
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClick activated protodrugs against cancer increase the therapeutic potential of chemotherapy through local capture and activation†
Kui
Wu‡
a,
Nathan A.
Yee‡
b,
Sangeetha
Srinivasan
b,
Amir
Mahmoodi
b,
Michael
Zakharian
b,
Jose M.
Mejia Oneto
*b and
Maksim
Royzen
 *a
*a
aUniversity at Albany, SUNY, 1400 Washington Ave., LS-1136, Albany, NY 12222, USA. E-mail: mroyzen@albany.edu
bShasqi, Inc., 665 3rd St., Suite 501, San Francisco, CA 94107, USA. E-mail: jose@shasqi.com
First published on 5th January 2021
Abstract
A desired goal of targeted cancer treatments is to achieve high tumor specificity with minimal side effects. Despite recent advances, this remains difficult to achieve in practice as most approaches rely on biomarkers or physiological differences between malignant and healthy tissue, and thus benefit only a subset of patients in need of treatment. To address this unmet need, we introduced a Click Activated Protodrugs Against Cancer (CAPAC) platform that enables targeted activation of drugs at a specific site in the body, i.e., a tumor. In contrast to antibodies (mAbs, ADCs) and other targeted approaches, the mechanism of action is based on in vivo click chemistry, and is thus independent of tumor biomarker expression or factors such as enzymatic activity, pH, or oxygen levels. The CAPAC platform consists of a tetrazine-modified sodium hyaluronate-based biopolymer injected at a tumor site, followed by one or more doses of a trans-cyclooctene (TCO)-modified cytotoxic protodrug with attenuated activity administered systemically. The protodrug is captured locally by the biopolymer through an inverse electron-demand Diels–Alder reaction between tetrazine and TCO, followed by conversion to the active drug directly at the tumor site, thereby overcoming the systemic limitations of conventional chemotherapy or the need for specific biomarkers of traditional targeted therapies. Here, TCO-modified protodrugs of four prominent cytotoxics (doxorubicin, paclitaxel, etoposide and gemcitabine) are used, highlighting the modularity of the CAPAC platform. In vitro evaluation of cytotoxicity, solubility, stability and activation rendered the protodrug of doxorubicin, SQP33, as the most promising candidate for in vivo studies. In mice, the maximum tolerated dose (MTD) of SQP33 in combination with locally injected tetrazine-modified biopolymer (SQL70) was determined to be 19.1-times the MTD of conventional doxorubicin. Pharmacokinetics studies in rats show that a single injection of SQL70 efficiently captures multiple SQP33 protodrug doses given cumulatively at 10.8-times the MTD of conventional doxorubicin with greatly reduced systemic toxicity. Finally, combined treatment with SQL70 and SQP33 (together called SQ3370) showed antitumor activity in a syngeneic tumor model in mice.
Introduction
Directing the activity of drugs to a location of the body where they are needed is a critical medicinal chemistry challenge, particularly with cytotoxic chemotherapy. Typically, only 1–2% of a systemically administered dose reaches the desired site i.e. the tumor, and as a result, these treatments are often associated with severe systemic adverse effects.1 This limits the amount of drug that can be given to a patient safely. Thus, a critical challenge associated with cytotoxic chemotherapies is their narrow therapeutic window. For example, doxorubicin (Dox) is a commonly used cytotoxic anthracycline drug with well-characterized and severe side effects. The antitumor activity of Dox is attributed to its interference of DNA replication by inhibiting topoisomerase II enzyme, and it has been used in the clinic to treat a variety of neoplastic conditions such as soft tissue sarcoma, acute lymphoblastic leukemia, acute myeloblastic leukemia, breast cancer, Kaposi's sarcoma and other types of cancer.2 Studies have shown that the efficacy of potent antitumor drugs like Dox increase with higher concentrations in vitro and in vivo.3,4 Despite its potent broad-spectrum antitumor activity, the clinical use of Dox is limited by life-threatening toxicities, with the most significant being largely irreversible and dose-dependent cardiotoxicity.5 The short- and long-term toxic effects in the heart range from alterations in myocardial structure and function, to congestive heart failure and severe cardiomyopathy. Congestive heart failure was reported in >4%, >18%, or 36% of patients who had received cumulative Dox doses of 500 to 550, 551 to 600, or >601 mg m−2, respectively.6 In response, the clinical lifetime maximum dose of Dox is limited to 400–450 mg m−2 in patients to minimize cardiotoxicity. However, this dose also limits the antitumor therapeutic benefit received by patients.7 Treatment of solid tumors is an important example of an unmet medical need for novel strategies to efficiently focus cytotoxic activity locally, while sparing the rest of the body of deleterious toxic effects.A number of drug delivery approaches have been developed in order to address these shortcomings. To improve the therapeutic window of Dox, targeted drug activation approaches have been developed in which Dox-based therapeutics with attenuated activity are administered systemically and activated in the body by inherent biological differences between cancerous and healthy tissue, such as pH,8 oxygen level,9 or protein expression.10 Several such therapeutics have been developed by modifying the aminoglycoside portion of Dox with peptides, designed to be cleaved by proteases elevated in cancerous cells: (1) L-377202 (Merck),11–13 (2) DTS-201 (or CPI-0004Na; Diatos),10 and (3) “Compound 5” (Bristol-Myers Squibb).14 The main benefit of cancer-activated therapeutics is that they may eliminate cancerous cells, while sparing healthy tissue from the cytotoxic effects. However, variability in the biological differences between cancerous and healthy tissue, as well as interpatient variabilities, are major limitations and current targeted activation approaches have failed to reach meaningful improvements to the safety profile of conventional Dox in the clinic.
In addition to dose-limiting toxicity, poor aqueous solubility is another obstacle in the systemic administration of well-established anticancer drugs, such as paclitaxel (PTX) and etoposide (ETP). Formulation of these drugs often requires organic co-solvents which restricts their utility in the clinic. PTX has been widely used to treat lung, breast, pancreatic and ovarian cancers.15 This anti-microtubule diterpenoid triggers cellular apoptosis by blocking the progression of mitosis. Due to poor solubility, PTX is typically formulated using ethanol or polyethylated castor oil, Cremophor EL. This formulation can cause serious side-effects, such as peripheral neuropathy, hypotension, and hypersensitivity.16,17ETP is frequently used in combination with other chemotherapeutic agents to treat refractory testicular tumors, small-cell lung cancer, lymphoma, non-lymphocytic leukemia and glioblastoma multiforme.18ETP inhibits DNA topoisomerase II, thereby inhibiting DNA synthesis at the pre-mitotic stage. Due to poor aqueous solubility, administration of high-doses of ETP requires formulation in ethanol, polysorbate 80, benzyl alcohol, and polyethylene glycol and can cause hypotension and allergic reaction.19,20
In order to address these challenges, we developed the modular, Click Activated Protodrugs Against Cancer (CAPAC) platform, in which cytotoxics are activated locally through the combination of systemically administered protodrugs with attenuated toxicity, and a locally injected protodrug-activating biopolymer. Click chemistry-based approaches involve highly reactive and mutually selective chemical groups that minimally interact with natural mammalian physiology – thus the site-specific targeting is achieved through chemistry rather than relying on the inherent biological properties of the tumor.21–24 The CAPAC platform is illustrated schematically in Fig. 1, and involves 4 steps: (1) a biopolymer modified with tetrazine (Tz) groups is injected directly to a tumor site. (2) A protodrug form of a cytotoxic, which has been chemically attenuated by modification with trans-cyclooctene (TCO), is administered systemically. (3) The protodrug is captured at the biopolymer injection site (the targeted region) and removed from circulation through a covalent inverse electron-demand Diels–Alder reaction between TCO and Tz, concentrating it to the desired site with precise spatial control. (4) A subsequent chemical reaction spontaneously releases the active drug. In this approach, the site-specific targeting is achieved through the chemical reactivity of Tz and TCO, rather than relying on the inherent biological properties of the tumor, resulting in a wider therapeutic window compared to other targeted approaches. Additionally, hydrophilic substituents can be incorporated to the TCO moiety, allowing for improved aqueous solubility of the protodrug over the parent compound.
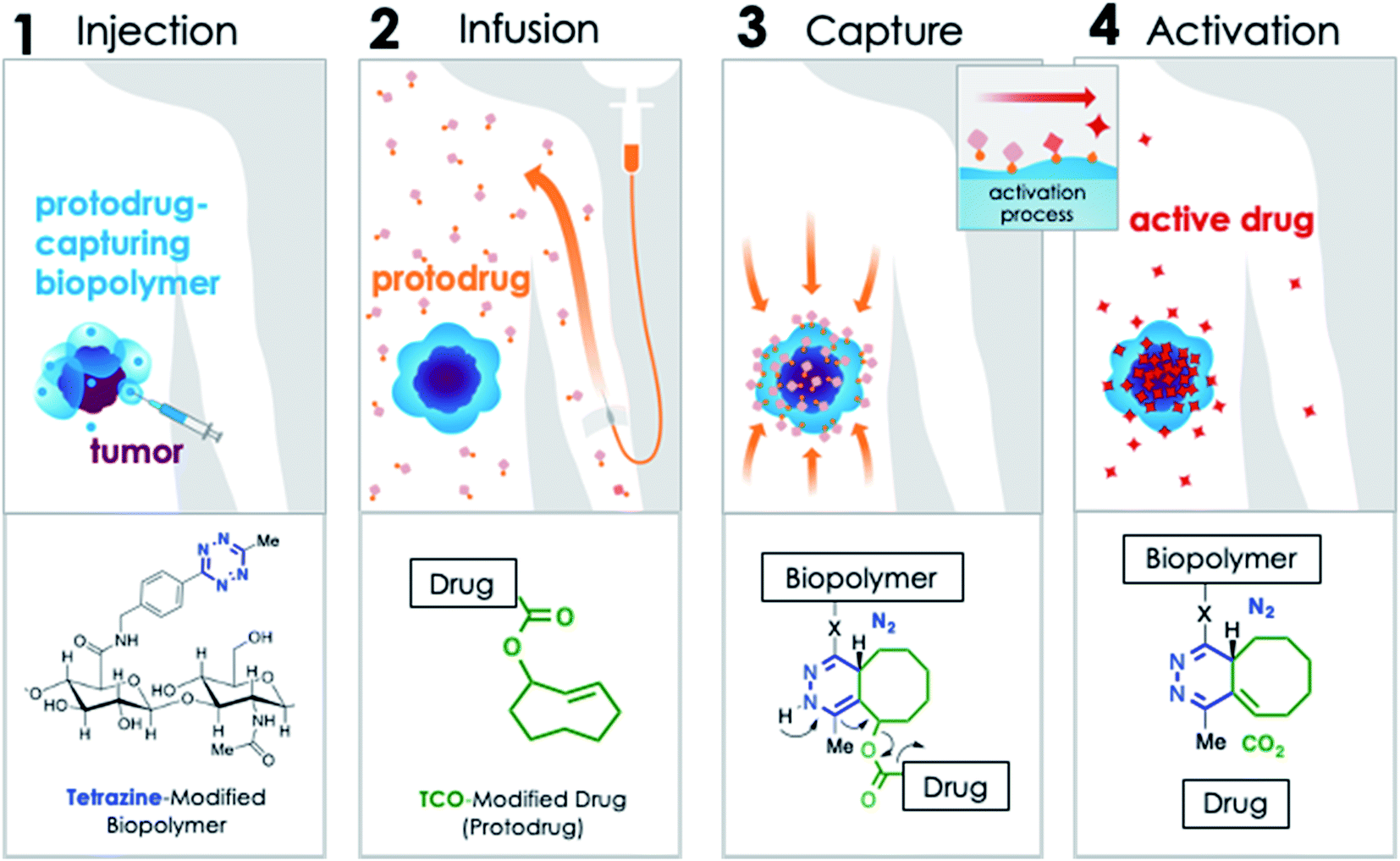 | ||
| Fig. 1 Mechanism of CAPAC platform: (1) Tz-modified biopolymer is locally injected at the pathological site. (2) A TCO-modified drug (protodrug) is infused systemically. (3) The protodrug is captured by the biopolymer at the desired site through a rapid covalent reaction between Tz and TCO moieties, followed by (4) chemical rearrangement to release active drug. | ||
A key aspect of the CAPAC platform is intratumoral/peritumoral injection of the Tz-modified biopolymer. Intratumoral therapy is an attractive alternative to conventional chemotherapy because it can allow safer and more aggressive administration of antineoplastic agents directly into a desired lesion. This modality can be used pre- and post-operatively and is especially beneficial for patients who have inoperable lesions, as well as those who are ineligible for surgery. Recently, intratumoral treatments have been shown to induce systemic antitumor responses in non-injected tumors.25,26 In this way, intratumoral therapies can be used to treat tumors that have advanced beyond a strictly local or regional site.
When the earliest clinical reports for direct intratumoral injection of chemotherapy were published more than 50 years ago,27,28 there were many issues such as local tissue damage from imprecise injections, rapid drug clearance, and the need for repeated in-patient injection procedures which prevented the technique from becoming a mainstream mode of chemotherapy administration.29 Since then, advances in material science and technology, such as the advent of image-guided techniques for biopsies and injections30 have addressed many of these issues. As a result, intratumoral injections are not only feasible, but increasingly common in clinical trials for cancer.31–33 In fact, the lead candidate of the CAPAC platform, SQ3370, is currently being evaluated in a Phase 1 clinical trial in patients with advanced solid tumors, in which the biopolymer is administered through peri/intratumoral injection.34
Results
We have previously demonstrated proof-of-concept and feasibility of the CAPAC platform in an animal model,24 and showed enhanced safety and efficacy with a cytotoxic protodrug in a mouse tumor model.35 The CAPAC approach allowed a higher amount of a TCO–Dox protodrug to be given intravenously (IV) without adverse effects as compared with conventional Dox treatment, and furthermore, it showed complete response when treating a local fibrosarcoma in mice. However, clinical translation of the previous construct was problematic due to poor aqueous solubility of the TCO–Dox protodrug, which considerably limited the amount of treatment that could be given IV. Described herein, we incorporated hydrophilic groups to the TCO moiety in order to improve aqueous solubility of the resulting protodrugs. We also highlight the modularity of the CAPAC platform through synthesis of novel protodrugs of small molecule chemotherapeutics like PTX, ETP and gemcitabine (GCB), in addition to Dox. We then evaluated the cytotoxicity, solubility, stability and activation of these protodrugs in vitro and identified the Dox protodrug, SQP33, as the best candidate for in vivo studies. SQP33 protodrug was tested in vivo with SQL70 biopolymer, which is composed of sodium hyaluronate (NaHA), a non-sulfated glycosaminoglycan, modified with Tz. IV administration of SQP33 following a subcutaneous (SC) injection of SQL70 (together called SQ3370 treatment) proved to be significantly less toxic than Dox hydrochloride (HCl), allowing markedly higher doses to be well-tolerated in mice and rats, with significantly reduced adverse cytotoxic exposure. Specifically, a cumulative SQP33 dose equivalent to 10.8-times the cumulative maximum tolerated dose (MTD) of Dox HCl was safely administered to rats resulting in a lower Dox exposure observed in their heart tissues as compared to rats treated with Dox HCl. The MTD of SQ3370 in mice was established at 19.1-times the single-dose MTD of Dox HCl. Furthermore, pharmacokinetic (PK) analysis in mice and rats confirmed the hypothesis that SQL70 biopolymer effectively captures SQP33 protodrug, resulting in release of active Dox at the desired site.Protodrug design, synthesis and in vitro evaluation
Recognizing that the TCO moiety is only present at the protodrug stage led to the design of a second generation of protodrugs with improved aqueous solubility. Structural elements of TCO were explored to introduce additional hydrophilic groups, as illustrated in Scheme 1. At the core of the protodrug structures is a heterobifunctional TCO36,37 bearing a carboxylic acid at a tertiary carbon, which was regioselectively modified with cytotoxic compounds on one end and functionalized with hydrophilic groups, such as glycine or aspartic acid on the other. Importantly, the TCO carboxylate used is 99% enantiomeric excess (ee) at that tertiary chiral carbon, so that only single diastereomers of the protodrugs are formed. Three general synthetic procedures have been developed that allow modular assembly of these constructs, while taking into account inherent sensitivity of Dox, PTX, ETP and GCB to various late-stage chemistries. In principle, other amino acids or hydrophilic groups could be installed as desired.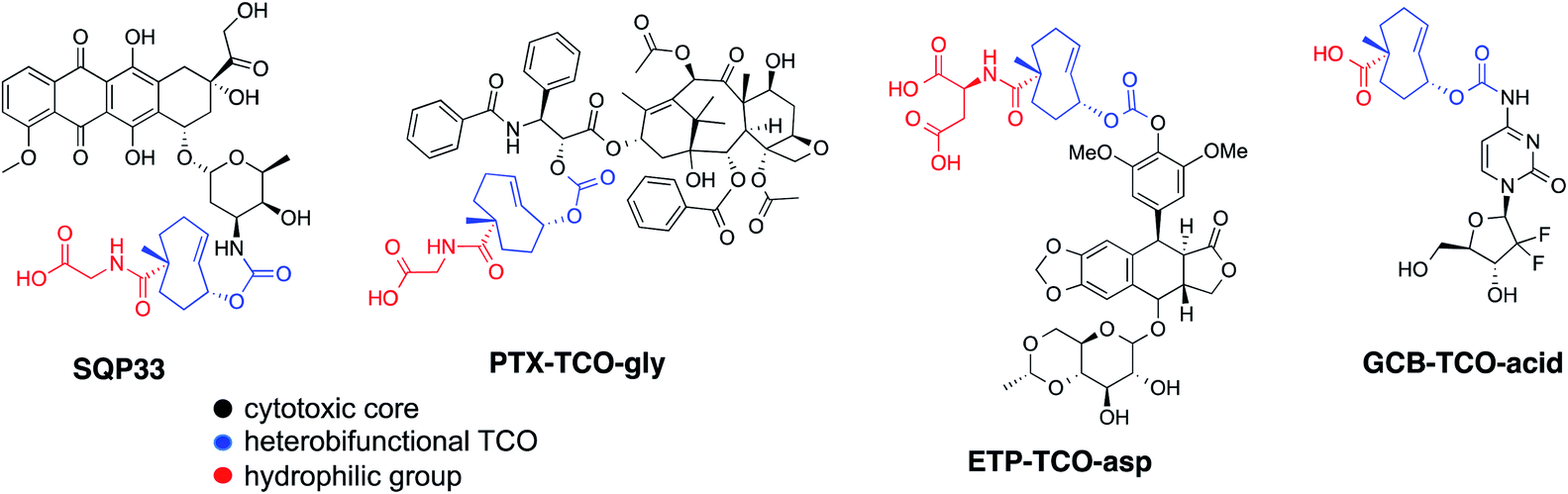 | ||
| Scheme 1 Chemical structures of TCO-protodrugs of various cytotoxics. | ||
The first strategy, utilized for the synthesis of Dox protodrug SQP33, entailed sequential coupling of the drug followed by the hydrophilic group to heterobifunctional TCO intermediate 1. Derivative 1 was prepared as a single diastereomer in 11 steps starting from 1,5-cyclooctadiene.36,37 The critical final steps are described in Scheme 2. Coupling of Dox to the more reactive NHS carbonate afforded compound 2. The final step proved to be challenging, as the direct coupling of glycine proceeded in low yield. Alternatively, coupling of glycine methyl ester failed to offer a path forward due to the challenges of subsequent saponification given the lability of Dox to basic hydrolysis of the methyl ester. Enzymatic ester hydrolysis was also unsuccessful due to poor ester solubility in phosphate-buffered saline (PBS). Moreover, addition of dimethyl sulfoxide (DMSO) as a co-solvent interfered with the esterase activity. We also considered acid-labile protecting groups, but, recognized that the sensitivity of the TCO group towards isomerization to the corresponding cis-isomer limited this strategy. Finally, we discovered that transient protection of the carboxylic acid moiety of glycine as a trimethylsilyl (TMS) ester allowed efficient coupling to 2. The TMS protecting group was found to be extremely moisture-sensitive and its deprotection was achieved upon aqueous workup of the reaction mixture.
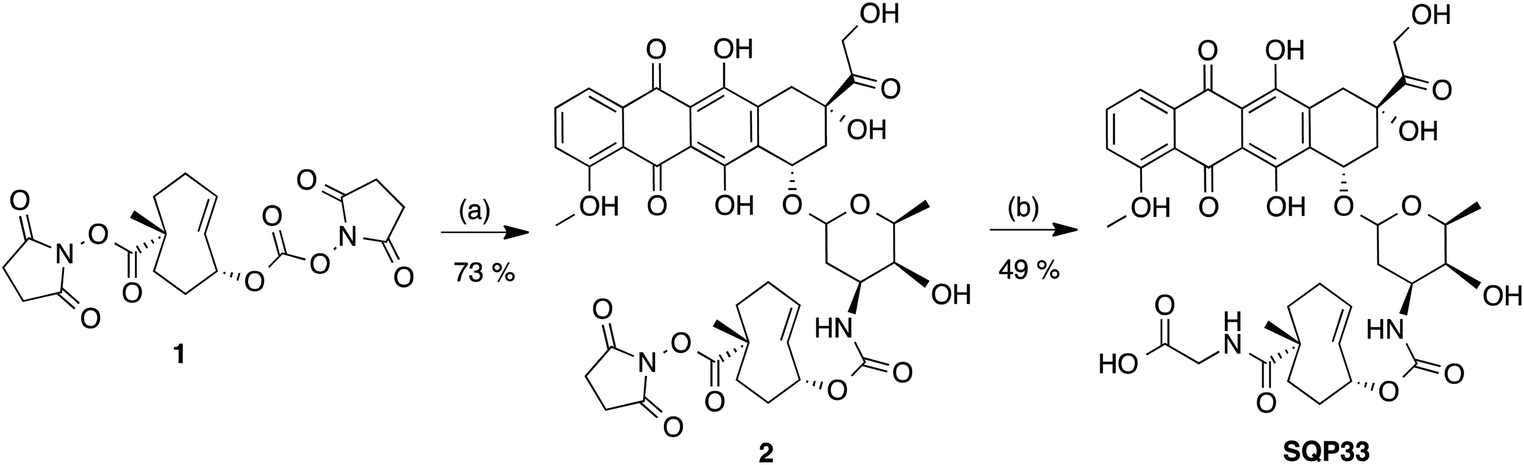 | ||
| Scheme 2 Synthesis of SQP33: (a) Dox, DIPEA, DMF; (b) glycine, TMS–Cl, DIPEA, CH2Cl2, CH3CN. | ||
Two different synthetic strategies, illustrated in Schemes 3A and B, were developed to synthesize the protodrugs of PTX and ETP. In each strategy, the heterobifunctional TCO derivative 3, was first coupled to hydrophilic groups containing protected carboxylic acids. As shown in the preparation of the PTX protodrug in Scheme 3A, Fmoc-protected glycine ester 4 was prepared, activated to p-nitrophenyl carbonate 5, and coupled with PTX at the side chain hydroxyl to give the Fmoc-protected intermediate 6. Then, PTX-TCO-gly was obtained by removal of the Fmoc protecting group using piperidine in DMF. The preparation of the ETP protodrug is shown in Scheme 3B. Briefly, TCO derivative 3 was modified with bis-2-(trimethylsilyl)ethyl aspartate to give intermediate 8, and the allylic alcohol group was converted to the p-nitrophenyl (PNP) carbonate 9. ETP-TCO-asp was obtained by coupling of 9 with ETP and subsequent deprotection of the bis-(trimethylsilyl)ethyl ester groups using TBAF.
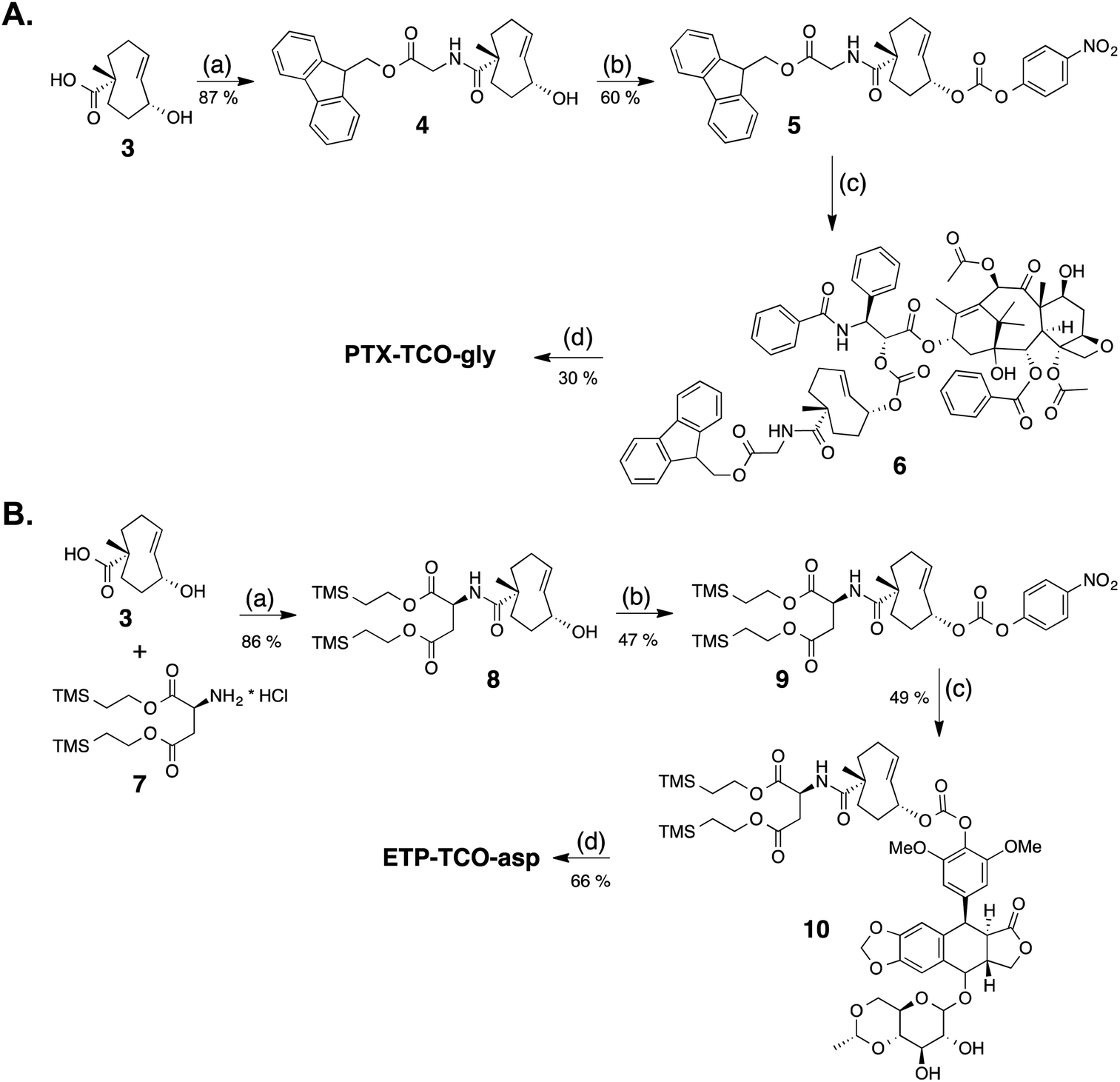 | ||
| Scheme 3 (A) Synthesis of PTX-TCO-gly: (a) Fmoc-O-glycine, HATU, DIPEA, DMF; (b) PNP–Cl, pyridine, CH2Cl2; (c) paclitaxel, DMAP, CH2Cl2; (d) piperidine, DMF. (B) Synthesis of ETP-TCO-asp: (a) HATU, DIPEA, DMF; (b) PNP–Cl, pyridine, CH2Cl2; (c) etoposide, Cs2CO3, DMF; (d) TBAF, THF. | ||
A third synthetic strategy was employed to access GCB-TCO-acid, as illustrated in Scheme 4. First, the carboxylic acid group of the TCO 3 was protected in two steps: (1) selective conversion to NHS ester 11 using N,N-disuccinimidyl carbonate (DSC), followed by (2) coupling with to 2-(trimethylsilyl)ethanol to give TMS ethyl ether 12. The allylic alcohol of 12 was subsequently converted into an active PNP carbonate, which reacted with the free amino group of tetraisopropyldisiloxane-protected gemcitabine 14 to give adduct 15. Global deprotection of the silyl groups with TBAF afforded GCB-TCO-acid.
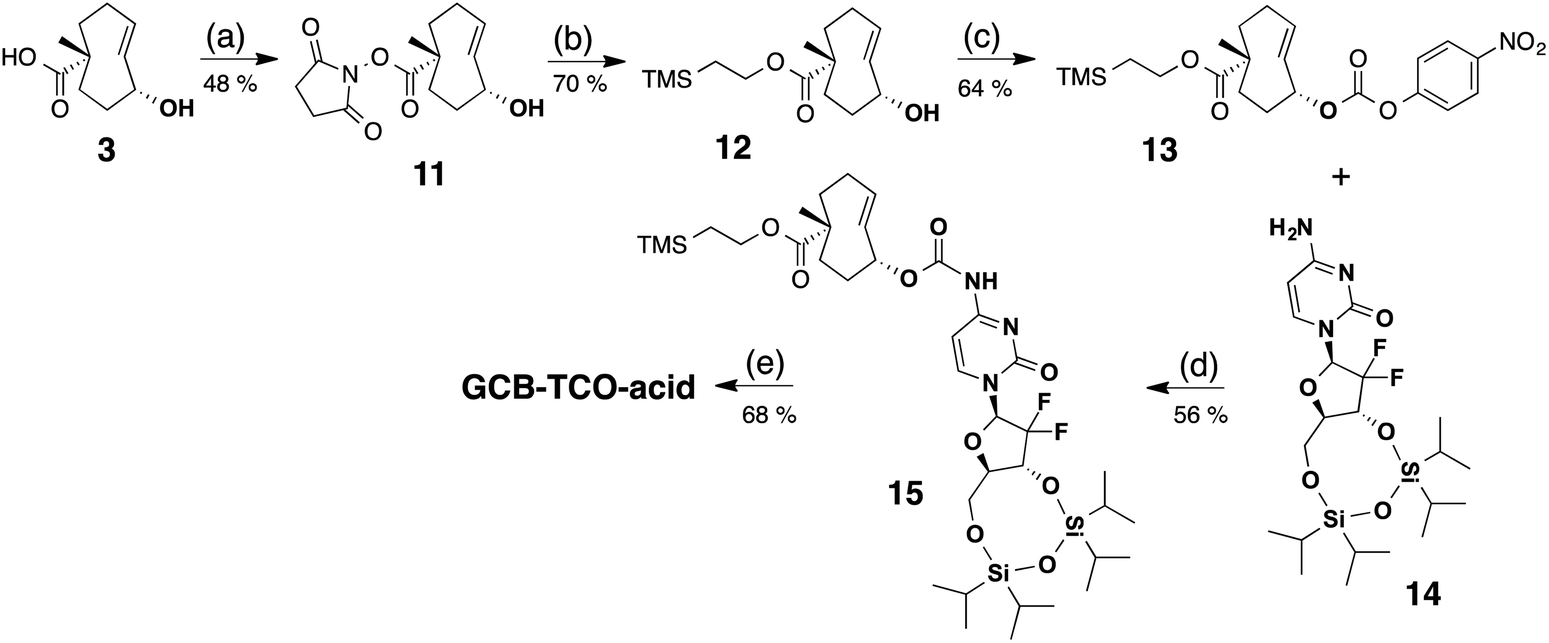 | ||
| Scheme 4 Synthesis of GCB-TCO-acid: (a) DSC, DIPEA, CH3CN; (b) TMS–ethanol, NaH, THF; (c) PNP–Cl, pyridine, CH2Cl2; (d) NaH, DMF; (e) TBAF, THF. | ||
Extensive in vitro evaluation of the protodrugs was carried out to choose a suitable candidate for in vivo studies. The goal was to identify protodrugs that would be at least ten-fold less cytotoxic than the parent drugs, and which were up to ten-fold more soluble in physiologically-relevant aqueous media. Both of these factors would allow safe administration of higher systemic doses. The stability of each protodrug was also tested in mouse blood plasma to exclude the possibility of non-specific protodrug activation. Lastly, it was confirmed that a Tz-modified biopolymer could both efficiently capture the protodrugs, and subsequently activate them into the active cytotoxic drugs.
Table 1 summarizes cytotoxicity, solubility, plasma stability and biopolymer-triggered activation of the protodrugs. TCO modification resulted in a pronounced lowering of cytotoxicity of the protodrugs of Dox and ETP. Specifically, SQP33 was 83-times less cytotoxic to murine colorectal MC38 cells than Dox (Fig. S1A–C†), while ETP-TCO-asp was 67-times less cytotoxic than ETP in this same assay (Fig. S3A–C†). In contrast, we found more modest reductions in cytotoxicity, less than 10-fold reduction, for the protodrugs of PTX and GCB, suggesting that the pharmacophore might not be fully masked in those derivatives (Fig. S2A–C and S4A–C†). In each case, the addition of the hydrophilic groups to the TCO moieties improved the solubility of each of the protodrugs, except for GCB, which is typically administered as an HCl salt. The most dramatic effect was again observed for SQP33, which was 7.3-times more soluble in PBS than the HCl salt of Dox. HPLC studies described in the ESI† confirmed that Tz-modified biopolymer can efficiently convert the protodrugs of Dox, PTX and ETP into the corresponding active cytotoxic agents over a 24 hour (h) period (Fig. S1D, S2D and S3D†). Activation of GCB-TCO-acid was studied using NMR (Fig. S4D†). Based on these results, SQP33 and ETP-TCO-asp appeared to be the best candidates for in vivo studies. To focus on a single agent for preclinical studies suitable for enabling an Investigational New Drug (IND) application, SQP33 was chosen on the basis of its plasma stability.
| Compound | Cytotoxicity (IC50) | Solubility in PBS (mg mL−1) | Protodrug plasma stability (%) |
|---|---|---|---|
| Dox HCl | 23 nM | 3.8 | |
| SQP33 | 1.9 μM (83-fold) | 28 | >99 |
| PTX | 32 nM | 0.1 | |
| PTX-TCO-gly | 80 nM (2.5-fold) | 1 | 96.5 |
| ETP | 160 nM | 2.8 | |
| ETP-TCO-asp | 10.8 μM (67-fold) | 5 | 65.8 |
| GCB HCl | 3 nM | 38.6 | |
| GCB-TCO-acid | 26 nM (8.7-fold) | 3 | >99 |
SQL70 biopolymer
SQL70 biopolymer is based on the non-sulfated glycosaminoglycan, NaHA, that is well tolerated in the body, and has been used clinically in unmodified, crosslinked, and derivatized form for over five decades.38,39 Commercially available NaHA was modified with Tz, as shown in Scheme 5. Briefly, NaHA was activated with NHS and the carbodiimide EDC-HCl in the presence of (4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine 16 in aqueous solution. Removal of reagents and byproducts by tangential flow filtration afforded the NaHA–Tz biopolymer. After assessment of different HA polymers and different degrees of Tz substitution, the NaHA–Tz biopolymer formed from 12 kDa HA and ∼19 weight% Tz modification was selected, henceforth called SQL70 biopolymer. When injected in mice, ∼50% of SQL70 of persisted at the SC injection site after 14 days (Fig. S5†). Due to this extended duration of residence, we anticipated that a single biopolymer injection could be used to activate multiple protodrug doses in vivo.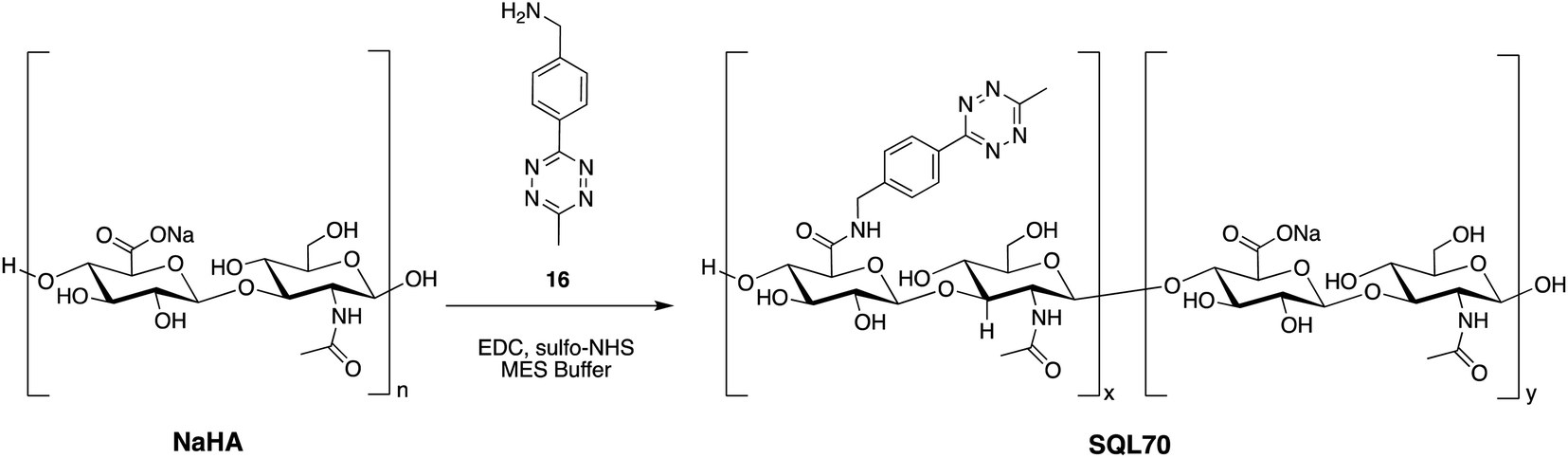 | ||
| Scheme 5 Structure and synthetic scheme of SQL70 biopolymer. | ||
Reduced systemic toxicity of SQ3370
The reduced toxicity of SQP33 (83-fold less toxic than Dox HClin vitro) translated to greater tolerability in vivo, enabling significantly higher doses to be administered in rodents compared to conventional Dox. To quantitatively assess the tolerability of SQ3370in vivo, a toxicity study was conducted in mice to determine the MTD compared to conventional Dox. C57BL/6 mice received varying dose levels of SQP33 (given IV once daily for 5 days) in the presence or absence of SC-injected SQL70 biopolymer, or received conventional Dox HCl as a single dose, using a 10% drop in body weight as the MTD threshold. A single dose of Dox HCl at 20 mg kg−1 induced a drop in body weight below 10% of baseline and was defined as the MTD (Fig. 2). In the presence of SQL70 biopolymer, SQP33 at a cumulative dose of 383 mg kg−1 (76.6 mg per kg per day), in Dox HCl equivalents (Dox eq.), resulted in a body weight drop slightly under 10%. The higher dose level tested (cumulative 487 mg kg−1 Dox eq.) caused a 15% drop in body weight (Fig. S7†). Thus, the MTD for SQ3370 was determined to be 383 mg kg−1 Dox eq., a 19.1-fold enhancement of tolerability over conventional Dox HCl. Notably, in the absence of SQL70 biopolymer, SQP33 at both 383 mg kg−1 and 487 mg kg−1 Dox eq. produced no decline in body weight, confirming in an animal model the safety of the protodrug itself.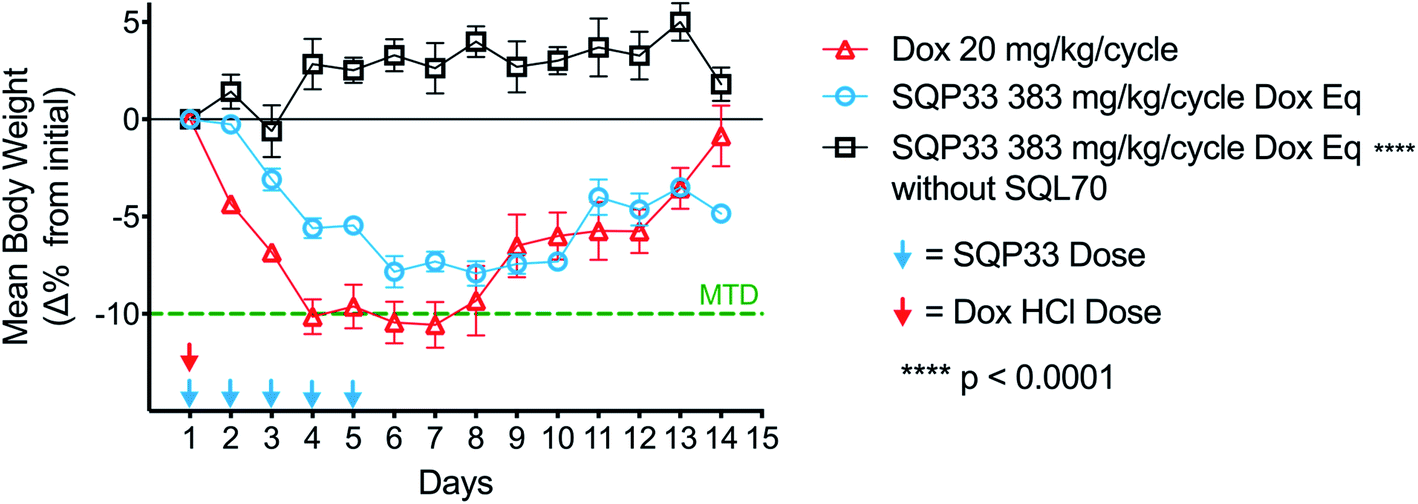 | ||
| Fig. 2 MTD of SQ3370vs. conventional Dox HCl in mice. C57BL/6 mice (n = 4–5 per group) were treated with one dose of Dox HCl at 20 mg kg−1 and compared to mice treated with 5 daily doses of SQP33 at 76.6 mg per kg per dose Dox eq. (cumulative 383 mg kg−1 Dox eq.), in the presence or absence of SQL70 biopolymer. Mice were weighed daily for up to 14 days once treatment started. A mean body weight loss ≥10% per group indicated the MTD. Data are mean ± SEM of percent change in body weights and significance was established by unpaired t-test with Welch's correction compared to the Dox HCl control group. | ||
Similarly, rats injected with SQL70 readily tolerated a cumulative IV dose of SQP33 equivalent to 10.8-times the cumulative MTD of Dox HCl without overt toxicities. These results represent a significant safety margin over existing clinically tested Dox-based therapeutics in both species. Fig. 3 compares the amount of Dox safely administered in one week viaSQ3370 to the MTD, given as a single dose or as the sum of multiple doses administered over a month, of conventional Dox HCl or Dox-based therapeutics that have undergone clinical evaluation. The tolerability of considerably higher doses in a shorter amount of time was attributed primarily to the attenuated systemic cytotoxicity of SQP33. Moreover, the ability of SQL70 biopolymer to efficiently sequester the protodrug out of circulation and concentrate the active Dox to a targeted site further contributed to higher dose tolerance. The efficiency of protodrug capture and activation in vivo were further investigated by PK analysis, as described below.
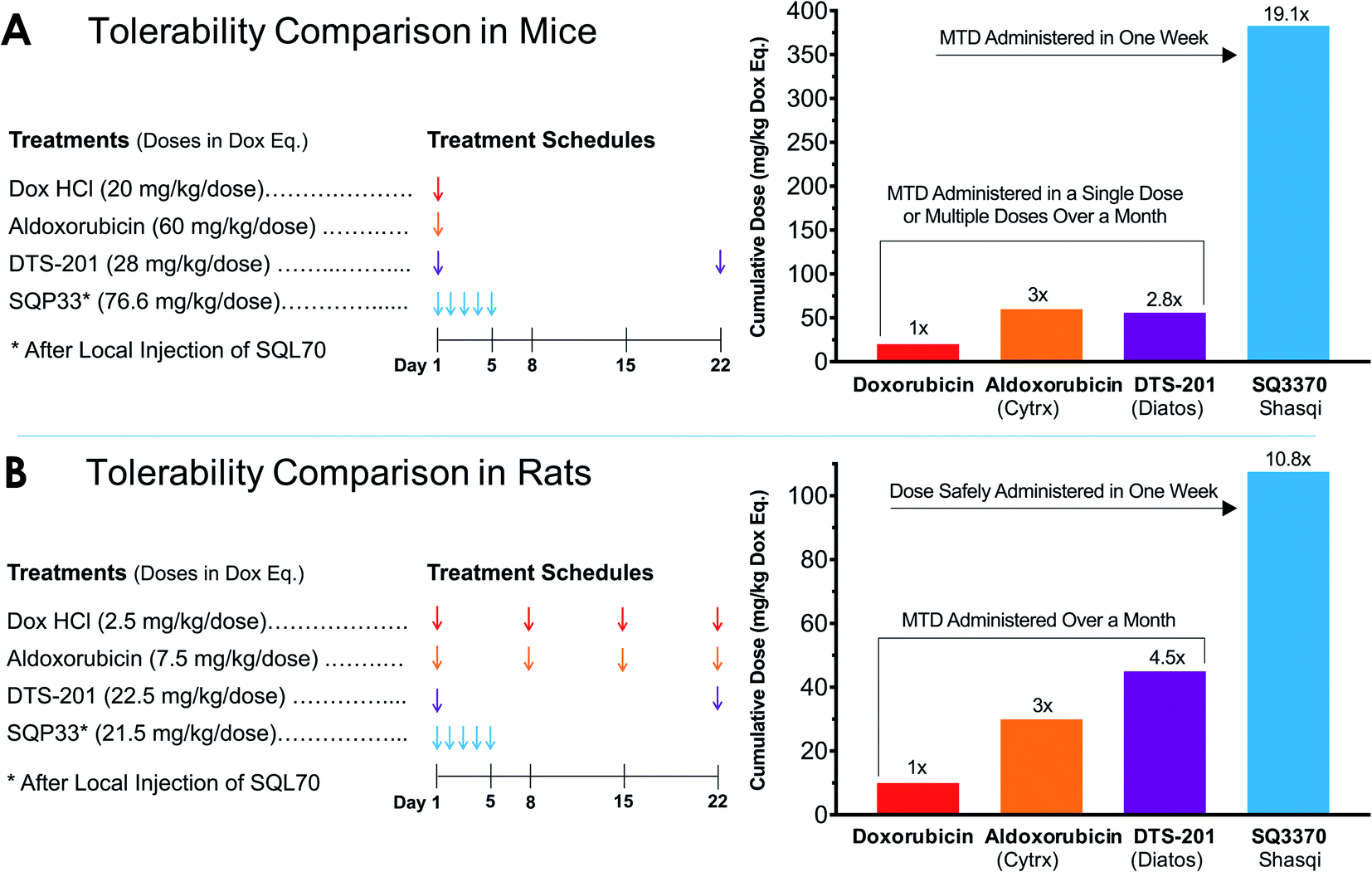 | ||
| Fig. 3 Tolerability of SQ3370 as compared to Dox and clinically tested Dox-based treatments in rodents. (A) The cumulative MTD of SQ3370 in mice was determined to be 383 mg kg−1 (76.6 mg per kg per day × 5 days), representing a 19.1-fold enhancement over the experimentally determined single-dose MTD of conventional Dox. (B) In rats, a cumulative dose of 107.5 mg kg−1 (21.5 mg per kg per day × 5 days) was tolerated without overt toxicities, a 10.8-fold margin over the conventional Dox MTD administered as 4 weekly doses.40 Previous Dox-based treatments have only been able to advance the MTD up to 60 mg kg−1 in mice40 and up to 45 mg kg−1, administered in two doses 22 days apart, in rats.41 All doses here are represented in Dox eq. | ||
In vivo pharmacokinetics
Studies in rodents were carried out to determine the plasma PK and tissue Dox exposure of treatment with SQ3370. To assess plasma PK, Sprague Dawley (SD) rats were split into two groups, one group receiving a SC SQL70 biopolymer injection 1 h before start of SQP33 dosing, and the other group receiving only SQP33 protodrug infusions without an SQL70 biopolymer injection. SQP33 was administered IV to both groups on Days 1, 2, 3, 4 and 5 at 21.5 mg per kg per day Dox eq. Each daily dose of SQP33 was the equivalent of 2.9-times the single-dose MTD of conventional Dox (7.4 mg kg−1),42 and the cumulative dose of SQP33 over the 5 days (107.5 mg kg−1 Dox eq.) was 10.8-times the multi-dose MTD of conventional Dox given over 22 days (10 mg kg−1),40 as discussed previously. No adverse effects were observed in animals receiving these high doses throughout the study. Blood was drawn 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h post-SQP33 injection on each day and liquid chromatography-mass spectrometry (LC-MS) was used to measure plasma SQP33 protodrug and active Dox concentrations. Day 1 plasma concentrations of protodrug and active Dox, following a single dose of SQP33 in the absence or presence of SQL70 biopolymer are shown in Fig. 4. When biopolymer was injected, within 30 minutes after dosing the plasma concentration of protodrug was below the limit of quantification (BLOQ; 1 ng mL−1), indicating rapid capture by the biopolymer. This was associated with an increase in Dox, confirming release of the active drug. Conversely, in the absence of SQL70 biopolymer, the administered protodrug was detected throughout the 24 h period, with significantly lower levels of Dox detected. Comparison of PK parameters (Tables S1 and S2†) in the absence and presence of SQL70 provide additional quantitative comparison. On Day 1, the plasma exposure of active Dox in the no-SQL70 group, as measured by area-under-the-curve (AUC), was only 2.2% of the value of the SQL70-injected group, suggesting that SQP33 protodrug was stable in vivo with minimal non-specific conversion to active Dox in the absence of SQL70 biopolymer. Accordingly, the Day 1 plasma exposure of SQP33 protodrug decreased by 2444-fold in the presence of SQL70 (compared to the no-SQL70 group), as rapid capture by the biopolymer rendered the protodrug undetectable in plasma following the 0.5 h timepoint.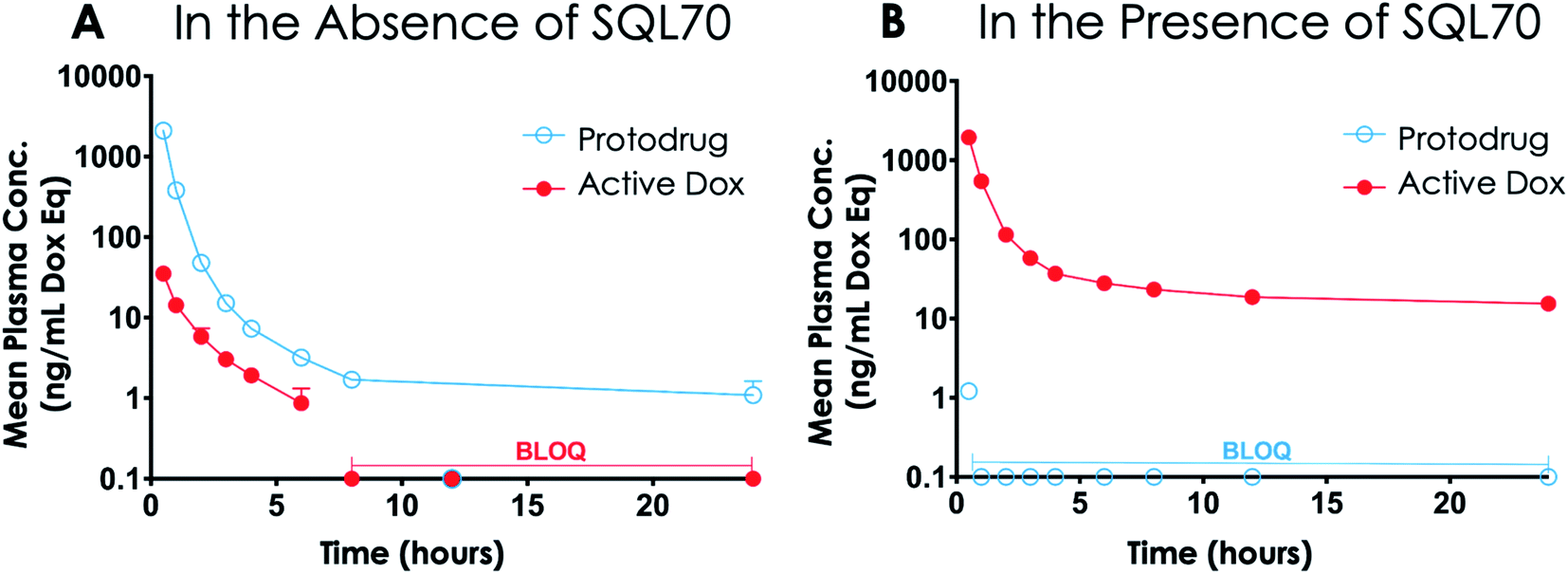 | ||
| Fig. 4 Nonspecific activation of SQP33 protodrug. SD rats were treated with an intravenous infusion of SQP33 protodrug at 21.5 mg kg−1 Dox eq. (2.9-times higher than the single-dose MTD of Dox HCl) in the absence (A) or presence (B) of SQL70 biopolymer (click activating group) injection. This figure shows the 24 h plasma concentrations of SQP33 protodrug and active Dox following treatment. Plasma concentrations of SQP33 protodrug and active Dox were measured using LC-MS at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after the SQP33 protodrug infusion. Concentrations are displayed on a log10 scale of the mean + SEM of n = 3 per timepoint, and measurements that were BLOQ (1 ng mL−1) are indicated. | ||
PK analysis also showed SQL70 biopolymer retains in vivo activity over the 5 day dosing period. The plasma concentrations of SQP33 protodrug and active Dox over the 5 day SQP33 dosing period following a single SQL70 biopolymer injection on Day 1 are shown in Fig. 5. An increase in the plasma concentration of active Dox was observed (compared to the no biopolymer control on Day 1) following each dose of SQP33 when SQL70 biopolymer was present, indicating a retention of the activating capabilities over the 5 day treatment period. We note that the maximum concentration (Cmax) of SQP33 protodrug increased with each additional dose, while the Cmax of active Dox decreased over the 5 days (Table S2†). The average plasma SQP33 protodrug Cmax on Day 5 with SQL70 biopolymer (Table S2†) was reduced by 289.7 ng mL−1 compared with the average Cmax (Table S1†) of the no-biopolymer treatment group from Day 1 to Day 5. This finding was correlated with a 74.9 ng mL−1 increase in the average plasma active Dox Cmax on Day 5 with SQL70 biopolymer (Table S2†) compared to the average without the biopolymer (Table S1†). This indicates that while activity is indeed retained over at least 5 days, there is a gradual decrease in the capture and activation properties. We hypothesize that this is due to multiple factors, including depletion of the number of active Tz sites on the biopolymer with repeated protodrug dosing, biopolymer clearance, or degradation of the Tz itself over time in vivo. Experiments in which a single dose of SQP33 was administered following SQL70 injection, with varying time intervals in between (Fig. S8†) show that activity diminishes even without repeated protodrug dosing, suggesting that Tz depletion is not the only factor.
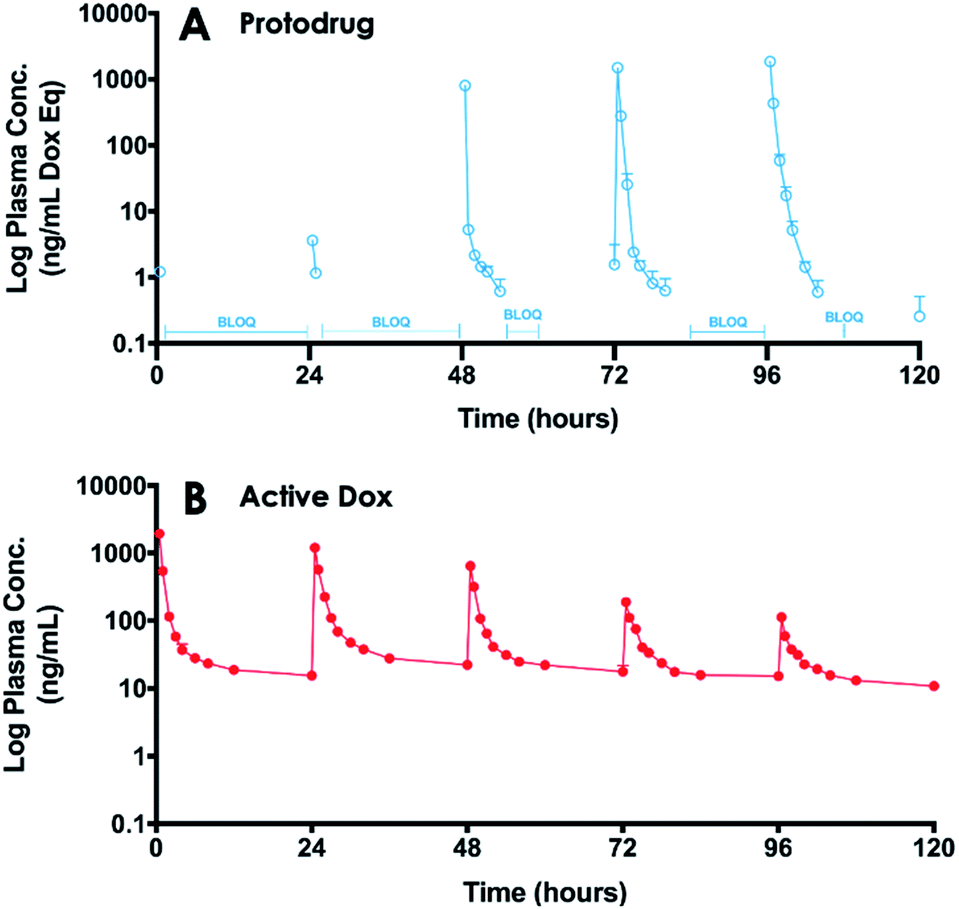 | ||
| Fig. 5 5 day plasma concentration–time curves of SQP33 protodrug and active Dox in rats treated with SQ3370. SD rats were given one SC SQL70 biopolymer injection 1 h before SQP33 protodrug dosing began. 21.5 mg per kg per dose Dox eq. of SQP33 protodrug was administered IV at time points 0, 24, 48, 72, and 96 h. The cumulative dose of SQP33 protodrug added up to 107.5 mg kg−1 Dox eq. (10.8-times higher than the multi-dose MTD of Dox HCl). Plasma concentrations of SQP33 protodrug and active Dox were measured using LC-MS at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after each SQP33 protodrug dose. Plasma concentrations over 120 h are displayed on a log10 scale of the mean + SEM of n = 3 per timepoint for both SQP33 protodrug (A) and active Dox (B). Plasma SQP33 protodrug curve (A) is disconnected between some measurements because concentrations between those measurements were BLOQ (1 ng mL−1). | ||
To explore the off-target active drug exposure of the CAPAC platform compared to conventional chemotherapy, we compared the tissue active Dox exposure from treatment with SQ3370 to that of treatment with conventional Dox HCl. Two groups of SD rats were treated with 5 daily doses SQP33 at 21.5 mg per kg per dose Dox eq. in the presence or absence of SQL70 biopolymer and compared to a control group that was injected IV with Dox HCl at 8.1 mg kg−1. Tissue samples were collected from the heart, kidney and liver 5 days after treatment, and analyzed using LC-MS. Fig. 6 shows the dose-adjusted tissue active Dox exposure for each group. In both groups that were treated with SQP33, adverse active Dox exposure to non-target tissues was significantly lower than the group treated with conventional Dox HCl relative to the dose administered. Importantly, the dose-adjusted heart Dox exposure was significantly lower in SQ3370 treatment, which may result in a reduction of Dox-induced cardiotoxicity. Notably, cumulative absolute Dox exposure to the heart was also lower with SQ3370 treatment (Fig. S9†), despite SQ3370 being administered at over 13-times the dose of conventional Dox.
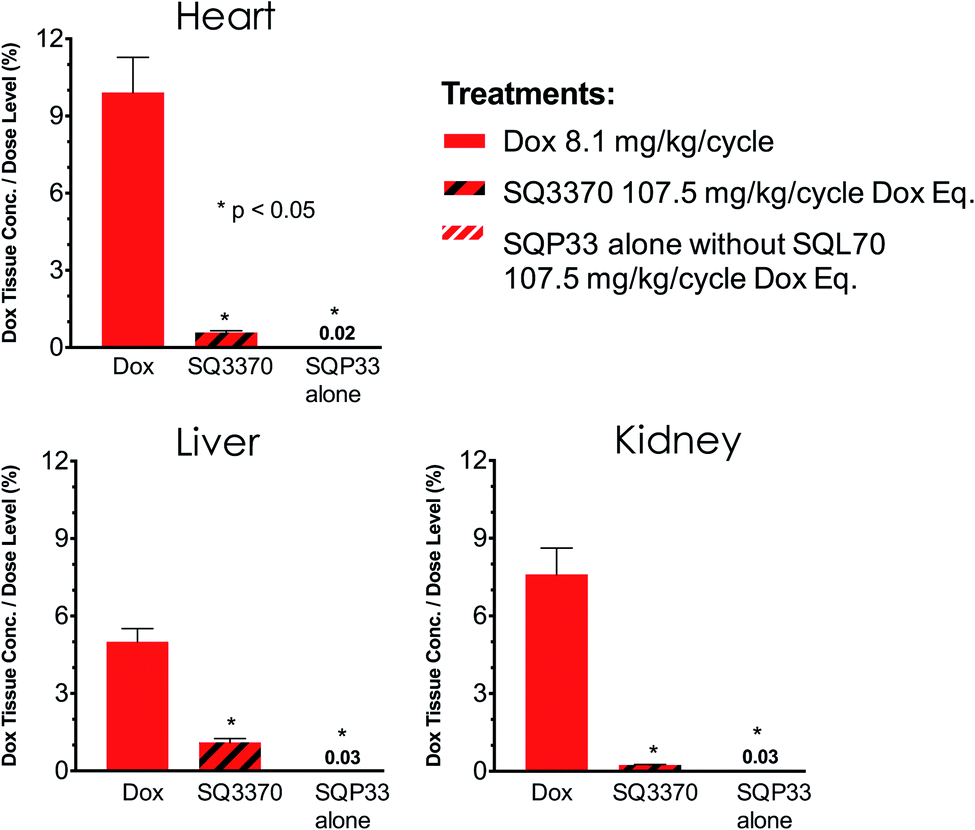 | ||
| Fig. 6 Active Dox exposure to non-target-tissues in SQ3370vs. conventional Dox HCl. SD rats (n = 3–6 per group) were treated with one dose of Dox HCl, at 8.1 mg kg−1, and compared to rats treated with 5 daily doses of SQP33, at 21.5 mg per kg per dose Dox eq., in the presence (+) or absence (w/o) of SQL70 biopolymer. Tissue samples were collected from the heart, kidney and liver 5 days after treatment, and analyzed using LC-MS. Tissue exposure is reported as a percent of the cumulative Dox eq. dose that was administered. Significance was established by unpaired t-test with Welch's correction. Data shows group mean + SEM. Active Dox exposure to non-target-tissues was significantly lower with SQ3370 treatment as compared to Dox HCl treatment. | ||
Lastly, we examined the PK and biodistribution of a protodrug dosing regimen longer than 5 days. Healthy female C57BL/6 mice were treated with a single SC biopolymer injection followed by 15 IV infusions of SQP33 protodrug at 43 mg per kg per dose Dox eq. into the tail vein over 3 weeks (5 daily doses per week on weekdays only). The dosage schedule is shown in Fig. S10A.† Blood samples were collected at 1 h (n = 3), 24 h (n = 2), and on Days 5, 12, and 19 (n = 5) following the first SQP33 protodrug injection. Biopolymer injection site tissue was collected on Days 19 (n = 1) and 26 (n = 4). Samples were analyzed by LC-MS for SQP33 protodrug and active Dox and are shown in Fig. S10B–D,† for plasma and targeted region (biopolymer injection site), respectively.
Consistent with results in rats, SQP33 was rapidly captured by the biopolymer on the first day of dosing. 1 h after the first SQP33 protodrug dose, plasma concentrations were almost exclusively active Dox, indicating efficient protodrug capture and activation. Within 24 h, there was no detectable SQP33 protodrug in plasma, and reduced levels of active Dox were observed compared with the 1 h time point – suggesting clearance of active Dox. Plasma analyses on Days 5, 12, and 19 showed increasing amounts of SQP33 protodrug 1 h after the injection. Conversely, plasma concentrations of active Dox decreased over the treatment course from 1257 ng mL−1 on Day 1 to 73.5 ng mL−1 on Day 5 and down to 37.7 ng mL−1 on Day 12. These trends are similar to those seen in rats and indicate the biopolymer activity gradually decreases over time in vivo.
Results from this experiment also show that on the last day of SQP33 protodrug dosing (Day 19), the average concentration of active Dox in plasma was smaller than that of SQP33; while, in notable contrast, concentration of active Dox at the biopolymer injection site was higher than that of SQP33 (Fig. S10C and Table S3†). Furthermore, on Day 26, 7 days after the final SQP33 dose, no SQP33 protodrug was detected at the biopolymer injection site, while active Dox levels were still more than 51% of the biopolymer injection site active Dox levels detected on Day 19 (Fig. S10D and Table S3†). Taken together, these results show a higher active drug exposure at the biopolymer injection area, as compared to plasma, and indicate that a detectable amount of active Dox remains at the biopolymer injection site for at least 7 days after the last SQP33 protodrug dose.
Antitumor efficacy in a mouse model
A study in tumor-bearing mice was conducted to evaluate if SQ3370 showed antitumor activity, as previously seen with the first-generation compounds.35 Immunocompetent C57BL/6 mice with syngeneic MC38 flank tumors were treated with either saline (IV) or SQ3370 (Fig. 7). For SQ3370 treatment, mice were first injected peritumorally with SQL70 biopolymer, followed by 5 daily doses of SQP33 protodrug IV (28.6 mg per kg per dose Dox eq.). On Day 38 post tumor cell inoculation, surviving animals in the SQ3370 treatment group received a second SQL70 injection and an additional 5 day regimen of SQP33 (11.9 mg per kg per dose Dox eq.). SQ3370 treatment significantly slowed tumor growth compared to control treatment, as well as prolonged median overall survival to 47 days, compared to 31 days for control treatment. These preliminary results confirm the antitumor activity of SQ3370. In-depth results of SQ3370 efficacy in tumor-bearing mice are being published separately.43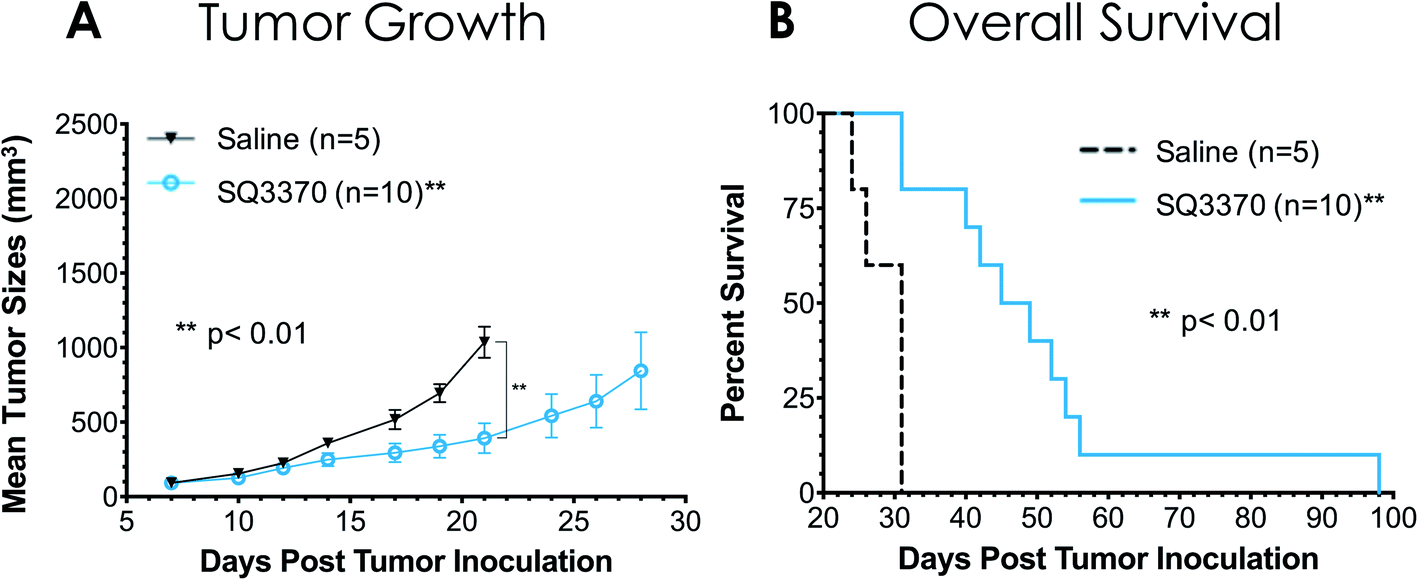 | ||
| Fig. 7 Antitumor activity of SQ3370 in mice. Two groups of immunocompetent C57BL/6 mice were inoculated SC in the right flank with 5 × 105 MC38 tumor cells on Day 0. Treatments started on Day 7 with a 100 μL peritumoral injection of SQL70 biopolymer to both groups, followed by 5 daily doses of saline (n = 5) or SQP33 protodrug at 28.6 mg per kg per dose Dox eq. (n = 10). On Day 38, the remaining animals, which were all from the SQ3370-treatment group, received a second 100 μL peritumoral injection of SQL70 biopolymer followed by another 5 daily doses of SQP33 protodrug at 11.9 mg per kg per dose Dox eq. Tumor growth curves (A) show mean ± SEM, and curves stopped after 1 or more mice in that group died or were sacrificed when tumor volume reached 2000 mm3. Tumor volumes of SQ3370-treated mice were significantly smaller (p < 0.01) as assessed by Welch's t test comparing Day 21 values (the last day all animals were alive in both groups). Kaplan–Meier survival curves (B) showed a significant improvement (p < 0.01) in overall survival in mice treated with SQ3370 as compared to saline, with median survival being 47 and 31 days respectively. Statistical significance in survival was determined by log-rank (Mantel–Cox) test. | ||
Discussion
We have previously demonstrated the proof-of-concept and feasibility of the technology in an animal model,24 and subsequently showed the enhanced safety and sustained antitumor responses in a mouse fibrosarcoma model with a single tumor.35 Prototypes of the protodrug and biopolymer were used for these early studies. Since then, we have made significant changes to optimize the treatment for clinical use.Here, hyaluronic acid (HA) was selected as the backbone in SQL70 biopolymer over alginate3 due to its greater tolerability and biocompatibility.44 Hyaluronic acid is a ubiquitous, non-sulfated glycosaminoglycan that is well tolerated in the body, and has been used clinically in unmodified, crosslinked, and derivatized form for over five decades.39SQL70 biopolymer was optimized for total tetrazine conjugation, pH and purity.
On the other hand, the TCO moiety of the protodrug was modified with a polar glycine group to improve its aqueous solubility. Our previous report used a protodrug lacking hydrophilic solubilizing groups on the TCO.35 While we were still able to demonstrate improved efficacy and safety, capping the amine in Dox with a hydrophobic TCO drastically reduced the aqueous solubility of the protodrug, limiting the amount of therapy that can be feasibly administered. To address this issue, we have developed a strategy to significantly improve the aqueous solubility of TCO protodrugs in order to enable higher dosing in the clinical setting and maximize the benefit of the reduced systemic toxicity of the CAPAC platform. Incorporating hydrophilic carboxylic acid groups into the TCO moiety was pursued as a way to improve solubility without affecting activity, as the TCO portion is released from the cytotoxic core upon activation. Utilizing the TCO group as a heterobifunctional scaffold proved to be highly versatile, allowing for the incorporation of solubilizing groups into each of the four protodrugs reported herein, overcoming the solubility limitations of not only the first-generation hydrophobic TCO, but also enhancing solubility compared to the parent drugs for Dox, PTX, and ETP. We expect this modular approach to be readily applicable to TCO modified drugs beyond the four reported here and would also allow the incorporation of various solubilizing groups to further increase the solubility as necessary.
To support the translational usage of CAPAC, specifically SQ3370, in the clinic, we have provided evidence from animal studies to highlight the safety and effectiveness compared to existing treatment options. When injected in mice, ∼50% of SQL70 of persisted at the subcutaneous injection site after 14 days. This extended duration enables a single biopolymer injection to activate multiple protodrug doses in vivo. SQ3370 treatment showed a significant reduction in toxicity, enabling doses of 19.1-fold and 10.8-fold the MTD of conventional Dox in mice and rats, respectively. PK results in rats demonstrate that the SQP33 protodrug is stable with minimal nonspecific conversion in the absence of biopolymer, and that the SQL70 biopolymer captures and activates measurable levels of drug for at least 5 days, as evidenced by plasma analysis. In mice, active Dox in the local tissue biopolymer injection site was detectable for at least 7 days after the last SQP33 protodrug dose and 26 days after the biopolymer injection, suggesting that CAPAC can continue to activate drug at the local site for extended periods following a single biopolymer injection. Preliminary results in tumor-bearing mice confirm the antitumor activity of SQ3370.
Collectively, our results demonstrate that the CAPAC platform represents a meaningful development in the progress of click chemistry-based therapeutics, with key advantages compared to other reported click-based approaches. Click chemistry has previously been used as a method for pretargeting, or tagging tumors with chemical handles that can be recognized and bound to click-reactive probes or therapeutics. In one general strategy, tumors in mice have been injected artificial azide-modified sugars (N-azidoacetylmannosamine and related derivatives), resulting in metabolic incorporation of azides into cell surface glycans, enabling targeting by cyclooctyne-modified fluorescence nanoparticles45 or a cyclooctyne conjugated to Dox through an enzymatically cleavable linker.46 While this work highlights the feasibility of using click chemistry to target tumors in vivo, it is unclear how metabolic incorporation of artificial sugars would translate clinically.
An alternative to metabolic incorporation is to use tumor-specific antibodies conjugated to a click handle to pretarget the tumor. Au and coworkers used azide modified anti-CD20 and anti-Lym1 to pretarget lymphoma tumors in mice, allowing for the delivery of cyclooctyne-nanoparticles containing a cargo the small molecule antitumor therapeutic BEZ235.47 Although the strategy did result in improved antitumor effectiveness over untargeted nanoparticle BEZ235, pretargeting with click-antibody conjugates faces practical obstacles for translational use in the clinic. Despite the high binding constant of antibodies for their targets, it has been challenging to incorporate enough click handles at the tumor to enable meaningful reaction with small molecule probes, even using TCO–tetrazine chemistry with orders of magnitude higher reaction kinetics than azide–cyclooctyne couplings.22 The CAPAC platform overcomes this limitation through injecting a high density of tetrazine sites conjugated to a biopolymer which remains at the tumor site for an extended duration. Additionally, the selectivity of both capture and drug activation stem directly from the click reaction itself. On the other hand, variability of enzyme and biomarker expression between patients, and even between tumor tissue and healthy tissue, interfere with the selectivity of antibody and enzyme-cleavable technologies. The CAPAC approach is completely independent from specific biomarkers or tumor properties, positioning it as a highly generalizable therapy against a broad array of solid tumor types.
Conclusions
We have described studies directed at the selection and preclinical development for targeted activation of systemically administered anticancer protodrugs using click chemistry. The CAPAC platform addresses critical limitations of conventional chemotherapy and targeted delivery approaches. Synthesis and in vitro evaluation of protodrugs of Dox, PTX, ETP and GCB have been described. The protodrug of Dox, SQP33, was found to be 83-times less cytotoxic than the parent drug and 7.3-times more soluble in PBS and was therefore selected as the best candidate for in vivo testing. SQP33 was found to be stable in plasma and was efficiently activated by a Tz-modified biopolymer. After injection of SQL70 biopolymer, SD rats tolerated cumulative SQP33 doses 10.8-times higher than the multi-dose MTD of Dox HCl, while mice tolerated doses up to 19.1-times the single-dose MTD of Dox HCl, making SQ3370 treatment notably less toxic than other clinically tested Dox-based therapeutics. Furthermore, SQ3370 showed significantly lower Dox exposure to the heart, thus, significantly diminishing the biggest limitation of treatment with conventional Dox HCl, and expanding the drug's therapeutic potential by allowing remarkably higher doses to be safely administered. The considerably wider therapeutic window of SQ3370 has been attributed to the greatly reduced systemic cytotoxicity of SQP33 protodrug combined with the ability of SQL70 biopolymer to effectively capture protodrug and concentrate it at a targeted site, and release active Dox locally – as shown by PK studies in mice and rats. Moreover, SQL70 biopolymer was shown to remain at the injection site for an extended duration to activate multiple IV doses of SQP33, allowing repeated dosing while maintaining a wide therapeutic window. Furthermore, the protodrug proved stable in vivo, with minimal nonspecific conversion. Finally, SQ3370 exhibited antitumor activity in a mouse model. The encouraging results presented herein have paved way for the clinical development of SQ3370. A first-in-human Phase I clinical trial of SQ3370 has been approved by the FDA and is currently enrolling patients with advanced solid tumors (http://ClinicalTrials.gov ID: NCT04106492).Live subject statement
The described animal studies were done in accordance with the NIH guidelines for care and use of laboratory animals (2011, 8th edition) and were approved by the Institutional Animal Care and Use Committee of Explora BioLabs (San Diego, USA) or BioDuro (Shanghai, China). No human subjects were involved in the studies presented here.Conflicts of interest
N. A. Yee, S. Srinivasan, A. Mahmoodi, M. Zakharian, and J. M. Mejía Oneto are paid employees and shareholders of Shasqi, Inc. (Shasqi). J. M. Mejía Oneto is the Founder and CEO of Shasqi. M. Royzen is an advisor and shareholder of Shasqi.References
- C. Li, D. F. Yu, T. Inoue, D. J. Yang, W. Tansey, C. W. Liu, L. Milas, N. R. Hunter, E. E. Kim and S. Wallace, Synthesis, biodistribution and imaging properties of indium-111-DTPA-paclitaxel in mice bearing mammary tumors, J. Nucl. Med., 1997, 38(7), 1042–1047 CAS.
- T. Cooley, D. Henry, M. Tonda, S. Sun, M. O'Connell and W. Rackoff, A randomized, double-blind study of pegylated liposomal doxorubicin for the treatment of AIDS-related Kaposi's sarcoma, Oncologist, 2007, 12(1), 114–123 CrossRef CAS.
- M. Hadla, S. Palazzolo, G. Corona, I. Caligiuri, V. Canzonieri, G. Toffoli and F. Rizzolio, Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models, Nanomedicine, 2016, 11(18), 2431–2441 CrossRef CAS.
- S. Napoli, M. A. Burton, I. J. Martins, Y. Chen, J. P. Codde and B. N. Gray, Dose response and toxicity of doxorubicin microspheres in a rat tumor model, Anticancer Drugs, 1992, 3(1), 47–53 CrossRef CAS.
- D. Jain, Cardiotoxicity of doxorubicin and other anthracycline derivatives, J. Nucl. Cardiol., 2000, 7(1), 53–62 CrossRef CAS.
- E. A. Lefrak, J. Pitha, S. Rosenheim and J. A. Gottlieb, A clinicopathologic analysis of adriamycin cardiotoxicity, Cancer, 1973, 32(2), 302–314 CrossRef CAS.
- R. A. Quintana, J. Banchs, R. Gupta, H. Y. Lin, S. D. Raj, A. Conley, V. Ravi, D. Araujo, R. S. Benjamin, S. Patel, S. Vadhan-Raj and N. Somaiah, Early Evidence of Cardiotoxicity and Tumor Response in Patients with Sarcomas after High Cumulative Dose Doxorubicin Given as a Continuous Infusion, Sarcoma, 2017, 2017, 7495914 CrossRef.
- M. M. Mita, R. B. Natale, E. M. Wolin, B. Laabs, H. Dinh, S. Wieland, D. J. Levitt and A. C. Mita, Pharmacokinetic study of aldoxorubicin in patients with solid tumors, Invest. New Drugs, 2015, 33(2), 341–348 CrossRef CAS.
- W. D. Tap, Z. Papai, B. A. Van Tine, S. Attia, K. N. Ganjoo, R. L. Jones, S. Schuetze, D. Reed, S. P. Chawla, R. F. Riedel, A. Krarup-Hansen, M. Toulmonde, I. Ray-Coquard, P. Hohenberger, G. Grignani, L. D. Cranmer, S. Okuno, M. Agulnik, W. Read, C. W. Ryan, T. Alcindor, X. F. G. Del Muro, G. T. Budd, H. Tawbi, T. Pearce, S. Kroll, D. K. Reinke and P. Schoffski, Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial, Lancet Oncol., 2017, 18(8), 1089–1103 CrossRef CAS.
- P. Schoffski, J. P. Delord, E. Brain, J. Robert, H. Dumez, J. Gasmi and A. Trouet, First-in-man phase I study assessing the safety and pharmacokinetics of a 1-hour intravenous infusion of the doxorubicin prodrug DTS-201 every 3 weeks in patients with advanced or metastatic solid tumours, Eur. J. Cancer, 2017, 86, 240–247 CrossRef CAS.
- D. DeFeo-Jones, S. F. Brady, D. M. Feng, B. K. Wong, T. Bolyar, K. Haskell, D. M. Kiefer, K. Leander, E. McAvoy, P. Lumma, J. M. Pawluczyk, J. Wai, S. L. Motzel, K. Keenan, M. Van Zwieten, J. H. Lin, V. M. Garsky, R. Freidinger, A. Oliff and R. E. Jones, A prostate-specific antigen (PSA)-activated vinblastine prodrug selectively kills PSA-secreting cells in vivo, Mol. Cancer Ther., 2002, 1(7), 451–459 CAS.
- D. DeFeo-Jones, V. M. Garsky, B. K. Wong, D. M. Feng, T. Bolyar, K. Haskell, D. M. Kiefer, K. Leander, E. McAvoy, P. Lumma, J. Wai, E. T. Senderak, S. L. Motzel, K. Keenan, M. Van Zwieten, J. H. Lin, R. Freidinger, J. Huff, A. Oliff and R. E. Jones, A peptide-doxorubicin 'prodrug' activated by prostate-specific antigen selectively kills prostate tumor cells positive for prostate-specific antigen in vivo, Nat. Med., 2000, 6(11), 1248–1252 CrossRef CAS.
- R. S. DiPaola, J. Rinehart, J. Nemunaitis, S. Ebbinghaus, E. Rubin, T. Capanna, M. Ciardella, S. Doyle-Lindrud, S. Goodwin, M. Fontaine, N. Adams, A. Williams, M. Schwartz, G. Winchell, K. Wickersham, P. Deutsch and S. L. Yao, Characterization of a novel prostate-specific antigen-activated peptide-doxorubicin conjugate in patients with prostate cancer, J. Clin. Oncol., 2002, 20(7), 1874–1879 CrossRef CAS.
- C. F. Albright, N. Graciani, W. Han, E. Yue, R. Stein, Z. Lai, M. Diamond, R. Dowling, L. Grimminger, S. Y. Zhang, D. Behrens, A. Musselman, R. Bruckner, M. Zhang, X. Jiang, D. Hu, A. Higley, S. Dimeo, M. Rafalski, S. Mandlekar, B. Car, S. Yeleswaram, A. Stern, R. A. Copeland, A. Combs, S. P. Seitz, G. L. Trainor, R. Taub, P. Huang and A. Oliff, Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity, Mol. Cancer Ther., 2005, 4(5), 751–760 CrossRef CAS.
- V. A. de Weger, J. H. Beijnen and J. H. Schellens, Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel--a review, Anticancer Drugs, 2014, 25(5), 488–494 CrossRef CAS.
- H. Gelderblom, J. Verweij, K. Nooter and A. Sparreboom, Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation, Eur. J. Cancer, 2001, 37(13), 1590–1598 CrossRef CAS.
- R. B. Weiss, R. C. Donehower, P. H. Wiernik, T. Ohnuma, R. J. Gralla, D. L. Trump, J. R. Baker Jr, D. A. Van Echo, D. D. Von Hoff and B. Leyland-Jones, Hypersensitivity reactions from taxol, J. Clin. Oncol., 1990, 8(7), 1263–1268 CrossRef CAS.
- K. R. Hande, Etoposide: four decades of development of a topoisomerase II inhibitor, Eur. J. Cancer, 1998, 34(10), 1514–1521 CrossRef CAS.
- S. Reif, D. Kingreen, C. Kloft, J. Grimm, W. Siegert, W. Schunack and U. Jaehde, Bioequivalence investigation of high-dose etoposide and etoposide phosphate in lymphoma patients, Cancer Chemother. Pharmacol., 2001, 48(2), 134–140 CrossRef CAS.
- N. Chao, A. Stein, G. Long, R. Negrin, M. Amylon, R. Wong, S. Forman and K. Blume, Busulfan/etoposide--initial experience with a new preparatory regimen for autologous bone marrow transplantation in patients with acute nonlymphoblastic leukemia, Blood, 1993, 81(2), 319–323 CrossRef CAS.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. Ten Hoeve, H. M. Janssen, A. van Onzen, P. J. Hudson and M. S. Robillard, Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice, Nat. Commun., 2018, 9(1), 1484 CrossRef.
- N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Reactive polymer enables efficient in vivo bioorthogonal chemistry, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(13), 4762–4767 CrossRef CAS.
- M. Czuban, S. Srinivasan, N. A. Yee, E. Agustin, A. Koliszak, E. Miller, I. Khan, I. Quinones, H. Noory, C. Motola, R. Volkmer, M. Di Luca, A. Trampuz, M. Royzen and J. M. Mejia Oneto, Bio-Orthogonal Chemistry and Reloadable Biomaterial Enable Local Activation of Antibiotic Prodrugs and Enhance Treatments against Staphylococcus aureus Infections, ACS Cent. Sci., 2018, 4(12), 1624–1632 CrossRef CAS.
- J. M. Mejia Oneto, M. Gupta, J. K. Leach, M. Lee and J. L. Sutcliffe, Implantable biomaterial based on click chemistry for targeting small molecules, Acta Biomater., 2014, 10(12), 5099–5105 CrossRef CAS.
- S. L. Hewitt, A. Bai, D. Bailey, K. Ichikawa, J. Zielinski, R. Karp, A. Apte, K. Arnold, S. J. Zacharek, M. S. Iliou, K. Bhatt, M. Garnaas, F. Musenge, A. Davis, N. Khatwani, S. V. Su, G. MacLean, S. J. Farlow, K. Burke and J. P. Frederick, Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs, Sci. Transl. Med., 2019, 11(477) CAS.
- A. Marabelle, L. Tselikas, T. de Baere and R. Houot, Intratumoral immunotherapy: using the tumor as the remedy, Ann. Oncol., 2017, 28(Supplement 12), xii33–xii43 CrossRef CAS.
- J. C. Bateman, Palliation of cancer in human patients by maintenance therapy with NN′N′′-triethylene thiophosphoramide and N-(3-oxapentamethylene)-N′N′′-diethylene phosphoramide, Ann. N. Y. Acad. Sci., 1958, 68(3), 1057–1071 CrossRef CAS.
- J. C. Bateman, Chemotherapy of solid tumors with triethylene thiophosphoramide, N. Engl. J. Med., 1955, 252(21), 879–887 CrossRef CAS.
- E. P. Goldberg, A. R. Hadba, B. A. Almond and J. S. Marotta, Intratumoral cancer chemotherapy and immunotherapy: opportunities for nonsystemic preoperative drug delivery, J. Pharm. Pharmacol., 2002, 54(2), 159–180 CrossRef CAS.
- R. A. Sheth, R. Murthy, D. S. Hong, S. Patel, M. J. Overman, A. Diab, P. Hwu and A. Tam, Assessment of Image-Guided Intratumoral Delivery of Immunotherapeutics in Patients With Cancer, JAMA Netw. Open, 2020, 3(7), e207911 CrossRef.
- O. Hamid, R. Ismail and I. Puzanov, Intratumoral Immunotherapy-Update 2019, Oncologist, 2020, 25(3), e423–e438 CrossRef.
- A. N. Gangopadhyay, R. Rajeev, S. P. Sharma, V. D. Upadhyaya, N. C. Arya, V. Kumar and S. C. Gopal, Anterior intratumoural chemotherapy: a newer modality of treatment in advanced solid tumours in children, Asian J. Surg., 2008, 31(4), 225–229 CrossRef CAS.
- A. Marabelle, R. Andtbacka, K. Harrington, I. Melero, R. Leidner, T. de Baere, C. Robert, P. A. Ascierto, J. F. Baurain, M. Imperiale, S. Rahimian, D. Tersago, E. Klumper, M. Hendriks, R. Kumar, M. Stern, K. Ohrling, C. Massacesi, I. Tchakov, A. Tse, J. Y. Douillard, J. Tabernero, J. Haanen and J. Brody, Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT), Ann. Oncol., 2018, 29(11), 2163–2174 CrossRef CAS.
- https://clinicaltrials.gov/ct2/show/NCT04106492, accessed December 10.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug To Treat Soft Tissue Sarcoma, ACS Cent. Sci., 2016, 2(7), 476–482 CrossRef CAS.
- I. Khan, L. M. Seebald, N. M. Robertson, M. V. Yigit and M. Royzen, Controlled in-cell activation of RNA therapeutics using bond-cleaving bio-orthogonal chemistry, Chem. Sci., 2017, 8(8), 5705–5712 RSC.
- N. M. Robertson, Y. Yang, I. Khan, V. E. LaMantia, M. Royzen and M. V. Yigit, Single-trigger dual-responsive nanoparticles for controllable and sequential prodrug activation, Nanoscale, 2017, 9(28), 10020–10030 RSC.
- G. D. Prestwich, Hyaluronic acid-based clinical biomaterials derived for cell and molecule delivery in regenerative medicine, J. Controlled Release, 2011, 155(2), 193–199 CrossRef CAS.
- J. A. Burdick and G. D. Prestwich, Hyaluronic acid hydrogels for biomedical applications, Adv. Mater., 2011, 23(12), H41–H56 CrossRef CAS.
- F. Kratz, G. Ehling, H. M. Kauffmann and C. Unger, Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin, Hum. Exp. Toxicol., 2007, 26(1), 19–35 CrossRef CAS.
- D. Ravel, V. Dubois, J. Quinonero, F. Meyer-Losic, J. Delord, P. Rochaix, C. Nicolazzi, F. Ribes, C. Mazerolles, E. Assouly, K. Vialatte, I. Hor, J. Kearsey and A. Trouet, Preclinical toxicity, toxicokinetics, and antitumoral efficacy studies of DTS-201, a tumor-selective peptidic prodrug of doxorubicin, Clin. Cancer Res., 2008, 14(4), 1258–1265 CrossRef CAS.
- C. Bertazzoli, C. Rovero, L. Ballerini, B. Lux, F. Balconi, V. Antongiovanni and U. Magrini, Experimental systemic toxicology of 4'-epidoxorubicin, a new, less cardiotoxic anthracycline antitumor agent, Toxicol. Appl. Pharmacol., 1985, 79(3), 412–422 CrossRef CAS.
- S. Srinivasan, N. A. Yee, K. Wu, M. Zakharian, A. Mahmoodi, M. Royzen and J. M. M. Oneto, SQ3370 Activates Cytotoxic Drug via Click Chemistry at Tumor and Elicits Sustained Responses in Injected & Non-injected Lesions, 2020, bioRxiv, 2020.10.13.337899.
- D. Jiang, J. Liang and P. W. Noble, Hyaluronan as an immune regulator in human diseases, Physiol. Rev., 2011, 91(1), 221–264 CrossRef CAS.
- H. Koo, S. Lee, J. H. Na, S. H. Kim, S. K. Hahn, K. Choi, I. C. Kwon, S. Y. Jeong and K. Kim, Bioorthogonal copper-free click chemistry in vivo for tumor-targeted delivery of nanoparticles, Angew. Chem., Int. Ed., 2012, 51(47), 11836–11840 CrossRef CAS.
- H. Wang, R. Wang, K. Cai, H. He, Y. Liu, J. Yen, Z. Wang, M. Xu, Y. Sun, X. Zhou, Q. Yin, L. Tang, I. T. Dobrucki, L. W. Dobrucki, E. J. Chaney, S. A. Boppart, T. M. Fan, S. Lezmi, X. Chen, L. Yin and J. Cheng, Selective in vivo metabolic cell-labeling-mediated cancer targeting, Nat. Chem. Biol., 2017, 13(4), 415–424 CrossRef CAS.
- K. M. Au, A. Z. Wang and S. I. Park, Pretargeted delivery of PI3K/mTOR small-molecule inhibitor-loaded nanoparticles for treatment of non-Hodgkin's lymphoma, Sci. Adv., 2020, 6(14), eaaz9798 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06099b |
| ‡ Co-first authorship. |
| This journal is © The Royal Society of Chemistry 2021 |