interest in this old antibiotic led to the discovery of its activity against Helicobacter pylori,6Plasmodium falciparum,11 and MRSA.12 For MRSA, indolmycin was found to be active against mupirocin- and fuscidic acid-resistant MRSA strains, with strains resistant to indolmycin emerging infrequently and with reduced fitness compared to sensitive strains.12 In addition, indolmycin has been shown to have minimal activity against common members of the human microbiota, suggesting that its narrow spectrum of activity is an asset.6 The first indole-substituted derivatives, 5-hydroxy and 5-methoxyindolmycin, were made by precursor-directed feeding of the indolmycin producer, Streptomyces griseus ATCC 12648, and they showed modest improvements in bioactivity against S. aureus and Escherichia coli.13 Two practical synthetic routes to indolmycin and some indole-substituted derivatives have been reported more recently,14,15 which enabled access to a small variety of indole-substituted derivatives. Additionally, a previous patent has described synthetic methods to produce a variety of derivatives; however, these methods do not appear to offer stereochemical control, and some require tailoring steps specific to each analog.16 Therefore, further development of indolmycin would benefit from a simpler diversification method that could be applied to produce a wider variety of analogs with stereochemical control.
Inspired by early biosynthetic studies,17,18 our group previously identified the indolmycin gene cluster and elucidated the biosynthetic pathway, demonstrating that indolmycin (1) is assembled from tryptophan, arginine and S-adenosylmethionine (SAM) in a three-part process (Fig. 1).19 In the first part, L-arginine is oxidized by Ind4 in an oxygen- and PLP-dependent reaction to 4,5-dehydro-2-iminoarginine, which is then enantioselectively reduced by imine reductase Ind5 and its chaperone Ind6 to 4,5-dehydro-D-arginine. In the second part, tryptophan (2) is deaminated by PLP-dependent transaminases, giving indole pyruvate (3). Compound 3 is then methylated by SAM-dependent C-methyltransferase Ind1 to 3-methyl-indolepyruvate (4) which is reduced by NADH-dependent ketone reductase Ind2 to form indolmycenic acid (5). Then, in the third part, 4,5-dehydro-D-arginine and 5 are coupled in an ATP-dependent fashion by Ind3 and Ind6, resulting in an oxazolinone-cyclized molecule, N-desmethyl-indolmycin, which is finally N-methylated by Ind7, a SAM-dependent N-methyltransferase, to form 1.
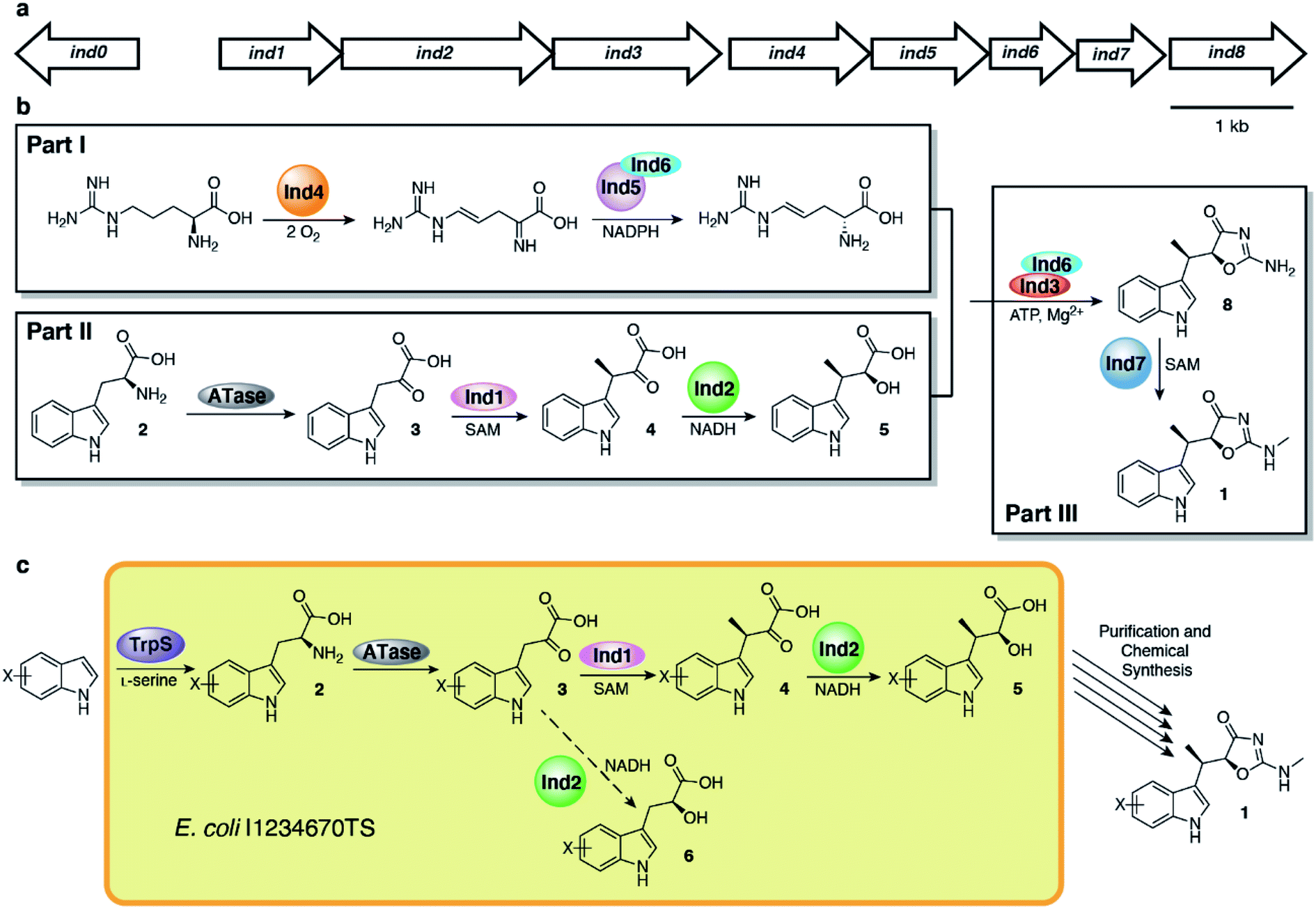 | ||
| Fig. 1 Indolmycin biosynthesis from Streptomyces griseus ATCC 12648. (a) Indolmycin biosynthetic gene cluster. (b) Indolmycin biosynthetic pathway from Streptomyces griseus ATCC 12648. (c) Semi-synthetic scheme towards indolmycin and derivatives using indolmycin biosynthetic genes. The dashed arrow indicates a predicted side-product based on LC-MS analysis. | ||
Armed with the elucidated biosynthetic pathway for 1, we set out to create an in vivo system to make 1 in E. coli. We first cloned all necessary genes into four plasmids and co-expressed the genes in E. coli (Fig. S1†), a strain which we named E. coli I1234670P5 (Table S1†). We found that the genes needed to produce indolmycin in E. coli were ind1, ind2, ind3, ind4, ind6, ind7, ind0 and pel5, a homologous gene of ind5 from Paenibacillus elgii B69 showing better production of active protein in E. coli.20,21 We also relied on the activity of endogenous E. coli aminotransferases to catalyze the initial tryptophan deamination step. However, only a small amount of 1 was produced (∼170 μg L−1 of bacterial culture) and the yield could not be improved despite our best efforts (Fig. 2a). However, we found that this construct produced substantial amounts of 5 ([M + H]+ = 220 m/z) at 40–50 mg L−1 of culture, along with a shunt product, C-desmethyl-indolmycenic acid (6; [M + H]+ = 206 m/z).
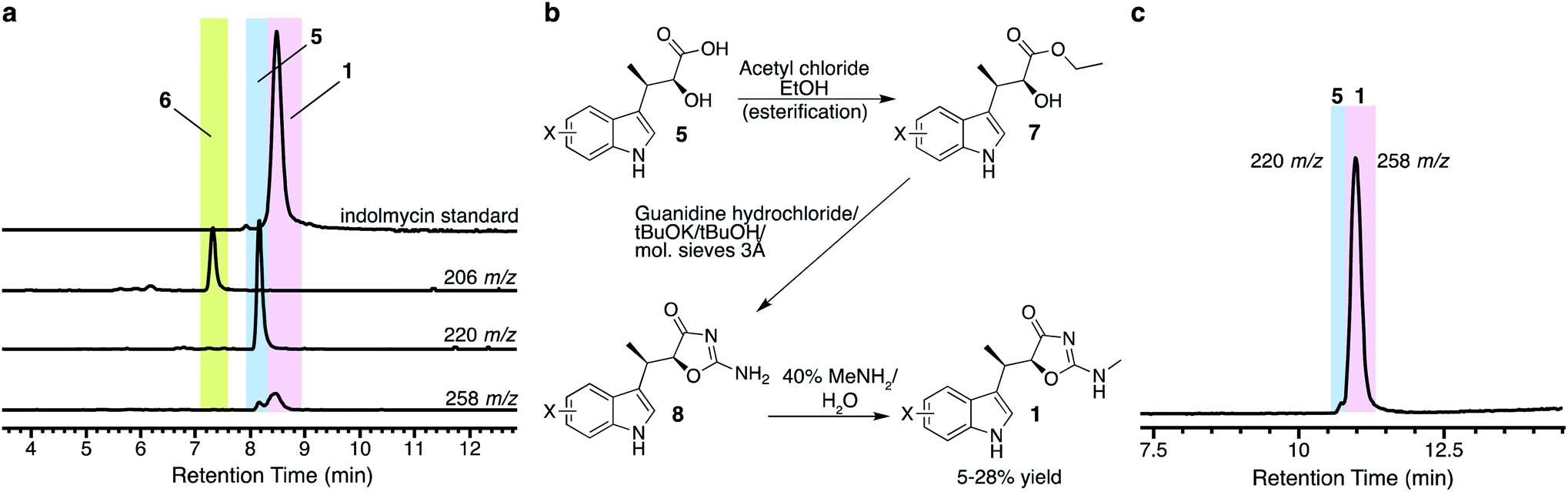 | ||
| Fig. 2 Biosynthetic production of 5 and semi-synthetic production of 1. (a) Extracted ion chromatograms show production of 5 with minimal production of 1 from E. coli I1234670P5. (b) Synthetic scheme to 1 from 5, adapted from literature methods.14,15 (c) Total ion chromatogram of compound 1 isolated after semi-synthesis and final purification by semi-preparative HPLC. Compounds are indicated with coloured boxes and numbered. | ||
Compound 5 itself has been a focus of total synthetic efforts toward 1, as it is the key chiral precursor.14,15,22–26 Since production of 5 was much higher than that of 1 from E. coli I1234670P5, we pursued a semi-synthetic method of obtaining 1 using our biosynthetic platform to access 5, combined with a three-step chemical transformation (Fig. 2b). We attempted to remove extraneous genes from our biosynthetic platform by only including ind0, ind1, and ind2, but the changes resulted in reduced amounts of 5 (Fig. S2†). At this time, it is unclear which of the other genes may be contributing to the production of compound 5. Therefore, we employed the full eight-gene construct toward synthesis of 5. We then adapted the three-step synthesis to make indolmycin,14,15 in which purified 5 was esterified to make the ethyl ester (7; [M + H]+ = 248 m/z; Fig. S3a†), cyclized to give N-desmethyl-indolmycin (8; [M + H]+ = 244 m/z; Fig. S3b and S4a†), and methylated at the exocyclic nitrogen to give 1 ([M + H]+ = 258 m/z; Fig. 2, S4b and Table S4†).
Then, we wanted to make derivatives of 1 from indole derivatives, which are more widely accessible than derivatives of 2. The tryptophan synthase (TrpS) from Salmonella enterica has been previously shown to couple a wide variety of indole derivatives to L-serine to generate derivatives of 2.27 We were able to replace pel5 with trpS in our biosynthetic platform without a reduction in the amount of 5 produced (Fig. S1b†), and we named the resulting strain E. coli I1234670TS. When we fed 5-fluoroindole to E. coli I1234670TS, we observed increased production of fluorinated metabolites, 5-fluoro-indolmycenic acid (5F-5; [M + H]+ = 238 m/z) and 5-fluoro-C-desmethyl-indolmycenic acid (5F-6; [M + H]+ = 224 m/z) (Fig. 3a). We then optimized the feeding conditions, finding that 5F-5 amounts were optimal when we fed E. coli I1234670TS with 0.5 mM 5-fluoroindole per day over two days (Fig. S5†).
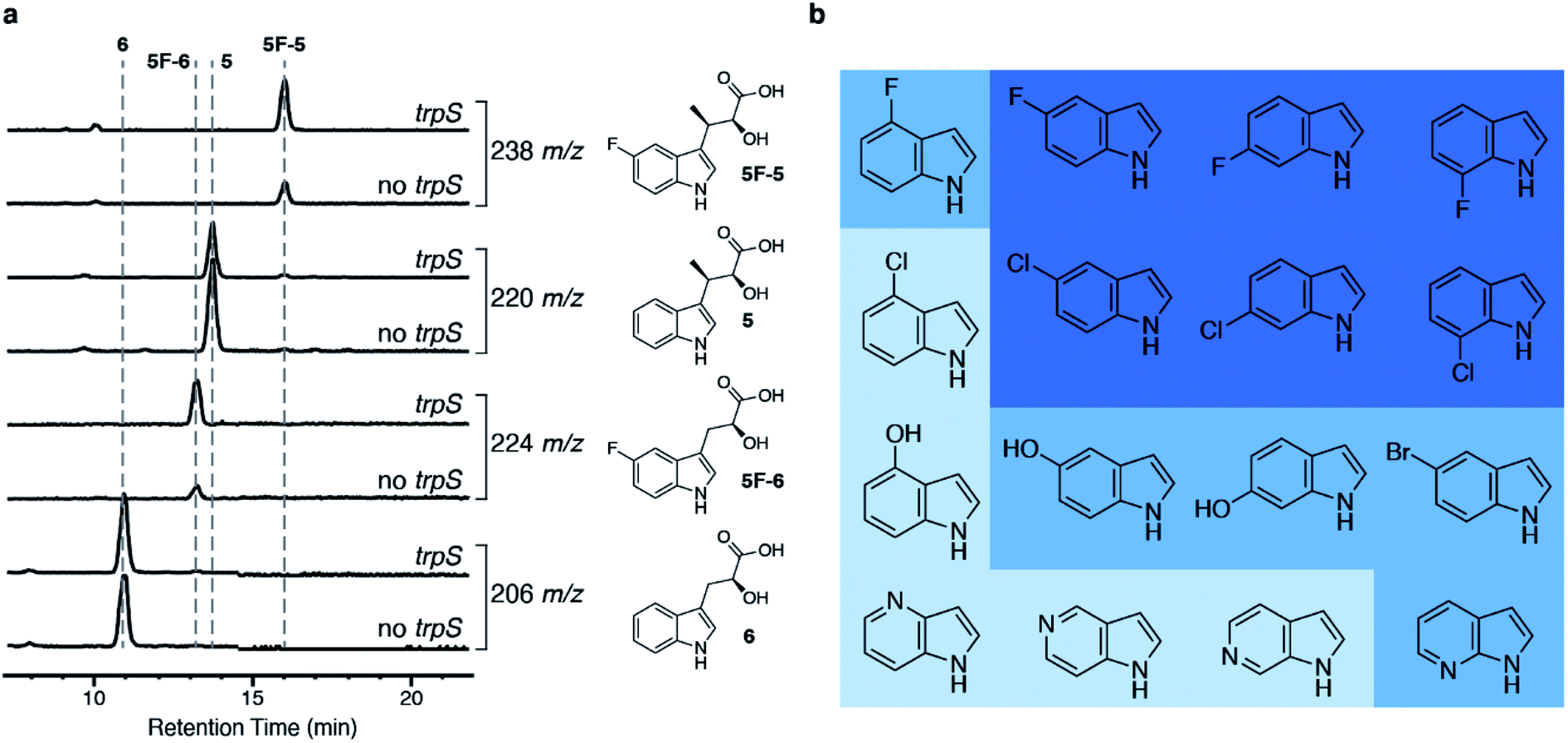 | ||
| Fig. 3 Addition of the trpS gene to the biosynthetic platform allows incorporation of substituted indoles into 5. (a) LC-MS analysis of strains with and without trpS (E. coli I1234670TS and E. coli I1234670P5, respectively) when fed 5-fluoroindole. Chemical structures of compounds with the corresponding extracted ion chromatogram are shown to the right of the traces. (b) Indole derivatives tested. Dark blue shows indoles that were incorporated into an analog of 5 (>48% of underivatized 5 by LC-MS analysis; Table S5†) and further purified; medium blue indicates indoles that were incorporated into an analog of 5 at lower levels (6–22% of underivatized 5 by LC-MS analysis; Table S5†) but were not further verified by purification; and light blue indicates indoles that did not show detectable incorporation into 5. | ||
To determine the scope of indole derivatives accepted by our biosynthetic platform, we fed a variety of indoles to E. coli I1234670TS and monitored the production of 5 and its derivatives by LC-MS. Out of the indoles tested, we found that fluorinated and chlorinated indoles substituted at the 5-, 6- and 7-positions were the best accepted by the biosynthetic platform (Fig. 3b, S6 and S7†). We predict that lower acceptance of indoles substituted at the 4-position may be due to steric hindrance, as 4-fluoroindole was moderately accepted, while 4-chloroindole was not observed at all. Although we observed LC-MS peaks consistent with conversion of some of the azaindoles and hydroxyindoles into 5 derivatives, further work is required to confirm, optimize and scale up the purification of these compounds (Fig. S7†). Cultures producing derivatives of 5, substituted at 5-, 6- and 7-positions, were further scaled up for purification of the 5-derivatives and downstream synthesis of 1-derivatives (Fig. S8–S10 and Table S4†). Each purified derivative of 5 and 1 was characterized by HR-MS and NMR (Table S3 and ESI Methods†). Overall, cultures fed with the fluorinated indoles produced a higher amount of 5-derivatives than the cultures fed with the chlorinated indoles.
1 and its derivatives were tested against MRSA (Fig. S11†). While the fluorinated derivatives showed bioactivity, the chlorinated derivatives of 1 did not show bioactivity at the maximum amount tested in the disk diffusion assay (30 μg). We determined MIC50 values for each fluorinated compound (Table 1). The MIC50 values demonstrate that 1 is a more potent inhibitor of MRSA than its derivatives, while 6F-1 showed the most potent inhibition of MRSA compared to any of the derivatives, followed by 7F-1 and 5F-1. The lack of bioactivity of the chlorinated compounds may be due to the bulky chlorinated substituent hindering the compounds' abilities to bind to the tryptophanyl-tRNA synthetase (TrpRS) target, which is supported by docking studies of the analogs into a bacterial TrpRS structure (Fig. S12†).
| MIC50 (μg mL−1) | |
|---|---|
| 1 | 1.21 ± 0.04 |
| 5F-1 | 32.5 ± 19.6 |
| 6F-1 | 6.49 ± 0.03 |
| 7F-1 | 16.7 ± 4.2 |
| 5Cl-1 | >200 |
| 6Cl-1 | >200 |
| 7Cl-1 | >200 |
Here we have shown that 5-, 6- and 7-fluorinated and chlorinated indole derivatives are accepted as substrates by S. enterica TrpS, E. coli tryptophan aminotransferase, Ind1 and Ind2. The fluorinated indoles, particularly 5-fluoroindole, showed greater incorporation into 5 than the chlorinated indoles, perhaps because the larger chlorine atom may have steric hindrance in some of the enzyme active sites, reducing the efficiency of turnover. This result is consistent with previous studies on TrpS from S. enterica, which shows the lowest acceptance for 4-chloro, 5-chloro and 5-bromo indoles.27 Other tested indoles, 5- and 6-hydroxyindoles and 7-azaindole, were only minimally converted to derivatives of 5 by this semi-synthetic platform, and 4-hydroxyindole and 4-, 5-, and 6-azaindole were not converted at all. Azatryptophans have been previously made using TrpS but required longer reaction times due to reduced nucleophilicity,28–30 which may explain our results. For 4-hydroxyindole, previous studies of hydroxytryptophan production by S. enterica TrpS are limited;29 however, E. coli TrpS was shown to accept 4-hydroxyindole during the production of psilocybin.31 It is unclear at this time why the 4-hydroxylated derivative of 5 was not observed. The substrate acceptance and 5-derivative production could be further improved with optimized reaction and purification conditions and engineering of strains and enzymes. For instance, TrpS has already been the target of many enzyme engineering studies focused on improving the substrate scope.32–38
Previous work by Demain and co-workers demonstrated successful production of 5-hydroxy and 5-methoxy derivatives of 1 from S. griseus fed with the corresponding derivatives of indole or 2; however, 6-substituted derivatives were not converted to derivatives of 1.13 By contrast, our system incorporated halogenated substituted indoles at the 6-position, meaning that the semi-synthetic method overcomes one limitation of the purely biosynthetic method. Development of this semi-synthetic method has also helped to identify some key hurdles to creating a purely biosynthetic method. These hurdles, such as accumulation of 5, an apparent role of proteins in Parts I or III (Fig. 1) in the formation of 5, and substrate scope bottlenecks, could be addressed in future attempts to produce 1 from E. coli. Furthermore, the biosynthetic portion of our system also overcomes challenges associated with synthetic methods. Synthetic studies were typically associated with making 5 through methods such as using a chiral auxiliary,25 epoxide ring-opening,14,24 lipase-assisted enantioselective acetylation,15 or a combination of these methods,26 and few attempts to synthesize halogenated derivatives have been reported, as the methods employed are likely to be incompatible with halogenated indole precursors. A patent has described methods to produce several derivatives of 1, including 4Cl-1 and 6F-1, but stereocenters were not controlled, to the best of our knowledge, and some analogs utilized additional tailoring steps or entirely unique routes.16 Therefore, these patented methods, individually, are not robust enough to enable production of a wide variety of derivatives, making a broad search for potentially bioactive candidates challenging. Our semi-synthetic method of obtaining 1 and its derivatives offers a simple and halogen-compatible alternative to purely synthetic methods that could be used to expand and diversify the accessible indolmycin molecules for broad bioactivity searches. Broad bioactivity searches could be followed by optimization of this platform, or creation of another, to produce enough amounts of the desirable compounds to continue work beyond the initial bioactivity testing. We note that this type of semi-synthetic method could also be applied to the diversification of other tryptophan-derived molecules. Moreover, the fluorinated derivatives of 1 showed bioactivity against MRSA, demonstrating that these compounds may serve as useful molecules for development of new antibiotics. Overall, this work lays a foundation for making derivatives of 1, while demonstrating the value of combining synthetic and biosynthetic methods to make and diversify natural product-derived compounds.
Author contributions
E. R. H. designed the project, completed most experimental work, and wrote the manuscript. S. K. synthesized some of the compounds. H.-Y. H. helped to develop synthetic methods, completed some of the synthetic work, and edited the manuscript. K. S. R. designed the project and wrote the manuscript.Conflicts of interest
E. R. H. and K. S. R. are on a provisional patent application, filed through the University of British Columbia based on the results described here.Acknowledgements
This work was supported by funding from Genome British Columbia (SOF148), the Canadian Institutes of Health Research (FDN-148381), the Natural Science and Engineering Research Council of Canada (RGPIN-2016-03778), and the Michael Smith Foundation for Health Research (Grant no. 16776). We thank Jessie Chen and Elena Polishchuk for assistance with disk diffusion assays and minimal inhibitory concentration determination, Maria Ezhova at the High-Resolution NMR Facility at the University of British Columbia for collecting NMR data, and the UBC Mass Spectrometry Centre for obtaining high-resolution mass spectrometry data.References
- G. A. Durand, D. Raoult and G. Dubourg, Int. J. Antimicrob. Agents, 2019, 53, 371–382 CrossRef CAS PubMed
.
- M. Hutchings, A. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed
- J. F. Prescott, Vet. Microbiol., 2014, 171, 273–278 CrossRef PubMed
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed
- W. S. Marsh, A. L. Garretson and E. M. Wesel, Antibiot. Chemother., 1960, 10, 316–320 CAS
- T. Kanamaru, Y. Nakano, Y. Toyoda, K.-I. Miyagawa, M. Tada, T. Kaisho and M. Nakao, Antimicrob. Agents Chemother., 2001, 45, 2455–2459 CrossRef CAS PubMed
- R. G. Werner, Antimicrob. Agents Chemother., 1980, 18, 858–862 CrossRef CAS PubMed
- G. H. M. Vondenhoff and A. Van Aerschot, Eur. J. Med. Chem., 2011, 46, 5227–5236 CrossRef CAS PubMed
- U. A. Ochsner, X. Sun, T. Jarvis, I. Critchley and N. Janjic, Expert Opin. Invest. Drugs, 2007, 16, 573–593 CrossRef CAS PubMed
- J. G. Hurdle, A. J. O'Neill and I. Chopra, Antimicrob. Agents Chemother., 2005, 49, 4821–4833 CrossRef CAS PubMed
- C. F. A. Pasaje, V. Cheung, K. Kennedy, E. E. Lim, J. B. Baell, M. D. W. Griffin and S. A. Ralph, Sci. Rep., 2016, 6, 27531 CrossRef CAS PubMed
- J. G. Hurdle, A. J. O'Neill and I. Chopra, J. Antimicrob. Chemother., 2004, 54, 549–552 CrossRef CAS PubMed
- R. G. Werner and A. L. Demain, J. Antibiot., 1981, 551–554 CrossRef CAS PubMed
- A. Hasuoka, Y. Nakayama, M. Adachi, H. Kamiguchi and K. Kamiyama, Chem. Pharm. Bull., 2001, 49, 1604–1608 CrossRef CAS PubMed
- N. Sutou, K. Kato and H. Akita, Tetrahedron: Asymmetry, 2008, 19, 1833–1838 CrossRef CAS
-
United States, US Pat., US6169102B1, 2001 Search PubMed
- U. Hornemann, L. H. Hurley, M. K. Speedie and H. G. Floss, J. Am. Chem. Soc., 1971, 93, 3028–3035 CrossRef CAS PubMed
- M. K. Speedie, U. Hornemann and H. G. Floss, J. Biol. Chem., 1975, 250, 7819–7825 CrossRef CAS
- Y.-L. Du, L. M. Alkhalaf and K. S. Ryan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2717–2722 CrossRef CAS PubMed
- J. Guo, M. A. Higgins, P. Daniel-Ivad and K. S. Ryan, J. Am. Chem. Soc., 2019, 141, 12258–12267 CrossRef CAS PubMed
- Y.-L. Du, R. Singh, L. M. Alkhalaf, E. Kuatsjah, H.-Y. He, L. D. Eltis and K. S. Ryan, Nat. Chem. Biol., 2016, 12, 194–199 CrossRef CAS PubMed
- M. S. von Wittenau and H. Els, J. Am. Chem. Soc., 1963, 85, 3425–3431 CrossRef
- M. N. Preobrazhenskaya, E. G. Balashova, K. F. Turchin, E. N. Padeiskaya, N. V. Uvarova, G. N. Pershin and N. N. Suvorov, Tetrahedron, 1968, 24, 6131–6143 CrossRef CAS PubMed
- T. H. Chan and K. K. Hill, J. Org. Chem., 1970, 35, 3519–3521 CrossRef CAS PubMed
- T. Takeda and T. Mukaiyama, Chem. Lett., 1980, 9, 163–166 CrossRef
- H. Akita, T. Kawaguchi, Y. Enoki and T. Oishi, Chem. Pharm. Bull., 1990, 38, 323–328 CrossRef CAS
- D. R. M. Smith, T. Willemse, D. S. Gkotsi, W. Schepens, B. U. W. Maes, S. Ballet and R. J. M. Goss, Org. Lett., 2014, 16, 2622–2625 CrossRef CAS PubMed
- M. J. Sloan and R. S. Phillips, Bioorg. Med. Chem. Lett., 1992, 2, 1053–1056 CrossRef CAS
- R. S. Phillips, Tetrahedron: Asymmetry, 2004, 15, 2787–2792 CrossRef CAS
- M. Winn, D. Francis and J. Micklefield, Angew. Chem., Int. Ed., 2018, 57, 6830–6833 CrossRef CAS PubMed
- A. M. Adams, N. A. Kaplan, Z. Wei, J. D. Brinton, C. S. Monnier, A. L. Enacopol, T. A. Ramelot and J. A. Jones, Metab. Eng., 2019, 56, 111–119 CrossRef CAS PubMed
- M. Herger, P. Van Roye, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, J. Am. Chem. Soc., 2016, 138, 8388–8391 CrossRef CAS PubMed
- D. Francis, M. Winn, J. Latham, M. F. Greaney and J. Micklefield, ChemBioChem, 2017, 18, 382–386 CrossRef CAS PubMed
- D. K. Romney, J. Murciano-calles, J. Wehrmüller and F. H. Arnold, J. Am. Chem. Soc., 2017, 23, 22–23 Search PubMed
- C. E. Boville, D. K. Romney, P. J. Almhjell, M. Sieben and F. H. Arnold, J. Org. Chem., 2018, 83, 7447–7452 CrossRef CAS
- A. R. Buller, P. Van Roye, J. Murciano-Calles and F. H. Arnold, Biochemistry, 2016, 55, 7043–7046 CrossRef CAS
-
J. Murciano-Calles, A. R. Buller and F. H. Arnold, in Directed Enzyme Evolution: Advances and Applications, Springer International Publishing, 2017, pp. 1–16 Search PubMed
- C. E. Boville, R. A. Scheele, P. Koch, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 14764–14768 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05843b |
| ‡ Current address: Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2021 |