
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed enantioselective olefinic C(sp2)–H allylic alkylation†
Rahul
Sarkar
 and
Santanu
Mukherjee
*
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@iisc.ac.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 15th January 2021
Abstract
The first iridium-catalyzed enantioselective olefinic C(sp2)–H allylic alkylation is developed in cooperation with Lewis base catalysis. This reaction, catalyzed by cinchonidine and an in situ generated cyclometalated Ir(I)/phosphoramidite complex, makes use of the latent enolate character of an α,β-unsaturated carbonyl compound, namely coumalate ester, to introduce an allyl group at its α-position in a branched-selective manner in moderate to good yield with good to excellent enantioselectivities (up to 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 er).
:2 er).
Introduction
Within the two decades since the seminal reports by Takeuchi1 and Helmchen,2 iridium-catalyzed allylic substitution has been established as an extremely powerful method for enantioselective synthesis.3 Remarkable advancements in catalyst design4 and mechanistic understanding5 have enabled enantioselective construction of a myriad of carbon–carbon and carbon–heteroatom bonds.3b Particularly in the realm of Ir-catalyzed allylic alkylation (AA), both stabilized and unstabilized carbon nucleophiles have been deployed, and led to numerous C(sp3)–H allylic alkylation reactions.6 In contrast, Ir-catalyzed enantioselective C(sp2)–H AA has so far been restricted to electron-rich (hetero)aromatic C(sp2)–H bonds,7 and allylic alkylation of olefinic C(sp2)–H bonds remains missing from this repertoire (Scheme 1A).8 Herein we report the first Ir-catalyzed enantioselective allylic alkylation of an olefinic C(sp2)–H bond, namely that of an α,β-unsaturated carbonyl compound.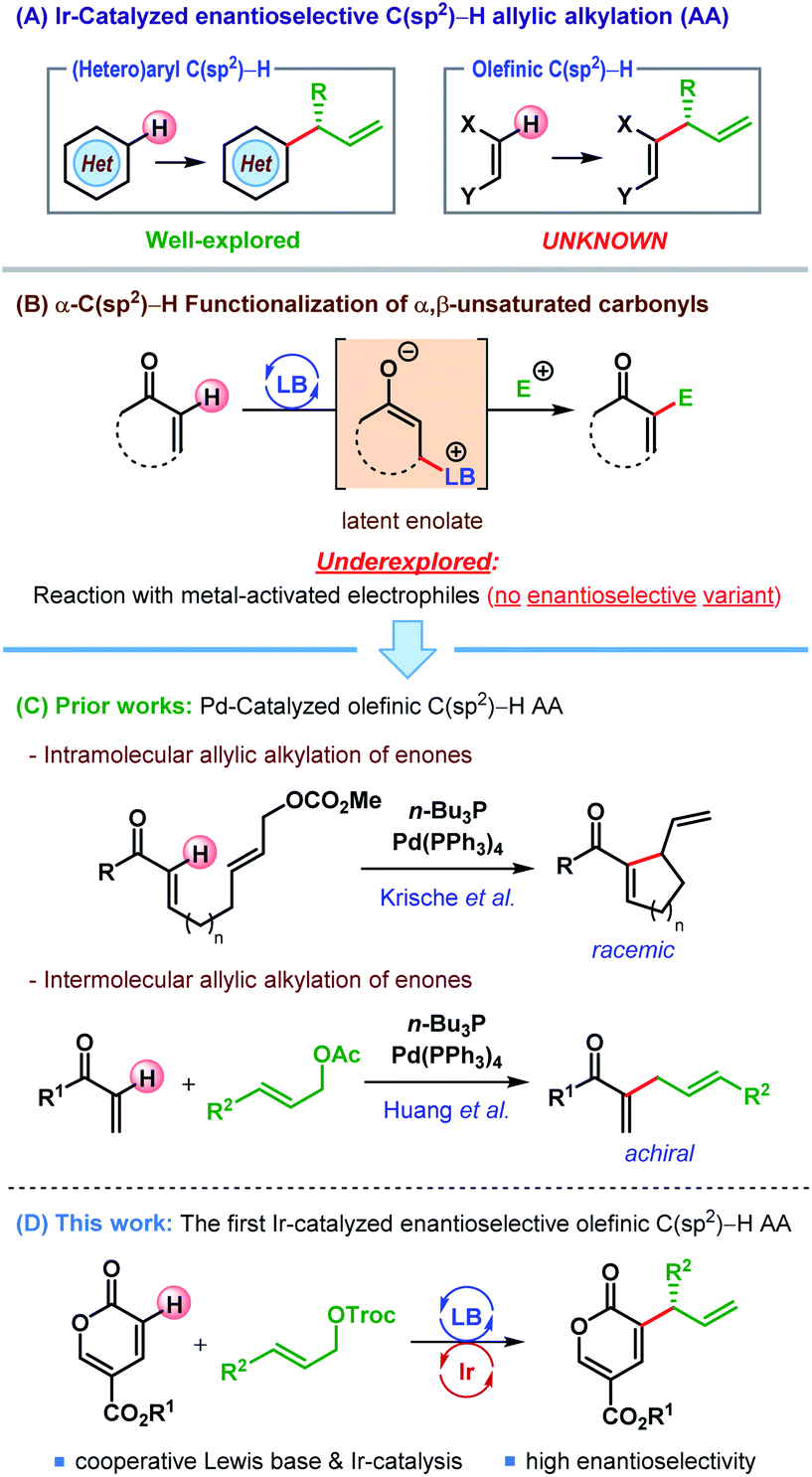 | ||
| Scheme 1 Catalytic C(sp2)–H allylic alkylation. | ||
α,β-Unsaturated carbonyl compounds can be visualized as latent enolates and thereby nucleophilic at their α-position, especially under Lewis base (LB) activation (Scheme 1B).9 The reactions of such latent enolates with a wide range of electrophiles, even in enantioselective fashion, have been well documented and lead to an overall α-C(sp2)–H functionalization of α,β-unsaturated carbonyls.9 However, the combinations of these latent enolates with metal-activated electrophiles are rare,10 and an enantioselective variant is still unknown.
In 2003, Krische and co-workers reported an intramolecular α-C(sp2)–H allylic alkylation of α,β-unsaturated ketones through the merger of palladium catalysis and Lewis base activation (Scheme 1C).10c The same strategy was later on extended to an intermolecular variant of the same reaction by Huang and co-workers (Scheme 1C).10a Despite the exciting potential of these reactions, an enantioselective α-C(sp2)–H allylic alkylation of α,β-unsaturated carbonyl compounds under transition metal catalysis is yet to be accomplished.11
With our interest in catalytic asymmetric AA reactions6c,12 we sought to address this long-standing problem. Since C(sp2)–H AA reactions do not generate any stereocenter at the nucleophilic site, an enantioselective version of this reaction must necessarily arise from the prostereogenicity of the electrophilic partner. As iridium-catalyzed AAS reactions with unsymmetrical allylic electrophiles typically lead to the formation of branched products predominantly,3 we surmised that π-allyl-Ir would be a suitable electrophilic partner for the latent enolate generated from α,β-unsaturated carbonyl compounds.
While contemplating the choice of α,β-unsaturated carbonyl compounds, we were attracted to the report by Liu and Zu on the enantioselective cross-vinylogous Rauhut–Currier (RC) reaction13 of methyl coumalate with α,β-unsaturated aldehydes under iminium activation.14 Coumalates are an interesting class of electron-deficient heterocycles, which can act as synthetic precursors for various useful frameworks including electron-deficient arenes and heterocycles.15 Whereas coumalates are well-known for their use as electron-deficient dienes in various cycloaddition reactions,16 their applications as latent enolate are scarce. Besides Zu's report, the only other example of the usage of coumalate esters as latent enolate was recently documented in the form of a Morita–Baylis–Hillman (MBH) reaction by Thorimbert, Dechoux and co-worker.17
Coumalates (1) are known to be electrophilic at their C6 position18 and could suffer nucleophilic addition by a Lewis base (LB) to generate a dienolate A (Scheme 2).14,17 Although A is nucleophilic both at C3 (α-) and C5 (γ-), attack to the in situ generated π-allyl-Ir intermediate B was expected to take place from the sterically favored C3 of A. This enantiodetermining carbon–carbon bond formation would result in the formation of the intermediate C. Subsequent removal of the α-proton and elimination of LB from C would furnish the desired α-allylic alkylation product 3.
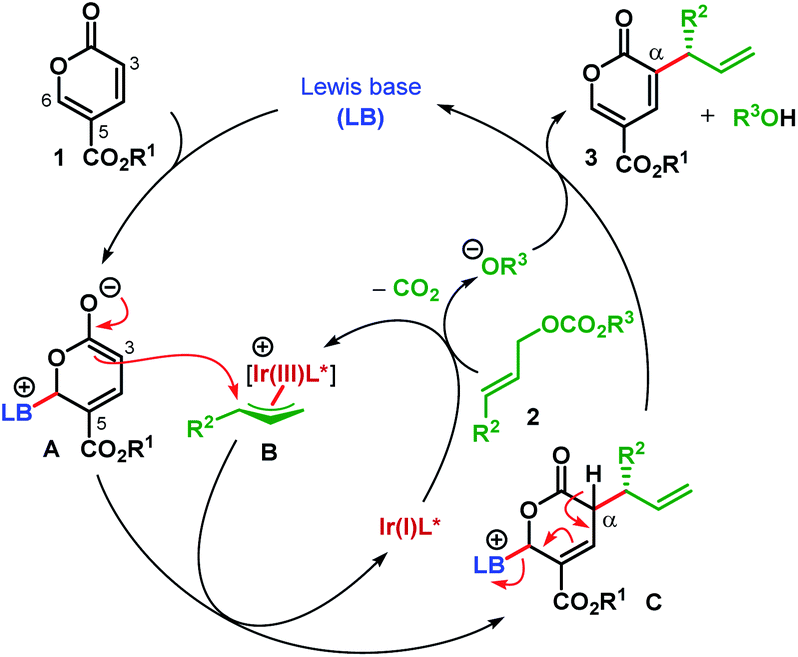 | ||
| Scheme 2 Cooperative catalytic hypothesis for the α-C(sp2)–H allylic alkylation of coumalates. | ||
We envisioned that the enantioinduction in this C(sp2)–H AA reaction could be achieved either by using a chiral Lewis base (LB) or a chiral ligand (L*) on Ir(I) or both. Combining two different catalytic modes in cooperative fashion has emerged as a very effective strategy for reaction development during the past decade19 and has also been applied to Ir-catalyzed AA reactions with great effect.20
Results and discussion
Accordingly, we began our investigation with the optimization of chiral ligand and Lewis base for the reaction between methyl coumalate 1a and tert-butyl cinnamyl carbonate 2a at 50 °C (Table 1).21 In the presence of a catalytic amount of DABCO (20 mol%) as the Lewis base, 3 mol% of [Ir(COD)Cl]2 along with 6 mol% of Feringa's phosphoramidite ligand L122 was first employed as the pre-catalyst for a reaction in EtOH. To our delight, the desired α-C(sp2)–H allylic alkylation was indeed found to take place to furnish 3aa with promising enantioselectivity (88:12 er), albeit in only 11% yield (entry 1). It is important to note that the use of EtOH as the solvent is crucial, as polar aprotic solvents such as THF and 1,2-dichloroethane failed to produce any 3aa.21 However, a significant increase in enantioselectivity was observed in a 1:1 mixture of EtOH and THF, although the yield remained low (entry 2). Changing the allylic electrophile to methyl cinnamyl carbonate 2b further improved the enantioselectivity (entry 3).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 1 | 2 | L | Lewis base | t [h] | 3 | Yieldb [%] | erc |
|
a The catalyst was prepared via n-PrNH2 activation.
b Yields were determined by 1H NMR spectroscopy with mesitylene as internal standard. Isolated yields are given in the parentheses.
c Enantiomeric ratios (er) were determined by HPLC analysis on a chiral stationary phase.
d EtOH was used as the solvent.
e With 1.5:1 ratio of 1b and 2c.
f With 2:1 ratio of 1b and 2c.
g With 2:1 ratio of 1b and 2c, and MeOCH2CH2OH/THF (1:1) as the solvent.
|
||||||||
| 1d | 1a | 2a | L1 | DABCO | 40 | 3aa | 11 | 88:12 |
| 2 | 1a | 2a | L1 | DABCO | 72 | 3aa | 10 | 92.5:7.5 |
| 3 | 1a | 2b | L1 | DABCO | 48 | 3aa | 16 | 95:5 |
| 4 | 1a | 2b | L1 | B1 | 48 | 3aa | 14 | 97:3 |
| 5 | 1a | 2c | L1 | B1 | 60 | 3aa | 32 | 97:3 |
| 6 | 1a | 2c | L1 | B2 | 72 | 3aa | 21 | 97:3 |
| 7 | 1a | 2c | L1 | B3 | 72 | 3aa | 42 | 98:2 |
| 8 | 1a | 2c | L1 | B4 | 72 | 3aa | 38 | 97:3 |
| 9 | 1a | 2c | L1 | B5 | 72 | 3aa | 25 | 97:3 |
| 10 | 1a | 2c | L1 | — | 48 | 3aa | <5 | — |
| 11 | 1b | 2c | L1 | B3 | 72 | 3bc | 44 | 98:2 |
| 12 | 1b | 2c | L2 | B3 | 72 | 3bc | 42 | 98:2 |
| 13 | 1b | 2c | L3 | B3 | 72 | 3bc | 40 | 3:97 |
| 14e | 1b | 2c | L1 | B3 | 72 | 3bc | 51 | 97.5:2.5 |
| 15f | 1b | 2c | L1 | B3 | 72 | 3bc | 54 | 97:3 |
| 16g | 1b | 2c | L1 | B3 | 72 | 3bc | 53(51) | 97:3 |

Cinchona alkaloids and their derivatives as Lewis base are known to promote MBH and RC reactions.9,23 This fact inspired us to use quinine B1 as a Lewis base catalyst. As anticipated, B1 facilitated the reaction to generate 3aa with improved er (entry 4). We realized that the low yield of the reaction, among other reasons, is due to the decomposition of 2b to cinnamyl alcohol under the reaction conditions. With 2,2,2-trichloroethyl cinnamyl carbonate 2c as an allylic electrophile, this decomposition was suppressed, and the product was obtained in 32% yield with the same level of enantioselectivity (entry 5).
A Lewis base screening at this point revealed cinchonidine B3 as the optimal both in terms of the reaction efficiency and enantioselectivity (entries 6–9). Please note that the same sense of stereochemical outcome as B1 and B3 was observed with pseudoenantiomeric quinidine (B4) and cinchonine (B5), which shows that the phosphoramidite ligand on Ir(I) is primarily responsible for enantioinduction. However, the presence of Lewis base is essential as no product formation was observed in the absence of any Lewis base (entry 10), thereby indicating the cooperative interplay between iridium and Lewis base. The use of tert-butyl coumalate 1b instead of methyl coumalate 1a, along with B3, resulted in a much cleaner reaction, delivering the product 3bc in 44% yield (entry 11). Efforts to ameliorate the reaction outcome using other phosphoramidite ligands (L2 and L3) met with failure (entries 12–13). Increasing the amount of 1b turned out to be beneficial as excess of allylic electrophile (2) led to considerable side reactions. A 2:1 ratio of 1b and 2c provided the best results, generating 3bc in 54% yield and with 97:3 er (entry 15). A 1:1 mixture of 2-methoxyethanol and THF as the solvent was proven to be equally effective as 1:1 EtOH/THF mixture and afforded the product 3bc with 51% isolated yield and 97:3 er (entry 16).
After optimizing the ligand, Lewis base and other parameters for the reaction between 1b and 2c (Table 1, entry 15), we chose to test the generality of our protocol for other substrate combinations. The Ir(I)/L1 catalyst system in cooperation with the Lewis base B3 was found to be rather general and catalyzes the enantioselective α-C(sp2)–H allylic alkylation of coumalates 1 with a large variety of allylic carbonates 2. As illustrated in Table 2A, tert-butyl coumalate 1b smoothly underwent allylic alkylation with cinnamyl carbonates (2c–o) bearing either electron-donating or electron-withdrawing substituent at various positions of the aryl ring. While the enantioselectivity of the reaction was found to be independent of the electronic nature of the substituents, highly electron-deficient aryl groups (e.g.2g–h) adversely affected the yield of the reaction. In the case of p-methoxyphenyl substituted allylic carbonate 2f, the product 3bf was obtained as an inseparable mixture with unreacted 1b.
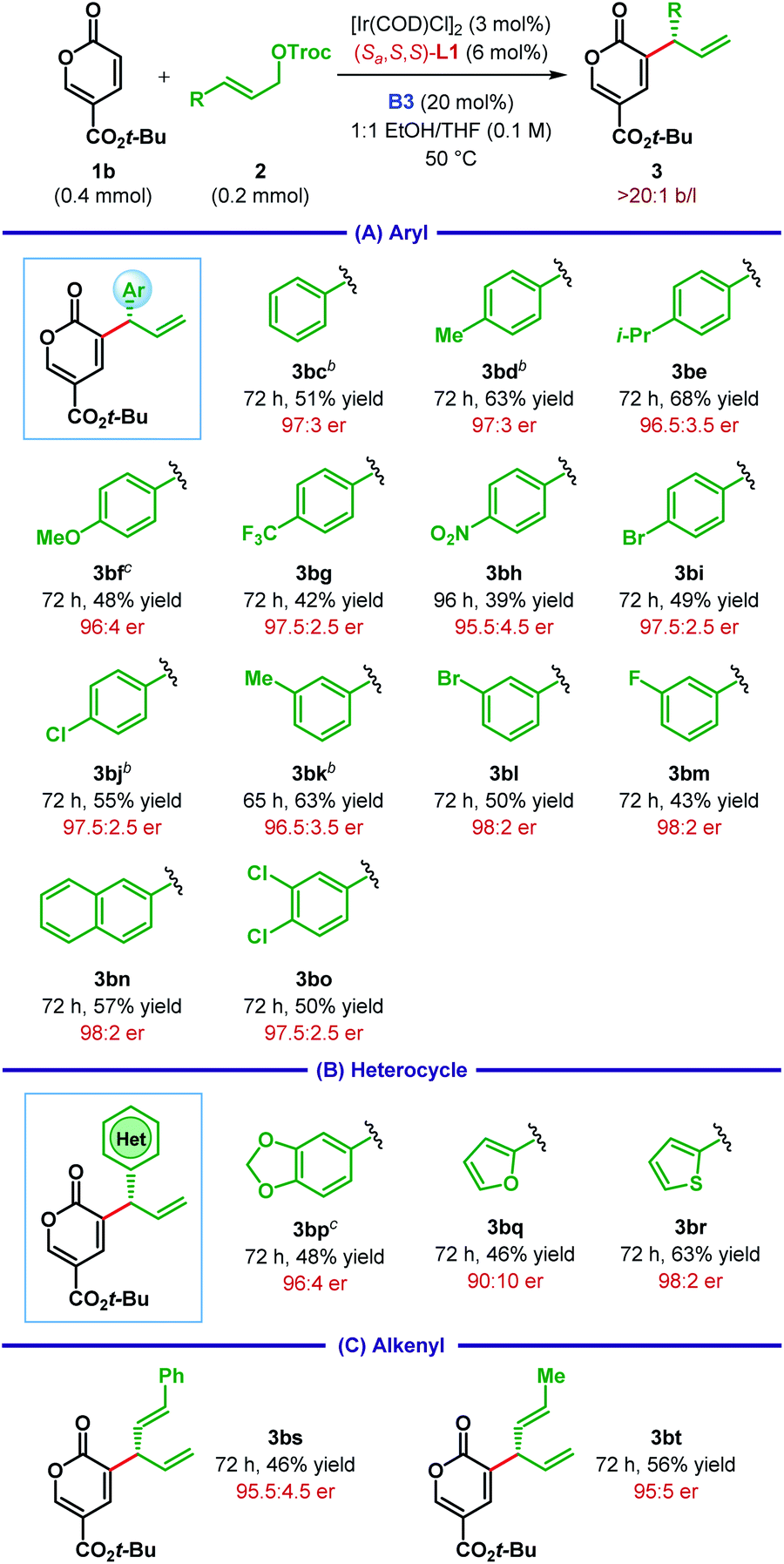
However, switching the stoichiometry of 1b and 2f from 2:1 to 1:1.2 facilitated the isolation of 3bf. For a particular substituent, very similar level of yield and enantioselectivity was observed, irrespective of its position (meta vs. para). Along the same line of observation, 3,4-dichlorocinnamyl carbonate 2o afforded the product 3bo with 97.5:2.5 er.
Apart from simple aryls, pharmaceutically relevant heterocycles can also be incorporated into the products. For example, dioxolane, furan and thiophene containing allylic carbonates (2p–r) reacted to give the corresponding products (3bp–br) with good to high enantioselectivity (Table 2B). In addition, alkenyl substituted allylic carbonates (2s–t) were well tolerated under the optimum reaction conditions to provide the products (3bs–bt) as a single regioisomer with high er (Table 2C).
The effect of the ester substituent of coumalate was next examined (Table 3). The bulky tert-butyl group can be replaced with simple methyl (1a), isopropyl (1c) and benzyl (1d) without affecting the enantioselectivity, even though the yield of these reactions remained relatively low compared to the tert-butyl coumalate 1b.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R (1) | t [h] | 3 | Yielda [%] | erb |
|
a Isolated yield after chromatographic purification.
b Er was determined by HPLC analysis on a chiral stationary phase.
c Reaction in MeOCH2CH2OH/THF (1:1).
d Reaction performed using 2:1 ratio of 1b:2c.
|
|||||
| 1c | Me (1a) | 96 | 3aa | 37 | 97:3 |
| 2c,d | t-Bu (1b) | 72 | 3bc | 51 | 97:3 |
| 3 | i-Pr (1c) | 72 | 3cc | 44 | 97:3 |
| 4 | Bn (1d) | 88 | 3dc | 40 | 97:3 |
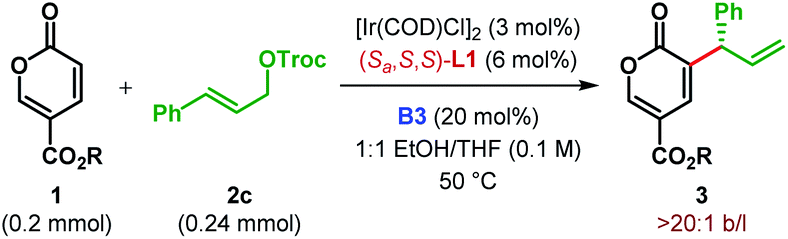
The scalability of our α-C(sp2)–H allylic alkylation protocol was showcased by performing the reaction between 1b and 2c on a 1.0 mmol scale (Scheme 3A). Under the optimum reaction conditions, product 3bc was isolated in a slightly improved yield with the same level of enantiopurity as the smaller scale reaction.
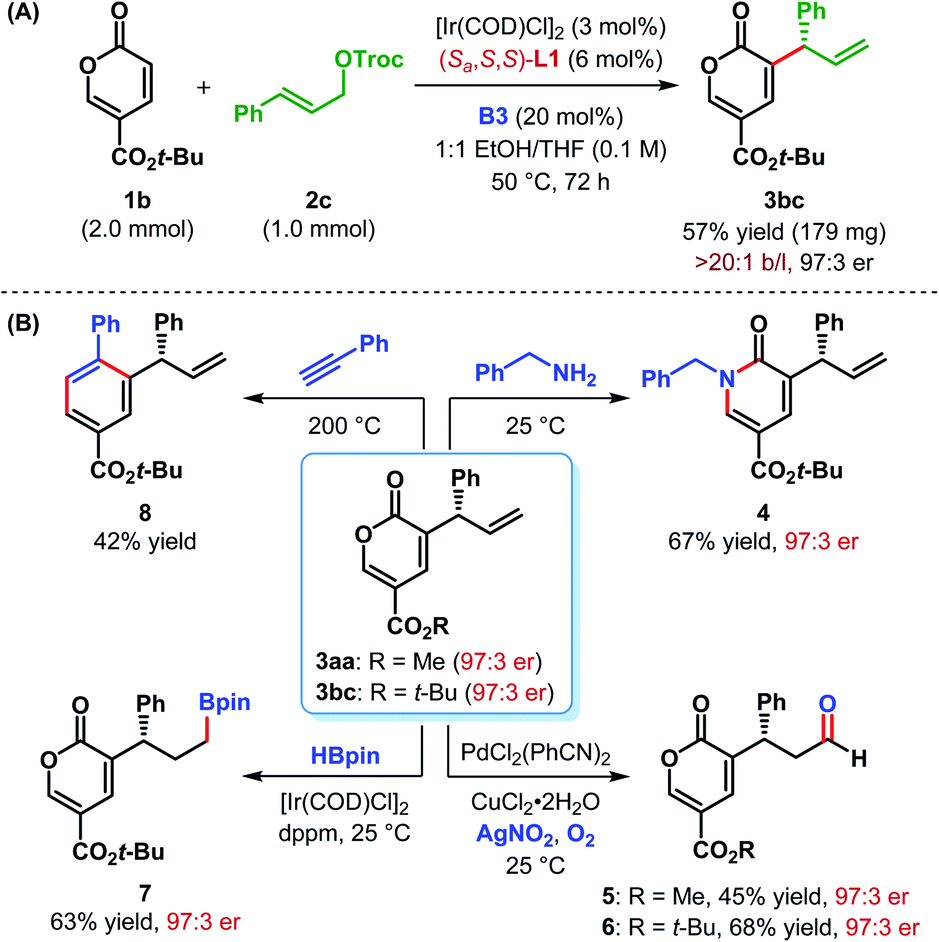 | ||
| Scheme 3 (A) Scale-up of α-C(sp2)–H allylic alkylation and (B) synthetic elaborations of α-allyl-coumalates. | ||
Although further investigation is required, the generally low yield of these C(sp2)–H allylic alkylation reactions may be attributed to the possible oligomerization of coumalates under Lewis basic conditions. In addition, 2H-pyran-2-ones are known to undergo ring-opening and decarboxylation.24
The densely functionalized enantioenriched α-allylic coumalates can serve as synthetically important building blocks for electron-deficient arenes and heterocycles. In addition, the newly installed allyl unit can be transformed into other useful functionalities. For example, treatment of 3bc with benzylamine in MeOH furnished 3-allyl-substituted 2-pyridone derivative 4 in 67% yield (Scheme 3B). The regioselective Wacker oxidation25 of 3aa and 3bc provided the corresponding aldehydes 5 and 6, respectively. The absolute configuration of 5 was previously established by Zu et al.14 The stereochemistry of 6 and the other allylated products 3, shown in Tables 2 and 3 were inferred in analogy with 5.21
Ir-catalyzed hydroboration26 of the terminal double bond of 3bc resulted in the formation of alkyl boronate 7 in 63% yield. The 2H-pyran-2-one core of the coumalate esters is known to participate in [4+2]-cycloaddition reactions.27 The 2H-pyran-2-one moiety present in compound 3bc was found to undergo [4+2]-cycloaddition/decarboxylative retro-cycloaddition cascade reaction when treated with phenylacetylene to give 3-allyl tert-butyl benzoate 8 as a single regioisomer in 42% yield (Scheme 3B). Direct synthesis of 8 would require an enantioselective allylic alkylation of an electron-deficient arene and would be electronically disfavoured. This strategy, therefore, compliments the usual Friedel–Crafts type C(sp2)–H allylic alkylation, which are generally favoured with electron-rich arenes.7 Enantiopurity of 3 was preserved during all these synthetic elaborations.
Conclusions
In conclusion, we have developed the first Ir-catalyzed enantioselective allylic alkylation of an olefinic C(sp2)–H bond – that of an α,β-unsaturated carbonyl compound, namely coumalate ester. Using linear allylic carbonates as the allylic electrophile, this reaction, cooperatively catalyzed by cinchonidine and an in situ generated cyclometalated Ir(I)/phosphoramidite complex, makes use of the latent enolate character of coumalate ester to introduce an allyl group at its α-position in a branched-selective manner with good to excellent enantioselectivities. This is also the first example of the enantioselective coupling of an α,β-unsaturated carbonyl-derived latent enolate with a metal-activated electrophile. The densely functionalized products allowed for the synthetic elaboration through functionalization of not only the newly installed allyl unit but also the 2H-pyran-2-one core of the coumalate ester. These maneuvers led to the formal C(sp2)–H allylic alkylation of electron-deficient arene – inaccessible by direct Friedel–Crafts type allylic alkylation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the Science and Engineering Research Board (SERB) [Grant No. EMR/2016/005045] and the Council of Scientific and Industrial Research (CSIR) [Grant No. 02(0385)/19/EMR-II] are gratefully acknowledged. R. S. thanks CSIR for a doctoral fellowship. High resolution mass spectra (HR-MS) were recorded on an equipment procured under the Department of Science and Technology (DST)-FIST grant [Grant No. SR/FST/CS II-040/2015].Notes and references
- R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed., 1997, 36, 263–265 CrossRef CAS.
- J. P. Janssen and G. Helmchen, Tetrahedron Lett., 1997, 38, 8025–8026 CrossRef CAS.
- For selected reviews, see: (a) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef; (b) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS; (c) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef CAS; (d) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, ACS Catal., 2016, 6, 6207–6213 CrossRef CAS; (e) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC; (f) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS.
- (a) W.-B. Liu, C. Zheng, C.-X. Zhuo, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812–4821 CrossRef CAS; (b) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139–3143 CrossRef CAS; (c) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef CAS.
- (a) S. L. Rössler, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 3603–3606 CrossRef; (b) J. A. Raskatov, S. Spiess, C. Gnamm, K. Brödner, F. Rominger and G. Helmchen, Chem.–Eur. J., 2010, 16, 6601–6615 CrossRef CAS; (c) S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228–7229 CrossRef CAS; (d) D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 11680–11681 CrossRef CAS . Also see 4a.
- For selected examples of C(sp3)–H allylic alkylation, see: (a) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 2039–2043 CrossRef CAS; (b) Y. Sempere, J. L. Alfke, S. L. Rössler and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 9537–9541 CrossRef CAS; (c) R. Sarkar and S. Mukherjee, Org. Lett., 2019, 21, 5315–5320 CrossRef CAS; (d) C.-Y. Shi, J.-Z. Xiao and L. Yin, Chem. Commun., 2018, 54, 11957–11960 RSC; (e) B.-B. Yue, Y. Deng, Y. Zheng, K. Wei and Y.-R. Yang, Org. Lett., 2019, 21, 2449–2452 CrossRef CAS; (f) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 CrossRef CAS; (g) X. Jiang and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 8887–8891 CrossRef CAS; (h) M. Bos and E. Riguet, Chem. Commun., 2017, 53, 4997–5000 RSC; (i) X. J. Liu and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 4002–4005 CrossRef CAS; (j) M. Zhan, R.-Z. Li, Z.-D. Mou, C.-G. Cao, J. Liu, Y.-W. Chen and D. Niu, ACS Catal., 2016, 6, 3381–3386 CrossRef CAS; (k) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 7644–7647 CrossRef CAS; (l) W. Chen, M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 15825–15828 CrossRef CAS; (m) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2068–2071 CrossRef CAS.
- (a) H. Tian, P. Zhang, F. Peng, H. Yang and H. Fu, Org. Lett., 2017, 19, 3775–3778 CrossRef CAS; (b) M. A. Schafroth, S. M. Rummelt, D. Sarlah and E. M. Carreira, Org. Lett., 2017, 19, 3235–3238 CrossRef CAS; (c) Z.-L. Zhao, Q. Gu, X.-Y. Wu and S.-L. You, Chem.–Asian J., 2017, 12, 2680–2683 CrossRef CAS; (d) Q.-L. Xu, C.-X. Zhuo, L.-X. Dai and S.-L. You, Org. Lett., 2013, 15, 5909–5911 CrossRef CAS; (e) Q.-L. Xu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 2579–2581 CrossRef CAS; (f) W.-B. Liu, H. He, L.-X. Dai and S.-L. You, Org. Lett., 2008, 10, 1815–1818 CrossRef CAS.
- For Ir-AAA of alkenyl trifluoroborate, see: J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef CAS.
- (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS; (c) R. R. Huddleston and M. J. Krische, Synlett, 2003, 12–21 CAS.
- (a) Y.-Q. Li, H.-J. Wang and Z.-Z. Huang, J. Org. Chem., 2016, 81, 4429–4433 CrossRef CAS; (b) P. K. Koech and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 5350–5351 CrossRef CAS; (c) B. G. Jellerichs, J.-R. Kong and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 7758–7759 CrossRef CAS.
- For a recent report on organocatalytic enantioselective α-C(sp2)–H allylic alkylation of allenyl ketones, see: (a) Y. Hu, W. Shi, B. Zheng, J. Liao, W. Wang, Y. Wu and H. Guo, Angew. Chem., Int. Ed., 2020, 59, 19820–19824 CrossRef CAS ; for an indirect approach using dienolsilane, see:; (b) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15249–15252 CrossRef CAS.
- (a) A. Chakrabarty and S. Mukherjee, Org. Lett., 2020, 22, 7752–7756 CrossRef CAS; (b) R. Sarkar, S. Mitra and S. Mukherjee, Chem. Sci., 2018, 9, 5767–5772 RSC; (c) S. Kayal and S. Mukherjee, Org. Lett., 2017, 19, 4944–4947 CrossRef CAS; (d) S. J. Singha Roy and S. Mukherjee, Chem. Commun., 2014, 50, 121–123 RSC.
- (a) L.-C. Wang, A. L. Luis, K. Agapiou, H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 2402–2403 CrossRef CAS; (b) S. A. Frank, D. J. Mergott and W. R. Roush, J. Am. Chem. Soc., 2002, 124, 2404–2405 CrossRef CAS . For a review, see:; (c) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS.
- Q. Liu and L. Zu, Angew. Chem., Int. Ed., 2018, 57, 9505–9509 CrossRef CAS.
- For selected examples, see: (a) G. A. Kraus and S. Wang, RSC Adv., 2017, 7, 56760–56763 RSC; (b) T. Pfennig, J. M. Carraher, A. Chemburkar, R. L. Johnson, A. T. Anderson, J.-P. Tessonnier, M. Neurock and B. H. Shanks, Green Chem., 2017, 19, 4879–4888 RSC; (c) K. Liu, H.-L. Teng and C.-J. Wang, Org. Lett., 2014, 16, 4508–4511 CrossRef CAS; (d) J. J. Lee and G. A. Kraus, Green Chem., 2014, 16, 2111–2116 RSC; (e) T. Guney, J. J. Lee and G. A. Kraus, Org. Lett., 2014, 16, 1124–1127 CrossRef CAS; (f) G. A. Kraus, S. Riley and T. Cordes, Green Chem., 2011, 13, 2734–2736 RSC; (g) P. M. Delaney, J. E. Moore and J. P. A. Harrity, Chem. Commun., 2006, 3323–3325 RSC.
- For selected examples, see: (a) C. J. F. Cole, L. Fuentes and S. A. Snyder, Chem. Sci., 2020, 11, 2175–2180 RSC; (b) M. Saktura, P. Grzelak, J. Dybowska and Ł. Albrecht, Org. Lett., 2020, 22, 1813–1817 CrossRef CAS; (c) X. Liang, W. Ye, W. Thor, L. Sun, B. Wang, S. He, Y.-K. Cheng and C.-S. Lee, Org. Chem. Front., 2020, 7, 840–845 RSC; (d) X. Gao, M. Xia, C. Yuan, L. Zhou, W. Sun, C. Li, B. Wu, D. Zhu, C. Zhang, B. Zheng, D. Wang and H. Guo, ACS Catal., 2019, 9, 1645–1654 CrossRef CAS; (e) S. Zheng and X. Lu, Org. Lett., 2009, 11, 3978–3981 CrossRef CAS; (f) R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849–1851 CrossRef CAS; (g) C.-H. Chen and C.-C. Liao, Org. Lett., 2000, 2, 2049–2052 CrossRef CAS; (h) B. M. Trost and S. Schneider, Angew. Chem., Int. Ed., 1989, 28, 213–215 CrossRef; (i) M. E. Jung, L. J. Street and Y. Usui, J. Am. Chem. Soc., 1986, 108, 6810–6811 CrossRef CAS.
- L. Chang, S. Thorimbert and L. Dechoux, Org. Biomol. Chem., 2019, 17, 2784–2791 RSC.
- (a) D. S. Ziegler, L. Klier, N. Müller, K. Karaghiosoff and P. Knochel, Synthesis, 2018, 50, 4383–4394 CrossRef CAS; (b) L. Chang, N. Klipfel, L. Dechoux and S. Thorimbert, Green Chem., 2018, 20, 1491–1498 RSC; (c) L. Chang, K. Plevová, S. Thorimbert and L. Dechoux, J. Org. Chem., 2017, 82, 5499–5505 CrossRef CAS; (d) K. Plevová, L. Chang, E. Martin, Q. Llopis, L. Dechoux and S. Thorimbert, Adv. Synth. Catal., 2016, 358, 3293–3297 CrossRef; (e) J. Agarwal, O. Bayounes, S. Thorimbert and L. Dechoux, RSC Adv., 2014, 4, 2772–2775 RSC.
- For selected reviews, see: (a) J. Fu, X. Huo, B. Li and W. Zhang, Org. Biomol. Chem., 2017, 15, 9747–9759 RSC; (b) G. Jindal, H. K. Kisan and R. B. Sunoj, ACS Catal., 2015, 5, 480–503 CrossRef CAS; (c) S. M. Inamdar, V. S. Shinde and N. T. Patil, Org. Biomol. Chem., 2015, 13, 8116–8162 RSC; (d) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS; (e) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC; (f) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC; (g) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC.
- For selected examples, see: (a) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48–54 CrossRef CAS; (b) Z.-C. Chen, Z. Chen, Z.-H. Yang, L. Guo, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2019, 58, 15021–15025 CrossRef CAS; (c) Z.-T. He, X. Jiang and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066–13073 CrossRef CAS; (d) C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2019, 58, 10521–10527 CrossRef CAS; (e) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef CAS; (f) X. Jiang, P. Boehm and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1239–1242 CrossRef CAS; (g) R. He, P. Liu, X. Huo and W. Zhang, Org. Lett., 2017, 19, 5513–5516 CrossRef CAS; (h) Y.-L. Su, Y.-H. Li, Y.-G. Chen and Z.-Y. Han, Chem. Commun., 2017, 53, 1985–1988 RSC; (i) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS; (j) L. Næsborg, K. S. Halskov, F. Tur, S. M. N. Mønsted and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 10193–10197 CrossRef; (k) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS.
- See the ESI† for details.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS.
- (a) L. A. Bryant, R. Fanelli and A. J. A. Cobb, Beilstein J. Org. Chem., 2016, 12, 429–443 CrossRef CAS; (b) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614–4628 CrossRef CAS; (c) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496–7504 CrossRef CAS.
- M. Chia, M. A. Haider, G. Pollock, G. A. Kraus, M. Neurock and J. A. Dumesic, J. Am. Chem. Soc., 2013, 135, 5699–5708 CrossRef CAS.
- (a) K. E. Kim, J. Li, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 13179–13182 CrossRef CAS; (b) Z. K. Wickens, B. Morandi and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 11257–11260 CrossRef CAS.
- Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695–10700 CrossRef CAS.
- (a) G. A. Kraus, G. R. Pollock Iii, C. L. Beck, K. Palmer and A. H. Winter, RSC Adv., 2013, 3, 12721–12725 RSC; (b) J. J. Lee and G. A. Kraus, Tetrahedron Lett., 2013, 54, 2366–2368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. See DOI: 10.1039/d0sc06208a |
| This journal is © The Royal Society of Chemistry 2021 |