
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced copper-catalyzed alkynylation and amination of remote unactivated C(sp3)-H bonds†
Zhusong
Cao
a,
Jianye
Li
b,
Youwen
Sun
a,
Hanwen
Zhang
a,
Xueling
Mo
a,
Xin
Cao
 *c and
Guozhu
Zhang
*ab
*c and
Guozhu
Zhang
*ab
aKey Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: guozhuzhang@sioc.ac.cn
bCollege of Chemistry, Central China Normal University (CCNU), 152 Luoyu Road, Wuhan, Hubei 430079, P. R. China
cZhongshan Hospital, Fudan University, 180 Fenglin Road, Shanghai 200032, P. R. China. E-mail: caox@fudan.edu.cn
First published on 16th February 2021
Abstract
A method for remote radical C–H alkynylation and amination of diverse aliphatic alcohols has been developed. The reaction features a copper nucleophile complex formed in situ as a photocatalyst, which reduces the silicon-tethered aliphatic iodide to an alkyl radical to initiate 1,n-hydrogen atom transfer. Unactivated secondary and tertiary C–H bonds at β, γ, and δ positions can be functionalized in a predictable manner.
Introduction
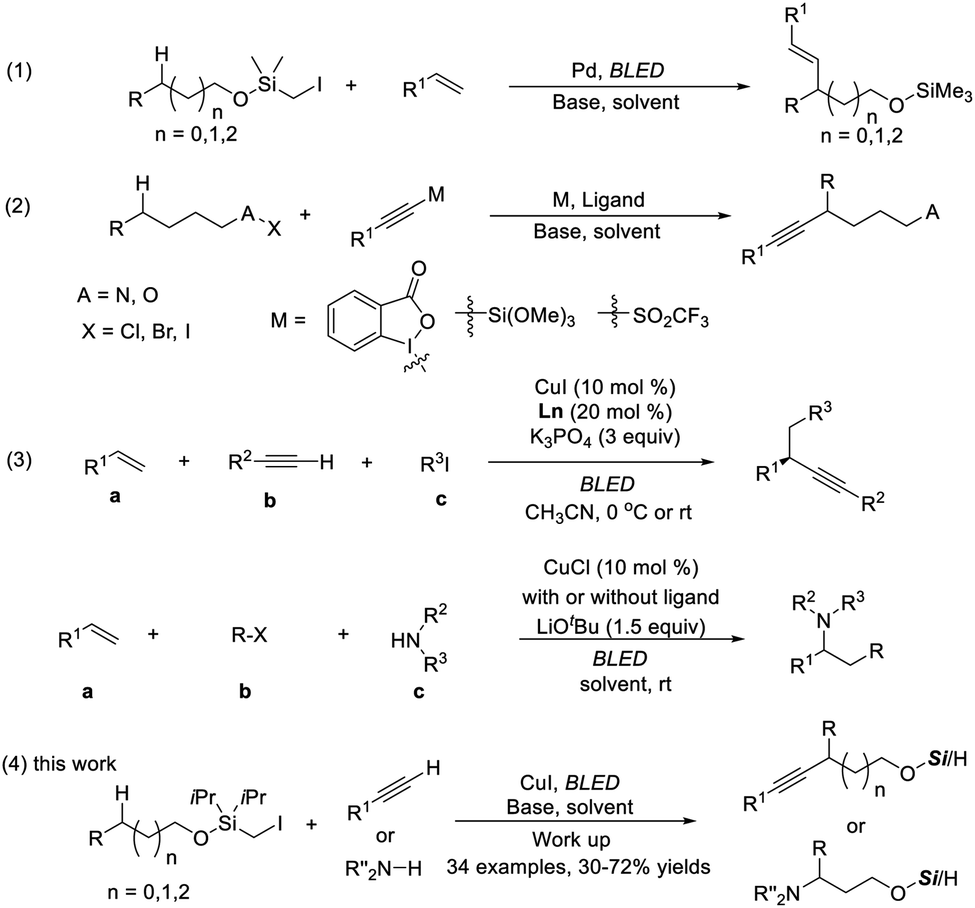
Direct functionalization of inert C(sp3)-H bonds provides an atom- and step-economic strategy for the rapid construction of valuable chemical frameworks.1 Along with transition-metal-mediated processes,2 radical-mediated hydrogen atom transfer (HAT) has emerged as an appealing and efficient strategy to activate and functionalize C(sp3)-H in a selective manner, and significant progress has been made in this field.3An easily installed and removable hydroxyl protecting group, the (halomethyl)silyl group, has been established as an excellent carbon radical precursor and widely applied in radical-type reactions.4 Gevorgyan pioneered remote aliphatic C–H functionalization through radical transposition processes initiated with silicon-tethered carbon radicals.5 Particularly inspiring is the photoinduced palladium-catalyzed remote C–H Heck reaction, allowing ready access to alkenyl alcohols (Scheme 1, eqn (1)).5a
 | ||
| Scheme 1 C–H alkynylation and amination of aliphatic alcohols. | ||
Benefitting from their electronic properties and various methods for further transformations, alkynes have been widely used as pivotal intermediates for the synthesis of complex biologically active or functional molecules.6 To our knowledge, remote C–H alkynylation at unactivated C–H bonds proceeding via intramolecular HAT has been explored only very recently, dominated by 1,5 transposition. Among the established methods, a polarized alkyne (X–Y) (e.g., X![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) R) is required and generally needs multistep synthesis (eqn (2)).7 Approaches allowing the use of a simple terminal alkyne in remote C–H bond alkynylation are limited and therefore highly desired.8
R) is required and generally needs multistep synthesis (eqn (2)).7 Approaches allowing the use of a simple terminal alkyne in remote C–H bond alkynylation are limited and therefore highly desired.8
Photocatalysis using inexpensive and readily available copper complexes experienced a significant growth, exhibiting highly tunable redox properties and diverse reactivity.9 Fu pioneered photo-induced copper-catalyzed C–N bond formation using carbazole as the reactant and photocatalyst,10 while Hwang and Lalic demonstrated that copper could catalyze the coupling of acetylene with aryl11 and alkyl halides12 under light irradiation, with copper-acetylide acting as the photo-excitable intermediate.
Results and discussion
Our group recently discovered a photo-catalytic system of copper(I) and realized the three-component carbon functionalization of alkenes (eqn (3)).13a,b The significance of this work lies in the following: (1) the identification of in situ-formed photoactive Cu(I) species with significantly enhanced reducing capability,13c,d and (2) the use of simple terminal alkynes and amines as coupling partners. Encouraged by the potential of C–H functionalization in synthesis, it was reckoned that carbon radical formation occurred from (halomethyl)silyl ether through Cu-acetylide photoreduction, followed by the selective 1,n-HAT. The newly formed radical could be recaptured by a Nu-copper species, which then furnished the remotely functionalized products.Herein, the successful implementation of this hypothesis is described. In the presence of a single Cu complex, various mono-substituted alkynes and carbazoles undergo a site-selective radical relay alkynylation and amination reaction of aliphatic alcohols (eqn (4)). The reaction proceeds under mild visible-light-induced conditions at room temperature, producing β-, δ-, and γ-functionalized products selectively without the use of exogenous photosensitizers or external oxidants.
Our attempt began by allowing a model substrate Si-tethered iodide 1a to react with 1-ethynyl-4-methylbenzene 2a in the presence of the 2,2′:6′,2′′-terpyridine copper catalyst under blue-light irradiation. After comprehensive investigation of the reaction conditions, it was pleasing to find that the translocated Sonogashira product 3a did form. Under the optimized conditions, 1a provided the desired product 3a in 60% yield after 32 h using CuI as the catalyst and 2,2′:6′,2′′-terpyridine as the ligand under blue-LED (BLED) irradiation (Table 1, entry 1). Under these conditions, the premature alkynylation at the Si-auxiliary site is suppressed to approximately 15%.
|
|
||
|---|---|---|
| Entry | Variation from the “standard conditions” | Yieldb% |
| a 1a (0.1 mmol), 2a (0.15 mmol), CuI (15 mol%), L1 (15 mol%) and K2CO3 (3 equiv.) in MeCN, under N2, rt, blue LEDs, 32 h. b Determined by 1H NMR analysis with internal standard (diethyl phthalate). c Isolated yield. d Pd(OAc)2 (10 mol%), xantphos (20 mol%) and Cs2CO3 (2 equiv.) in PhH, under N2, rt, blue LEDs, 32 h. | ||
| 1 | None | 62(60c) |
| 2 | Without CuI, base or light | 0 |
| 3 | L2 instead of L1 | 41 |
| 4 | L3 or L4 instead of L1 | 0 |
| 5 | K3PO4 | 39 |
| 6 | Cs2CO3 or Na2CO3 instead of K2CO3 | 0 |
| 7 | CuBr instead of CuI | 35 |
| 8 | CuTc instead of CuI | 52 |
| 9 | Cu(OTf)2 instead of CuI | 32 |
| 10 | DMF instead of MeCN | 35 |
| 11 | DCM instead of MeCN | 38 |
| 12 | Et2O or THF instead of MeCN | Trace |
| 13 | Reaction was performed at 10 °C | 20 |
| 14d | Pd(OAc)2, xantphos and Cs2CO3 in PhH | 0 |
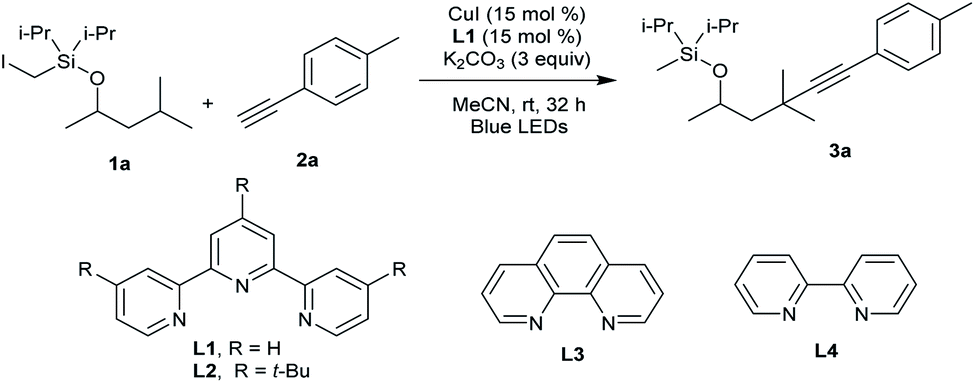
Control experiments determined that product 3a was not produced in the absence of a copper salt, ligand, or base (entry 2). It was found that a structurally similar ligand with tBu substitution on the terpyridine L2 led to decreased yield (entry 3). Bidentate ligands L3 and L4 proved to be inefficient in this transformation (entry 4). Switching the base from K2CO3 to Cs2CO3, Na2CO3, or K3PO4 was detrimental to the reaction, and traces to small amounts of product were observed (entries 5 and 6). The performance of other copper salts such as CuBr and CuTc was also briefly examined, but they were less efficient for this reaction (entries 7–9). Further reaction optimization experiments identified MeCN as the best solvent (entries 10–12). The reaction running at lower temperature decreased the yield (entry 13). The use of the Pd catalyst gave no 3a, highlighting the unique role of Cu in this tandem HAT and coupling process (entry 14).
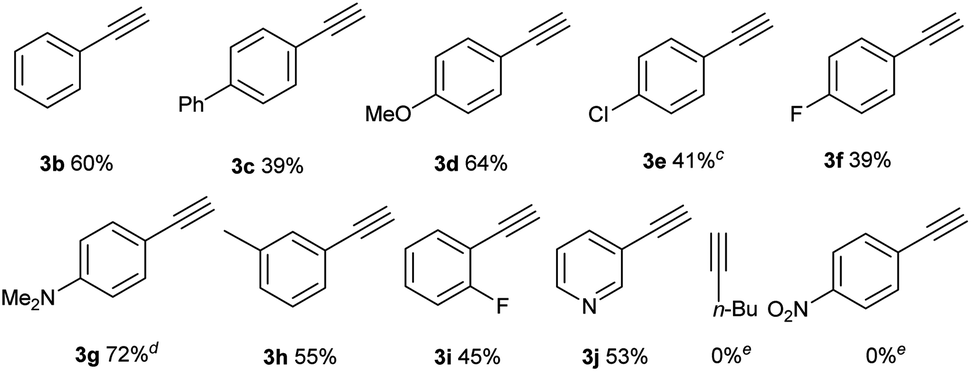
After the optimal reaction conditions were established, the substrate scope with respect to alkynes was first investigated, keeping iodide 1a as the substrate (Table 2). For aryl-substituted alkynes, various electronically different para-substituted phenylalkynes reacted to provide the corresponding C–H alkynylation products 3b–3g in 39%–72% yields. Similar yields were obtained for alkynes bearing ortho- and meta-substituted aryl groups (3h–3i). 3-Ethynylpyridine was a competent substrate as well (3j). Notably, aliphatic and electron-deficient alkynes did not provide the desired product under current reaction conditions.
|
|
|---|
| a 1a (0.1 mmol), 2 (0.15 mmol), CuI (15 mol%), L1 (15 mol%), and K2CO3 (3 equiv.) in MeCN (0.05 M), under N2, rt, blue LEDs, 32 h. b Different deprotection procedures were applied depending on the products: TBAF in THF; or AcCl and montmorillonite K10 in CH2Cl2, See the ESI for details. c 64 h. d R′ = Ac. e 1a remained. |
|
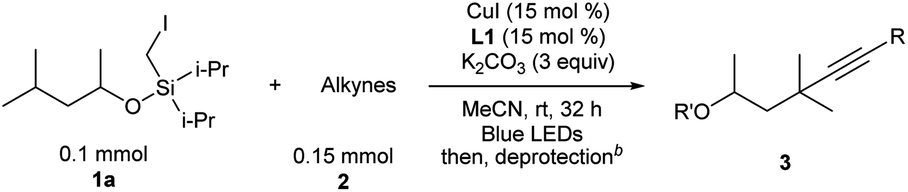
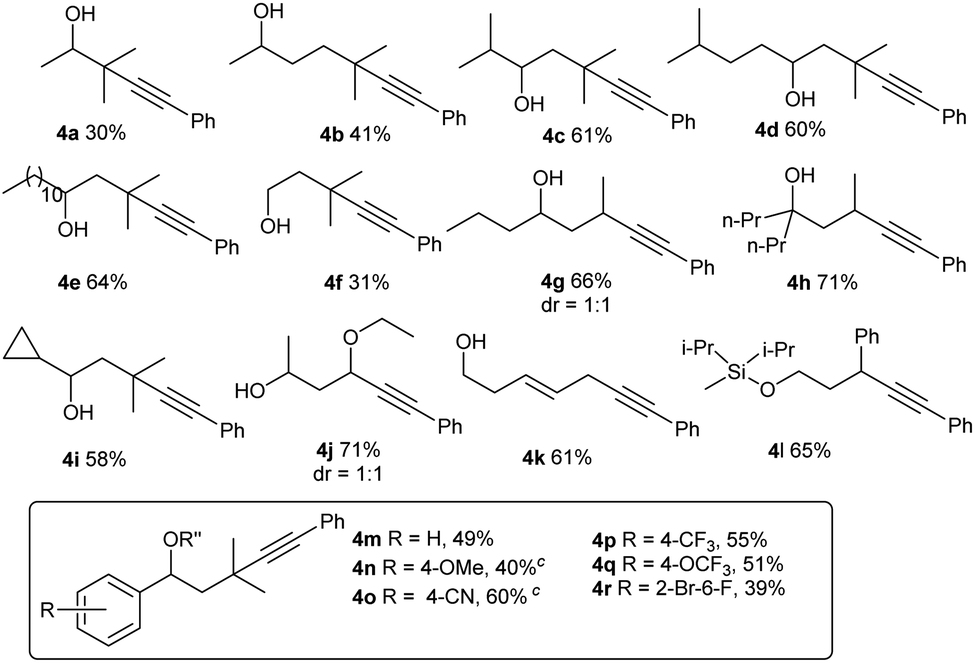
For Si-tethered alcohols (Table 3), although 1,5-HAT is kinetically less favorable than 1,6-HAT,4d,14 product 4a was isolated in 30% yield. Next, the possibility of achieving a δ-Sonogashira reaction was examined; remarkably, selective δ-alkynylation of alcohols proceeded well (4b). Substrates containing competitive tertiary C–H sites (β- vs. γ-, and γ- vs. δ) were tested, and, as expected, γ-functionalized alkenols were obtained as the sole regioisomers (4c and 4d).
|
|
|---|
| a 1a (0.1 mmol), 2b (0.15 mmol), CuI (15 mol%), L1 (15 mol%), and K2CO3 (3 equiv.) in MeCN (0.05 M), under N2, rt, blue LEDs, 32 h. R = iPr for 4a–4g, 4i–4r; R = Me for 4h. b Different deprotection procedures were applied depending on the products: TBAF in THF; or AcCl and montmorillonite K10 in CH2Cl2. See the ESI for details. c R′′ = Ac. |
|
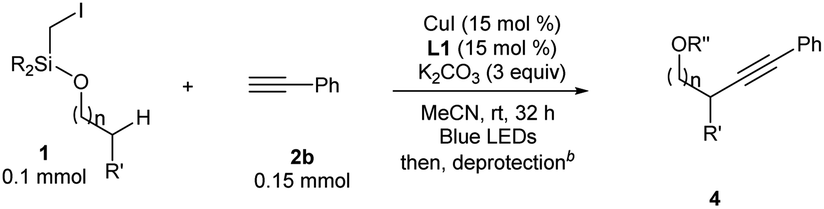
In Gevorgyan's remote Heck reaction, the γ-benzylic C–H alkenylation under Pd catalysis leads to the de-saturated by-product exclusively; in contrast, Cu-catalyzed γ-benzylic C–H alkynylation worked well (4l). Benzylic alcohols are capable substrates, and a range of functional groups were tolerated on the phenyl ring (4m–4r).
This photo-induced Cu-catalyzed remote C–H functionalization strategy could be expanded to C–H amination. The site-selective amination could readily take place at the tertiary and secondary C–H sites using 9H-carbazole as the reactant (5a and 5b).10,13b A benzylic position could be aminated as well (5c). The functional-group compatibility was investigated briefly, and Cl, MeO, and tBu are readily allowed on the carbazole ring (5d–5f) (Table 4). The carbazole motif frequently occurs in natural products, drug molecules and chiral ligands, and the alkylated carbazoles obtained by this method are not easily accessed through other approaches.15
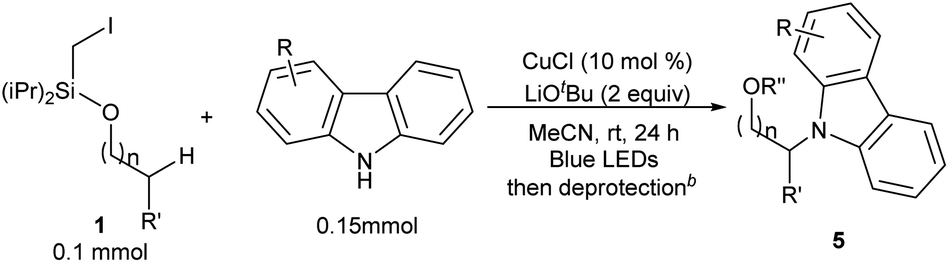
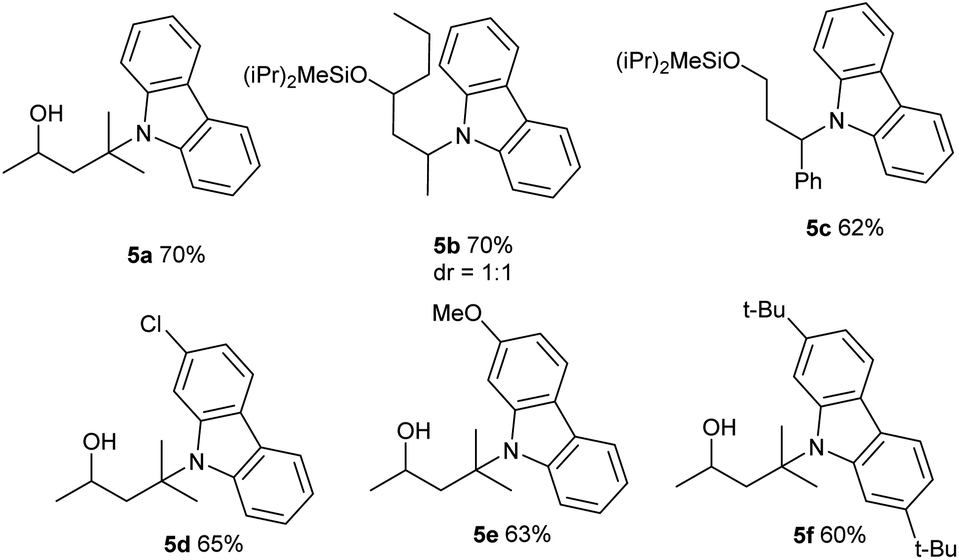
In an effort to understand the mechanism of this transformation, some preliminary control experiments were conducted. With addition of TEMPO, the reactions were inhibited and no desired product 3a was observed (Scheme 2, eqn (5)). Furthermore, to identify the possible intermediate, iodide 6 was independently synthesized and smoothly converted into 4h under standard conditions (eqn (6)).
 | ||
| Scheme 2 Mechanistic investigation. | ||
The UV-vis spectra of individual reagents or complexes were recorded at the reaction concentration in MeCN. L1-Cu-alkyne (should form in situ in the reaction) shows absorption in the range 380–500 nm.13a In addition, the Stern–Volmer experiment indicated that the excited state of the L1-Cu-Nu complex formed in situ could be quenched by Si-iodide. These results suggest that a complex of nucleophile, copper, and base accounts for the photoactive species under BLED irradiation. Moreover, the quantum yield (Φ = 0.75%) suggested that a radical-chain process might not be involved (see the ESI† for details).
Based on the literature5c,12,13a and these findings, a reaction mechanism was proposed (Fig. 1). In the presence of a base, the [L1Cu(I)(CCR′′)] formed in situ serves as the photoactive species to undergo photoexcitation to generate [L1Cu(I)(CCR′′)]* (B). This intermediate delivers an electron to the alkyl halide, leading to [L1Cu(II)(CCR′′)] (C) and an alkyl radical (R˙) (D). Subsequently, the latter undergoes a 1,n-HAT process, generating the translocated radical species (E). The radical species E could reversibly form the intermediate F either by a direct atom transfer from the CuI species or by recombination with the Cu complex followed by reductive elimination. Alternatively, instead of iodide, an alkyne could be delivered to afford the alkynylated product and regenerate the Cu catalyst. Related examples of this so-called radical relay strategy have been extensively studied by Liu,3g Xiao,16 and Liu.17
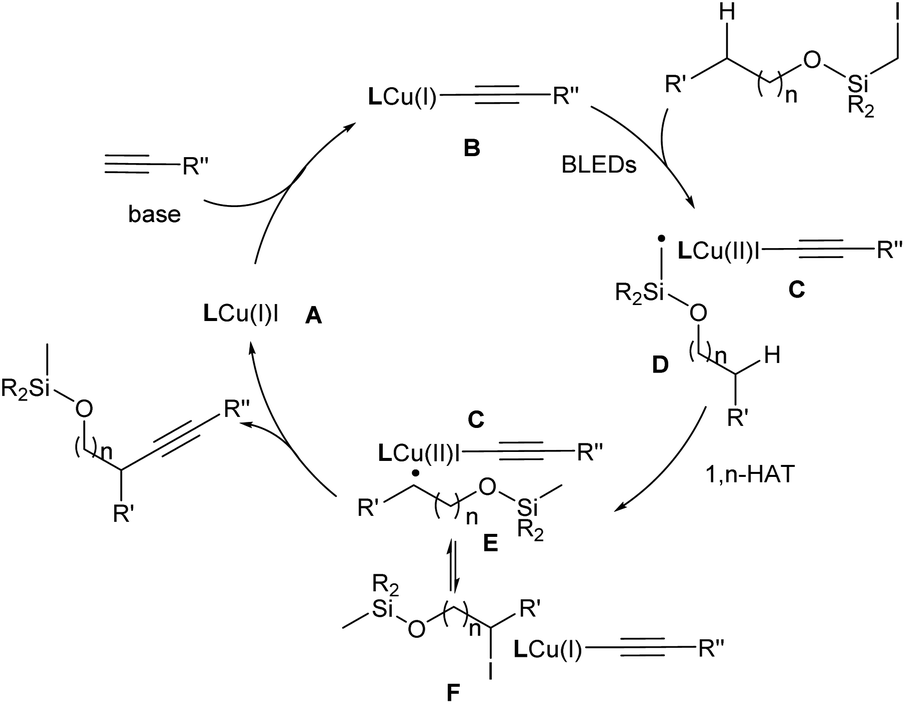 | ||
| Fig. 1 Proposed reaction mechanism. | ||
γ-Alkynyl alcohols are valuable intermediates in organic synthesis, and they can participate in a wide range of transformations at triple bonds or hydroxyl groups. Thus, it can be shown that the γ-alkynylated product can be readily transformed into cis-tetrahydropyran through hydro hydroalkoxylation mediated by a Lewis acid (Scheme 3, eqn (7)). Only one more salicylaldehyde was added under the same conditions, and a cascade hydroalkoxylation–formal [4 + 2] cycloaddition reaction of the alkyne took place to construct tetrahydrofurano/pyrano chromene (eqn (8)).18
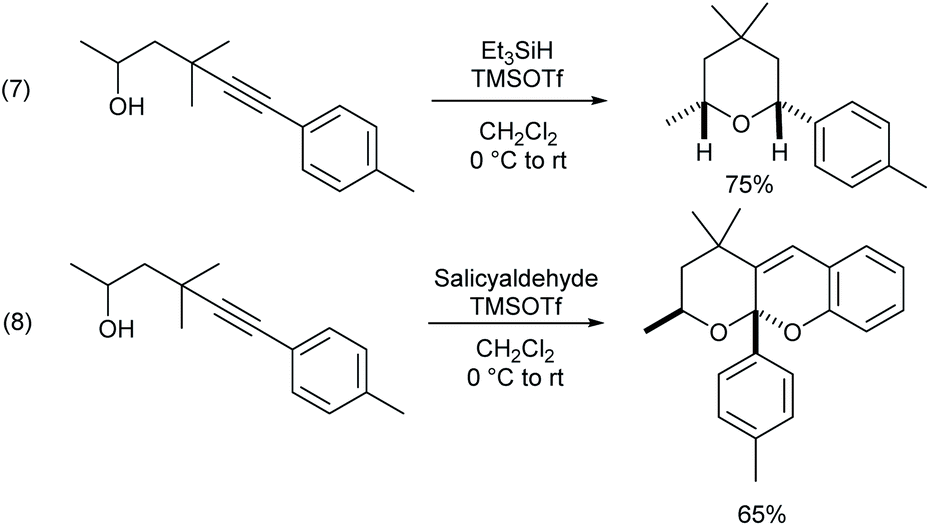 | ||
| Scheme 3 Transformations of γ-alkynyl alcohols. | ||
Conclusions
In summary, in this work, a mild approach for visible-light-promoted, copper-catalyzed remote functionalization of C–H bonds of aliphatic alcohols at β, γ, and δ via a radical 1,n-HAT process is described. These reactions proceed at low temperature and are compatible with a range of functional groups. This system introduces the idea of merging visible-light photoredox catalysis with copper-catalyzed C–H functionalization. A single copper nucleophile complex serves as the photo-coupling catalyst and reactant as well. It is anticipated that this new methodology will find application in the C–H functionalization reactions directly using Nu–H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSFC-22071073, 21772218, and 21821002, XDB20000000, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, and Chinese Academy of Sciences.Notes and references
- For selected reviews, (a) H. M. L Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS; (d) J. F Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS; (f) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS; (g) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS.
- (a) H. Park and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142(39), 16552 CrossRef CAS; (b) Y. Tan, S. Chen, Z. Zhou, Y. Hong, S. Ivlev, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 21706 CrossRef CAS; (c) C. Wang, N. A. Naren, P. Zheng and G. Dong, J. Am. Chem. Soc., 2020, 142, 8962 CrossRef CAS; (d) J. Rodrigalvarez, M. Nappi, H. Azuma, N. J. Floden, M. E. Burns and M. J. Gaunt, Nat. Chem., 2020, 12, 76 CrossRef CAS; (e) Y.-Q. Han, Y. Ding, T. Zhou, S.-Y. Yan, H. Song and B.-F. Shi, J. Am. Chem. Soc., 2019, 141, 4558 CrossRef CAS; (f) C. C. Roberts, E. Chong, J. W. Kampf, A. J. Canty, A. Ariafard and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 19513 CrossRef CAS; (g) S. Greßies, F. Klauck, J. H. Kim, C. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 9950 CrossRef.
- (a) W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847 CrossRef CAS; (b) V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389 CrossRef CAS; (c) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS; (d) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS; (e) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef; (f) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77 CrossRef CAS; (g) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014 CrossRef CAS; (h) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS; (i) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS; (j) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974 RSC.
- (a) H. Nishiyama, T. Kitajima, M. Matsumoto and K. Itoh, J. Org. Chem., 1984, 49, 2298 CrossRef CAS; (b) J. W. Wilt, Tetrahedron, 1985, 41, 3979 CrossRef CAS; (c) G. Stork and M. J. Kahn, J. Am. Chem. Soc., 1985, 107, 500 CrossRef CAS; (d) J. W. Wilt, J. Lusztyk, M. Peeran and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 281 CrossRef.
- (a) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340 CrossRef CAS; (b) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857 CrossRef CAS; (c) P. Chuentragool, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794 CrossRef CAS; (d) D. Kurandina, D. Yadagiri, M. Rivas, A. Kavun, P. Chuentragool, K. Hayama and V. Gevorgyan, J. Am. Chem. Soc., 2019, 141, 8104 CAS.
- (a) Chemistry of Triple-Bonded Functional Groups, ed. S. Patai, Wiley, New York, 1994 Search PubMed; (b) H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2010, 39, 264 RSC; (c) J. Chen, X. Cao, Q. An, Y. Zhang, K. Li, W. Yao, F. Shi, Y. Pan, Q. Jia, W. Zhou, F. Yang, F. Wei, N. Wang and B. Yu, Nat. Commun., 2018, 9, 1406 CrossRef.
- (a) D. Bafaluy, Z. Georgieva and K. Muñiz, Angew. Chem., Int. Ed., 2020, 59, 14241 CrossRef CAS; (b) N. Herron, D. Liu, G. Xia and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 2766 CrossRef; (c) Z. Zhang, X. Zhang and D. A. Nagib, Chem, 2019, 5, 3127 CrossRef CAS; (d) A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405 CrossRef CAS; (e) Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207 RSC; (f) Z. Liu, H. Xiao, B. Zhang, H. Shen, L. Zhu and C. Li, Angew. Chem., Int. Ed., 2019, 58, 2510 CrossRef CAS; (g) Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288 CrossRef CAS; (h) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940 CrossRef CAS; (i) C. G. Na and E. J. Alexanian, Angew. Chem., Int. Ed., 2018, 57, 13106 CrossRef CAS; (j) L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817 CrossRef CAS; (k) T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306 CrossRef CAS; (l) J. Richers, M. Heilmann, M. Drees and K. Tiefenbacher, Org. Lett., 2016, 18, 6472 CrossRef CAS; (m) E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef CAS; (n) T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871 CrossRef CAS; (o) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef.
- Z.-H. Zhang, X.-Y. Dong, X.-Y. Du, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Nat. Commun., 2019, 10, 5689 CrossRef.
- (a) A. C. Hernandez-Perez and S. K. Collins, Acc. Chem. Res., 2016, 49, 1557 CrossRef CAS; (b) O. Reiser, Acc. Chem. Res., 2016, 49, 1990 CrossRef CAS; (c) C. Minozzi, A. Caron, J.-C. Grenier-Petel, J. Santandrea and S. K. Collins, Angew. Chem., Int. Ed., 2018, 57, 5477 CrossRef CAS; (d) C. Deldaele, B. Michelet, H. Baguia, S. Kajouj, E. Romero, C. Moucheron and G. Evano, Chimia, 2018, 72, 621 CrossRef CAS; (e) A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, 450 CrossRef; (f) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227 RSC.
- (a) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS; (b) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681 CrossRef CAS.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS.
- A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492 CrossRef CAS.
- (a) Y. Zhang, Y. Sun, B. Chen, M. Xu, C. Li, D. Zhang and G. Zhang, Org. Lett., 2020, 22, 1490 CrossRef CAS; (b) Y. Xiong, X. Ma and G. Zhang, Org. Lett., 2019, 21, 1699 CrossRef CAS; (c) X. Mo, B. Chen and G. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13998 CrossRef CAS; (d) C. Li, B. Chen, X. Ma, X. Mo and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2130 CrossRef CAS.
- M. Koreeda and L. G. Hamann, J. Am. Chem. Soc., 1990, 112, 8175 CrossRef CAS.
- (a) W. A. L. van Otterlo and C. B. de Koning, Chem. Rev., 2009, 109, 3743 CrossRef CAS; (b) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS; (c) M. Inoue, T. Suzuki and M. Nakada, J. Am. Chem. Soc., 2003, 125, 1140 CrossRef CAS.
- F. Lu, D. Liu, L. Zhu, L. Lu, Q. Yang, Q. Zhou, Y. Wei, Y. Lan and W. Xiao, J. Am. Chem. Soc., 2019, 141, 6167 CrossRef CAS.
- (a) J. Lin, F. Wang, X. Dong, W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 14841 CrossRef; (b) Y.-X. Cao, X.-Y. Dong, J. Yang, S.-P. Jiang, S. Zhou, Z.-L. Li, G.-Q. Chen and X.-Y. Liu, Adv. Synth. Catal., 2020, 362, 2280 CrossRef CAS.
- (a) S. J. Gharpure, D. S. Vishwakarma and S. K. Nanda, Org. Lett., 2017, 19, 6534 CrossRef CAS; (b) S. J. Gharpure, S. K. Nanda, Padmaja and Y. G. Shelke, Chem.–Eur. J., 2017, 23, 10007 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05883a |
| This journal is © The Royal Society of Chemistry 2021 |