
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmon-assisted click chemistry at low temperature: an inverse temperature effect on the reaction rate†
Olga
Guselnikova
 ab,
Jiří
Váňa
c,
Linh Trinh
Phuong
a,
Illia
Panov
d,
Lubomír
Rulíšek
e,
Andrii
Trelin
a,
Pavel
Postnikov
ab,
Václav
Švorčík
a,
Erik
Andris
*e and
Oleksiy
Lyutakov
*ab
ab,
Jiří
Váňa
c,
Linh Trinh
Phuong
a,
Illia
Panov
d,
Lubomír
Rulíšek
e,
Andrii
Trelin
a,
Pavel
Postnikov
ab,
Václav
Švorčík
a,
Erik
Andris
*e and
Oleksiy
Lyutakov
*ab
aDepartment of Solid State Engineering, University of Chemistry and Technology, 166 28 Prague, Czech Republic. E-mail: oleksiy.lyutakov@vscht.cz
bResearch School of Chemistry & Applied Biomedical Sciences, National Research Tomsk Polytechnic University, Lenin Avenue 30, Tomsk 634050, Russia
cInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
dGroup of Advanced Materials and Organic Synthesis, Institute of Chemical Process Fundamentals, Czech Academy of Sciences, Rozvojová 1/135, 165 02 Prague, Czech Republic
eInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, 166 10 Prague 6, Czech Republic. E-mail: erik.andris@uochb.cas.cz
First published on 15th March 2021
Abstract
Plasmon assistance promotes a range of chemical transformations by decreasing their activation energies. In a common case, thermal and plasmon assistance work synergistically: higher temperature results in higher plasmon-enhanced catalysis efficiency. Herein, we report an unexpected tenfold increase in the reaction efficiency of surface plasmon-assisted Huisgen dipolar azide–alkyne cycloaddition (AAC) when the reaction mixture is cooled from room temperature to −35 °C. We attribute the observed increase in the reaction efficiency to complete plasmon-induced annihilation of the reaction barrier, prolongation of plasmon lifetime, and decreased relaxation of plasmon-excited-states under cooling. Furthermore, control quenching experiments supported by theoretical calculations indicate that plasmon-mediated substrate excitation to an electronic triplet state may play the key role in plasmon-assisted chemical transformation. Last but not least, we demonstrated the possible applicability of plasmon assistance to biological systems by AAC coupling of biotin to gold nanoparticles performed at −35 °C.
Introduction
The illumination of metal nanostructures at wavelengths corresponding to the plasmon resonance maximum leads to collective excitation of free electron oscillations and generation of hot electrons.1,2 The excited electrons quickly reach non-equilibrium Fermi energy distribution through electron–electron scattering and then lose energy at a lower rate due to electron–phonon scattering.3,4 Within this time-window, a direct injection of electrons from the metal nanostructure or intramolecular electron excitation may occur in nearby organic molecules possessing suitable acceptor molecular orbitals.5–12 Excited organic molecules are then involved in (plasmon-assisted) chemical transformations.8–14According to current theories, such plasmon-assisted reaction pathways include several events: (i) plasmon excitation of organic molecules by external electron injection or HOMO–LUMO intramolecular electron transition, (ii) vibrational relaxation of molecules in the excited state, and (iii) relaxation of an injected or excited electron, resulting in an “activated” molecule, which has a lower residual barrier for the desired chemical transformation.15 This residual barrier is then overcome by conventional heating.6 Under these assumptions, temperature and light act in synergy – the light excites plasmons, which activates the nearby molecule, while the temperature is responsible for overcoming the residual thermodynamic barrier.4,16,17 The relative importance of non-thermal and thermal effects in plasmon nanocatalysis is, however, far from being fully understood.18
The temperature dependence of plasmon-assisted reaction kinetics could differ significantly, depending on the value of the residual activation barrier and temperature impact on different stages of plasmon-assisted transformation(s).19–25 This may even result in the non-monotonic temperature dependence of the reaction rate as has been recently demonstrated for CO oxidation.26 In the special case of a completely vanished residual barrier, temperature effects on the lifetimes of excited electrons or molecular excited states can be expected to lead to inverse temperature dependence of the reaction rates: firstly, higher temperature could increase the electron–photon scattering and thus decrease the plasmon lifetime and probability of the reactant excitation. Secondly, it could decrease the lifetime of the electronically excited molecules, making the desired reaction less probable. To the best of our knowledge, this phenomenon has not been demonstrated yet.
Despite the fact that organic plasmon nanocatalysis is a rapidly evolving field, detailed mechanisms of the initial stages of plasmon nanocatalysis remain elusive.27–29 We previously demonstrated that plasmon assistance can induce the azide–alkyne cycloaddition (AAC) reaction.30 AAC is an important biochemical coupling reaction performed either under heating or with the utilization of metal catalysts31,32 (Cu, Ru, etc.). Carrying out the AAC reaction at low temperatures could make it cleaner and accessible to more delicate substrates. In this study, we report unprecedented inverse temperature dependence observed for the plasmon-assisted AAC reaction and attempt to explain its origin by exploring the reaction mechanisms using the combination of theoretical and experimental approaches.
Experimental procedures
Used materials, preparation of initial reagents and grafted nanoparticles, measurement techniques and control experiments are described in the ESI.†Plasmon-driven azide–alkyne cycloaddition
1 ml of suspension of AuNPs with attached 4-ethynylphenyl groups (further designated as AuNPs–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) in acetonitrile was mixed with 2 ml of 1 mM solution of 4-azidobenzoic acid, and the resulting mixture was illuminated for 3 hours with a light-emitting diode (LED) (660 nm emission wavelength, irradiance on the first reactor window – 20 mW mm−2) in a controlled temperature chamber under magnetic stirring. The intensity of the LED irradiation was measured by using Photodiode Power Sensors (Thorlabs, S142C). After the reaction, the modified AuNPs were separated by centrifugation (7000 rpm, 20 min), washed with acetonitrile and methanol, and dispersed in methanol to a final volume of 0.5 ml. 0.1 ml of the resulting suspension was deposited on a silica substrate (0.5 cm × 0.5 cm), dried at room temperature and subjected to further SERS analysis. The AAC experiments were performed separately five times for each temperature. In turn all SERS measurements were also repeated 5 times at different spots on the sample surface. The obtained SERS results were evaluated as averaged peak intensity with error bars corresponding to the deviation of characteristic signals from separated samples and uncertainty in the framework of single samples. Spectra were plotted according to ref. 33.
CH) in acetonitrile was mixed with 2 ml of 1 mM solution of 4-azidobenzoic acid, and the resulting mixture was illuminated for 3 hours with a light-emitting diode (LED) (660 nm emission wavelength, irradiance on the first reactor window – 20 mW mm−2) in a controlled temperature chamber under magnetic stirring. The intensity of the LED irradiation was measured by using Photodiode Power Sensors (Thorlabs, S142C). After the reaction, the modified AuNPs were separated by centrifugation (7000 rpm, 20 min), washed with acetonitrile and methanol, and dispersed in methanol to a final volume of 0.5 ml. 0.1 ml of the resulting suspension was deposited on a silica substrate (0.5 cm × 0.5 cm), dried at room temperature and subjected to further SERS analysis. The AAC experiments were performed separately five times for each temperature. In turn all SERS measurements were also repeated 5 times at different spots on the sample surface. The obtained SERS results were evaluated as averaged peak intensity with error bars corresponding to the deviation of characteristic signals from separated samples and uncertainty in the framework of single samples. Spectra were plotted according to ref. 33.
Plasmon-driven azide–alkyne cycloaddition with biotin azide
1 ml of AuNPs–CCH suspension in acetonitrile was mixed with 0.5 ml of 1.6 mM solution of biotin azide and the suspension was illuminated for 3 hours with LEDs (660 nm, 20 μW mm−2) at −35 °C in a controlled temperature chamber. After the reaction modified AuNPs were separated by centrifugation (7000 rpm, 20 min), washed with acetonitrile, methanol and dispersed methanol to a final volume 0.5 ml and 0.1 ml was deposited on a silica substrate (0.5 cm × 0.5 cm) and dried at room temperature for further analysis.
Computational methods
The reaction pathways were calculated with the Gaussian 16 rev. A.03 program,34–38 employing density functional theory (DFT). Specifically, we used the B3LYP39 functional with D3 Grimme's dispersion correction40,41 and the 6-311++G(d,p) basis set. All reported minima and transition state structures were confirmed by the calculation and diagonalization of their Hessian matrices. The reported energies are Gibbs free energies calculated with the SMD method in acetonitrile solvent42 at 298.15 K and the 1 bar standard state. The singlet excited state energies were calculated employing the TD-DFT method.43 The energy of S0heat was calculated with the non-equilibrium geometry and is therefore only a crude estimate.Results and discussion
Efficiency of plasmon-assisted azide–alkyne cycloaddition increases at lower temperature
The schematic representation of our experimental setup is shown in Fig. 1A. To perform the plasmon-assisted AAC, we grafted the alkyne reagent onto the surface of spherical gold nanoparticles (AuNPs). We chose the “gold standard” spherical AuNPs as a plasmonic support because they are resistant to oxidation, show only insignificant catalytic activity and exhibit homogeneous distribution of plasmon evanescent wave around their surfaces. The nanoparticles were grafted through the diazonium approach with 4-ethynylphenyl groups (Fig. 1A), which keeps these chemical moieties near the plasmonic evanescent wave.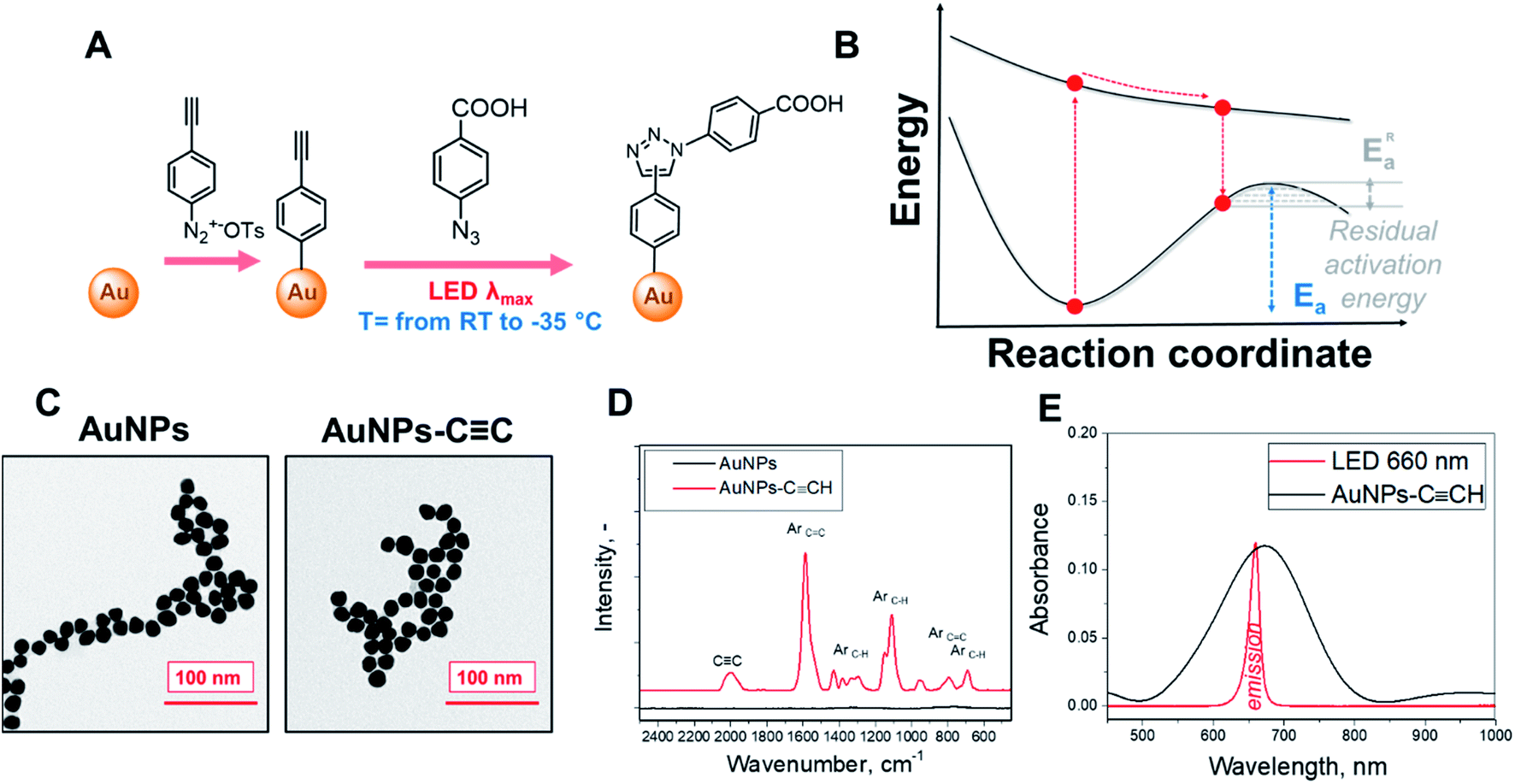 | ||
| Fig. 1 (A) Schematic representation of the used experimental concept–grafting of the AuNP surface with 4-ethynylphenyl groups and the subsequent plasmon-induced click reaction on the AuNP surface; (B) proposed principle of the decrease of the AAC residual activation energy; (C) TEM images of AuNPs before and after grafting with 4-ethynylphenyl groups; (D) SERS spectra of pristine AuNPs and AuNPs–CCH; (E) UV-Vis spectra of AuNPs–CCH in acetonitrile and the emission spectrum of the LED source further used for plasmon excitation. | ||
The immobilization of 4-ethynylphenyl units was confirmed by Raman measurements (Fig. 1C and S1† – appearance of characteristic and pronounced SERS vibrational bands; assignment of individual bands is given in Table S1†), whereas the conservation of the AuNP morphology and size after the grafting procedure was confirmed by TEM (Fig. 1D). The UV-Vis spectra of the grafted AuNPs in acetonitrile exhibit a pronounced plasmon absorption band near the 650 nm wavelength. To maximize the absorption efficiency, we utilized LEDs with a 660 nm emission wavelength for plasmon assistance (Fig. 1E, absorption spectra in Fig. S2† show that the 660 nm light is not absorbed by other compounds added to the reaction mixture).
The modified AuNPs were dispersed in acetonitrile solution with a large excess of 4-azidobenzoic acid and subjected to plasmon triggering under different temperatures between room temperature and the solvent melting point. After the reaction, the AuNPs were removed from the reaction mixture by centrifugation, and washed and dried to remove any absorbed azide. The AuNP sediment was then subjected to SERS measurements (Table S1†) in the dry phase – such a route allows us to exclude the side effects of solvent or Raman laser illumination of the reaction mixture. The reaction progress was tracked by SERS measurements through the decrease of the CC characteristic SERS band (at 2000 cm−1 – the maximum peak intensity in the 1990–2050 cm−1 spectral area was considered) and appearance of the triazole ring characteristic band (at 2235 cm−1). In addition, the Raman band from the benzene ring (which is not involved in the reaction) was used as the intrinsic SERS marker, i.e. the reaction progress was monitored (semiquantitative) by employing the ratios of the peaks at 2000/1590 cm−1 and 2235/1590 cm−1. For the cross-check, an Au–C related SERS band was used as a control (I2235/I401 instead of I2235/I1590 – results are given in Fig. S3†). It should also be noted that two triazole isomers (1,4 and 1,5) could form during the AAC; however, Raman spectroscopy does not allow us to distinguish between them.
The reaction kinetics of the AAC at room temperature (RT) is presented in Fig. S4† and related discussion in the ESI.† At RT, the reaction takes place during the first 12 hours and is finished after 20 hours, where the consumption of 4-ethynylphenyl groups significantly decelerates. To simplify further analysis, we monitored the reaction conversion at 3 hours, which should correspond to the initial stages of the reaction, where the conversion should depend on the reaction rate rather than the availability of surface attached reaction centres.
In the next step, we examined the temperature effect on the AAC plasmon-assisted reaction. Experiments were performed in the +25 to −35 °C temperature range (melting point of acetonitrile is −45 °C, whereas viscosity at −35 °C is 0.9 mPa s and at +25 °C 0.4 mPa s). The SERS spectra recorded after the reaction performed at different temperatures are shown in Fig. 2A. The appearance of characteristic triazole ring SERS peaks and the simultaneous decrease of acetylene peak intensity are well evident at all temperatures. In turn, Fig. 2B and S3† show the evaluation of the characteristic SERS band ratio, corresponding to the consumption of acetylene groups and formation of triazole rings. The amounts of reacted acetylene groups as well as formed triazole rings slightly increase during the gradual reduction of the reaction temperature up to −10 °C. A further decrease in the reaction mixture temperature leads to a rapid increase of observed conversion, which becomes especially pronounced in the −20 °C to −30 °C temperature range. At temperatures near −30 °C the increase of the reaction rate reaches a plateau. Thus, we observed almost one order of magnitude increase of the reaction rate upon cooling the reaction from RT up to −35 °C. Control measurements indicate the absence of 4-ethynylphenyl group degradation as well as no formation of side-products (Fig. S5† and related discussion). In addition, the illumination of the reaction mixture, performed in the absence of plasmon-active nanoparticles, does not indicate any chemical conversion (see Fig. S6 and S7† and related discussion). Therefore, our results clearly show the increase of the AAC rate under the plasmon excitation and the fact that the decrease in temperature leads to a larger conversion of the alkyne reactant.
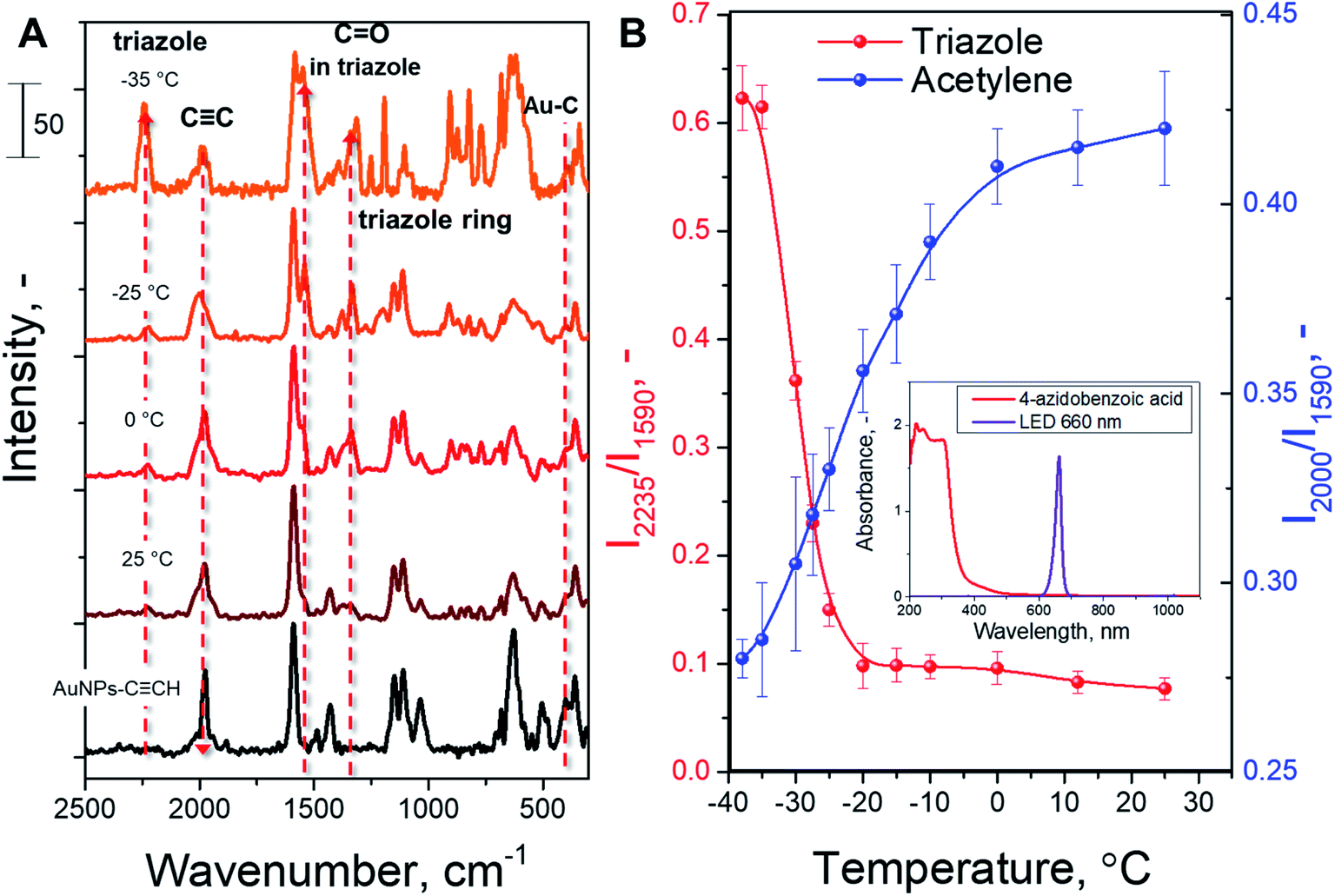 | ||
| Fig. 2 (A) SERS spectra, recorded on AuNP arrays after a plasmon-induced click reaction on the nanoparticle surface performed at different temperatures; (B) dependence of reaction conversion (estimated as the increase of I2235/I1590 and the decrease of I2000/I1590 SERS peak ratios) on the temperature. | ||
Mechanistic studies
We postulate that the plasmon assistance could significantly decrease the residual activation energy of AAC according the mechanism presented in Fig. 1B – i.e. via electronic excitation of reactant(s), their vibrational relaxation in an electronic excited state, and product formation through the residual activation barrier, overcoming of which often requires heating.44,45 However, heating can also increase the electron–phonon scattering and shorten the lifetime of plasmon-excited molecular states. Therefore, the heating of the reaction mixture can negatively affect the reaction rate, if quantum effects prevail. So, the main question is which stage limits the reaction progress – probability of plasmon-assisted excitation of molecules, lifetime of excited molecules or overcoming the residual potential barrier?To find the dominant rate-limiting effects in our case, we assessed the potential impact of viscosity increase, plasmon heating (which can be locally enhanced due to nanoparticle agglomeration with temperature decrease), damping of electron scattering, and prolongation of hot-electron lifetime at low temperatures (Fig. S8–S10,† and related discussion), as well as performed DFT calculations of potential reaction pathways. First, experimental and theoretical estimations of nanoparticle agglomeration and related plasmonic local heating indicate the absence of apparent nanoparticle agglomeration and a moderate (approximately 1 K) increase of temperature in the close vicinity of metal nanoparticles under our experimental conditions (Fig. S9 and S10†). Indeed, the collective heating of the suspension of AuNPs, as well as a large temperature gradient near AuNP surface,46,47 does not allow us to completely exclude contributions from plasmon heating. At the same time, the inverse temperature dependence observed in our case is not consistent with plasmon heating as the main driving force of this plasmon-assisted chemical transformation.
Secondly, control experiments with the addition of PEG, performed at RT, do not result in comparable reaction acceleration, which could prove negligible viscosity influence on AAC progress (Fig. S11 and S12,† and related discussion). Thirdly, the prolongation of the plasmon-excited electron lifetime was estimated from the temperature dependency of the Drude scattering parameter, taking into account the electron–phonon scattering in the bulk of AuNPs.48 Simple estimation of excited electron relaxation, performed on the basis of Holstein approximation, indicates the increase of their lifetime with temperature decrease by ∼25%. Thus, the prolongation of the plasmonic excitation lifetime can explain only a fraction of the observed reaction speedup.
In DFT calculations, aimed at exploring potential AAC pathways, we focused on comparing the reactivity of differently activated phenylacetylene (Fig. 3A, reaction coordinates are given in the ESI†). Since the acetylene moieties were spatially separated from AuNPs (Fig. 1), we did not consider their interactions with the gold surface. Two common modes of plasmonic activation were considered: injection of hot electrons from metal nanoparticles or intramolecular HOMO–LUMO electron excitation. As a baseline, we show the thermal pathway of the AAC reaction (designated as PW0) in Fig. 3A. This reaction type represents Huisgen pericyclic [3 + 2] dipolar cycloaddition, where the product is formed via a single cyclic transition state TS0 through an energy barrier of 124 kJ mol−1 in a concerted manner.
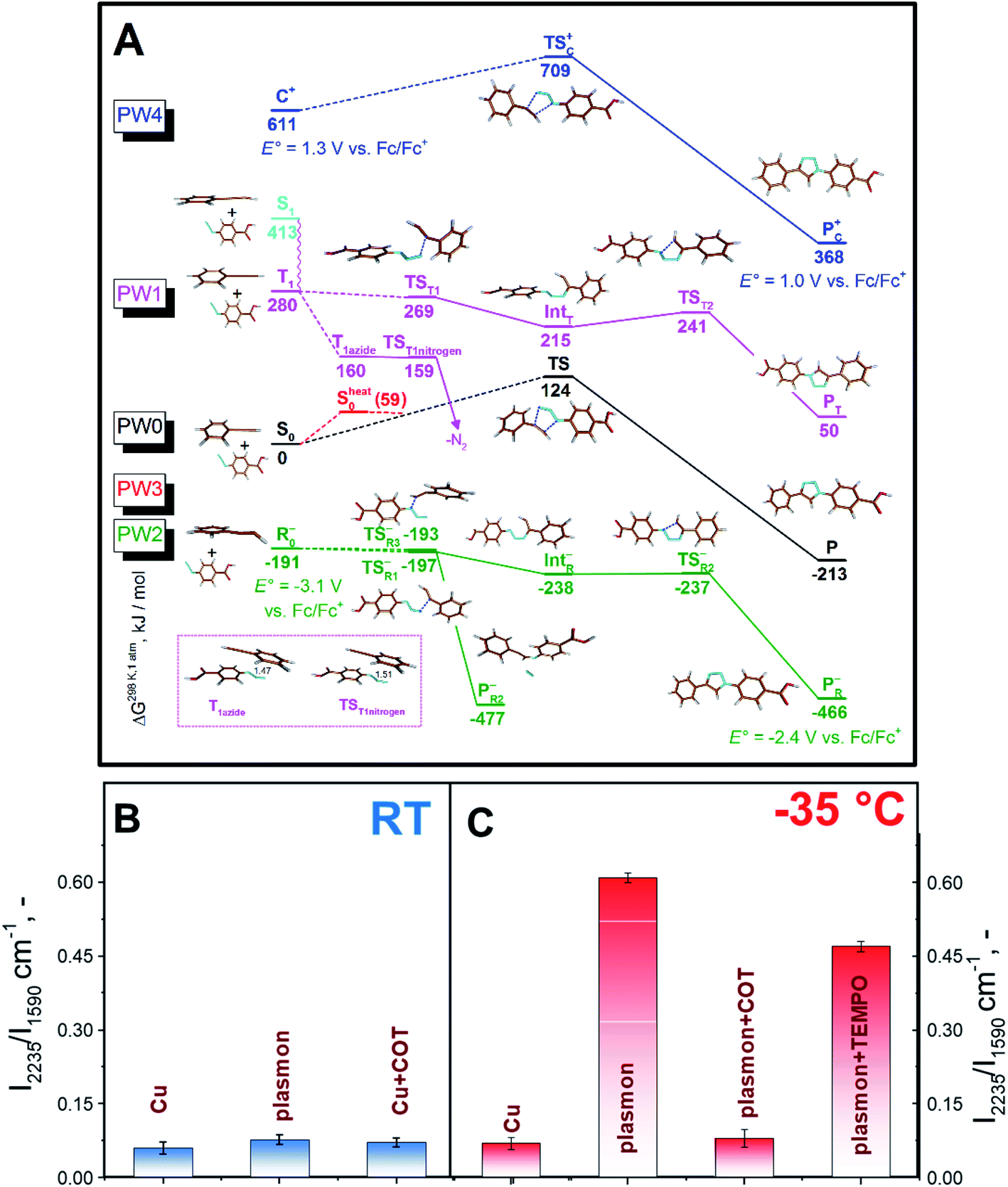 | ||
| Fig. 3 (A) DFT-calculated (B3LYP-D3/6-311++G(d,p)) reaction pathways under plasmon assistance and related transition states. E°′ values indicate calculated redox potentials vs. Fc/Fc+. (B and C) Conversion rate of AAC on the AuNP surface at room (B) or lower (C) temperatures, performed under different conditions – by Cu-catalysis (Cu) without plasmon assistance, under plasmon assistance (plasmon) or in the presence of triplet state (cyclooctatetraene – COT) or radical (TEMPO) scavengers. | ||
Next, we considered the plasmon-assisted electron excitation of the alkyne to a triplet T1 state (PW1), which corresponds to energy-transfer catalysis. The energy needed to form the T1 state is larger than the energy of a single 660 nm photon (181 kJ mol−1). Indeed, the dependence of the reaction rate on the laser power was found to be nonlinear (Fig. S13†), which indicates the contribution from multiphoton absorption processes. This contribution becomes even more pronounced at −35 °C. Product formation from the T1 (Fig. 3A) state would start by C–N bond formation viaTST1, accompanied by the spin density transfer to the azide unit. IntT formed in this process can undergo ring closure viaTST2 to give a triplet state of the product, which finally undergoes intersystem crossing (ISC) to P. The ISC might also take place in earlier steps in the reaction path, for example after the formation of TST1, because they are all higher in energy than the TS in the ground state and would thus have sufficient energy to overcome the barrier leading to P. If the triplet excitation is transferred to the azide molecule via the Dexter mechanism without the concomitant C–N bond formation, it would lead to instant elimination of the nitrogen molecule and formation of triplet nitrene (TST1nitrogen), which would react with the acetonitrile solvent and would not stay bound to the nanoparticles. If the PW1 reaction coordinate is indeed operational, it could contribute to the observed reaction rate temperature dependence, since decreasing temperature, besides the prolongation of the plasmon lifetime, slows down the relaxation of the triplet state T149 and therefore increases the probability that it encounters a suitably positioned azide reactant.
In the case of plasmon assistance through hot electron injection (photoredox catalysis), we considered two possibilities. The first one (PW2 in Fig. 3A) involves the reaction of the thus formed phenylacetylene radical-anion with azide in a stepwise manner. We found different transition states for the formation of the first bond differing in the orientation of reactants in the TSR1− and TSR3− transition states. Subsequently, we were able to find the remaining intermediate IntR and transition state TSR2− leading to the observed product from TSR1−. However, the almost isoenergetic TSR3− leads to the expulsion of the nitrogen molecule, which indicated that the reaction pathway PW2 should lead to the formation of sizeable amounts of by-products.
The second possible reaction pathway (PW3 in Fig. 3A) considers the activation of the phenylacetylene molecule by first forming the phenylacetylene radical-anion and subsequent loss of the injected electron and reaction of vibrationally excited phenylacetylene S0heat with azide. In this case, a vibrational energy of 59 kJ mol−1 could lower the reaction barrier of the concerted process, but since it is much less than 124 kJ mol−1 required to reach TS, and this path cannot be responsible for the observed temperature dependence. We also briefly considered the activation of the phenylacetylene substrate with hot holes,19 analogous to the recently proposed photoredox AAC mechanism50 (PW4 in Fig. 3A), and this pathway also did not lead to a substantial decrease of the reaction barrier and is thus likely not operative.
To get further insight, and see whether PW1 or PW2 are in operation, we performed control experiments with the addition of TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, a scavenger of radicals that should block both PW2 and PW3, and addition of cyclooctatetraene (COT), a scavenger of the triplet excited state,51 that should block PW1 (Fig. 3B and C). As can be seen in Fig. 3C, the addition of both compounds decreases the conversion at lower temperature. The effect of COT, however, was found to be significantly higher (Fig. 3 and S14†). Even the addition of a negligible amount of COT (10−6 M) results in almost complete suppression of triazole formation at RT and at −35 °C. Additional control experiments involving cyclohexene and 4-cyanobenzoic acid (see Fig. S15 and S16,† and related discussion) indicate that the impact of COT should be attributed mostly to excited state quenching. However, we also observed moderate inhibition of the reaction with 4-cyanobenzoic acid, which may be attributed to the partial blockage of surface reaction sites. Overall, the results of plasmon-assisted experiments performed with scavengers of radicals or excited states do not exclude the hot-electron injection related reaction pathway but indicate that PW1 should be viewed as the dominant mechanism. In addition, the different effects of the individual additives also run counter to the dominant effect of viscosity or the local heating effect.
We also checked the ability of the “reverse” AAC initiation, through the grafting of azide chemical moieties onto the AuNP surface in reaction with phenylacetylene dissolved in acetonitrile under plasmon excitation at −35 °C (Fig. S16†). In this case, we did not detect the appearance of a reaction product. The singlet–triplet gap in the azide is much smaller than in the alkyne (ca. 150 kJ mol−1) and thus triplet azide does not have enough energy to overcome the energy barrier of TST1.
Finally, we compared our results with the catalytic efficiency of common Cu catalysts (Fig. 2B, C, and S7B†). It can be seen that plasmon assistance has similar efficiency to the Cu catalyst at RT and significantly greater efficiency at −35 °C. Moreover, control experiments with COT addition and Cu-based catalyst do not indicate any changes in the reaction rate. Therefore, the measured effect of reaction quenching by COT points to the involvement of the plasmon-excited molecular triplet state (or triplet state transfer).
Taken together, we find that the PW1 mechanism is the most plausible, because it couples the release of a large amount of thermal energy (up to 280 kJ mol−1) from the triplet state of the alkyne with the physical proximity of the other reacting partner (azide). As a result, there is effectively no residual barrier, which is fully consistent with the observed reverse temperature behaviour of the reaction rate.
AAC coupling of biotin
High reaction temperatures or utilization of toxic catalysis leads to lower reaction selectivity, restricts the reaction scope, and thus restricts the applicability for bioorganic transformations.52–54 To demonstrate the advantages of plasmon-assisted AAC at low temperature, we performed the coupling of biotin azide to AuNPs–CCH. Biotin is widely used for the detection of cellular alkyne cholesterol,55 preparation of a novel multifunctional benzophenone linker for pull-down assays,56 and photoaffinity labelling57 and double-click stapling techniques.58 Classically, the AAC of biotin is performed with copper-based catalysts, which can sometimes be undesirable due to the presence of traces of copper in reaction products.
The schematic representation of biotin coupling with 4-ethynylphenyl moieties on AuNPs is presented in Fig. 4 (top) and the results of plasmon-assisted AAC performed at −35 °C without the addition of catalysts are shown in Fig. 4 (bottom). The SERS spectrum of biotin-azide is also presented for comparison (peak assignment is given in Table S2†). The comparison of the Raman spectra of biotin-azide powder and AuNPs coupled with biotin by plasmon-induced AAC shows that the reaction proceeds with a high yield (estimated as the decrease of the CC vibration band and well visible appearance of a biotin-related SERS band) at sub-zero temperature under copper-free conditions. This shows that our protocol is applicable to the AAC reactions of more complex substrates without the addition of metal catalysts or heating.
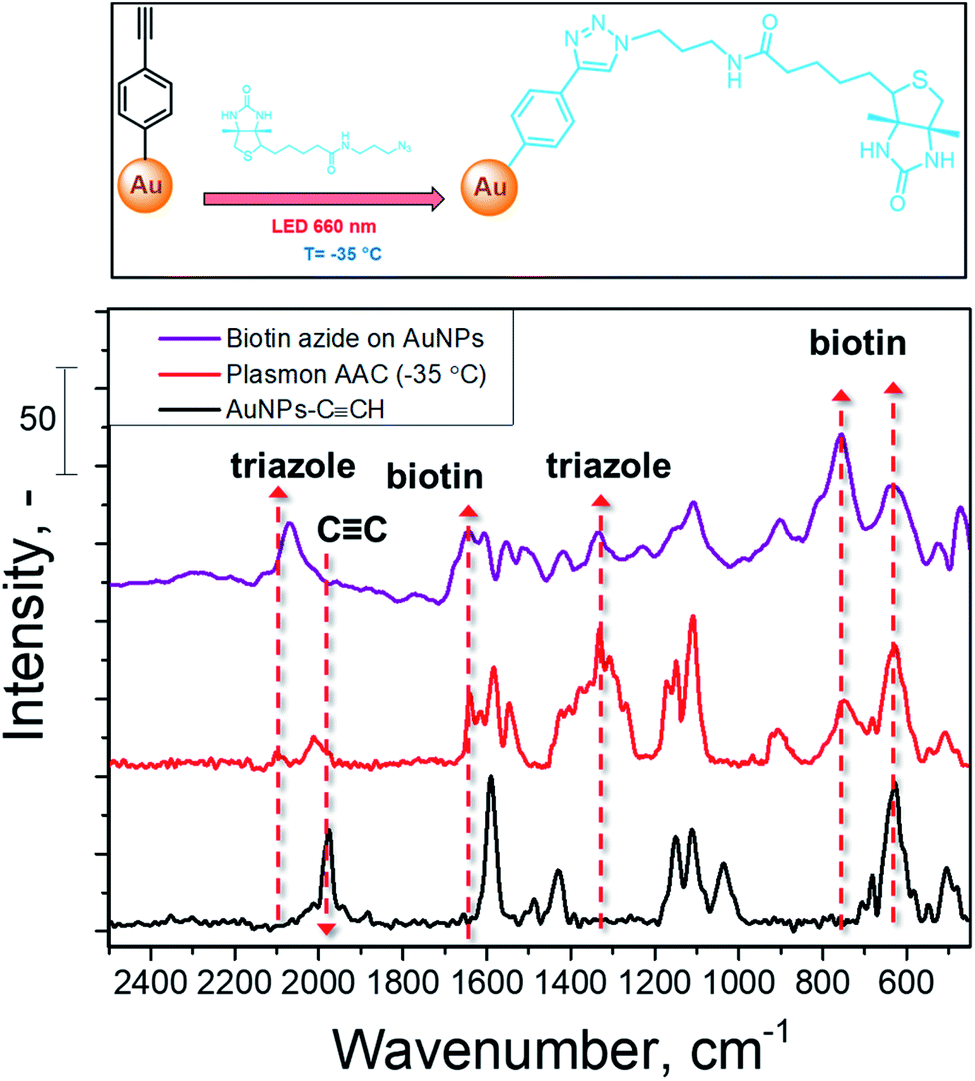 | ||
| Fig. 4 SERS spectrum of biotin-azide and SERS spectra recorded on AuNPs–CCH before and after conjugation with biotin-azide, performed at −35 °C with plasmon assistance. | ||
Conclusions
Plasmon assistance promotes a range of chemical transformations through lowering of the activation energy and the synergy between the heating of the reaction mixture and plasmon assistance is well described in the field of plasmonic chemistry. Here, we theoretically and experimentally demonstrated the opposite situation, where the decrease of reaction temperature led to a significant increase of plasmon-assisted catalytic efficiency. To model and explain the details of the complex process, we utilized an AAC reaction on the surface of spherical gold nanoparticles. The maximum reaction rate is observed at temperatures significantly below zero and it significantly exceeds the efficiency of common Cu-based catalysis. Density functional theory calculations identify several barrierless AAC pathways under plasmon assistance. Control experiments with cyclooctatetraene (triplet quencher) point to the involvement of the plasmon excited triplet state in the mechanism of the AAC reaction. The observed reverse dependence of the reaction rate on temperature can be explained by the decrease of electron-phonon scattering and retardation of organic molecule relaxation after the plasmon-induced excitation. Finally, we carried out a plasmon-assisted AAC reaction at −35 °C employing a bio-relevant compound – biotin. The presented results demonstrate the potential of plasmonic chemistry in the synthesis of complex biomolecules and hybrid compounds.Author contributions
O. G. performed the experiments; J. V. performed the quantum calculations; L. T. P. performed the experiments; I. P. designed the experiments; L. R. designed the calculations; A. T. performed the numerical calculations; P. P. analyzed the results; V. S. designed the experiments; E. A. analyzed the theoretical results and wrote the manuscript; O. L. analyzed the experimental results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grant Agency of the Czech Republic (project 18-26170S), the Czech Ministry of Education, Youth and Sports (LTAUSA19148), the Tomsk Polytechnic University Competitiveness Enhancement Program.References
- J. Szczerbiński, L. Gyr, J. Kaeslin and R. Zenobi, Nano Lett., 2018, 18, 6740–6749 CrossRef PubMed.
- A. Manjavacas, J. G. Liu, V. Kulkarni and P. Nordlander, ACS Nano, 2014, 8, 7630–7638 CrossRef CAS PubMed.
- M. Li, P. Yuan, Q. Q. Chen, L. H. Lin, P. M. Radjenovic, Y. L. He, J. Y. Wang, F. L. Zhang, S. Y. Luo, N. F. Zheng, S. J. Zhang, Z. Q. Tian and J. F. Li, Anal. Chem., 2020, 92, 7146–7153 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- H. Zhang, J. Wei, X. G. Zhang, Y. J. Zhang, P. M. Radjenovica, D. Y. Wu, F. Pan, Z. Q. Tian and J. F. Li, Chem, 2020, 6, 689–702 CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- H. Wei, S. K. Loeb, N. J. Halas and J. H. Kim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15473–15481 CrossRef CAS PubMed.
- L. Liu, S. Ouyang and J. Ye, Angew. Chem., 2013, 125, 6821–6825 CrossRef.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- B. Wu, J. Lee, S. Mubeen, Y.-S. Jun, G. D. Stucky and M. Moskovits, Adv. Opt. Mater., 2016, 4, 1041–1046 CrossRef CAS.
- R. Wang, J. Li, J. Rigor, N. Large, P. Z. El-Khoury, A. Y. Rogachev and D. Kurouski, J. Phys. Chem. C, 2020, 124, 2238–2244 CrossRef CAS.
- P. Christopher and S. Linic, ChemCatChem, 2010, 2, 78–83 CrossRef CAS.
- E. Kazuma and Y. Kim, Angew. Chem., Int. Ed., 2019, 58, 4800–4808 CrossRef CAS PubMed.
- E. G. Maksimov and G. P. Mofulevich, Determination of the electron–phonon coupling constant from optical measurements, 1972 Search PubMed.
- C. Y. Young and L. J. Sham, Phys. Rev., 1969, 188, 1108–1110 CrossRef CAS.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS PubMed.
- A. Gellé, T. Jin, L. De La Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 120, 986–1041 CrossRef PubMed.
- N. Zou, G. Chen, X. Mao, H. Shen, E. Choudhary, X. Zhou and P. Chen, ACS Nano, 2018, 12, 5570–5579 CrossRef CAS PubMed.
- Y. Yu, V. Sundaresan and K. A. Willets, J. Phys. Chem. C, 2018, 122, 5040–5048 CrossRef CAS.
- J. Biener, M. M. Biener, R. J. Madix and C. M. Friend, ACS Catal., 2015, 5, 6263–6270 CrossRef CAS.
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42–43 CrossRef CAS PubMed.
- K. M. Kosuda, A. Wittstock, C. M. Friend and M. Bäumer, Angew. Chem., Int. Ed., 2012, 51, 1698–1701 CrossRef CAS PubMed.
- P. Novello, C. V. Varanasi and J. Liu, ACS Catal., 2019, 9, 578–586 CrossRef CAS.
- A. Gellé, T. Jin, L. De La Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 120, 986–1041 CrossRef PubMed.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- Y. Sivan, J. Baraban, I. W. Un and Y. Dubi, Science, 2019, 364 Search PubMed.
- O. Guselnikova, P. Postnikov, M. M. Chehimi, Y. Kalachyovaa, V. Svorcik and O. Lyutakov, Langmuir, 2019, 35, 2023–2032 CrossRef CAS PubMed.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed.
- C. Wang, D. Ikhlef, S. Kahlal, J. Y. Saillard and D. Astruc, Metal-catalyzed azide–alkyne “click” reactions: Mechanistic overview and recent trends, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- B. Schrader and D. S. Moore, Pure Appl. Chem., 1997, 69, 1451–1468 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16 Revision A.03, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- Y. Dubi, I. W. Un and Y. Sivan, Chem. Sci., 2020, 11, 5017–5027 RSC.
- S. W. Lee, J. W. Hong, H. Lee, D. H. Wi, S. M. Kim, S. W. Han and J. Y. Park, Nanoscale, 2018, 10, 10835–10843 RSC.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 2047–7538 Search PubMed.
- H. R. De Barros, I. García, C. Kuttner, N. Zeballos, P. H. C. Camargo, S. I. C. De Torresi, F. López-Gallego and L. M. Liz-Marzán, ACS Catal., 2021, 11, 414–423 CrossRef CAS.
- T. Itoh, T. Asahi and H. Masuhara, Appl. Phys. Lett., 2001, 79, 1667–1669 CrossRef CAS.
- M. Terazima, S. Yamauchi and N. Hirota, J. Phys. Chem., 1986, 90, 4294–4297 CrossRef CAS.
- Z. G. Wu, X. J. Liao, L. Yuan, Y. Wang, Y. X. Zheng, J. L. Zuo and Y. Pan, Chem.–Eur. J., 2020, 26, 5694–5700 CrossRef CAS PubMed.
- W. Gong, P. Das, S. Samanta, J. Xiong, W. Pan, Z. Gu, J. Zhang, J. Qu and Z. Yang, Chem. Commun., 2019, 55, 8695–8704 RSC.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS.
- Y. Le, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winansl, W. Elám, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlö’Gl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef PubMed.
- J. Lu, J. J. Bravo-Suárez, A. Takahashi, M. Haruta and S. T. Oyama, J. Catal., 2005, 232, 85–95 CrossRef CAS.
- A. Gaebler, A. Penno, L. Kuerschner and C. Thiele, J. Lipid Res., 2016, 57, 1934–1947 CrossRef CAS PubMed.
- Y. Wu, L. B. Olsen, Y. H. Lau, C. H. Jensen, M. Rossmann, Y. R. Baker, H. F. Sore, S. Collins and D. R. Spring, ChemBioChem, 2016, 17, 689–692 CrossRef CAS PubMed.
- D. P. Murale, S. C. Hong, M. M. Haque and J. S. Lee, Proteome Sci., 2017, 15, 1–34 Search PubMed.
- C. M. Grison, G. M. Burslem, J. A. Miles, L. K. A. Pilsl, D. J. Yeo, Z. Imani, S. L. Warriner, M. E. Webb and A. J. Wilson, Chem. Sci., 2017, 8, 5166–5171 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05898j |
| This journal is © The Royal Society of Chemistry 2021 |