
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective cleavage of unactivated arene ring C–C bonds by iridium: key roles of benzylic C–H activation and metal–metal cooperativity†
Yancong
Tian
,
Martin
Jakoobi
 ,
Roman
Boulatov
* and
Alexey G.
Sergeev
*
,
Roman
Boulatov
* and
Alexey G.
Sergeev
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: boulatov@liverpool.ac.uk; sergeev@liverpool.ac.uk
First published on 12th January 2021
Abstract
The cleavage of aromatic C–C bonds is central for conversion of fossil fuels into industrial chemicals and designing novel arene functionalisations through ring opening, expansion and contraction. However, the current progress is hampered by both the lack of experimental examples of selective oxidative addition of aromatic C–C bonds and limited understanding of the factors that favour insertion into the C–C rather than the C–H bonds. Here, we describe the comprehensive mechanism of the only reported chemo- and regioselective insertion of a transition metal into a range of substituted arene rings in simple iridium(I) complexes. The experimental and computational data reveal that this ring cleavage requires both reversible scission of a benzylic C–H bond and cooperativity of two Ir centres sandwiching the arene in the product-determining intermediate. The mechanism explains the chemoselectivity and scope of this unique C–C activation in industrially important methylarenes and provides a general insight into the role of metal–metal cooperativity in the cleavage of unsaturated C–C bonds.
Introduction
Arene functionalisations are a common route to many indispensable building blocks for organic synthesis.1 Most of these functionalisations rely on well-established activation of aromatic C–H bonds by metal complexes, which leave the aromatic system intact.1–6 In contrast, functionalisations that involve breaking the aromatic ring are rare despite their tremendous synthetic potential to provide convenient access to a range of ring opening, contraction and expansion products from cheap hydrocarbons.7–10 The main challenge in developing such transformations is the slow and unselective oxidative addition of aromatic C–C bonds due to their higher kinetic and thermodynamic stability compared to that of C–H bonds.9,10 As a result, insertion of metal complexes into aromatic C–H bonds is overwhelmingly more common that insertion into aromatic C–C bonds.Of the seven examples of arene ring scission by a well-defined metal complexe reported to date,11,12 only cleavage of C6(CF3)6 by a Pt complex,13 and benzene and biphenylene by Al complexes14,15 proceed selectively. In all other cases,16–19 including reactions of Al complexes with alkylarenes,19 C–H scission competes with or even dominates the reaction.
To develop synthetic applications of aromatic C–C activation, factors that control the reactivity and selectivity and hence the substrate scope must be elucidated. Such understanding is currently lacking. The reported mechanistic studies of observed arene C–C cleavage are limited to benzene16,20–22 or quinoxaline23–25 and are exclusively computational. These DFT calculations focused primarily on C–C scission following reduction or dehydrogenation of the aromatic ring20,21,23–25 instead of the more fundamentally and synthetically important but little-understood C–C scission in intact arenes.14,22,26 Two known computationally identified examples of direct aromatic C–C activation in substituted arenes have never been realized experimentally, illustrating the challenges of integrating experimental and computational approaches in this area.22,26 As a result, a critical question of the role of substituents on the rate of aromatic C–C cleavage and the selectivity of C–C vs. C–H activation (and hence the scope) remains completely unexplored.
We recently reported that simple Cp* iridium complexes cleave the arene ring of a range of industrially important unactivated arenes, including mesitylene, o-, m-, p-xylenes and toluene (Fig. 1A), but surprisingly not benzene. This C–C activation yields diiridium metallacycles with excellent yields and high regioselectivity without observable C–H activation products.11,12 Consequently, these Cp*Ir complexes provide the best starting point found so far both for developing practical strategies of arene functionalisations and for understanding the mechanism of C–C vs. C–H selectivity and the role of the substituents in enabling selective arene cleavage.
 | ||
| Fig. 1 (A) Arene ring C–C scission in Cp*Ir(η4-methylarene) complexes. (B) The key diiridium intermediate. (C) Mesitylene ring scission in 1. | ||
Here we describe the first mechanism of cleavage of arene ring C–C bonds in unactivated arenes of industrial importance that is consistent with both experimental (kinetic, isotope labelling, intermediate interception) and quantum-chemical data. Our study reveals the key role of benzylic C–H activation and metal–metal cooperativity for enabling this rare C–C oxidative addition. Analysis of the main and higher-energy reaction paths, including competing C–H activation steps, explains the unparalleled chemoselectivity and offers a straightforward model for explaining the scope of this C–C activation. Our results also provide an insight into the role of metal–metal cooperativity in activation of arene ring C–C bonds by highlighting the importance of anti- rather than syn-arrangement of the two cooperating metal centres. In a wider context, this geometrical requirement improves the understanding of a number of known cooperative C–C activations in other unsaturated molecules, where anti-arrangement was postulated, but not rationalised.11,12,16,18,27–33
Results and Discussion
Experimental kinetics and mechanistic observations
We previously observed that C–C bond activation in all Cp*Ir(η4-methylarene) complexes reported to date has a similar kinetics, which suggests a common mechanism.11,12 Among all these complexes, as a starting point for our experimental studies we chose Cp*Ir(η4-mesitylene) as a model substrate because this complex and the metallacycle product, 2, each exists as a single isomer (Fig. 1C).Kinetic measurements of C–C scission in 1 as 0.01–0.1 M solutions in cyclohexane-d12 at 40, 50, 60, 70 and 80 °C revealed first order in 1 and zero order in mesitylene during at least three reaction half times (Fig. S2 and S3†), with ΔH≠o = 25.1 ± 2.1 kcal mol−1 and ΔS≠o = −1.0 ± 6.5 cal (mol−1 K−1) derived from the Eyring plot, corresponding to ΔG≠ = 25.4 ± 3.0 kcal mol−1 at 50 °C (Fig. S5†). The observed first order rate law and a small ΔS≠o are consistent with unimolecular rate-determining step (RDS) that does not involve dissociation of mesitylene. When 1 is heated in the presence of excess of mesitylene-d3, (CH3)3C6D3, no deuterium incorporation into 1 or the metallacycle product 2 is observed, which implies practically irreversible dissociation of mesitylene.
Several observations indicate the likely importance of benzylic C–H bond activation in conversion of 1 to 2. First, heating 1 in benzene-d6 at 50 °C yielded metallacycle 2-d (64%) with partially deuterated methyl groups of the broken arene ring and non-deuterated mesitylene (51%) as the main products after 24 h (Fig. 2A). Deuteration of 1 during this reaction was undetectable. Likewise, heating 2 in C6D6 at 50 °C for 24 h yielded no detectable amount of 2-dn. Note that in neither experiment we observed deuteration of methyl groups of Cp* ligands. Second, thermolysis of 1 in the presence of excess PMe3 generated benzylic Ir hydride 4 as the main product (Fig. 2B) with no trace of metallacycle 2. Such selective benzylic C–H bond scission in the presence of aromatic C–H bonds is unusual. Indeed, the C–H bond oxidative addition in alkylarenes typically affects aromatic C–H bonds34–36 and exclusive benzylic C–H cleavage mainly occurs in radical processes.37 Third, arene complexes lacking benzylic C–H bonds, e.g. Cp*Ir(η4-benzene), 5, does not undergo C–C cleavage under similar conditions.11 Finally, a rare example38 of an arene tautomer (3 in Fig. 2A), generated as a minor product of thermolysis of 1 in non-alkane solvents, is consistent with transient benzylic C–H bond activation. However, the negligible KIEs (1.06 ± 0.09 and 1.09 ± 0.09) measured in separate thermolysis experiments of 1 and its deuterated analogues 1-d3 and 1-d9 (Fig. 2C, S9, S10, Table S9†), suggest that C–H bonds are not cleaved in the RDS. The lack of detectable H/D scrambling between benzylic and arene ring hydrogens in the starting mesitylene complexes (1-d3 and 1-d9), the metallacycle products (2-d3 and 2-d9) or eliminated mesitylene, and the absence of C–C bond activation in Cp*Ir(η4-benzene) argue against activation of aromatic C–H bonds during conversion of 1 to metallacycle 2.
 | ||
| Fig. 2 (A) H/D exchange and the formation of tautomer 3 upon heating 1 in C6D6. (B) Benzylic C–H activation upon heating of 1 in the presence PMe3. (C) The lack of KIE and intramolecular H/D scrambling upon cleavage of 1-d3 and 1-d9. *KIEs were calculated from initial rates for separate reactions of the deuterated and non-deuterated species. | ||
DFT calculations of the reaction mechanisms
The obtained experimental observations led us to three hypotheses, which we used for our computational search for the reaction mechanism:A triple-decker anti-(Cp*Ir)2(μ,η4:η2-methylarene) intermediate involved in isomerization of Ir2-metallacycles (Fig. 1B)12 is also an intermediate in the conversion of 1 to 2.
This diiridium species is formed from a coordinatively unsaturated mononuclear Cp*Ir(η2-arene) intermediate resulting from η4 → η2 sliding of the arene ligand.
The reaction requires cleavage of a benzylic, but not an aromatic C–H bond after the RDS to account for: (a) the generation of benzylic Ir hydride Cp*Ir(H)(η1-(CH2)C6H3Me2)(PMe3), 4 upon heating of 1 in the presence of PMe3 (Fig. 2B); (b) the lack of KIEs and H/D scrambling between benzylic and aromatic hydrogens; (c) the inertness of Cp*Ir(η4-benzene).
We performed all geometry optimizations, reaction path calculations and calculations of thermodynamic corrections with the B3LYP functional and a mixed basis set of LANL2DZ for Ir and 6-31G(d) for all other atoms, as recommended for calculations of activation barriers in reactions involving Ir–Ir and Ir–C bonds.39–44 To test the suitability of this model chemistry, we also reoptimized the lowest-energy conformers of the starting complex 1, the final product 2, the three highest-energy transition states and the two intermediates immediately preceding them in the main mechanisms (Fig. 3) at B3LYP-D3/def2SVP.45 This model chemistry likely yields a more realistic description of the electronic structure of organometallic Ir complexes, albeit at the considerable additional computational cost that precluded its use for all computations in this work. All relative electronic energies at B3LYP-D3/def2SVP were within 3 kcal mol−1 of those at B3LYP/(6-31G(d)+ LANL2DZ), Table S10.† The good agreement between the two sets of energies confirms that B3LYP/(6-31G(d)+ LANL2DZ) provides an appropriate balance of accuracy and performance to allow detailed enumeration of multiple reaction paths in multiple Cp*Ir(ηn-arene) complexes, which distinguishes our current work from computational studies of arene C–C bond scission in the literature.20,21,23–25 We computed enthalpies and free energies by adding the thermodynamic corrections to the single-point energies calculated at the M06-L/(6-311+G(d)+LANL2TZ) level with a conductor polarizable continuum model (CPCM) of the reaction solvent.
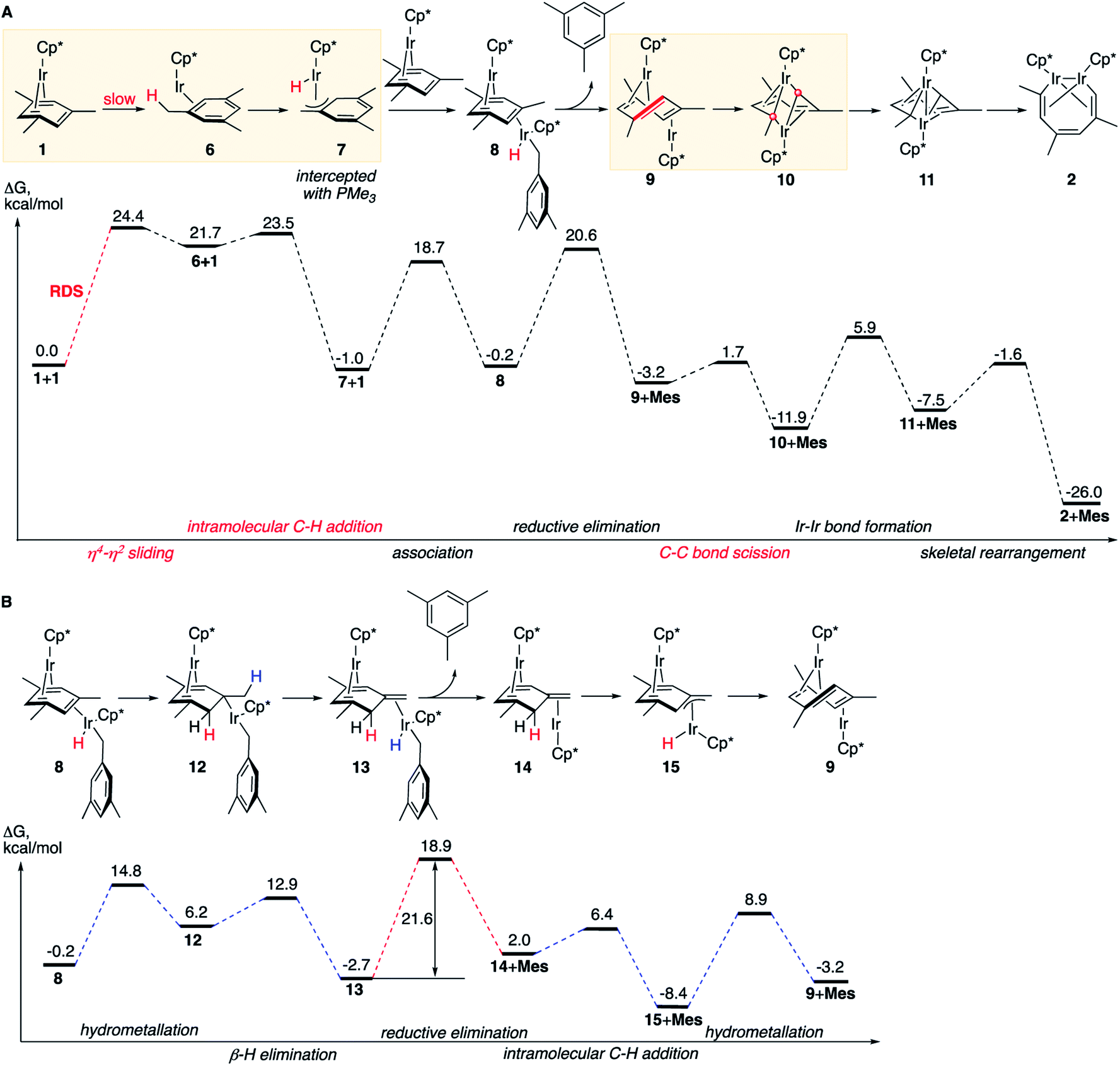 | ||
| Fig. 3 The calculated dominant reaction mechanisms and the corresponding free energy diagrams. (A) The simplest mechanism. (B) A competing path of the conversion of 8 to 9. All free energies are relative to 2 moles of 1, at M06-L/(6-311+G(d)+LANL2TZ)//B3LYP/(6-31G(d)+LANL2DZ), 1 M concentrations and 50 °C. Mes denotes mesitylene. Two red round legends indicate two carbon atoms involved in the C–C oxidative addition. | ||
Our calculations led to one lowest energy (dominant) mechanism that involves reversible benzylic C–H activation, and three higher energy mechanisms that occur via (a) aromatic C–H activation, (b) double benzylic and aromatic C–H activation and (c) without C–H activation. Comparison of these mechanisms presented below explains the observed scope of C–C scission in arenes, the role of metal–metal cooperativity and the observed exclusive formation of C–C, but not C–H activation products.
The dominant reaction mechanism
The lowest energy mechanism is shown in Fig. 3A. The first step is the rate-determining isomerization of the starting complex 1 by η4 → η2 sliding of the mesitylene ligand with ΔG≠ of 24.4 kcal mol−1. The resulting high-energy coordinatively unsaturated intermediate Cp*Ir(η2-mesitylene), 6 undergoes facile Ir insertion into benzylic C–H bond of the coordinated mesitylene yielding hydride Cp*Ir(H)(η3-(CH2)C6H3Me2), 7, with free-energy barrier of just 1.8 kcal mol−1. This complex binds to the uncoordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of starting complex Cp*Ir(η4-mesitylene), 1, at its least-hindered face to form mixed-valent diiridium hydride 8 with the two Ir atoms on the opposite faces of the bridging mesitylene. Note that here the η4-coordinated arene acts as a strained cycloalkene ligand. This propensity of η4-arenes has been documented, e.g. in ring-opening metathesis.46 Conversion of 8 to the key diiridium sandwich intermediate anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9, requires reductive elimination of mesitylene, which proceeds over nearly identical barriers in 8 and in its tautomer 13 (20.8 vs. 21.6 kcal mol−1, respectively). The latter forms rapidly from 8 by sequential hydrometallation (8 to 12) and β-hydrogen elimination (12 to 13, Fig. 3B) highlighting the ability of η4-arenes to mimic reactivity of strained alkenes such as norbornene and norbornadiene.46
C bond of starting complex Cp*Ir(η4-mesitylene), 1, at its least-hindered face to form mixed-valent diiridium hydride 8 with the two Ir atoms on the opposite faces of the bridging mesitylene. Note that here the η4-coordinated arene acts as a strained cycloalkene ligand. This propensity of η4-arenes has been documented, e.g. in ring-opening metathesis.46 Conversion of 8 to the key diiridium sandwich intermediate anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9, requires reductive elimination of mesitylene, which proceeds over nearly identical barriers in 8 and in its tautomer 13 (20.8 vs. 21.6 kcal mol−1, respectively). The latter forms rapidly from 8 by sequential hydrometallation (8 to 12) and β-hydrogen elimination (12 to 13, Fig. 3B) highlighting the ability of η4-arenes to mimic reactivity of strained alkenes such as norbornene and norbornadiene.46
At present we lack experimental estimates of the relative contributions to the reaction rate of the direct and stepwise (Fig. 3B) conversions of 8 to anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9. As described in the next section, undetectable H/D scrambling in partially deuterated reactants (Fig. 2C) is consistent with both mechanisms, whereas observation of metallacycle 2 with partially deuterated Me groups upon heating 1 in C6D6 and the generation of tautomer 3 in non-alkane solvents (Fig. 2A) suggest the intermediacy of 13.
The resulting diiridium(I) sandwich intermediate 9 undergoes dinuclear oxidative addition of an arene ring C–C bond in the bent bridging mesitylene ligand to give Ir(II) complex 10 over the barrier of just 4.9 kcal mol−1. The subsequent formation of an Ir–Ir bond 10 and backbone reorganization in the resulting flyover complex 11 yields the product, 2. Notably, C–C bond scission (9 → 10) involves one of the lowest activation barriers of the mechanism (4.9 kcal mol−1), considerably lower than those involving the formation of the Ir–Ir bond (17.8 kcal mol−1) or mesitylene elimination (>20 kcal mol−1), and is the lowest among reported calculated metal insertions into an arene (benzene) ring.16,20–22 Note that in contrast to what was proposed earlier47 neither η4-arene complex 1, nor η2-arene complex 6 undergo direct insertion of iridium into the arene ring to give the corresponding iridacycloheptatriene as this insertion is kinetically prohibited under the reaction conditions (ΔG≠ > 40 kcal mol−1, Table S12†).
Experimental validation of the computed mechanism
The first (unimolecular) step of the lowest energy reaction mechanism (Fig. 3) is rate determining, which tentatively agrees with the observed first order rate law. However, such direct comparison might be potentially misleading as the mechanism includes bimolecular and unimolecular steps, as well as competing paths for conversion of intermediate 8 to 9 with barriers of some steps only 3 kcal mol−1 lower than the barrier of RDS (24.4 kcal mol−1). To probe validity of the proposed mechanism, we calculated the time-dependent concentrations of all species in Fig. 3 by numerical integration of the underlying differential rate law using calculated barrier heights at starting concentrations of 1 of 0.01–0.1 M and of free mesitylene of 0–1 M, and the reaction temperatures of 50–150 °C. The results confirmed that the mechanisms in Fig. 3 reproduce the key kinetic observations: (a) the reaction rate is first order in 1 (Fig. S14†) and 0th order in mesitylene (Fig. S15†); (b) The apparent activation parameters, from the Eyring plot of the calculated rate constants for the depletion of 1vs. the reaction time, are ΔH≠o = 24.0 kcal mol−1 and ΔS≠o = 3.1 cal (mol−1 K−1) vs. measured 25.1 ± 2.1 kcal mol−1 and ΔS≠o = −1.0 ± 6.5 cal (mol−1 K−1) (Tables S7 and S20†); (c) no intermediate accumulates to a fraction that would make it detectable by 1H NMR (Fig. S13†).The mechanisms in Fig. 3 also accommodate all observed isotope effects. First, consistent with the negligible experimental KIEs all steps involving the formation or scission of a C–H bond or Ir–H bond occur after the rate-limiting barrier. Second, the observed incorporation of D in metallacycle 2 during thermolysis of 1 in benzene-d6 is consistent with a kinetic competition of two reactions of arene–tautomer complex 14: isomerisation to 9 and oxidative addition of C6D6 (Fig. 4A) to yield an IrIII(D)(C6D5) adduct, 16. Retrotautomerisation of 16 to 17 deuterates the bridging mesitylene. Intermediate 17 then yields deuterated intermediate 9-d by two competing mechanisms with loss of C6D5H (Fig. 4A; brown and blue paths). Third, the calculated high face-selectivity of C–H tautomerization (e.g., 8 → 12 → 13, Fig. 4B) ensures that the same H atom is transferred from Ir to a mesitylene sp2–C atom and back and prevents H/D scrambling in 1-d3 or 1-d9 (Fig. 2C) as observed. The lowest-energy path for the exchange of an aryl and a benzylic H atom requires rotation around the exocyclic CC bond in 14-dn (Fig. 4B) over a prohibitively high free energy barrier of 38 kcal mol−1. Finally, the lack of incorporation of D during thermolysis of 1 in the presence of mesitylene-d3, C6D3Me3, reflects the irreversible formation of anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9 (ΔG≠9→8 is 18.9 kcal mol−1 larger than ΔG≠9→10, Fig. 3A).
 | ||
| Fig. 4 Calculated mechanisms of the observed isotope effects. (A) A competing path responsible for deuteration of 2 and the generation of a complex of a mesitylene tautomer, 3 upon thermolysis of 1 in C6D6. (B) The mechanistic origin of the high face-selectivity of H atom transfer between Ir and a Me group of the bridging mesitylene. All free energies are relative to 2 moles of 1, at M06-L/(6-311+G(d)+LANL2TZ)//B3LYP/(6-31G(d)+LANL2DZ), 1 M concentrations and 50 °C. | ||
The formation of mesitylene tautomer complex 3, a side product of thermolysis of 1 in non-alkane solvents, including benzene (Fig. 2A), is explained by facile and mildly endergonic dissociation of 16 and its isotopomer, 19 (ΔG≠ = 17.5 kcal mol−1, ΔG° = 4.1 kcal mol−1, Fig. 4A).
Finally, the formation of Cp*Ir(H)(η1-(CH2)C6H3Me2)(PMe3), 4 (Fig. 2B) instead of metallacycle 2 in the presence of PMe3 is consistent with the lower calculated barrier for the reaction of intermediate 7, Cp*Ir(H)(η3-(CH2)C6H3Me2), with PMe3 (14.6 kcal mol−1) as opposed to the reaction of 7 with 1 (19.7 kcal mol−1). This suggests that PMe3 binds to 7 more than 104-fold faster than to 1 and hence blocks the formation of diiridium intermediate 8 and subsequent C–C bond cleavage. Although an equimolar mixture of Cp*Ir(H)(η1-(CH2)C6H3Me2)(PMe3), 4, and 1 is thermodynamically less stable by 4.1 kcal mol−1 than that of metallacycle 2, PMe3 and mesitylene, such conversion is too slow (overall ΔG≠ = 35.5 kcal mol−1) to occur on the experimental timescale at 50 °C.
Alternative reaction paths
We found it beneficial to systematically search for alternative paths connecting 1 and 2, for four reasons: (1) to confirm that the reaction mechanisms discussed above (Fig. 3A and B) comprise the lowest-energy paths; (2) to understand why paths occurring through transient benzylic C–H activation, rather than aromatic C–H activation or without C–H bond scission are uniquely efficient at arene C–C cleavage; (3) to explain the observed exclusive C–C vs. C–H chemoselectivity; (4) to identify other factors that promote or block arene C–C bond scission at metal complexes and hence could help predict the feasibility of the cleavage.We identified the next lowest-barrier path for arene ring cleavage (Fig. 5, blue sequence), which occurs via a double C–H activation and has the highest barrier of only 3.3 kcal mol−1 higher than that in the main mechanism (Fig. 3A and B). This path also involves benzylic C–H activation, but the resulting benzylic hydride Cp*Ir(H)(η3-(CH2)C6H3Me2), 7, isomerises to an aryl hydride, Cp*Ir(H)(η1-C6H3Me2), 21, by surprisingly facile intramolecular oxidative addition of an arene C–H bond to yield an IrV dihydride 20 followed by the rate-limiting C–H bond forming reductive elimination. Subsequent binding of 1 and reductive elimination of mesitylene yields the key intermediate anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9, with overall ΔG≠ of 27.7 kcal mol−1 (Fig. 5) vs. 24.4 kcal in the main mechanism (Fig. 3A and B). The minor contribution of this path to the overall kinetics is consistent with its high estimated KIE (∼1.6) not being observed experimentally.
 | ||
| Fig. 5 Comparison of the lowest energy path (in black; see Fig. 3 for details) and less favourable reaction paths for arene ring cleavage with and without C–H activation (in blue and red, respectively). All free energies are relative to 2 moles of 1, at M06-L/(6-311+G(d)+LANL2TZ)//B3LYP/(6-31G(d)+LANL2DZ), 1 M concentrations and 50 °C. Mes denotes mesitylene. | ||
Preferential formation of benzylic hydride 7 as compared to aryl hydride 21 illustrates the unusual reactivity of the Cp*Ir moiety towards C–H bonds. First, oxidative addition of the benzylic C–H bond in Cp*Ir(η2-mesitylene), 6 → 7, is considerably faster than direct oxidative addition of an aromatic C–H bond, 6 → 21, because it requires traversal of the barrier of 1.8 kcal mol−1vs. 21.7 kcal mol−1. Second, the benzylic adduct Cp*Ir(H)(η3-(CH2)C6H3Me2), 7, is >12 kcal mol−1 more stable than the aryl analogue 21. This selectivity is orthogonal to that of a more common Cp*Ir(PMe3) fragment34 and is rare in C–H activation,36,48–51 but appears to be essential for the observed reactivity. It is important to note that both benzylic and aromatic C–H activation intermediates (7 and 21) are not kinetically stable under reaction conditions and convert into products of C–C cleavage as shown in Fig. 3 and 5. That is, C–H activation here facilitates, rather than overrides arene ring C–C activation, which contrasts to what is typically seen in classical organometallic chemistry.1–6 For example, in the recently reported Al(I)-mediated arene activation, the metal smoothly inserts into ring C–C bond in reaction with benzene via a transient η2-arene complex,14 while with xylenes C–H activation occurs exclusively and completely blocks the C–C cleavage.19
We also identified one lower-energy reaction path that does not involve C–H activation and does not cause the C–C scission. Consideration of this unproductive path is important for predicting the feasibility of C–C cleavage as shown in the following section. This path instead generating the reactive anti-(Cp*Ir)2(μ,η4:η2-mesitylene) intermediate 9 gives its inert syn-(Cp*Ir)2(μ,η3:η3-mesitylene) isomer 25 (Fig. 5, red sequence). This route starts with association of 1 and its unsaturated isomer Cp*Ir(η2-mesitylene), 6, over the free energy barrier of 4.0 kcal mol−1 to give syn-Cp*Ir(μ,η4:η2-mesitylene)Ir(η2-mesitylene)Cp* (23), in which both Ir atoms are on the same face of the bridging mesitylene. Facile elimination of η2-mesitylene generates syn-(Cp*Ir)2(μ,η4:η2-mesitylene), 24, which is in a rapid equilibrium with the Ir–Ir bonded complex syn-(Cp*Ir)2(μ,η3:η3-mesitylene), 25 (ΔG≠ = 4.7 kcal mol−1, ΔG° = −7.3 kcal mol−1). Our reaction path calculations suggest that the two syn-diiridium isomers 24 and 25 and the product of C–C bond scission, 26, are connected by a single ambimodal52 transition state with an additional, lower-energy, transition state separating 24 and 25. The very high barriers separating 25 from either 1 or 26 (46.6 and 49.1 kcal mol−1, respectively) means that 25 is a kinetic trap preventing arene cleavage. The calculated high kinetic and thermodynamic stability of 25 is consistent with that of other group 9 syn bridging arene complexes being sufficiently stable to be isolated.53,54
Note that in thermolysis of Cp*Ir(η4-mesitylene), 1, syn-(Cp*Ir)2(μ,η3:η3-mesitylene), 25, is not observed, despite the rate-limiting barrier for its formation (6 → 23, red path, Fig. 5) being only 1.3 kcal mol−1 higher than that of the main mechanism (1 → 6). The reason is that unimolecular isomerization of 6 to 7 over an 1.8 kcal mol−1 barrier is >104-faster than diffusion-limited bimolecular binding of 6 to 1. As mentioned above, the latter is necessary to yield the inert syn diiridium isomer 25via the red path (Fig. 5). This “dead-end” path appears to explain the inertness of arene complexes lacking benzylic C–H bonds.11 In particular, our detailed calculations suggest that this path has the lowest energy for thermolysis of benzene complex 5 (Fig. 5 and S12†).
Factors enabling cleavage of arene ring C–C bonds
The obtained data, summarised in Fig. 6, suggest that susceptibility of Cp*Ir(η4-arene) to arene cleavage is determined by the kinetic competition between paths leading to the syn and anti isomers of bridging arene complexes (Cp*Ir)2(μ-arene). Only the anti isomer enables arene cleavage and it does this at least in two ways.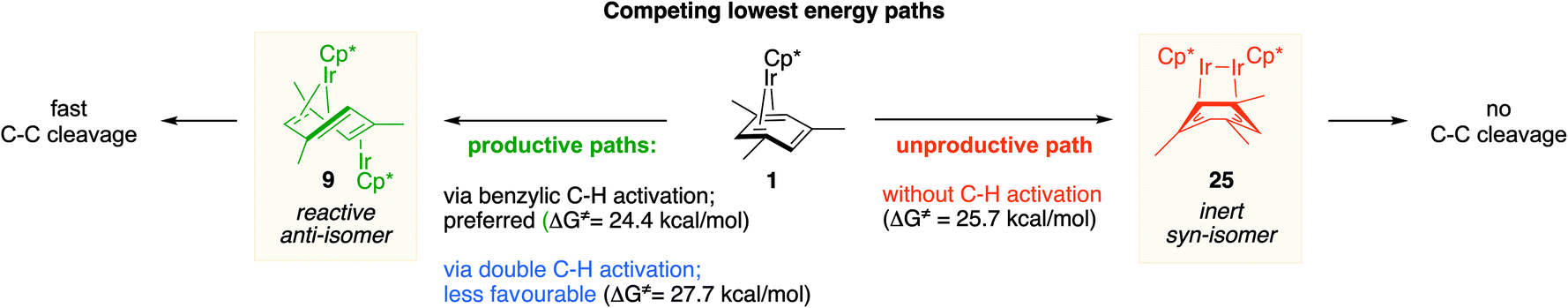 | ||
| Fig. 6 Summary of the lowest energy paths for thermolysis of 1. | ||
First, sandwiching the arene ring between two Ir atoms appears to enable cooperative C–C scission. The barrier for such scission in anti-(Cp*Ir)2(μ,η4:η2-mesitylene), 9, 4.5 kcal mol−1, is considerably lower than that in the syn analogue, 24, (41.8 kcal mol−1) or the barrier separating Cp*Ir(η4-mesitylene) from the corresponding iridacyloheptatriene (46.1 kcal mol−1) (Table S12†). The relative closeness of the last two numbers suggests that coordination of two Ir atoms to the same arene by itself labilises the arene C–C bonds negligibly. A similar difference is calculated in benzene and m-xylene complexes (Table S11†). The origin of this diiridium cooperativity remains to be established, but the anti geometry appears to enable more bonding Ir–C contacts (Ir–C distance <2.1 Å) in the transition state of C–C bond scission (two per each Ir) than either the syn analogue or Cp*Ir(η4-mesitylene) (two in each). Metal–metal cooperativity is increasingly recognized as a key factor in enabling difficult organic transformations at discrete metal complexes.55,56 Although the role of metal–metal cooperativity in arene C–C bond activation has received little attention, previously reported C–C oxidative additions in benzene,16 biphenylene,15 cyclopentadienyl18 and cyclooctatetraene ligands28,29 suggested the involvement of reactive intermediates with the anti arrangement of the two metal centers. The difference in reactivities of anti and syn isomers of these intermediates, however, have not been studied, and the role of the anti geometry in enabling cooperativity in C–C bond scission remains to be enumerated.
Second, the anti geometry prevents the formation of the Ir–Ir bond prior to scission of the arene C–C bond: when this Ir–Ir bond forms with intact arene, as occurs in the syn isomer, the resulting intermediate (e.g., 25 in Fig. 5 and S11†) is too stable to react further. In contrast, the formation of metal–metal bond is usually thought to facilitate rather than hamper the metal-assisted C–C cleavage in arenes,11,12,16,18 and other aromatic and unsaturated hydrocarbons (biphenylene,27 cyclooctatetraene,28,29,57,58 and cyclopentadienyl18 anions, cycloalkenes30 and alkynes31–33).
These roles of the anti-Ir2 intermediate in determining the outcome of thermolysis of Cp*Ir(arene) complexes appears to be general, as suggested by our calculations on benzene and m-xylene analogues of 1 (Table S12†). In other words, the observed cleavage of methylarenes reflects the kinetic selectivity for the formation of the anti isomers, which occurs after the RDS. To understand factors that determine the anti/syn selectivity and hence the range of cleavable arenes, we compare in Fig. 7 two paths leading to each anti- and syn-(Cp*Ir)2(μ-arene) for mesitylene, which is cleaved, and benzene, which is not. Four of these eight paths involved association of reactant Cp*Ir(η4-arene) with its coordinatively unsaturated Cp*IrI(η2-arene) isomer (Fig. 7A and B). The other four proceeded by association of Cp*Ir(η4-arene) with the corresponding product of oxidative addition of the C–H bond, Cp*Ir(aryl)(H) (Fig. 7C and D).
 | ||
| Fig. 7 Calculated competing mechanisms of the formation of syn and anti diiridium intermediates by association of reactant Cp*Ir(η4-arene) with either IrI (A and B) or IrIII (C and D) intermediate in thermolysis of Cp*Ir(η4-mesitylene) (A and C) or Cp*Ir(η4-benzene) (B and D). The free energies are relative to two moles of Cp*Ir(η4-arene) in all schemes. Pathways in green and red lead to the reactive and unreactive intermediates, respectively. The boxed structures are the predicted intermediates. All free energies are at M06-L/(6-311+G(d)+LANL2TZ)//B3LYP/(6-31G(d)+LANL2DZ), 1 M concentrations and 50 °C, relative to 2 moles of 1 (A and C) or of Cp*Ir(η4-C6H6) (B and D). | ||
Comparison of these mechanisms for the cleavage of mesitylene and benzene highlights the key role that facile activation of benzylic C–H bond plays in arene scission in Cp*Ir(η4-arene) complexes. Fig. 7 shows that only association of Cp*Ir(η4-mesitylene), 1, with IrIII intermediate, Cp*Ir(H)(η3-CH2C6H3Me3), 7, yields reactive anti-(Cp*Ir)2(μ-mesitylene), 9 by way of intermediate 8 (Fig. 7C). In all other scenarios, the formation of syn-diiridium intermediates is favoured both kinetically and thermodynamically, e.g., 1 + 6 → 23vs.27 (Fig. 7A), 5 + 28 → 31vs.29 (Fig. 7B) and 5 + 35 → 34vs.36 (Fig. 7D). Because intermediate 7 is formed from intermediate 6, the productive path (7 → 9) is only accessible if conversion of 6 to 7 is faster than addition of 6 to 1. In other words, the unusually fast intramolecular oxidative addition of a benzylic C–H bond in Cp*Ir(η2-methylarene) enables C–C bond cleavage by outcompeting bimolecular addition of the same intermediate to Cp*Ir(η4-arene).59
Model for predicting the scope of the C–C cleavage
Fig. 7, which summarises calculated reaction paths for the arene cleavage in mesitylene 1 and benzene 5 complexes, demonstrates that an arene C–C bond is cleaved only if the mixed-valence Ir(I)Ir(III) anti-intermediate (e.g., 8 for mesitylene and 36 for benzene) is formed faster than any of its three analogues (syn Ir(I)Ir(III) and syn or anti Ir(I)Ir(I)). In each case, the most stable of the diiridium intermediates also forms fastest. For example, in C–C cleaving thermolysis of 1 (Fig. 7A and C), productive intermediate 8 (anti-Ir(I)Ir(III)) is both most stable and is formed via the lowest barrier. Conversely, in thermolysis of the benzene analogue 5 (Fig. 7B and D), 36, syn-Ir(I)Ir(III), is the most stable and fastest forming diiridium intermediate which gives unproductive complex 32 inert towards C–C cleavage. In either case, anti- or syn-Ir(I)Ir(III) intermediates form faster than their Ir(I)Ir(I) isomers.We hypothesised that calculation of relative energies of anti- and syn-Ir(I)–Ir(III) intermediates (Fig. 8; 37 and 38) can be used to predict the feasibility of the arene C–C cleavage. To test this, we first calculated the relative energy of 37 and 38 for two additional arenes, toluene and m-xylene, earlier shown to undergo cleavage (Fig. 8A).11 In both cases, anti isomers were found to be most stable, which agrees with experimental data (Table 1). Next, we first predicted and then experimentally tested the reactivity of three new experimentally untested unactivated arenes: fully methylated benzene as well as naphthalene and 2,6-dimethylnaphthalene (Fig. 8B). For all these arenes unproductive syn isomers 38 had lower energy than productive anti37 (Table 1) suggesting inertness toward C–C scission. We prepared the corresponding Cp*Ir(η4-arene) complexes 39–41 and characterised the C6Me6 (39) and naphthalene (40) complexes by X-ray (Fig. 9). Thermolysis of 39–41 at 150 °C for 24–36 h led to consumption of the starting complexes and expected release of some free arene, but no arene C–C cleavage was observed. 1H NMR and HR-MS spectra of the products indicated formation of inert syn-(Cp*Ir)2(arene) and (Cp*Ir)3(arene) complexes, which is in agreement with our theoretical predictions. The lack of C–C bond scission in 39 and 41 suggests that the presence of benzylic C–H bonds alone is insufficient to enable C–C bond scission and highlights the validity of the proposed model. Although the exact factors that determine the relative stabilities of the syn- and anti-diiridium intermediates have yet to be identified,60 steric effects likely to play an important role here. With the exception of the two naphthalenes, the less-stable Ir(I)Ir(III) intermediates have more short nonbonding H⋯H contacts (<2.60 Å) than their more stable analogues (Table 1).
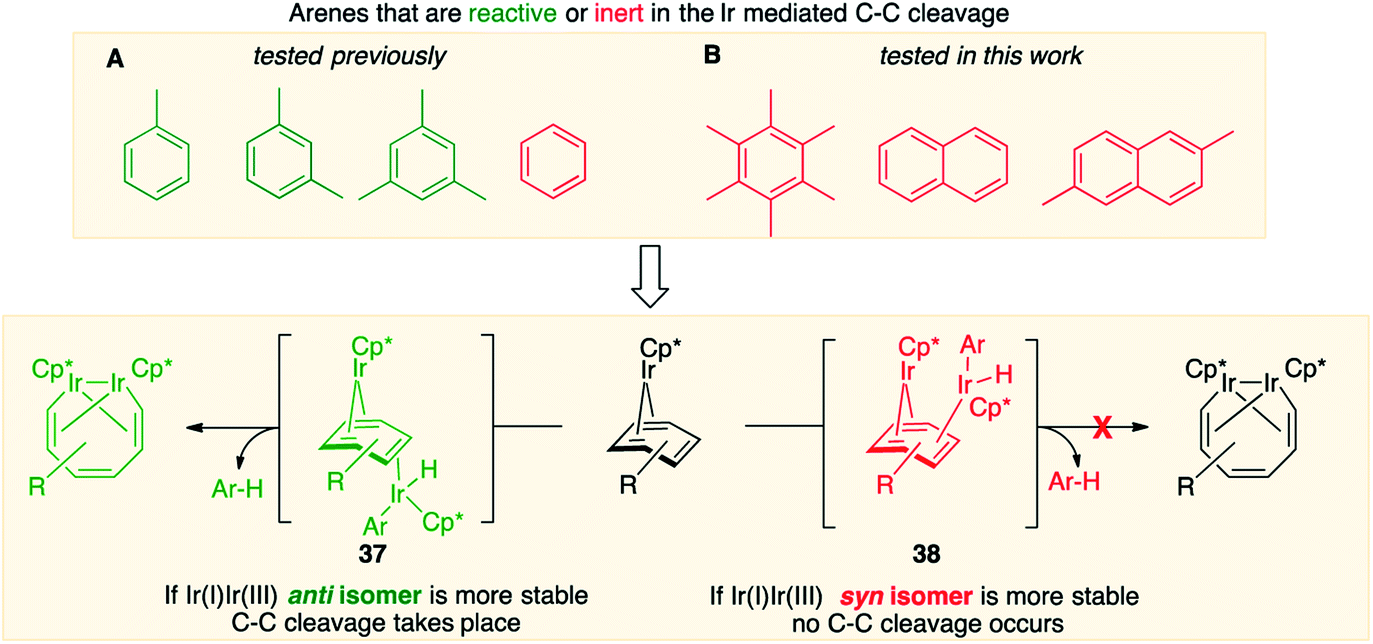 | ||
| Fig. 8 Computational model for predicting the feasibility Ir mediated arene C–C cleavages reported previously (A) and in this work (B). | ||
| Ar |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
| a The electronic energy of syn-Ir(I)Ir(III) intermediate 38 relative to that of anti-Ir(I)Ir(III) analogue, 37 at M06-L/6-311+G(d)+LANL2TZ)//B3LYP/(6-31G(d)+LANL2DZ)). b All short (<2.60 Å) interactions between any two hydrogen atoms located in different ligands (e.g. between Ar and Cp*, Ar and ArH etc.). | |||||||
| ΔE(syn/anti), kcal mol−1a | 2.5 | 9.7 | 12.3 | −1.6 | −0.3 | −2.0 | −9.7 |
| Excess of short H⋯H contactsb in syn (38) vs. anti (37) intermediates | 1 | 2 | 4 | −1 | −3 | 4 | 1 |
| Arene cleavage expected? | Yes | Yes | Yes | No | No | No | No |
| Observed? | Yes | Yes | Yes | No | No | No | No |

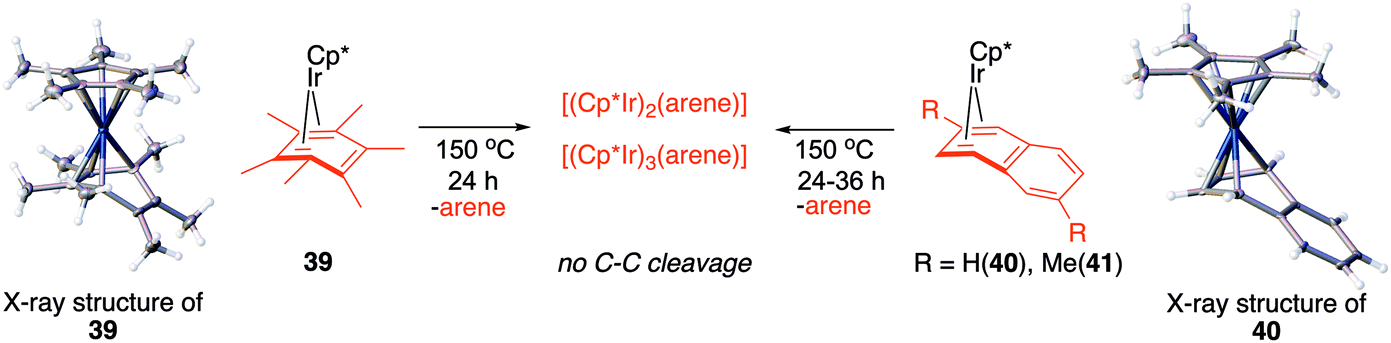 | ||
| Fig. 9 Attempted Ir-mediated cleavage of hexamethylbenzene, naphthalene and 1,6-dimethylnaphthalene complexes confirming the feasibility of the proposed model. | ||
Conclusions
In summary, we presented here a comprehensive mechanistic analysis of the most selective and general oxidative addition of arene ring C–C bonds known to date, so far observed only in Cp*Ir(η4-arene) complexes. The obtained results suggest that the unique capacity of the Cp*Ir system to cleave methylated arene rings selectively results from remarkably facile reversible scission of a benzylic C–H bond. This favours the formation of the anti-(Cp*Ir)2(μ-methylarene) intermediate that undergoes fast ring C–C scission over its more thermodynamically stable and inert syn isomer. The higher reactivity of the anti-intermediate results from the cooperative action of the two Ir atoms that lowers the barrier for the C–C bond scission to less than 5 kcal mol−1 compared to more than 40 kcal mol−1 in the syn isomer. The analysis of a range of reaction pathways in the mesitylene and benzene complexes suggests that the occurrence of C–C scission in a specific arene can be predicted by comparing the electronic energies of just two isomeric intermediates for each arene. These energies correlate with the relative rates for the formation of the reactive anti and inert syn diiridium intermediates. Because these energies are easily calculated, they provide a useful model for predicting the scope of the process. Application of this approach successfully explained the experimentally observed C–C scission in mesitylene, m-xylene and toluene and the lack of C–C scission in benzene, hexamethylbenzene, naphthalene and 2,6-dimethylnaphthalene. The rate-determining step for this cleavage is η4 to η2 sliding of the arene ligand in the starting complex and further investigation of this step will provide further means to control this process.The high reactivity of the resulting Cp*Ir(η2-arene) intermediate in C–H oxidative addition discovered during our mechanistic studies offers additional opportunities for developing novel functionalisation methods based both on selective arene C–C and C–H bond scissions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Leverhulme Trust (Grant RPG-2018-406 to A.G.S.) EPSRC Early Career Fellowship (EP/ L000075/1 to R.B.) and Petroleum Research Fund (58885-ND7 to RB) for financial support. Computations reported here were performed in the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562, with computational resources provided by the SDSC under allocation TG-CHE140039. HR-MS analyses were performed by EPSRC UK National Mass Spectrometry Facility at Swansea University. We thank Dr Nathan Halcovitch at Department of Chemistry, Lancaster University for X-ray measurements of 39 and 40.Notes and references
- Arene chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, Wiley, Hoboken, 2016 Search PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed. Engl., 2012, 51, 10236–10254 CrossRef CAS.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- J. F. Hartwig and M. A. Larsen, ACS Cent Sci., 2016, 2, 281–292 CrossRef CAS.
- P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS.
- S. Kancherla, K. B. Jørgensen and M. A. Fernández-Ibáñez, Synthesis, 2019, 51, 643–663 CrossRef CAS.
- S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 2437–2442, DOI:10.1055/s-0031-1289520.
- P. H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS.
- M. Jakoobi and A. G. Sergeev, Chem.–Asian J., 2019, 14, 2181–2192 CrossRef CAS.
- A. P. Y. Chan and A. G. Sergeev, Coord. Chem. Rev., 2020, 413, 213213 CrossRef CAS.
- M. Jakoobi, N. Halcovitch, G. F. S. Whitehead and A. G. Sergeev, Angew. Chem., Int. Ed., 2017, 56, 3266–3269 CrossRef CAS.
- M. Jakoobi, Y. C. Tian, R. Boulatov and A. G. Sergeev, J. Am. Chem. Soc., 2019, 141, 6048–6053 CrossRef CAS.
- J. Browning, M. Green, A. Laguna, L. E. Smart, J. L. Spencer and F. G. A. Stone, J. Chem. Soc., Chem. Commun., 1975, 723–724 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS.
- R. Y. Kong and M. R. Crimmin, Angew. Chem., Int. Ed., 2021, 60 DOI:10.1002/anie.202011594.
- D. Ellis, D. McKay, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2010, 49, 4943–4945 CrossRef CAS.
- A. Sattler and G. Parkin, Nature, 2010, 463, 523–526 CrossRef CAS.
- S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS.
- X. H. Kang, G. Luo, L. Luo, S. W. Hu, Y. Luo and Z. M. Hou, J. Am. Chem. Soc., 2016, 138, 11550–11559 CrossRef CAS.
- B. Zhu, W. Guan, L. K. Yan and Z. M. Su, J. Am. Chem. Soc., 2016, 138, 11069–11072 CrossRef CAS.
- J. J. Cabrera-Trujillo and I. Fernandez, Chem.–Eur. J., 2020, 26, 11806–11813 CrossRef CAS.
- G. P. Miscione, M. A. Carvajal and A. Bottoni, Organometallics, 2011, 30, 4924–4934 CrossRef CAS.
- J. Li and K. Yoshizawa, Chem.–Eur. J., 2012, 18, 783–787 CrossRef.
- Y. Liu, D. Zhang, J. Gao and C. Liu, Chem.–Eur. J., 2012, 18, 15537–15545 CrossRef CAS.
- X. Wen, X. Wu and J. Li, Org. Lett., 2018, 20, 1505–1508 CrossRef CAS.
- C. Perthuisot, B. L. Edelbach, D. L. Zubris and W. D. Jones, Organometallics, 1997, 16, 2016–2023 CrossRef CAS.
- W. Geibel, G. Wilke, R. Goddard, C. Kruger and R. Mynott, J. Organomet. Chem., 1978, 160, 139–147 CrossRef CAS.
- O. T. Summerscales, J. O. C. Jimenez-Halla, G. Merino and P. P. Power, J. Am. Chem. Soc., 2011, 133, 180–183 CrossRef CAS.
- Y. Ohki and H. Suzuki, Angew. Chem., Int. Ed. Engl., 2000, 39, 3463–3465 CrossRef CAS.
- R. B. King and C. A. Harmon, Inorg. Chem., 1976, 15, 879–885 CrossRef CAS.
- G. G. Cash, R. C. Pettersen and R. B. King, J. Chem. Soc., Chem. Commun., 1977, 30–31 RSC.
- E. Cabrera, J. C. Daran and Y. Jeannin, J. Chem. Soc., Chem. Commun., 1988, 607–608, 10.1039/c39880000607.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3929–3939 CrossRef CAS.
- W. D. Jones and F. J. Feher, J. Am. Chem. Soc., 1984, 106, 1650–1663 CrossRef CAS.
- P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 10462–10463 CrossRef CAS.
- R. Yazaki and T. Ohshima, Tetrahedron Lett., 2019, 60, 151225 CrossRef.
- V. G. Shubin, R. N. Berezina and V. N. Piottukh-Peletski, J. Organomet. Chem., 1973, 54, 239–246 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- P. Liu, X. F. Xu, X. F. Dong, B. K. Keitz, M. B. Herbert, R. H. Grubbs and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1464–1467 CrossRef CAS.
- M. Lin, G. Y. Kang, Y. A. Guo and Z. X. Yu, J. Am. Chem. Soc., 2012, 134, 398–405 CrossRef CAS.
- D. G. Gusev, Organometallics, 2013, 32, 4239–4243 CrossRef CAS.
- K. H. Hopmann, Organometallics, 2016, 35, 3795–3807 CrossRef CAS.
- S. Y. Hong, Y. Park, Y. Hwang, Y. B. Kim, M. H. Baik and S. Chang, Science, 2018, 359, 1016–1021 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- P. D. Zeits, T. Fiedler and J. A. Gladysz, Chem. Commun., 2012, 48, 7925–7927 RSC.
- Q. H. Luu, T. Fiedler and J. A. Gladysz, Angew. Chem., Int. Ed., 2017, 56, 5664–5666 CrossRef CAS.
- B. P. Cleary and R. Eisenberg, Organometallics, 1992, 11, 2335–2337 CrossRef CAS.
- B. P. Cleary and R. Eisenberg, J. Am. Chem. Soc., 1995, 117, 3510–3521 CrossRef CAS.
- W. H. Lam, K. C. Lam, Z. Lin, S. Shimada, R. N. Perutz and T. B. Marder, Dalton Trans., 2004, 1556–1562, 10.1039/b402632b.
- Y. Zhu, L. Fan, C. H. Chen, S. R. Finnell, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6701–6703 CrossRef CAS.
- H. V. Pham and K. N. Houk, J. Org. Chem., 2014, 79, 8968–8976 CrossRef CAS.
- J. Müller, P. E. Gaede and K. Qiao, J. Organomet. Chem., 1994, 480, 213–220 CrossRef.
- J. Müller, P. E. Gaede and K. Qiao, Angew. Chem., Int. Ed. Engl., 1993, 32, 1697–1699 CrossRef.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- N. Xiong, G. Zhang, X. Sun and R. Zeng, Chin. J. Chem., 2020, 38, 185–201 CrossRef CAS.
- J. Edwin, W. E. Geiger, A. Salzer, U. Ruppli and A. L. Rheingold, J. Am. Chem. Soc., 1987, 109, 7893–7894 CrossRef CAS.
- W. E. Geiger, A. Salzer, J. Edwin, W. Vonphilipsborn, U. Piantini and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 7113–7121 CrossRef CAS.
- Other means of driving association of two mono-Ir complexes towards the kinetic product, anti-(Cp*Ir)2(μ-arene) may exist but Fig. 7 suggests that neither steric (Avs.B and Cvs.D) nor electronic (Ir(I) vs. Ir(III)) factors alone may suffice..
- Our computations of the relative stabilities of syn- and anti-Ir(I)Ir(III) diiridium intermediates of few experimentally untested arenes suggest that in some cases the anti-isomers have higher stability even for arenes without benzylic C–H bonds (Table S15†)..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2040076 and 2040077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05900e |
| This journal is © The Royal Society of Chemistry 2021 |