
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein†
Karan
Kapoor‡
 ,
Shashank
Pant‡
and
Emad
Tajkhorshid
*
,
Shashank
Pant‡
and
Emad
Tajkhorshid
*
NIH Center for Macromolecular Modeling and Bioinformatics, Beckman Institute for Advanced Science and Technology, Department of Biochemistry, Center for Biophysics and Quantitative Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: emad@illinois.edu
First published on 9th April 2021
Abstract
P-glycoprotein (Pgp) is a major efflux pump in humans, overexpressed in a variety of cancers and associated with the development of multi-drug resistance. Allosteric modulation by various ligands (e.g., transport substrates, inhibitors, and ATP) has been biochemically shown to directly influence structural dynamics, and thereby, the function of Pgp. However, the molecular details of such effects, particularly with respect to the role and involvement of the surrounding lipids, are not well established. Here, we employ all-atom molecular dynamics (MD) simulations to study the conformational landscape of Pgp in the presence of a high-affinity, third-generation inhibitor, tariquidar, in comparison to the nucleotide-free (APO) and the ATP-bound states, in order to characterize the mechanical effects of the inhibitor that might be of relevance to its blocking mechanism of Pgp. Simulations in a multi-component lipid bilayer show a dynamic equilibrium between open(er) and more closed inward-facing (IF) conformations in the APO state, with binding of ATP shifting the equilibrium towards conformations more prone to ATP hydrolysis and subsequent events in the transport cycle. In the presence of the inhibitor bound to the drug-binding pocket within the transmembrane domain (TMD), Pgp samples more open IF conformations, and the nucleotide binding domains (NBDs) become highly dynamic. Interestingly, and reproduced in multiple independent simulations, the inhibitor is observed to facilitate recruitment of lipid molecules into the Pgp lumen through the two proposed drug-entry portals, where the lipid head groups from the cytoplasmic leaflet penetrate into and, in some cases, translocate inside the TMD, while the lipid tails remain extended into the bulk lipid environment. These “wedge” lipids likely enhance the inhibitor-induced conformational restriction of the TMD leading to the differential modulation of coupling pathways observed with the NBDs downstream. We suggest a novel inhibitory mechanism for tariquidar, and potentially for related third-generation Pgp inhibitors, where lipids are seen to enhance the inhibitory role in the catalytic cycle of membrane transporters.
Introduction
ATP-binding cassette (ABC) transporters form one of the largest transporter superfamilies expressed in cellular and intracellular membranes.1–3 They use ATP as a source of energy to actively transport various substrates between the two sides of the membrane. A group of ABC transporters, known as ABC exporters, provide a major mechanism for active efflux of small molecules out of the cell.4,5 ABCB1, or P-glycoprotein (Pgp), a prominent member of this superfamily, is ubiquitously expressed in different tissues6 where it exports a wide range of charged and neutral hydrophobic molecules.7 This promiscuous quality of Pgp is exploited by, e.g., cancer cells that over-express the transporter in their cellular membranes, and thereby acquire resistance to chemotherapeutic reagents and other drug molecules.8 In this context, a major drug development effort has been aiming at inhibiting Pgp and therefore achieving more effective chemotherapeutic treatments from the currently used anticancer drugs. In order to make such efforts more effective, it is imperative to understand the mechanism of the transporter in its physiological context, i.e., the cellular membrane. Such understanding will also provide deeper insights into the fundamental questions about the function of ABC transporter and how it may be modulated by the main constituents of the cellular membrane, i.e., lipids, which are present at the immediate vicinity of the protein in a very dense arrangement and can interact with the protein in many different ways.Structurally, Pgp shows a pseudo two-fold symmetry, folding into a single-chain heterodimeric structure containing two transmembrane domains (TMDs), each consisting of a six-helical transmembrane leaflet associated with a nucleotide-binding domain (NBD).9–12 Somewhat common in structurally known ABC exporters, two prominent entry portals exist within the transmembrane region of Pgp, one on either side of the TMDs, which in principle can allow access not only to small molecules to diffuse from within the membrane into the large, central, semi-hydrophobic lumen of the transporter but also to lipids that are frequently observed to bind to these portal regions in the resolved structures of Pgp10 and other ABC transporters.13
Substrate translocation in ABC transporters like Pgp involves complex structural transitions between inward-facing (IF) and outward-facing (OF) states of the transporter, alternatively exposing the substrate binding site/region to the inside and outside of the cell, a model coined as the “alternate access” mechanism.14–16 These conformational changes in ABC transporters are closely coupled to and fueled by binding and hydrolysis of ATP in the NBDs. In Pgp and other ABC exporters, this mechanism facilitates the export of substrate molecules out of the cell.16–23 The conformational coupling between the NBDs and the TMDs is key to the ATP-driven transport in Pgp and other ABC transporters. Without getting into specifics, in this transport scheme, ATP binding to the two NBDs promotes their dimerization, with the resulting conformational changes directly coupled to the TMDs and their subsequent transition into the OF state.18,19 Hydrolysis of ATP, on the other hand, releases the dimerized NBDs, allowing the transporter to reset to the IF state.24–26 Through these general couplings, ATP binding and hydrolysis in the NBDs mechanically control the interconversion between the IF and OF, and therefore, the accessibility of the substrate between the two sides of the membrane.
As a multi-domain protein, Pgp exhibits a complex behavior, with the binding of ATP, substrates and inhibitors directly modulating the conformational states and characteristics of the transporter.27 Pgp crystal structures, which have been resolved in a non-membrane environment, have mostly found the transporter in excessively open, IF(-like) states, characterized by large separations between the two TMD leaflets on the cytoplasmic side, and between the two NBDs.9–12 Similarly, other experimental biophysical studies conducted in non-native environments such as micelles have reached comparable conclusions regarding the open IF state and its highly dynamic nature.28–31 On the other hand, biochemical studies performed in more realistic and physiological environments (e.g., nanodiscs or other lipid bilayers), have presented an alternate view of the transporter's mode of action, where the APO (nucleotide-free) and ATP-bound transporter states are present in a thermodynamic equilibrium capable of sampling similar conformations,32–34 highlighting the importance of the lipid bilayer in regulating Pgp's conformational dynamics.
A number of studies have also aimed at developing novel inhibitors for targeting this biomedically important transporter.35 Out of these, third-generation inhibitors, displaying higher binding affinity and specificity for Pgp, provide the most promising candidates. Tariquidar (TAR), a large (∼650 Da), positively charged, amphiphilic compound, is one such high-affinity third-generation inhibitor that binds to the central cavity of Pgp, and it has been suggested to function by destabilizing NBD conformations necessary for ATP hydrolysis.31 Recent structural studies have provided additional static information on the location and binding mode of other third-generation inhibitors,36 but the molecular details of the mechanism of action of these molecules remain unclear.
Here, we present a molecular dynamics (MD) study providing an atomic-level description of the conformational dynamics of Pgp in the APO, ATP-bound, and TAR-bound states in a natural environment of a lipid bilayer, and propose a model connecting these changes to the possible inhibition mechanisms of Pgp. Our simulations capture a dynamic equilibrium between the open and closed IF conformations in APO Pgp, with ATP binding observed to promote NBD dimerization and closing of the cytoplasmic gate. Compared to the APO and ATP-bound states, TAR-bound Pgp shows highly dynamic NBDs and TMD portal regions, sampling more widely open IF conformations. We also observe a substantially enhanced inhibitor-mediated recruitment of the membrane lipids into the central lumen of the transporter, which together with the inhibitor binding itself result in a differential allosteric coupling between TMD and NBDs, highlighting an important role of lipids in modulating the conformational landscape of Pgp and its inhibition. While the study here focuses on a Pgp inhibitor, the findings regarding the role of lipids can be expected to exist for other types of ligands binding Pgp including the transported substrates, though the mode and degree of lipid interaction with the ligand and the protein can be different in each case.
Methods
In order to characterize the conformational dynamics of Pgp in the presence and absence of the third-generation inhibitor TAR, we modeled and performed atomistic simulations of 3 different systems, namely, APO (nucleotide-free, drug-free), ATP (ATP/Mg2+-bound), and TAR (ATP/Mg2+-bound, TAR-bound). We discuss the system construction and the MD methodology in the following sections.System preparation
A recent crystal structure of Pgp (PDB: 4M1M)12 in the IF state was used as the starting point for constructing APO, ATP, and TAR systems. For the generation of the APO system, only chain B was retained from the PDB structure, containing residues 34–626 and 696–1271. The missing region (residues 627–695) corresponds to a flexible linker region connecting NBD1 to the TMD2, not resolved in any available structure of Pgp and was not included in the simulations. For the generation of the ATP system, ATP and Mg2+ were docked into their respective binding sites in the two NBDs using a rigorous protocol described by Wen et al.,37 which reproduces the conserved nucleotide binding characteristics observed in higher-resolution structures of ABC transporters (for this study, the structure of HlyB38 was used as the template).For the generation of the TAR system, we used a variant of ensemble docking, which we term as extended-ensemble docking,39 that first samples the conformations of the transporter along its global transition pathway and then utilizes a collection of these conformations representing the entire transition pathway for docking. Briefly, the transition pathway between the ATP/Mg2+-bound IF state (modeled above) and the OF structure of Pgp (described in Verhalen et al.23) was generated using steered molecular dynamics (SMD) along specific collective variables optimized for successfully describing the alternate-access mechanism in ABC-transporters.40,41 We note that in doing so, we make the assumption that ATP/Mg2+ binding to the IF state is sufficient for the protein to transition to the OF state. The SMD simulation resulted in intermediate structures and conformations along the transition pathway that were then used for our extended-ensemble docking approach. Subsequently, docking of TAR, allowing flexibility around all rotatable bonds, was carried out in the extended-ensemble of the protein containing 50 structures generated along the transition pathway. Finally, the docked structure showing the strongest predicted binding affinity (highest docking score) was selected as the starting structure for the TAR systems. This TAR-bound structure shows a Cα root mean squared deviation (RMSD) of 4.95 Å with respect to the starting structure of the ATP system. Of importance to our comparative study here, the initial TAR-bound structure is also in an IF state, similar to the APO and ATP initial systems simulated. It should also be noted that a recent DEER study showed that inhibitor might have different effects on the conformational dynamics of the transporter depending on the presence or absence of ATP/Mg2+ in the NBDs31 (with the former case studied in the present study).
Membrane embedding
To study the conformational dynamics of Pgp in APO, ATP/Mg2+, and inhibitor-bound states, we carried out multiple all-atom MD simulations for the three systems in explicit membranes including anionic lipids, mimicking conditions used in recent FRET and DEER studies on Pgp reconstituted in nanodiscs.31,33 Five independent lipid bilayers containing anionic phospholipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC 40%), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE 43%), 1-hexadecanoyl-2-(9Z-hexadecenoyl)-glycero-3-phospho-(1′-sn-glycerol) (PYPG 14%), and 1-palmitoyl-2-myristoyl-1,3-bis(sn-3′-phosphatidyl)-sn-glycerol (PMCL 3%) were constructed using the Membrane Builder module of CHARMM GUI.42 Subsequently, each state of Pgp was inserted into the membrane independently and the overlapping lipids removed from the bilayer (Fig. 1). | ||
| Fig. 1 Simulation systems: APO, ATP/Mg2+-bound (ATP), and TAR-bound (TAR) IF conformations of Pgp used in the simulations, with the two TMD leaflets, each connected to an NBD, colored in red and blue, respectively (see Methods for details on system construction). All the bound states were inserted in lipid bilayers containing: POPC = 40%, POPE = 43%, PYPG = 14%, and PMCL = 3% lipids matching experimental conditions in recent FRET and DEER studies on Pgp reconstituted in nanodiscs,31,33 shown in different colors. See text for chemical details of the tails. | ||
It is to be noted that in order to remove any bias generated due to the initial distribution of lipids, all the lipids molecules occupying the central cavity of Pgp and/or penetrating the two TMD portals were specifically removed. Thus, two strategies were employed for sampling the conformational space of the studied systems: (A) randomizing the initial distribution of lipids around Pgp, and (B) randomizing the initial velocities of the atoms (Table 1).
In addition to the above-described three systems, we also performed simulations of the starting TAR-bound Pgp conformation after removing the inhibitor. This was done so as to check the stability of the starting TAR-bound Pgp conformation, as well as a point of comparison to the starting conformations of the APO and ATP-bound systems. We term this structure as the “TAR-less” system or the “control” simulation.
MD simulations
All the simulations were performed using NAMD2.43,44 CHARMM36m protein and lipid force field45,46 and TIP3P water47 were used. The inhibitor molecule (TAR) was parametrized using the force field ToolKit (ffTK) plugin48 of VMD.49 Briefly, the geometry optimization was performed using second order Møller–Plesset (MP2) theory50 and the 6-31G(d) basis set in Gaussian09.51 After geometry optimization, partial charges were assigned using quantum mechanical calculations. The bonds and angular terms were calculated directly from the Hessian, and the dihedral parameters were fit to the QM potential energy landscape.The protonation states of the ionizable residues of Pgp were calculated using the PropKa server.52 The C-terminal carboxylate capping group, N-terminal ammonium capping group, and all the hydrogen atoms were added to the structures using the PSFGEN plugin of VMD.49 All systems were solvated and neutralized with 0.15 M NaCl. The final systems contained ∼250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms each with approximate dimensions of 135 × 135 × 190 Å3.
000 atoms each with approximate dimensions of 135 × 135 × 190 Å3.
All the simulations were performed as NPT ensembles, at a temperature of 310 K maintained using Langevin thermostat with a damping coefficient of γ = 1 ps−1. Constant pressure was maintained at 1 bar using the Nosé–Hoover Langevin piston method.53 The particle mesh Ewald (PME) method54 was used in calculating the long-range electrostatic forces. A 12 Å cutoff distance and a 10 Å switching distance were used in calculating the non-bonded interactions. All the simulations were performed with the keyword vdwForceSwitching, in order to use force-based cutoffs. An integration timestep of 2 fs was used in all simulations. The system was initially energy-minimized and equilibrated for 10 ns with the Cα atoms restrained to their initial positions with a force constant k = 1 kcal mol−1 Å−2. All restraints were then released in the following 500 ns production run. Additionally, a 200 ns control simulation of the TAR-less system was also carried out (Table 1).
The initial and final structures for all the simulations, topology/parameter files, and configuration files for the runs are all made available in Open Science Framework, https://osf.io/3kyx2/?view_only=54eccc30d54347b0b857d18f7d402df9. Trajectory files will be made available upon request.
Analysis
The conformational dynamics of Pgp were characterized in terms of the large-scale motions of the different domains of the transporter, as well as secondary structure changes in the different simulated systems. As the surrounding lipid environment can play a significant role in modulating the conformational ensemble of membrane proteins, we characterized the lipid density and penetration inside the protein in the different systems, as well. Additionally, we also characterized the potential conformational coupling between the TMD and NBDs of Pgp, as the structural changes observed in the different systems may differentially affect the communication between these domains. Analysis on the conformational changes in the protein and calculation of lipid properties around the protein were carried out on all the simulation runs. All analyses were performed using our in-house scripts and/or VMD plugins. | ||
| Fig. 2 Conformational dynamics of Pgp in different bound states: (A) schematic representation of Pgp depicting the geometric parameters used for monitoring its global conformational dynamics. The two halves of the protein are colored in red and blue. Conformational dynamics in all the simulated systems were evaluated by analyzing: (B) dNBD: the center of mass distance between the two NBDs, (C) γ: relative twist of the two NBDs, (D) dcyt: the distance between Cα atoms of K145 and M787 on the cytoplasmic side of the TMDs, (E) α: the angle describing relative orientation of the TMD helices measuring the degree of cytoplasmic opening, and, (F) dext: the distance between Cα atoms of S92 and R745 on the extracellular end of the TMDs. The histograms in (B–F) are calculated using the entire data set and indicate the presence of more widely open IF conformations in the TAR-bound system (shown in blue), compared to APO (green) and ATP (orange) systems. Dotted vertical lines in each panel mark the values in the recent structures for an inhibitor (zosuquidar)-bound Pgp (PDB: 6QEE)36 (black) and for APO Pgp (PDB: 4M1M)12 (purple). Time series of the distances shown in the histograms are provided in Fig. S1.† (G) Representative snapshots for ATP, TARclosed, TARwide, and APO are shown. | ||
Results
Multiple all-atom MD simulations of Pgp in APO, ATP- and inhibitor-bound states allowed us to characterize major structural and dynamical differences between these states, also highlighting the influential and differential role played by the lipids in the dynamics of the transporter. The results presented here are primarily divided into three sections describing: (1) global and local conformational dynamics of Pgp in different bound states, (2) lipid-mediated structural modulation of Pgp, and, (3) allosteric coupling between the TMDs and NBDs.Conformational dynamics of different bound states of Pgp
Similar to NBD distances, a significant degree of cytoplasmic TMD closing (dcyt) is observed for the ATP systems, whereas the APO systems show relatively more open cytoplasmic TMD conformations (Fig. 2A and D and S1†). The TAR systems sample a broader range of dcyt, lying on either side of the starting crystal structure. In terms of the cytoplasmic opening, α, the ATP systems show smaller TMD angles as compared to APO (Fig. 2E). The TAR systems show TMD cytoplasmic angles similar to the APO systems, as well as a large population similar to the starting crystal structure and the recent cryoEM structure of zosuquidar-bound Pgp.58 We note that in all the simulated systems, including TARwide and TARclosed, TAR remains stably bound inside the central cavity of Pgp (Fig. S2†). Additionally, comparison of the TAR-bound simulations with the zosuquidar-bound Pgp structure shows a large similarity between the protein residues involved in interactions with the two inhibitor molecules. Pgp may thus display a shared (conserved) binding site for these two molecules, and possibly other third-generation inhibitors in general.
Given that during the simulations, Pgp does not sample conformations in the occluded or OF states, no significant changes are noticeable in the extracellular opening, as measured by dext (Fig. 2F). The control (TAR-less) simulation (the TAR-bound conformation after removing the inhibitor) displays a dynamical behavior similar to the ATP-bound Pgp, supporting the structural similarity of the two protein conformations used to initiate these two simulations (Fig. S3†).
 | ||
| Fig. 3 Differences in secondary structure: (A) comparison of the degree of helicity in the portal helix TM10 (residues 876–889) in APO, ATP, and TAR Pgp with the zosuquidar-bound (ZOS) Pgp cryoEM structure.58 The analysis was performed combining all the simulations for each state. The structure of TM10 in a representative trajectory snapshot from: (B) TAR-bound (orange), (C) ATP (yellow), and, (D) APO Pgp (red) aligned with zosuquidar-bound Pgp (ZOS)58 (blue). Helicity of other portal helices (TM4, TM6, and TM12) is shown in Fig. S5.† A p value of less than 0.001 was obtained on comparing the degree of helicity in the different systems using one-way ANOVA test, pointing to statistical significance of the results. | ||
Lipid-mediated structural modulation of Pgp
The role of membrane lipids in modulating the structural properties of Pgp was further evaluated by calculating the lipid distribution and packing around the protein as well as lipid penetration/translocation inside the central cavity of Pgp. | ||
| Fig. 4 Lipid packing around and inside Pgp: the cytoplasmic-leaflet lipid density profile for (A) TAR, (B) APO, and (C) ATP Pgp simulations was calculated over all the simulated replicas for each system. The molecular systems are aligned along the Z-axis and viewed from the extracellular side of the protein, with the XY indicating the plane of the membrane. All histograms are normalized the same way, that is, with respect to the average bulk density of lipids. The lipid molecules access the central cavity through the entry portals located on either side of the TMD. TAR-bound and APO systems show enhanced lipid densities inside the central cavity of Pgp, while a low density was observed in the case of ATP-bound systems. In each panel, a part of Pgp spanning the cytoplasmic membrane leaflet (taken at the end of one of the 500 ns simulations for each system) is shown in cartoon representation with the two Pgp halves colored in green and blue, respectively. | ||
 | ||
| Fig. 5 Lipid access into the lumen of Pgp: (A) a lipid access event was counted if the entire head-group phosphate of any lipid accessed the central cavity of Pgp, defined by a cylinder (shown in blue) with a cross-sectional area of 300 Å2 and a height of 25 Å (see Methods for the criteria used to define the boundaries of the central cavity). Pgp is shown in a translucent surface representation. (B) Count of lipids accessing the central cavity of Pgp through the entry portals in APO (green), ATP (orange), and TAR (red) simulations, calculated by counting the number of phospholipid head groups accessing the central cavity over the course of the 500 ns simulations. Two TAR-bound conformations (TARwide and TARclosed) are defined based on the dNBD separation (Fig. 2B). In comparison to the APO and ATP systems, more lipid access events are observed in the TAR systems, especially for the TARwide conformations. A p value of less than 0.001 was obtained on comparing the lipid access events for the different systems using one-way ANOVA test, pointing to statistical significance of the results. | ||
The key Pgp residues involved in lipid recruitment through Portals 1 and 2 were determined by monitoring the portal residues which make contacts with the lipid head groups (Fig. S6A–C†). Both entry portals are observed to be rich in basic residues that directly interact with the negatively charged phosphate groups of lipids: R355, K238, and K230 at Portal 1, and K881 and K996 at Portal 2 (Fig. S6D–G†). Interestingly, we also observed system-dependent opening and re-arrangement of the basic residues in Portal 2, which may play a role in the differential lipid recruitment observed in the different systems.
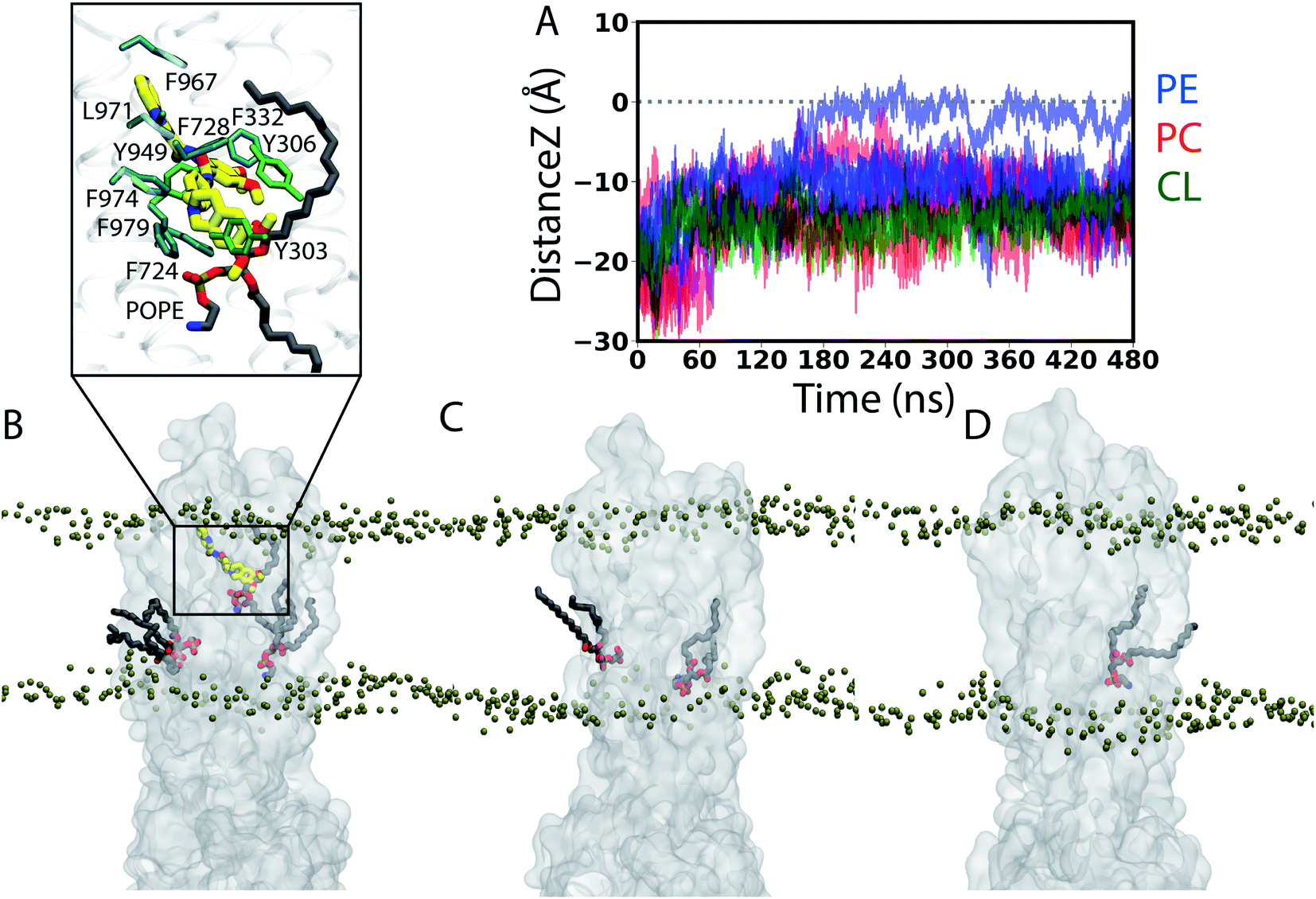 | ||
| Fig. 6 Trans-leaflet lipid translocation inside the lumen of Pgp: (A) translocation of lipids in the central cavity of Pgp that might result in their flipping was monitored by calculating the z component of the center of mass of the lipid head groups in all the simulated systems. In total, we captured 12 lipid molecules from the cytoplasmic (lower) leaflet that undergo partial flipping during the simulations. Data for the APO Pgp are shown in Fig. S7.† (B–D) Lipids (licorice representation) penetrating into the central cavity of (B) TAR-bound, (C) APO and (D) ATP Pgp (translucent surface representation) from representative snapshots of the simulations. Phosphorus atoms from the lipid head groups are shown as tan beads to delineate the membrane boundaries. Inset in (B) shows the closeup view of the interactions between the bound TAR (thick sticks) and the flipping lipid (PE) molecule showing the largest translation (black trace in (A)) into the other side of the membrane, as well as the surrounding Pgp residues within 3.5 Å of either TAR or the head group of the translocating (PE) lipid. A 2D representation of the TAR binding pocket along with the flipping PE molecule is shown in Fig. S2B.† | ||
Conformational coupling between TMDs and NBDs
Dynamic network analysis was employed to evaluate the allosteric coupling between the NBDs and TMDs in the different simulated systems. The coupling pathway was calculated between TM10 in the TMD, which shows major conformational differences between the three systems (as described above), and the conserved Q-loop in the NBD, which is important for coupling conformational changes between the NBDs and TMDs and subsequent conformational transition between the IF and OF states.57,60The APO and ATP systems predominantly show short and direct coupling pathways connecting the TM10 with the Q-loop (Fig. 7A and B). In the case of the ATP systems, the pathway connects the TMD with the NBD directly through the coupling helices located at the interface of TMD and NBD, whereas in the APO systems, the pathway connects the two domains without directly passing through the coupling helices.
 | ||
| Fig. 7 Allosteric coupling between TMDs and NBDs: The coupling pathway calculated for a representative 500 ns long simulation of: (A) ATP, (B) APO, and (C) TAR-bound Pgp, between S988 in TMD (the source) and Q1114 in the NBD (the sink), is shown in green. Q-loop (containing conserved Q1114) is shown in yellow, coupling helices in purple, A-loop (containing conserved Y1040) in orange, and the rest of the protein in a transparent cartoon representation. All the residues that form the allosteric pathway are highlighted in the representative replica for each system. In the majority of TAR-bound simulations, extended coupling pathways passing primarily through the coupling helices and the Q-loop in the NBDs are observed. Shorter and more direct pathways connecting the TMD and the NBD are observed in the ATP and APO simulations. | ||
In the case of the inhibitor-bound systems, on the other hand, elongated pathways are predicted passing through additional portions of the NBD (Fig. 7C). In some cases, the pathway connects the coupling helices with the A-loop, which contains a conserved aromatic residue (Y1040) critical for ATP binding.61 As a result, the pathway transverses a larger portion of the NBD before reaching the Q-loop. In addition, by employing dynamic network analysis, we also investigated the communities, i.e., the sets of residues that move in concert with each other during the simulations. Similar communities were observed in the TMDs and the NBDs for all three systems (Fig. S8†).
Discussion
Pgp is a biochemically well studied ABC exporter that actively extrudes certain xenobiotics and other amphiphiles across the plasma membrane. The transporter is overexpressed in a variety of cancer cells where it is closely associated with the development of multidrug resistance by interfering with the therapeutic accumulation of the drugs inside the affected cells. As such, a major effort is invested in developing novel strategies for targeting and inhibiting Pgp. In the present study, we have carried out an extensive set of all-atom MD simulations to characterize the conformational dynamics of Pgp in a number of native states arising during the transport cycle (APO and ATP-bound), as well as in the presence of a third-generation inhibitor, TAR. We capture differential conformational dynamics, both globally and locally, associated with the different bound states of Pgp, and, furthermore, we propose a novel lipid contribution to the mechanism of action of the inhibitor, which might also accompany the binding of other ligand types to Pgp and other ABC transporters, though with different modes and in different extents.The global orientational dynamics captured in our MD simulations of Pgp embedded in multi-component lipid bilayers, each starting from a different lipid distribution around Pgp, show a dynamic equilibrium between open and closed (dimer-like) NBD conformations of the transporter in the APO system, with the NBD twist angles showing large variations around the crystal structure values (Fig. 2 and S1†). Along with NBD dynamics, the transporter also samples distances corresponding to both more open and more closed TMDs in comparison to the starting crystal structure used in the simulation, but it remains relatively open with respect to the ATP-bound state. The APO systems also show a higher lipid density inside the binding cavity of Pgp as well as a greater number of lipid access events than the ATP systems, which both can be attributed to the more open configuration of the TMDs on the cytoplasmic side (Fig. 4 and 5). As the substrates are believed to access the central binding cavity of Pgp largely from the lipid bilayer,32 the larger cytoplasmic TMD opening in the APO state is expected to provide an ideal pathway for this process. In this context, the pathway observed in our simulations for the penetrating lipid molecules accessing the central cavity of Pgp may also represent a potential route for the lateral translocation and diffusion of the substrate molecules into the central cavity. The excessively open, IF-like structures of APO Pgp that have been observed in structural studies using reconstituted Pgp in detergent or micellar environments,9–12 confer it with no added physiological function and may portray artifacts of the non-physical (non-membrane) environments used in these experiments. The more closed Pgp conformations captured in the APO simulations, on the other hand, have been recently characterized in cryoEM,34 luminescence resonance energy transfer (LRET),33 as well as atomic-force microscopy experiments (AFM),32 when Pgp was reconstituted in lipid nanodiscs or in liposomes, constituting a more natural membrane environment for the transporter.
In the ATP-bound systems, the conformational equilibrium is shifted towards closed-IF like states characterized by closure of the TMD cytoplasmic gate in addition to dimerization of the NBDs (Fig. 2 and S1†). The ATP systems in our simulations lie close to the recent ATP-bound dimerized structure of human Pgp,55 adopting a closed NBD conformation coupled to a closed TMD. As expected, a low lipid density is observed within the central cavity of Pgp in the ATP systems which can be attributed to the partial closure of the cytoplasmic side of the TMDs in these systems (Fig. 4 and 5). Substrate binding and subsequent binding of ATP to the NBDs in the APO systems may concurrently drive occlusion of the substrate-bound TMD and the complete closure of the inter-NBD gap,36 in line with the conclusions of earlier mutagenesis and biochemical studies.33,62 Alternatively, ATP binding prior to the substrate entry may drive the transporter to a substrate-free occluded state, responsible for the known basal ATPase activity of the transporter.63,64 A close interaction between the coupling helices in the TMDs and the Q-loop in the NBDs, observed more in the ATP-bound systems than the APO systems (Fig. 7), may allow fine-tuning of the coupled movements of different domains of the transporter necessary for the transition to the OF state and completion of the transport cycle, events which are not fully observed here due to the limited timescale of the simulations.
From our systematic MD study, clear differences are observed in the conformational ensemble of Pgp in the presence of an inhibitor bound to the central cavity. In the presence of the inhibitor, TAR, Pgp becomes conformationally more dynamic, both in the TMDs and the NBDs, sampling two major conformations, termed TARwide and TARclosed, with different degrees of NBD dimerization (Fig. 2 and S1†). Two major Pgp conformations, characterized by distinct degrees of NBD opening, were also observed from a recent inhibitor-bound cryoEM structures,36,58 with one of these structure showing an NBD distance within the range observed in our simulations for the TARclosed conformations. One major finding of the present study is the lateral penetration of lipid molecules into the luminal region of Pgp, primarily from the cytoplasmic openings provided by the TMD portals. Lipid penetration into the lumen of Pgp takes place to different degrees in different bound states of Pgp, with the ATP systems showing the least and the TAR systems showing the largest number of such events. The differential lipid occupancy observed in the TARclosed and TARwide (Fig. 5) may contribute to the small energy barrier between these two states that may be readily overcome through the diffusion of lipids from or into the binding cavity of Pgp.
Introduction of the inhibitor in the central cavity of Pgp, lying at the apex of the TMDs, leads to an increase in the fluctuations of neighboring protein residues (Fig. S4†), as well as local unfolding of the portal helix TM10 (Fig. 3), one of the helices present at the drug entry Portal 2 and involved in forming direct interactions with the inhibitor. The zosuquidar-bound cryoEM structure shows a similar disorder in TM10 in the presence of the inhibitor,36 although local unfolding in Portal 1 helices (TM4 and TM6) is also observed in this structure. This unfolding of the Portal 1 helices, which is not observed in other Pgp structures,9,21,22 has been suspected by a recent computational study65 to be an artifact of the small size of the nanodisc used in structural determination.
Compared to the transported substrates, Pgp inhibitors have been suggested to function by filling up the drug-binding cavity more completely, while at the same time, forming a larger number of contacts with the binding residues.36,66 Although the binding stoichiometry of TAR to Pgp is not well established, given its larger volume9 and flexibility (more than twice rotatable bonds as compared to zosuquidar for which an inhibitor-bound cryoEM structure exists36), only one TAR molecule may suitably fit inside the central cavity of Pgp. The single TAR molecule may instead successfully fill the binding cavity through recruiting lipids, as seen from the increased lipid density and lipid access events inside the central cavity of Pgp in our TAR-bound simulations compared to APO and ATP-bound systems (Fig. 4, 5 and ESI Video 1†). Additionally, these “wedge-lipids”, where the lipid head groups from the cytoplasmic leaflet translate towards the membrane center while the lipid tails remain extended into the bulk membrane, are only observed in the inhibitor-bound systems (Fig. 6). The inhibitor may promote the recruitment of these wedge-lipids inside Pgp through a combination of conformational changes in the protein (larger TMD cytoplasmic portal/gate opening and local unfolding of TM10), encouraging lipid diffusion and translation, and then directly forming interactions with the lipid head groups inside the central cavity. It is possible that lipids binding to the central cavity of Pgp (in the absence of the inhibitor) may have similar effects on the dynamics of the transporter but to a lesser extent; in the absence of the inhibitors, lipid are presented less opportunities to penetrate the protein (e.g., due to more closed portals) and they may more readily diffuse out of the pocket.
A recent study using DEER spectroscopy suggested that high-affinity, third generation inhibitors like TAR can differentially modulate the interactions between the A-loop and ATP compared to substrate-bound Pgp, in turn inhibiting ATP hydrolysis.31 A previous cysteine-scanning mutagenesis study has shown that the long-range conformational changes in the NBDs can be regulated by inhibitors/substrates.62 Additionally, lipid-binding sites observed at the regions formed by the kinking of the portal helices in response to inhibitor binding have been suggested to play a direct role in the modulation of Pgp function.36 Inhibitor-mediated lipid diffusion resulting in wedge-lipids inside the central cavity of Pgp, as observed in our simulations, may contribute to the differential coupling observed in the inhibitor-bound simulations compared to the APO and ATP systems (Fig. 7), modulating the transfer of signal between the TMDs and NBDs. This may in turn disrupt the natural dynamics of the NBDs, preventing their optimal interactions and dimerization and consequently inhibiting ATPase activity. In addition to directly restricting access to the substrate binding site, competitive inhibitors like TAR may thus successfully inhibit Pgp by stabilizing alternate occluded conformations, halting or shifting the equilibrium away from the OF states of the transporter.
The RMSF calculations evaluating differential fluctuations of Pgp in different bound states also highlighted that NBD2 is relatively more dynamic compared to NBD1 in the ATP-bound system (Fig. S4†). This asymmetry in NBD dynamics, not observed in the APO system, is further amplified upon TAR binding. An asymmetric behavior in the NBDs of Pgp has also been reported in DEER studies of both ATP/substrate-bound and ATP/inhibitor-bound systems,23,31 where the asymmetry has been proposed to be a constitutive property of Pgp and directly involved in its asymmetric ATP hydrolysis and two-stroke transport mechanism of the OF state. Whether the conformational changes observed in our study in the IF state are correlated to the proposed transport mechanism in the OF state of Pgp remains to be further investigated. Additionally, as it is not possible to deconvolute the effect of ATP binding and TAR binding on the observed differential dynamics of the two NBDs in our simulations (both systems included ATP), we cannot determine whether these changes can be attributed to the chemical nature of the TAR molecule itself.
Conclusion
As a multi-domain protein, Pgp displays a complex dynamical behavior, with the binding of ATP, substrates, inhibitors and surrounding lipids potentially modulating its conformational ensemble in many different ways. By employing extensive all-atom MD simulations, we show that a third generation, high-affinity inhibitor, TAR, allosterically modulates the conformational dynamics of both TMDs and NBDs, and their coupling, in Pgp, an additional mechanism that may contribute to its overall inhibitory effect on the transporter (Fig. 8). This additional mode of inhibition, which is complementary to the inhibitor's occupation of the substrate binding site (orthosteric inhibition), becomes amplified due to the inhibitor-mediated recruitment of wedge-lipids inside the central cavity of Pgp. | ||
| Fig. 8 Modulation of the conformational dynamics of Pgp by the lipidic environment and bound inhibitor: In nucleotide-free (APO) state, Pgp exists in a dynamic equilibrium between open and closed IF conformations. ATP binding in the NBDs results in shifting of the equilibrium towards predominantly closed IF conformations of Pgp. The presence of a third-generation inhibitor (TAR) leads to increased lipid recruitment into the central cavity of Pgp, thereby, imposing a stronger inhibitory effect on NBD dimerization and closure of the cytoplasmic side of the TMDs. | ||
Furthermore, we show that ATP binding to the NBDs (in the absence of inhibitor) modifies the conformational dynamics of the transporter, favoring closed IF conformations, which exist in a dynamic equilibrium with more open IF conformations. The open IF conformations captured for the APO simulations show large cytoplasmic opening that may allow active recruitment of substrate molecules, whereas the closed IF conformations favor the closure of cytoplasmic elements and, thereby, promote ATP binding and NBD dimerization.
To achieve the alternating access mechanism, membrane transporters are designed to undergo relatively larger conformational changes than other membrane proteins during their function. It is not therefore surprising that their structural dynamics and conformational changes can be significantly affected by membrane lipids. This mode of lipid effects might be more pronounced in ABC exporters such as Pgp in which large portals form by the extended helices in their structure, specially on the cytoplasmic side and in their IF state. In fact, alternate engagement and disengagement of lipids with the transporter should be considered almost critical parts/steps of the transport mechanism in these molecular machines. The coupling and interplay of these lipid events with ligand binding effects, reported in this study for the case of an inhibitor, can be expected to also exist for other modes of ligand binding and modulation in ABC transporters. One should expect ligand-mediated modulation of lipid–protein interactions even for the transported substrate, though in order for that transport mechanism to succeed the mode and extent of the interactions in this case has to be different from what we report here for a Pgp inhibitor.
From a pharmacological perspective, the additional role of lipids in inhibiting Pgp may also serve as a novel molecular strategy for the development of more potent and more specific inhibitors. The results presented in the study will provide another example of diverse mechanisms by which membrane lipids can modulate the function of complex membrane proteins such as Pgp.
Author contributions
K. K., S. P., and E. T. designed the research. K. K. and S. P. performed and analyzed the simulations. The manuscript was written by all the authors.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under awards P41-GM104601 (to ET) and R01-GM123455 (to ET). We also acknowledge computing resources provided by Blue Waters at National Center for Supercomputing Applications, and Extreme Science and Engineering Discovery Environment (Grant MCA06N060 to ET). S. P. would like to acknowledge Beckman Institute Graduate Fellowship for funding.References
- M. Dean, Y. Hamon and G. Chimini, The human ATP-binding cassette (ABC) transporter superfamily, J. Lipid Res., 2001, 42, 1007–1017 CrossRef CAS.
- V. Vasiliou, K. Vasiliou and D. W. Nebert, Human ATP-binding cassette (ABC) transporter family, Hum. Genomics, 2009, 3, 281 CrossRef CAS PubMed.
- K. P. Locher, Mechanistic diversity in ATP-binding cassette (ABC) transporters, Nat. Struct. Mol. Biol., 2016, 23, 487 CrossRef CAS PubMed.
- M. Horio, M. M. Gottesman and I. Pastan, ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3580–3584 CrossRef CAS PubMed.
- W. Busch and M. H. Saier, The transporter classification (TC) system, Crit. Rev. Biochem. Mol. Biol., 2002, 37, 287–337 CrossRef CAS PubMed.
- F. J. Sharom, The P-glycoprotein multidrug transporter, Essays Biochem., 2011, 50, 161–178 CrossRef CAS PubMed.
- I. B. Holland and M. A. Blight, ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans, J. Mol. Biol., 1999, 293, 381–399 CrossRef CAS PubMed.
- F. J. Sharom, Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function, Front. Oncol., 2014, 4, 1–19 Search PubMed.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Structure of P-Glycoprotein reveals a molecular basis for poly-specific drug binding, Science, 2009, 323, 1718–1722 CrossRef CAS PubMed.
- M. S. Jin, M. L. Oldham, Q. Zhang and J. Chen, Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans, Nature, 2012, 490, 566–569 CrossRef CAS PubMed.
- M. F. Rosenberg, R. Callaghan, S. Modok, C. F. Higgins and R. C. Ford, Three-dimensional Structure of P-glycoprotein: the transmembrane regions adopt an asymmetric configuration in the nucleotide-bound state, J. Biol. Chem., 2005, 280, 2857–2862 CrossRef CAS PubMed.
- J. Li, K. F. Jaimes and S. G. Aller, Refined structures of mouse P-glycoprotein, Protein Sci., 2014, 23, 34–36 CrossRef CAS PubMed.
- J. Neumann, D. Rose-Sperling and U. A. Hellmich, Diverse relations between ABC transporters and lipids: an overview, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 605–618 CrossRef CAS PubMed.
- P. Mitchell, A general theory of membrane transport from studies of bacteria, Nature, 1957, 180, 134–136 CrossRef CAS PubMed.
- O. Jardetzky, Simple allosteric model for membrane pumps, Nature, 1966, 211, 969–970 CrossRef CAS PubMed.
- T. Jiang, P.-C. Wen, N. Trebesch, Z. Zhao, S. Pant, K. Kapoor, M. Shekhar and E. Tajkhorshid, Computational Dissection of Membrane Transport at a Microscopic Level, Trends Biochem. Sci., 2020, 45, 202–216 CrossRef CAS PubMed.
- R. J. Dawson and K. P. Locher, Structure of a bacterial multidrug ABC transporter, Nature, 2006, 443, 180–185 CrossRef CAS PubMed.
- M. L. Oldham, A. L. Davidson and J. Chen, Structural insights into ABC transporter mechanism, Curr. Opin. Struct. Biol., 2008, 18, 726–733 CrossRef CAS PubMed.
- K. P. Locher, Structure and mechanism of ATP-binding cassette transporters, Philos. Trans. R. Soc. London, Ser. B, 2009, 364, 239–245 CrossRef CAS PubMed.
- M. Hohl, C. Briand, M. G. Grütter and M. A. Seeger, Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation, Nat. Struct. Mol. Biol., 2012, 19, 395–402 CrossRef CAS PubMed.
- R. J. Dawson and K. P. Locher, Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP, FEBS Lett., 2007, 581, 935–938 CrossRef CAS PubMed.
- A. Ward, C. L. Reyes, J. Yu, C. B. Roth and G. Chang, Flexibility in the ABC transporter MsbA: alternating access with a twist, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19005–19010 CrossRef CAS PubMed.
- B. Verhalen, R. Dastvan, S. Thangapandian, Y. Peskova, H. A. Koteiche, R. K. Nakamoto, E. Tajkhorshid and H. S. Mchaourab, Energy Transduction and Alternating Access of the Mammalian ABC Transporter P-glycoprotein, Nature, 2017, 543, 738–741 CrossRef CAS PubMed.
- S. V. Ambudkar, C. Kimchi-Sarfaty, Z. E. Sauna and M. M. Gottesman, P-glycoprotein: from genomics to mechanism, Oncogene, 2003, 22, 7468–7485 CrossRef CAS PubMed.
- Z. E. Sauna and S. V. Ambudkar, About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work, Mol. Cancer Ther., 2007, 6, 13–23 CrossRef CAS PubMed.
- S. Dehghani-Ghahnaviyeh, K. Kapoor and E. Tajkhorshid, Conformational changes in the nucleotide-binding domains of P-glycoprotein induced by ATP hydrolysis, FEBS Lett., 2021, 595, 735–749 CrossRef CAS PubMed.
- D. Szöllősi, D. Rose-Sperling, U. A. Hellmich and T. Stockner, Comparison of mechanistic transport cycle models of ABC exporters, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 818–832 CrossRef PubMed.
- Q. Qu and F. J. Sharom, FRET Analysis Indicates That the Two ATPase Active Sites of the P-Glycoprotein Multidrug Transporter Are Closely Associated, Biochemistry, 2001, 40, 1413–1422 CrossRef CAS PubMed.
- S. Shukla, B. Abel, E. E. Chufan and S. V. Ambudkar, Effects of a detergent micelle environment on P-glycoprotein (ABCB1)–ligand interactions, J. Biol. Chem., 2017, 292, 7066–7076 CrossRef CAS PubMed.
- M. J. Li, M. Guttman and W. M. Atkins, Conformational dynamics of P-glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale, J. Biol. Chem., 2018, 293, 6297–6307 CrossRef CAS PubMed.
- R. Dastvan, S. Mishra, Y. B. Peskova, R. K. Nakamoto and H. S. Mchaourab, Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors, Science, 2019, 364, 689–692 CrossRef CAS PubMed.
- K. Sigdel, L. Wilt, B. Marsh, A. Roberts and G. King, The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy, Biochem. Pharmacol., 2018, 156, 302–311 CrossRef CAS PubMed.
- M. E. Zoghbi, L. Mok, D. J. Swartz, A. Singh, G. A. Fendley, I. L. Urbatsch and G. A. Altenberg, Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis, J. Biol. Chem., 2017, 292, 20412–20424 CrossRef CAS PubMed.
- G. A. Frank, S. Shukla, P. Rao, M. J. Borgnia, A. Bartesaghi, A. Merk, A. Mobin, L. Esser, L. A. Earl and M. M. Gottesman, et al., Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle, Mol. Pharmacol., 2016, 90, 35–41 CrossRef CAS PubMed.
- A. Palmeira, M. H. Vasconcelos, A. Paiva, M. X. Fernandes, M. Pinto and E. Sousa, Dual inhibitors of P-glycoprotein and tumor cell growth: (re)discovering thioxanthones, Biochem. Pharmacol., 2012, 83, 57–68 CrossRef CAS PubMed.
- A. Alam, J. Kowal, E. Broude, I. Roninson and K. P. Locher, Structural insight into substrate and inhibitor discrimination by human P-glycoprotein, Science, 2019, 363, 753–756 CrossRef CAS PubMed.
- P.-C. Wen, B. Verhalen, S. Wilkens, H. S. Mchaourab and E. Tajkhorshid, On the origin of large flexibility of P-glycoprotein in the inward-facing state, J. Biol. Chem., 2013, 288, 19211–19220 CrossRef CAS PubMed.
- J. Zaitseva, S. Jenewein, T. Jumpertz, I. B. Holland and L. Schmitt, H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB, EMBO J., 2005, 24, 1901–1910 CrossRef CAS PubMed.
- K. Kapoor, S. Thangapandian and E. Tajkhorshid, Extended-ensemble docking to probe evolution of ligand binding sites in proteins undergoing large-scale structural changes for their function, bioRxiv, 2021 DOI:10.1101/2020.11.15.383794.
- M. Moradi and E. Tajkhorshid, Mechanistic picture for conformational transition of a membrane transporter at atomic resolution, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18916–18921 CrossRef CAS PubMed.
- M. Moradi and E. Tajkhorshid, Computational recipe for efficient description of large-scale conformational changes in biomolecular systems, J. Chem. Theory Comput., 2014, 10, 2866–2880 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. Radak, R. D. Skeel and A. Singharoy, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- C. G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid and J. C. Gumbart, Rapid parameterization of small molecules using the force field toolkit, J. Comput. Chem., 2013, 34, 2757–2770 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- C. Møller and M. S. Plesset, Note on an Approximation Treatment for Many-Electron Systems, Phys. Rev., 1934, 46, 618–622 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian and A. F. Izmaylov, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant pressure molecular dynamics algorithms, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- Y. Kim and J. Chen, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation, Science, 2018, 359, 915–919 CrossRef CAS PubMed.
- A. Sethi, J. Eargle, A. A. Black and Z. Luthey-Schulten, Dynamical networks in tRNA:protein complexes, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6620–6625 CrossRef CAS PubMed.
- A. L. Davidson, E. Dassa, C. Orelle and J. Chen, Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems, Microbiol. Mol. Biol. Rev., 2008, 72, 317–364 CrossRef CAS PubMed.
- A. Alam, R. Kung, J. Kowal, R. A. McLeod, N. Tremp, E. V. Broude, I. B. Roninson, H. Stahlberg and K. P. Locher, Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1973–1982 CrossRef PubMed.
- E. Barreto-Ojeda, V. Corradi, R.-X. Gu and D. P. Tieleman, Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein, J. Gen. Physiol., 2018, 150, 417–429 CrossRef CAS PubMed.
- S. Wilkens, Structure and mechanism of ABC transporters, F1000Research, 2015, 7, 14 Search PubMed.
- S. V. Ambudkar, I.-W. Kim, D. Xia and Z. E. Sauna, The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding, FEBS Lett., 2006, 580, 1049–1055 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Simultaneous Binding of Two Different Drugs in the Binding Pocket of the Human Multidrug Resistance P-glycoprotein, J. Biol. Chem., 2003, 278, 39706–39710 CrossRef CAS PubMed.
- M. K. Al-Shawi, M. K. Polar, H. Omote and R. A. Figler, Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein, J. Biol. Chem., 2003, 278, 52629–52640 CrossRef CAS PubMed.
- L. Esser, F. Zhou, K. M. Pluchino, J. Shiloach, J. Ma, W.-k. Tang, C. Gutierrez, A. Zhang, S. Shukla, J. P. Madigan, T. Zhou, P. D. Kwong, S. V. Ambudkar, M. M. Gottesman and D. Xia, Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity, J. Biol. Chem., 2017, 292, 446–461 CrossRef CAS PubMed.
- C. A. Bonito, M. J. U. Ferreira, R. Ferreira and D. J. dos Santos, Does human P-glycoprotein efflux involve transmembrane alpha helix breakage?, bioRxiv, 2019, 692988 Search PubMed.
- T. W. Loo and D. M. Clarke, Mapping the binding site of the inhibitor tariquidar that stabilizes the first transmembrane domain of P-glycoprotein, J. Biol. Chem., 2015, 290, 29389–29401 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06288j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |