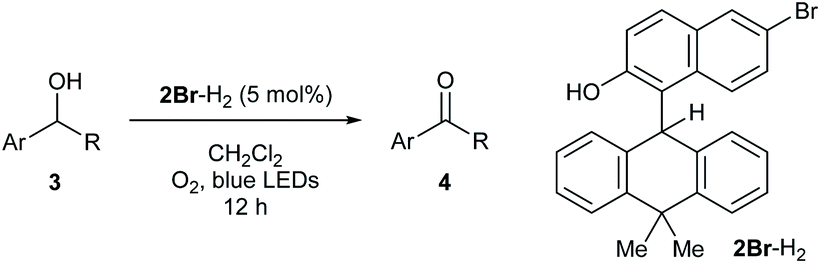
DOI:
10.1039/D0SC06240E
(Edge Article)
Chem. Sci., 2021,
12, 2778-2783
o-Quinone methide with overcrowded olefin component as a dehydridation catalyst under aerobic photoirradiation conditions†
Received
12th November 2020
, Accepted 10th January 2021
First published on 15th January 2021
Abstract
An o-quinone methide (o-QM) featuring an overcrowded olefinic framework is introduced, which exhibits dehydridation activity owing to its enhanced zwitterionic character, particularly through photoexcitation. The characteristics of this o-QM enable the operation of dehydridative catalysis in the oxidation of benzylic secondary alcohols under aerobic photoirradiation conditions. An experimental analysis and density functional theory calculations provide mechanistic insights; the ground-state zwitterionic intermediate abstracts a hydride and proton simultaneously, and the active oxygen species facilitate catalyst regeneration.
Introduction
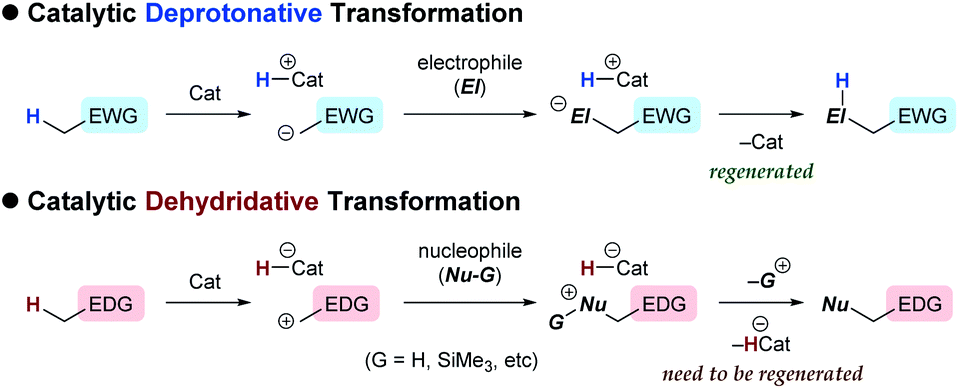
In contrast to the catalytic transformations that proceed through deprotonative generation of an anion as a nucleophile, alternative reactions triggered by hydride abstraction to generate a cationic intermediate as an electrophile have garnered less attention in organic chemistry, as a chemical term “dehydridation” sounds unusual. This is probably because of a deficit in effective catalysts for dehydridation, which could be ascribed to the intrinsic problems associated with the dehydridative transformations.1,2 While the deprotonative system commonly relies on base catalysis (proton-transfer catalysis) and involves deprotonation and protonation to complete a catalyst turnover,3 the dehydridative system generally lacks the catalyst regeneration step, rendering the development of the dehydridation catalyst difficult (Fig. 1). Mechanistically, initial dehydridation from a substrate generates a cationic intermediate and hydridated catalyst (H-Cat−), and subsequent bond formation furnishes a cationic product precursor. The precursor is prone to liberate a cationic group (G), such as proton and trialkylsilylium ions, to yield a stable product rather than accepting a hydride for the catalyst regeneration. Therefore, the dehydridation catalyst often requires an independent oxidative regeneration process, in which the terminal oxidant should selectively react with H-Cat− without degradating the substrate. These features constitute inherent impediments to eliciting the full synthetic potential of dehydridative catalysis.4
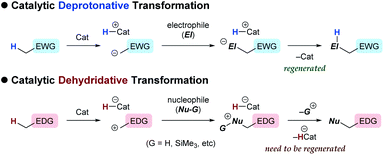 |
| | Fig. 1 General schemes of catalytic deprotonative and dehydridative transformations. | |
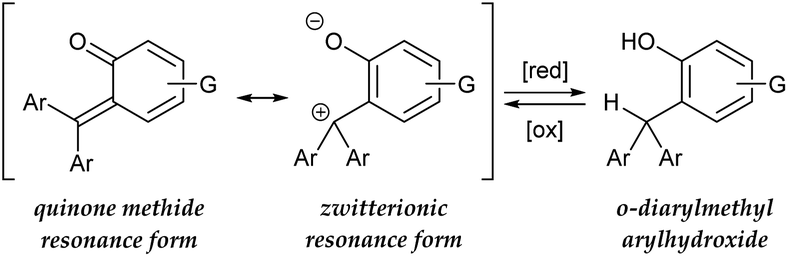
In view of designing a dehydridation catalyst under these circumstances, we became interested in one of the canonical structures of o-quinone methide (o-QM), which could be regarded as a zwitterionic triarylmethilide (Scheme 1).5 As triarylmethilide (triarylcarbenium) ion, exemplified by tritylium ion, has been used as a powerful dehydridating reagent,4 we envisioned that the zwitterionic form of o-QM could have an ability to engage in dehydridation, being transformed to its hydrogenated (reduced) form, o-diarylmethyl arylhydroxide. However, although o-QMs undergo 1,4-addition of various nucleophiles, their capability of accepting a hydride from simple organic molecules, 1,4-reduction reactivity, remains obscure.6 On the other hand, o-diarylmethyl arylhydroxide is known to be converted to o-QM under mild oxidative conditions,7 which implies the feasibility of in situ regeneration of o-QM after dehydridation event. These characteristics of o-QMs and their redox behavior prompted us to explore the possibility of imparting reactivity as a dehydridation catalyst to o-QMs through pertinent structural modifications and optimization of reaction conditions. Here, we report the implementation of this approach, revealing the catalytic performance of novel o-QM featuring an overcrowded olefinic core in the dehydridative oxidation of secondary benzylic alcohols under aerobic photoirradiation conditions.
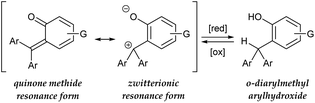 |
| | Scheme 1 Hypothetical basis of potential hydride-abstracting capability of o-quinone methide. | |
Results and discussion
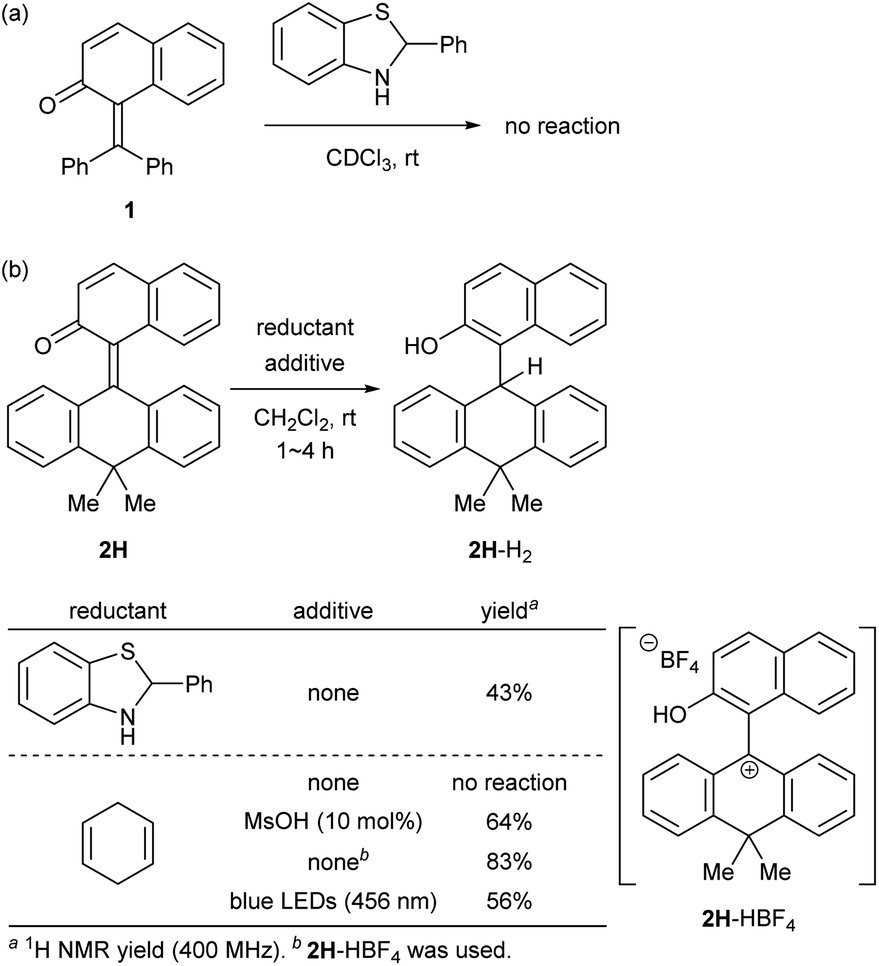
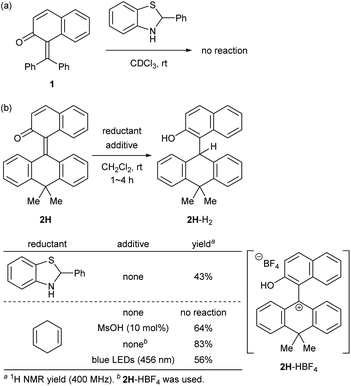
At the outset, we selected a representative isolable o-QM 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 (Fig. 2a), and evaluated its intrinsic reactivity toward an organic reductant, 2-phenylbenzothiazoline,9 in CDCl3 by 1H NMR (400 MHz) monitoring experiment, which showed that conversion of 1 to the reduced form 1-H2via 1,4-reduction did not occur (Fig. 3a). We inferred that the observed low reactivity could be ascribed to the insufficient contribution of the zwitterionic canonical form to the resonance structures of 1. To increase the zwitterionic character of o-QM, we devised an o-QM 2H featuring 10,10-dimethyl-9(10H)-anthracenylidene framework with expectation that 2H would be compelled to form a twisted structure due to the steric repulsion between the substituents on the olefinic carbons at the fjord region. However, X-ray diffraction analysis of an orange crystal of 2H revealed its distorted but non-twisted (anti-folded) three-dimensional structure having a nearly identical α,β-unsaturated core to that of 1 (Fig. 2b). The burden of the distortion is mainly localized on the ring systems. Specifically, the mean plane dihedral angle of the benzocyclohexadienone subunit of 2H (142.21°) is markedly smaller than that of 1 (153.68°), and the dihydroanthracene subunit is also significantly bent (121.53°). In silico structural optimization of 2H and 1 using density functional theory (DFT) calculations (SMD(CH2Cl2)-CAMB3LYP/6-31+G(d) level)10 derived nearly identical structures to those obtained from the X-ray analysis, suggesting that the observed skeletal distortion was not affected by the crystal packing force (see ESI†). In addition, a comparison of the maximum absorption wavelengths of 2H (381 nm) with that of 1 (363 nm) supports a similar effective conjugation length (α,β-unsaturated character) (Fig. 2c). These observations implied a negligible difference in the cationic character of their benzylic carbons. However, 2H reacted with 2-phenylbenzothiazoline with higher efficiency to give the reduced form 2H-H2 at ambient temperature (43%), suggesting the enhanced cationicity (Fig. 3b). Although 2H did not undergo 1,4-reduction with less reactive cyclohexa-1,4-diene as a hydride donor, its dehydridation activity could be enhanced by adding an acidic additive to effectively increase the participation of the zwitterionic structure as its protonated, carbocationic form. For instance, the reaction of 2H with cyclohexa-1,4-diene proceeded smoothly in the presence of 10 mol% of methanesulfonic acid (MsOH) to afford 2H-H2 in 64% yield. Protonation of the carbonyl oxygen of o-QM induces the generation of the protonated form of the zwitterionic resonance structure of 2H, 2H-HOMs, as evident from the indicative change in solution color from pale yellow to deep red. For precise structural elucidation of the protonated form of 2H, 2H-HBF4 was separately prepared by the treatment of 2H-H2O with HBF4 in toluene. The subsequent recrystallization from the hexane/CH2Cl2 solvent system gave deep red crystals, which were subjected to X-ray diffraction analysis. As shown in Fig. 2d, 2-naphthol and dihydroanthracene subunits connected perpendicularly (torsion angle: 78.1°), and the benzylic carbon of the connecting bond adopted an almost complete sp2-hybridized planar structure (sum of the angles: 360°), accounting for its cationic character. Notably, the reduction of 2H-HBF4 to 2H-H2 with cyclohexa-1,4-diene was even faster (83%), demonstrating its high reactivity. These observations corroborate the correlation between the cationicity of the benzylic carbon atom of o-QM 2H and its dehydridation activity.
8 (Fig. 2a), and evaluated its intrinsic reactivity toward an organic reductant, 2-phenylbenzothiazoline,9 in CDCl3 by 1H NMR (400 MHz) monitoring experiment, which showed that conversion of 1 to the reduced form 1-H2via 1,4-reduction did not occur (Fig. 3a). We inferred that the observed low reactivity could be ascribed to the insufficient contribution of the zwitterionic canonical form to the resonance structures of 1. To increase the zwitterionic character of o-QM, we devised an o-QM 2H featuring 10,10-dimethyl-9(10H)-anthracenylidene framework with expectation that 2H would be compelled to form a twisted structure due to the steric repulsion between the substituents on the olefinic carbons at the fjord region. However, X-ray diffraction analysis of an orange crystal of 2H revealed its distorted but non-twisted (anti-folded) three-dimensional structure having a nearly identical α,β-unsaturated core to that of 1 (Fig. 2b). The burden of the distortion is mainly localized on the ring systems. Specifically, the mean plane dihedral angle of the benzocyclohexadienone subunit of 2H (142.21°) is markedly smaller than that of 1 (153.68°), and the dihydroanthracene subunit is also significantly bent (121.53°). In silico structural optimization of 2H and 1 using density functional theory (DFT) calculations (SMD(CH2Cl2)-CAMB3LYP/6-31+G(d) level)10 derived nearly identical structures to those obtained from the X-ray analysis, suggesting that the observed skeletal distortion was not affected by the crystal packing force (see ESI†). In addition, a comparison of the maximum absorption wavelengths of 2H (381 nm) with that of 1 (363 nm) supports a similar effective conjugation length (α,β-unsaturated character) (Fig. 2c). These observations implied a negligible difference in the cationic character of their benzylic carbons. However, 2H reacted with 2-phenylbenzothiazoline with higher efficiency to give the reduced form 2H-H2 at ambient temperature (43%), suggesting the enhanced cationicity (Fig. 3b). Although 2H did not undergo 1,4-reduction with less reactive cyclohexa-1,4-diene as a hydride donor, its dehydridation activity could be enhanced by adding an acidic additive to effectively increase the participation of the zwitterionic structure as its protonated, carbocationic form. For instance, the reaction of 2H with cyclohexa-1,4-diene proceeded smoothly in the presence of 10 mol% of methanesulfonic acid (MsOH) to afford 2H-H2 in 64% yield. Protonation of the carbonyl oxygen of o-QM induces the generation of the protonated form of the zwitterionic resonance structure of 2H, 2H-HOMs, as evident from the indicative change in solution color from pale yellow to deep red. For precise structural elucidation of the protonated form of 2H, 2H-HBF4 was separately prepared by the treatment of 2H-H2O with HBF4 in toluene. The subsequent recrystallization from the hexane/CH2Cl2 solvent system gave deep red crystals, which were subjected to X-ray diffraction analysis. As shown in Fig. 2d, 2-naphthol and dihydroanthracene subunits connected perpendicularly (torsion angle: 78.1°), and the benzylic carbon of the connecting bond adopted an almost complete sp2-hybridized planar structure (sum of the angles: 360°), accounting for its cationic character. Notably, the reduction of 2H-HBF4 to 2H-H2 with cyclohexa-1,4-diene was even faster (83%), demonstrating its high reactivity. These observations corroborate the correlation between the cationicity of the benzylic carbon atom of o-QM 2H and its dehydridation activity.
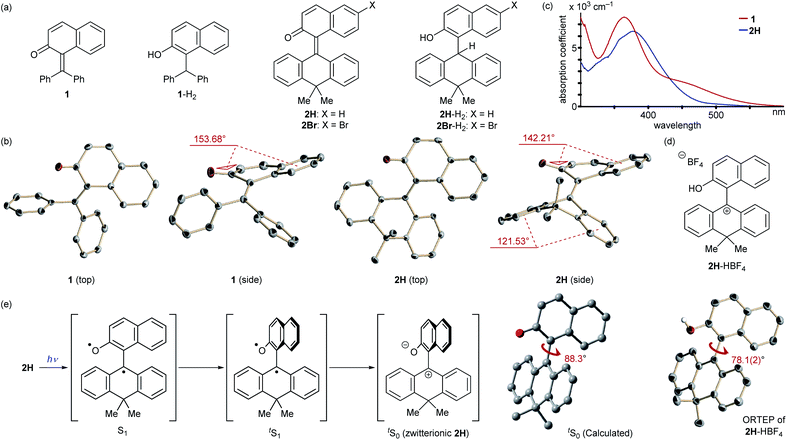 |
| | Fig. 2 (a) Molecular structures of o-QMs and their reduced forms. (b) ORTEP diagrams of o-QMs. (c) UV spectra of o-QMs. (d) Structure and ORTEP diagram of protonated o-QM, 2H-HBF4 (BF4− was omitted for clarity). (e) Expected molecular motion of 2H upon photoexcitation and calculated structure of tS0 (calculation was performed at SMD(CH2Cl2)-TD-CAMB3LYP/6-31+G(d) level). | |
 |
| | Fig. 3 Reduction of o-QM 1 (a) and 2H (b). | |
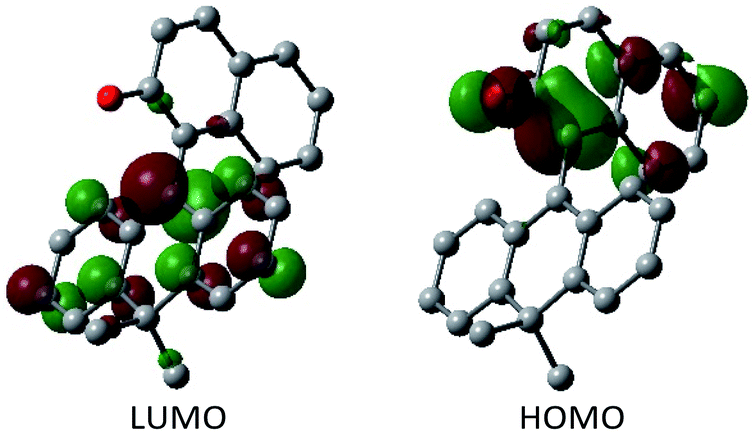
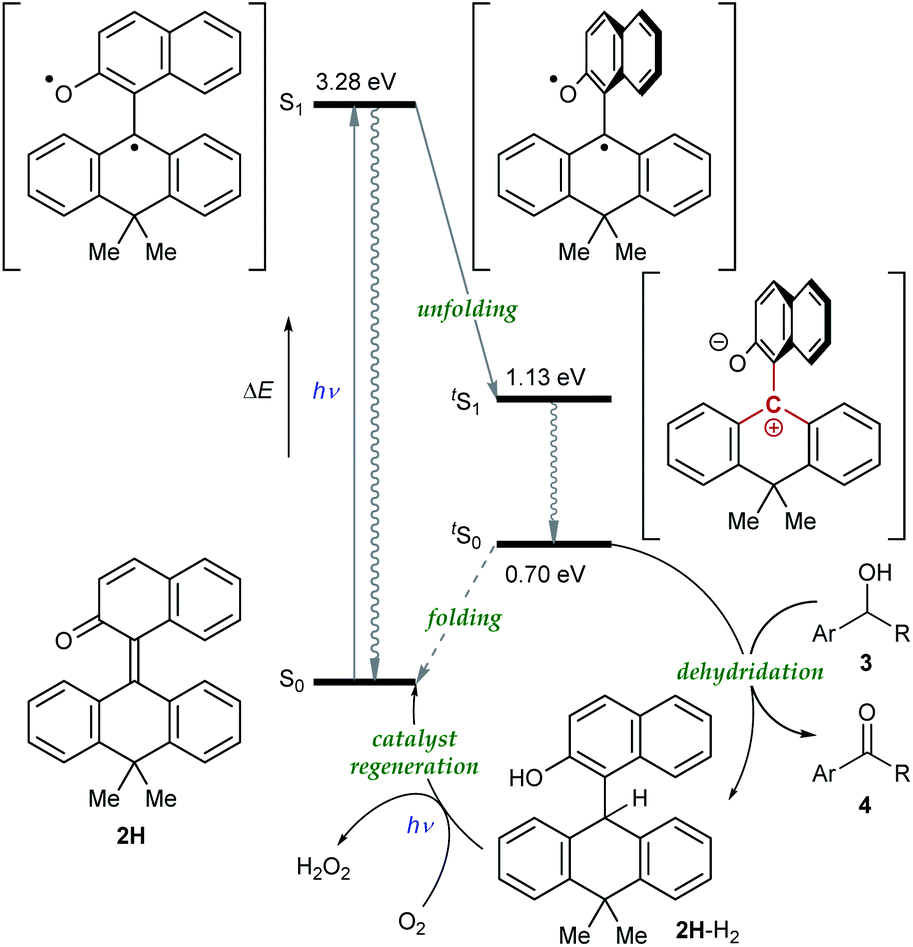
Having verified the structure–activity relationship of o-QM 2H in the reduction with the hydride donors, we were intrigued with the possibility of increasing the reactivity of 2H by other means, preferably by taking advantage of its inherent structural attributes, without the addition of acidic additives. Toward this end, we paid our attention to the push–pull-type structure of the tetrasubstituted olefinic component of 2H and envisaged that its local excitation by photoirradiation would cause twisting around the C–C axis at the singlet excited state (S1) of the folded conformer to give stabilized twisted S1 (tS1), in analogy with the behavior of the overcrowded ethylenes (Fig. 2e).11 Further relaxation with preservation of the twisted structure furnishes ground-state intermediate tS0 of distinct zwitterionic character, which would exert enhanced dehydridation activity. This hypothesis was substantiated by attempting the reaction of 2H with cyclohexa-1,4-diene in CH2Cl2 under irradiation with blue LEDs (456 nm), which exhibited smooth conversion into 2H-H2 (56% for 1 h, Fig. 3b). To obtain support for the origin of the increased hydride-abstracting ability of 2H, we calculated the structure of tS0 at the SMD(CH2Cl2)-TD-CAMB3LYP/6-31+G(d) level and confirmed its similarity to the structure of 2H-HBF4 in accordance with the presumed predominant contribution of the zwitterionic form (Fig. 2dvs.2e). Molecular orbital of tS0 revealed intramolecular charge separation and the largest orbital lobe in LUMO of the resultant zwitterionic structure resided at the benzylic carbon in good agreement with its expected carbocationic character (Fig. 4).
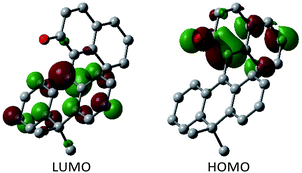 |
| | Fig. 4 Molecular orbital of tS0 of 2H. | |
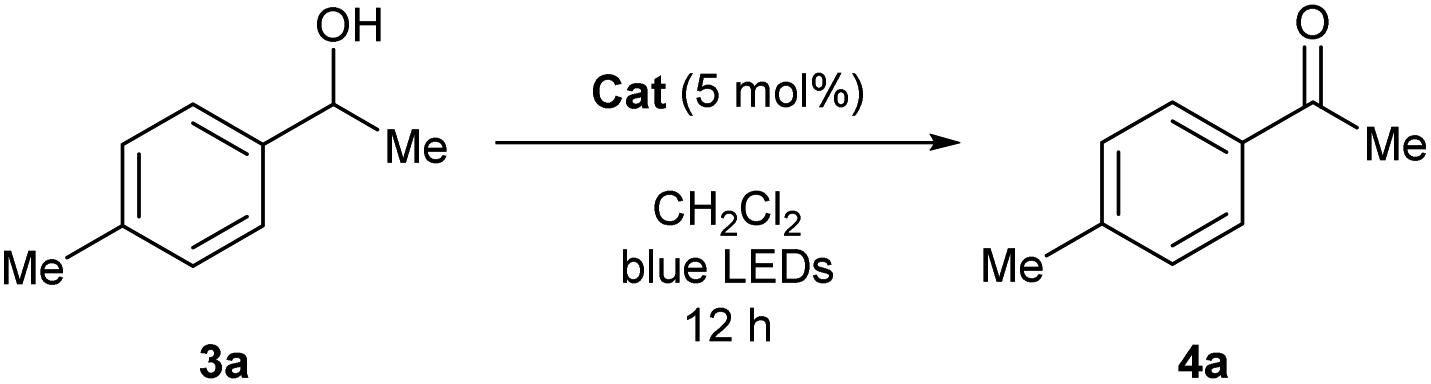
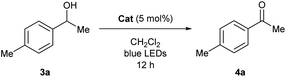
To experimentally explore the feasibility of the dehydridative catalysis of o-QMs, we chose the oxidation of benzylic secondary alcohols as a model reaction system. Prior to assessing the viability of the regeneration of o-QM with various oxidants, we sought to evaluate the intrinsic reactivity of 2H as a dehydridation catalyst with 1-(4-methylphenyl)ethyl alcohol (3a) as a representative substrate (Table 1). Thus, 3a was treated with each 5 mol% of 2H and the co-catalyst MsOH in CH2Cl2 at ambient temperature for 12 h, which produced 4′-methylacetophenone (4a) in 4% 1H NMR yield (400 MHz, trimethylsilylbenzene as the internal standard), an amount less than the loading of 2H (entry 1). Considering the beneficial effect of photoirradiation on the reactivity of 2H, we also conducted the reaction with irradiation of blue LEDs (456 nm) in the absence of MsOH under otherwise similar conditions, which exhibited a higher conversion to afford 4a in 17% yield (entry 2). While this unexpected outcome suggested the possibility of regenerating 2H with photoirradiation, close examination of the reaction conditions revealed that traces of O2 acted as an oxidant to regenerate 2H under light irradiation. Indeed, performing the reaction under air and O2 atmospheres further improved conversion to 51% and 59%, respectively, whereas only a trace amount of 4a was detected under a thoroughly inert atmosphere (entries 3–5). These profiles led us to attempt the reaction with the reduced form 2H-H2 as a precatalyst, assuming that active catalyst 2H could be generated in situ under aerobic photoirradiation conditions (entry 6). The observed high conversion of 3a validated the hypothesis and the operation of the dehydridative catalysis of 2H in this oxidation. This finding allowed us to continue further investigation with the use of more stable and easy-to-handle 2H-H2 as a precatalyst, and we found that introduction of a bromo functionality to the 6-position of the 2-naphthol subunit (2Br-H2) delivered a critical enhancement in the catalytic activity, enabling a complete conversion of 3a within 12 h (entry 7). It should be noted that the importance of the overcrowded alkene component of 2H for exerting efficient dehydridative catalysis was confirmed by comparing the reactivity with that of 1 (entry 8).
|
|
| Entry |
Catalyst |
Yieldb (%) |
Note |
|
Unless otherwise noted, reaction was performed with 0.1 mmol of 3a in the presence of 5 mol% of catalyst in CH2Cl2 under blue LEDs irradiation (456 nm) at ambient temperature for 12 h.
1H NMR yield (400 MHz) determined by trimethylsilylbenzene as the internal standard.
|
| 1 |
2H
|
4 |
MsOH (5 mol%) without photoirradiation |
| 2 |
2H
|
17 |
Under Ar |
| 3 |
2H
|
51 |
Under air |
| 4 |
2H
|
59 |
Under O2 |
| 5 |
2H
|
4 |
Under Ar (degassed) |
| 6 |
2H-H2 |
76 |
Under O2 |
| 7 |
2Br-H2 |
97 |
Under O2 |
| 8 |
1
|
8 |
Under O2 |
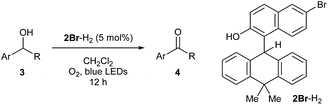
With the optimized conditions in hand, we turned our attention to examine the substrate scope of the dehydridative catalysis (Table 2). Generally, 5 mol% of 2Br-H2 was sufficient for smooth conversion of alcohols 3 to afford the corresponding ketones 4 in high chemical yield. Incorporation of various substituents of different electronic properties to an arbitrary position of the aromatic nuclei was tolerated (entries 1–10). The alkyl substituent on the hydroxy-bearing carbon could also be varied and not only linear but also branched chains were well accommodated (entries 11–15). As demonstrated in the reaction with a diol, chemoselective oxidation of benzylic alcohols over aliphatic alcohols was feasible (entry 16).12 In addition, scalability of this protocol was confirmed with 3a as a substrate (entry 17).
Table 2 Substrate generalitya
|
|
| Entry |
Ar |
R |
Yieldb (%) |
|
Unless otherwise noted, reaction was conducted on 0.1 mmol scale with 5 mol% of 2Br-H2 in CH2Cl2 under photoirradiation (456 nm) for 12 h.
Isolated yield was indicated.
5 mmol scale reaction.
|
| 1 |
4-CF3C6H4 |
Me |
85 |
| 2 |
4-NCC6H4 |
Me |
94 |
| 3 |
4-ClC6H4 |
Me |
84 |
| 4 |
4-BrC6H4 |
Me |
97 |
| 5 |
C6H5 |
Me |
90 |
| 6 |
3-ClC6H4 |
Me |
86 |
| 7 |
3-MeC6H4 |
Me |
86 |
| 8 |
3-MeOC6H4 |
Me |
95 |
| 9 |
2-ClC6H4 |
Me |
89 |
| 10 |
2-MeC6H4 |
Me |
72 |
| 11 |
C6H5 |
Et |
98 |
| 12 |
C6H5 |
n
Pr |
99 |
| 13 |
C6H5 |
i
Bu |
84 |
| 14 |
C6H5 |
i
Pr |
87 |
| 15 |
C6H5 |
c
Hex |
94 |
| 16 |
C6H5 |
(CH2)2OH |
77 |
| 17c |
4-MeC6H4 |
Me |
74 |
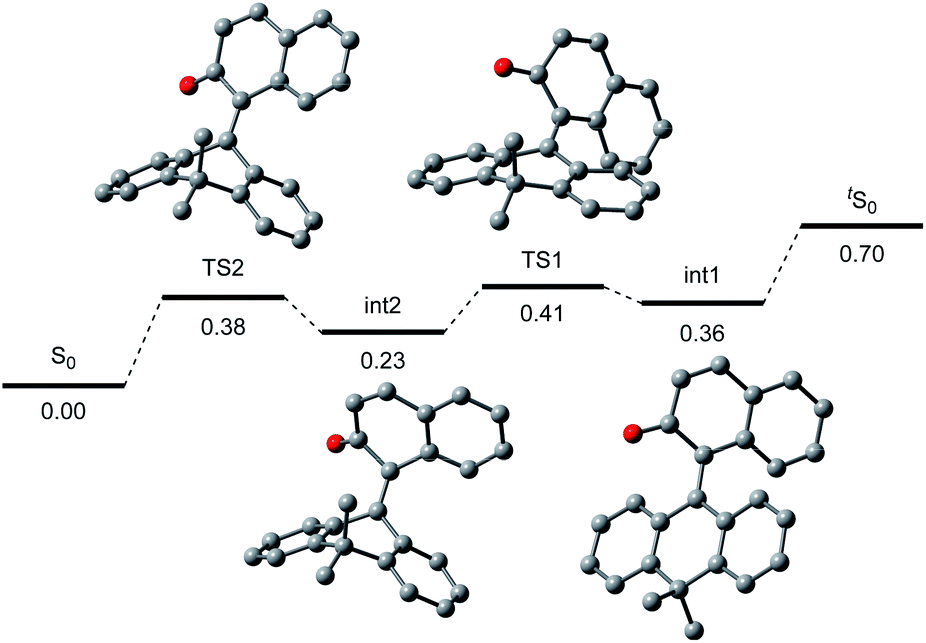
To gain further insight into the reaction mechanism, we simulated the molecular motion of the postulated excitation process of 2H using time-dependent density functional theory (TD-DFT) calculations (SMD(CH2Cl2)-TD-CAMB3LYP/6-31+G(d) level). As illustrated in Fig. 5, the calculated energy difference between the folded S1 and tS1 (ΔE = 2.15 eV) signified that photoexcitation of 2H to S1 and spontaneous unfolding furnishes tS1. The subsequent rapid relaxation to the ground-state zwitterionic intermediate tS0 may well occur owing to the small energy gap (ΔE = 0.43 eV), while this process was estimated to be a forbidden transition. Further simulation of the folding pathway from tS0 to S0 to estimate the lifetime of tS0 uncovered the intervention of a two-step transition process (Fig. 6). Following the relaxation from tS1, tS0 spontaneously undergoes conformational change to give thermodynamically more stable int1. Then, increase in the double-bond character of the bridging C–C bond with concurrent bending of the dihydroanthracene subunit along the C9–C10 axis affords int2 via TS1. Subsequent rotation around the bridging C–C bond eventually forms S0via TS2. These computational analyses imply that int1 would have a certain lifetime, although both of the two energetic barriers of the folding process are very small. Accordingly, the triarylmethilide moiety of tS0 would engage in dehydridation from the methine carbon of alcohol 3 to give the corresponding ketone 4 with concomitant generation of 2H-H2. During this process, the aryloxide moiety of tS0 would simultaneously accept a proton from the hydroxy functionality of 3. The regeneration of 2H from 2H-H2 is facilitated likely by active oxygen species, such as singlet oxygen or superoxide, generated through excitation of O2 by sensitization with the 2-naphthol subunit of the catalyst, as confirmed by the detection of H2O2 in the reaction mixture (see ESI†).13–15 UV-vis spectrum of 2H-H2 indicated that its absorption terminus reaches 470 nm and thus, 2H-H2 could be excited by irradiation of blue LEDs (Fig. S2†). The estimated energy of 2H-H2 at the excited state was 2.17 eV, which is sufficient to activate the ground-state triplet O2, allowing the regeneration of 2H through rapid abstraction of the neighboring benzylic hydrogen by the resulting activated O2. Although the TD-DFT analysis also suggested the feasibility of an alternative process involving an electron-donor–acceptor (EDA) complex of 2H-H2 and O2 followed by its photoexcitation, careful examination of the UV-vis spectroscopy of 2H-H2 under Ar and O2 atmospheres showed no evidence of the formation of EDA complex.
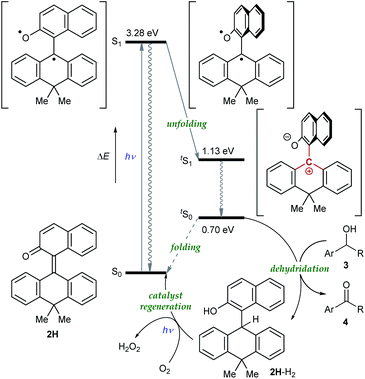 |
| | Fig. 5 Plausible mechanism. | |
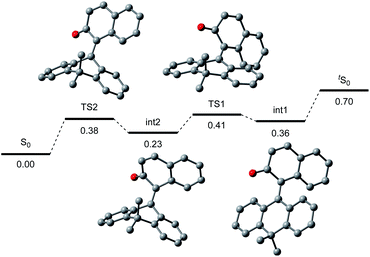 |
| | Fig. 6 Folding process from tS0 (upper right) to S0 (lower left). | |
Conclusions
We have demonstrated the feasibility of eliciting the latent reactivity of o-QMs as a dehydridation catalyst. The introduction of an overcrowded olefinic framework is a key for enhancing the contribution of the zwitterionic resonance form of o-QM and its dehydridation activity can be fully realized by photoexcitation. This unique property of the novel o-QM is exploited to establish dehydridative catalysis in the oxidation of benzylic secondary alcohols under aerobic photoirradiation conditions, where the ground-state zwitterionic intermediate acts as an actual reactive species and the catalyst is regenerated by either singlet oxygen or superoxide as supported by experimental and theoretical analyses. We anticipate that the present study stimulates further research endeavor for exploring synthetic potential of dehydridative catalysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was provided by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis” (No. 17H06444, to TO), and Grants of JSPS for Scientific Research. This work was also supported by Program for Leading Graduate Schools “Integrative Graduate Education and Research Program in Green Natural Sciences” in Nagoya University and WISE Program (Doctoral Pro-gram for World-leading Innovative & Smart Education) “Graduate Program of Transformative Chem-Bio Research” in Nagoya University. We appreciate Prof. Daisuke Yokogawa (Univ. of Tokyo) and Prof. Takeshi Yanai (Nagoya Univ.) for insightful discussion on the theoretical analysis.
Notes and references
-
(a) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS;
(b) A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638 CrossRef CAS.
- J.-J. Tian, N.-N. Zeng, N. Liu, X.-S. Tu and X.-C. Wang, ACS Catal., 2019, 9, 295 CrossRef CAS.
- N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS.
-
(a) M. E. Jung and L. M. Speltz, J. Am. Chem. Soc., 1976, 98, 7882 CrossRef CAS;
(b) J. M. Gil-Negrete, J. P. Sestelo and L. A. Sarandeses, J. Org. Chem., 2019, 84, 9778 CrossRef CAS;
(c) W. Chen, Z. Xie, H. Zheng, H. Lou and L. Liu, Org. Lett., 2014, 16, 5988 CrossRef CAS;
(d) M. E. Jung, J. Org. Chem., 1976, 41, 1479 CrossRef CAS;
(e) M. E. Jung and R. W. Brown, Tetrahedron Lett., 1978, 31, 2771 CrossRef;
(f) M. E. Jung, Y.-G. Pan, M. W. Rathke, D. F. Sullivan and R. P. Woodbury, J. Org. Chem., 1977, 42, 3961 CrossRef CAS;
(g) M. Wan, Z. Meng, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 13845 CrossRef CAS;
(h) X. Liu, Z. Meng, C. Li, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 6012 CrossRef CAS;
(i) T. Katsina, L. Clavier, J.-F. Giffard, N. M. Portela da Silva, J. Fournier, R. Tamion, C. Copin, S. Arseniyadis and A. Jean, Org. Process Res. Dev., 2020, 24, 856 CrossRef CAS.
-
(a) H. Sugimoto, S. Nakamura and T. Ohwada, Adv. Synth. Catal., 2007, 349, 669 CrossRef CAS;
(b) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS;
(c)
M. M. Toteva and J. P. Richard, in Advances in Physical Organic Chemistry, ed. J. P. Richard, Elsevier, 2011, vol. 45, pp. 39–91 Search PubMed;
(d) Đ. Škalamera, K. Mlinarić-Majerski, I. M. Kleiner, M. Kralj, J. Oake, P. Wan, C. Bohne and N. Basarić, J. Org. Chem., 2017, 82, 6006 CrossRef.
-
(a) R. Chen, Y. Liu and S. Cui, Chem. Commun., 2018, 54, 11753 RSC;
(b) M.-M. Chu, S.-S. Qi, W.-Z. Ju, Y.-F. Wang, X.-Y. Chen, D.-Q. Xu and Z.-Y. Xu, Org. Chem. Front., 2019, 6, 1140 RSC and references therein.
- T. Inoue, S. Inoue and K. Sato, Bull. Chem. Soc. Jpn., 1990, 63, 1062 CrossRef CAS.
- T. W. Lewis, E. N. Duesler, R. B. Kress, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1980, 102, 4659 CrossRef CAS.
-
(a) H. Chikashita, M. Miyazaki and K. Itoh, Synthesis, 1984, 308 CrossRef CAS;
(b) H. Chikashita, M. Miyazaki and K. Itoh, J. Chem. Soc., Perkin Trans. 1, 1987, 699 RSC;
(c) C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388 CrossRef CAS.
-
(a) T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS;
(b)
M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed;
(c) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS.
- T. Nishiuchi, R. Ito, E. Stratmann and T. Kubo, J. Org. Chem., 2020, 85, 179 CrossRef CAS.
- Under the present reaction conditions, oxidation of aliphatic primary and secondary alcohols hardly proceeded (Ph(CH2)3OH: <5%, cyclohexanol: 5%). In the case of allylic and propargylic alcohols, unavoidable side reactions afforded complex mixtures.
-
(a) W. M. Draper and D. G. Crosby, J. Agric. Food Chem., 1983, 31, 734 CrossRef CAS;
(b) M. Hayyan, M. Ali Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS.
-
(a) C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC;
(b) D. Uraguchi, M. Torii and T. Ooi, ACS Catal., 2017, 7, 2765 CrossRef CAS.
- Although singlet oxygen has been known to directly oxidize benzylic alcohols in polar solvent,16 the oxidation did not occur in the presence of methylene blue as a catalytic sensitizer in CH2Cl2 under blue LED irradiation.
-
(a) Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035 CrossRef CAS;
(b) N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kohsuke
Kato
a,
Kohsuke
Kato