DOI:
10.1039/D0SC06341J
(Edge Article)
Chem. Sci., 2021,
12, 2225-2230
A Giese reaction for electron-rich alkenes†‡
Received
18th November 2020
, Accepted 17th December 2020
First published on 17th December 2020
Abstract
A general method for the hydroalkylation of electron-rich terminal and non-terminal alkenes such as enol esters, alkenyl sulfides, enol ethers, silyl enol ethers, enamides and enecarbamates has been developed. The reactions are carried out at room temperature under air initiation in the presence of triethylborane acting as a chain transfer reagent and 4-tert-butylcatechol (TBC) as a source of hydrogen atom. The efficacy of the reaction is best explained by very favorable polar effects supporting the chain process and minimizing undesired polar reactions. The stereoselective hydroalkylation of chiral N-(alk-1-en-1-yl)oxazolidin-2-ones takes place with good to excellent diastereocontrol.
Introduction
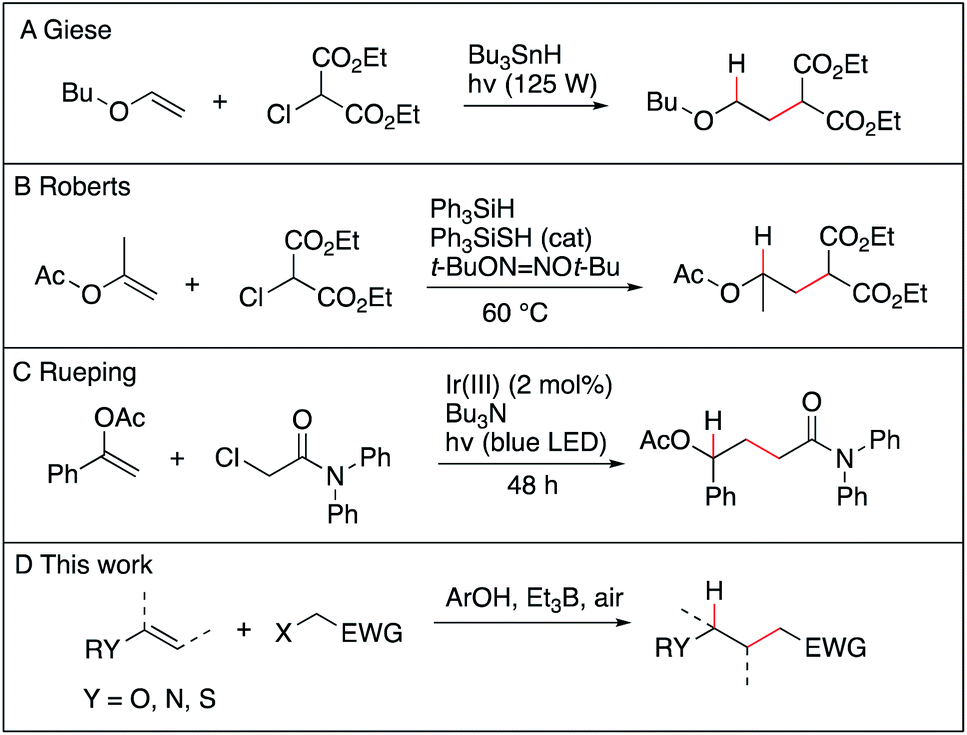
The anti-Markovnikov selective hydroalkylation of heteroatom-substituted electron-rich alkenes such as enol esters, enol ethers, thioenol ethers and enamides is an attractive process for the preparation of a variety of functionalized building blocks used for the synthesis of natural products and analogues. The well-established transition metal catalyzed hydroformylation reaction represents an effective approach to introduce one carbon atom1 and some promising results, such as the iridium catalyzed hydroalkylation of terminal alkenes with ureas,2 may emerge in the future. However, a general solution allowing to introduce a broad range of functionalized alkyl groups remains still greatly needed. Radical chemistry has been proved during the last 40 years to be one of the mildest method to achieve C–C bond formation.3–7 As for the hydroalkylation process, most of the reported methods described the addition of nucleophilic radicals to electron-poor olefins (the classical Giese reaction),8–12 the reversed process, i.e., addition of electrophilic radical to electron-rich olefins, remains scarce. The addition of diethyl chloromalonate to vinyl ethers and silyl enol ethers using tributyltin hydride as the hydrogen source was reported by Giese et al. (Scheme 1A),13 followed a few years later by Renaud et al. who reported the hydroalkylation of enamines with sulfinylated and sulfonylated carbon-centered radicals in the presence of tributyltin hydride.14–16 Examples of two-step procedures involving a xanthate group transfer reaction followed by a reduction step have been reported by Zard.17,18 Roberts et al. reported triphenylsilane-mediated hydroalkylation of enol esters with electrophilic radicals in the presence of a thiol catalyst (Scheme 1B).19 Recently, Ryu et al. reported the hydroalkylation of butyl vinyl ether with ethyl 2-bromoacetate using in situ generated HBr as a source of hydrogen atom.20 Rueping et al. reported recently photoredox-catalyzed hydroalkylation of styrene derivatives and related olefins with α-halo amides (Scheme 1C)21 that was later extended to cyclization of enamides.22 These methods, however, suffer from serious limitations, such as limited scope, competing direct reduction of the halide, toxicity of reagents such as tin hydride, use of expensive catalyst, and long reaction time. The hydroalkylation of enol ethers, vinyl sulfides, and enamides with Markovnikov regioselectivity has been reported recently by Baran and Shenvi using an elegant metal-hydride hydrogen atom transfer process.23–25 Developing a general, mild and environmentally friendly method for the hydroalkylation of electron-rich alkenes with anti-Markovnikov regioselectivity remains to date an unsolved problem.
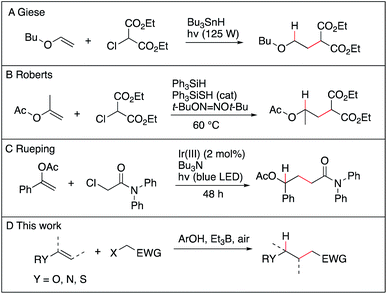 |
| | Scheme 1 Hydroalkylation of electron-rich alkenes with anti-Markovnikov regioselectivity. | |
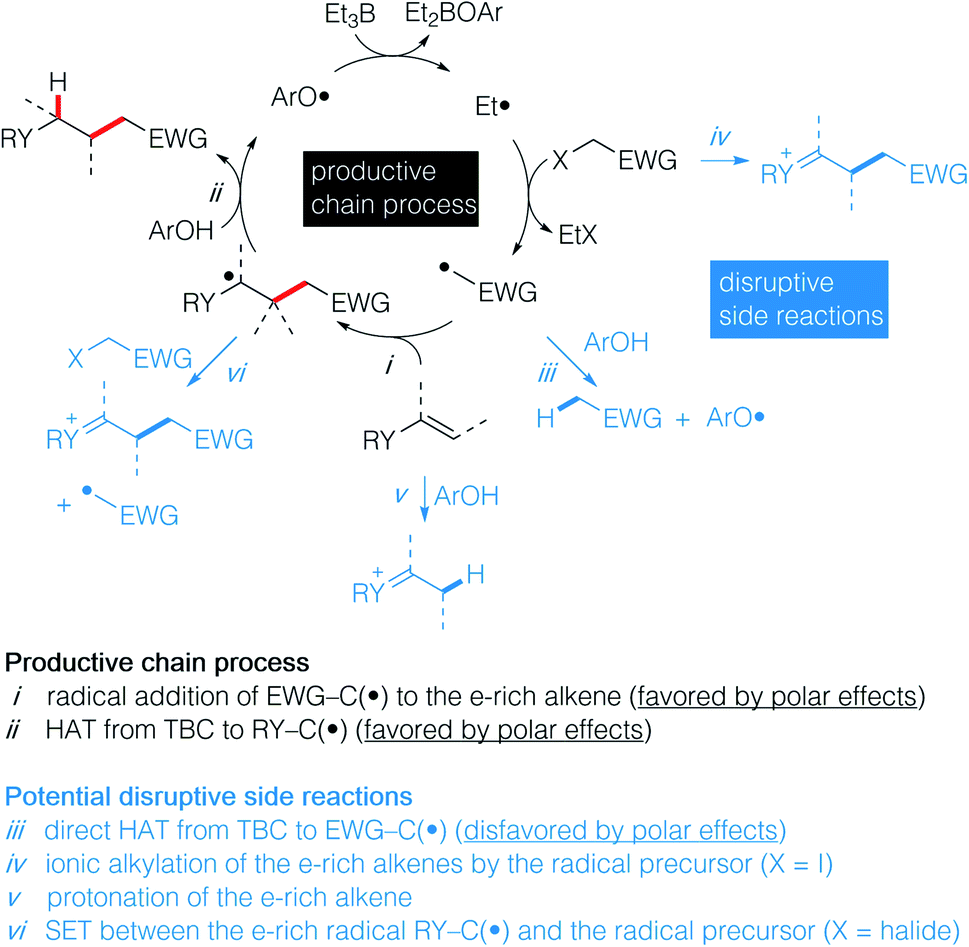
Recently, we have reported the hydroalkylation of mono- and polysubstituted unactivated alkenes with activated alkyl iodides by using 4-tert-butylcatechol (TBC) as the hydrogen source and triethylborane as an initiator and chain transfer reagent.26,27 The high efficiency of this reaction was attributed to strong polar effects, the catechol being a source of electrophilic hydrogen atoms, and to a unique repair mechanism, the system of catechol/Et3B being able to annihilate and repair undesired hydrogen atom transfer process involving the starting alkenes. Encouraged by these results, we decided to investigate the challenging hydroalkylation of electron-rich alkenes such as enol esters, enol ethers, enamides and related compounds. We described here a particularly general and simple approach to achieve this goal using TBC, a well-known biomimetic and non-toxic phenolic source of hydrogen atom (Scheme 1D). This reaction was expected to be strongly favored by polar effects since the electron-poor alkyl radicals add rapidly to the electron-rich alkenes (Scheme 2i). Moreover, the unique protic character of the OH group of TBC favors the fast reduction of the electron-rich radical adducts (Scheme 2ii) and disfavors the reduction of the initial electrophilic radicals (Scheme 2iii). Potential undesired chain disruptive side reactions such as ionic alkylations, protonation of the electron-rich alkenes (Scheme 2iv and v), and single electron transfer (SET) between the electron-rich radical adduct and the starting radical precursor (Scheme 2vi) do not take place under our reaction conditions.
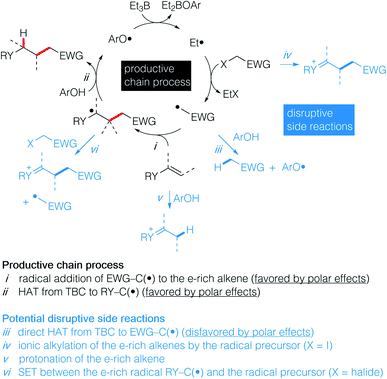 |
| | Scheme 2 Proposed strategy for a general approach to hydroalkylated electron-rich alkenes showing the productive chain process (in black) and potential disruptive side reactions (in blue). | |
Results and discussion
Hydroalkylation of enol esters and alkenyl sulfides
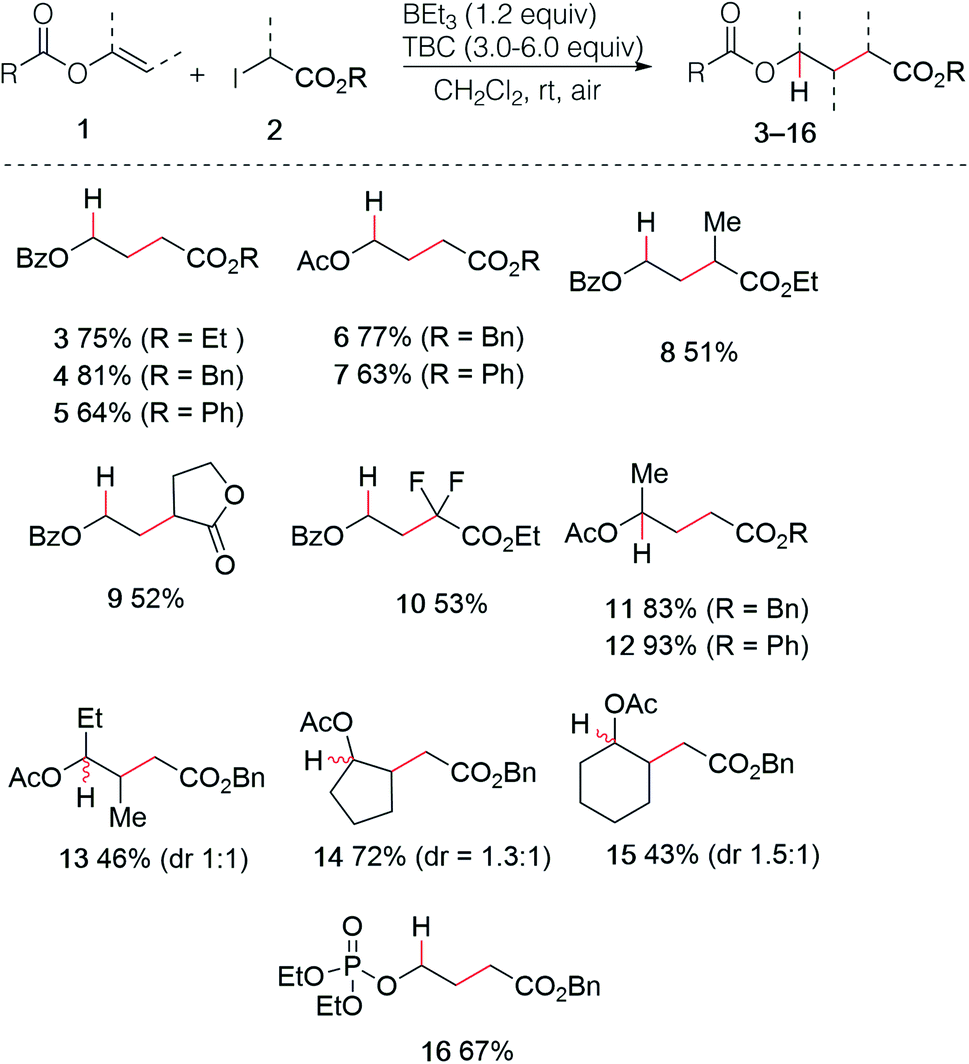
The use of enol esters was tested first. Simple mixing vinyl benzoate (5.0 equiv.), ethyl iodoacetate (1.0 equiv.), TBC (3.0 equiv.) and triethylborane (1 M in hexane, 1.2 equiv.) in dichloromethane under nitrogen atmosphere followed by stirring the reaction mixture open to air afforded the desired hydroalkylated product 3 in 75% yield (Scheme 3). Various di- and trisubstituted enol esters were tested using different electrophilic radical precursors. The method worked efficiently with terminal (3–12) as well as non-terminal enol esters (13–15) and can be also extended to the phosphate ester (16). A broad range of 2-iodoesters such as simple iodoacetates (3–7, 11–16), 2-iodopropionates (8), the iodolactone (9) and the difluoroiodoacetate (10) were all found to react cleanly under these reaction conditions.
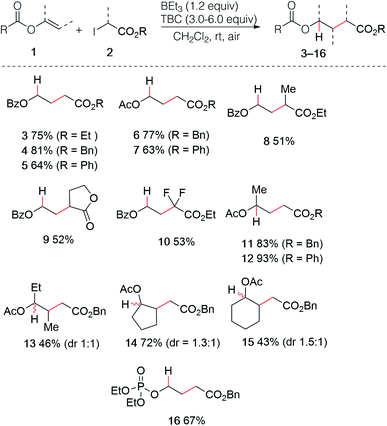 |
| | Scheme 3 Radical hydroalkylation of enol esters and vinyl phosphate. | |
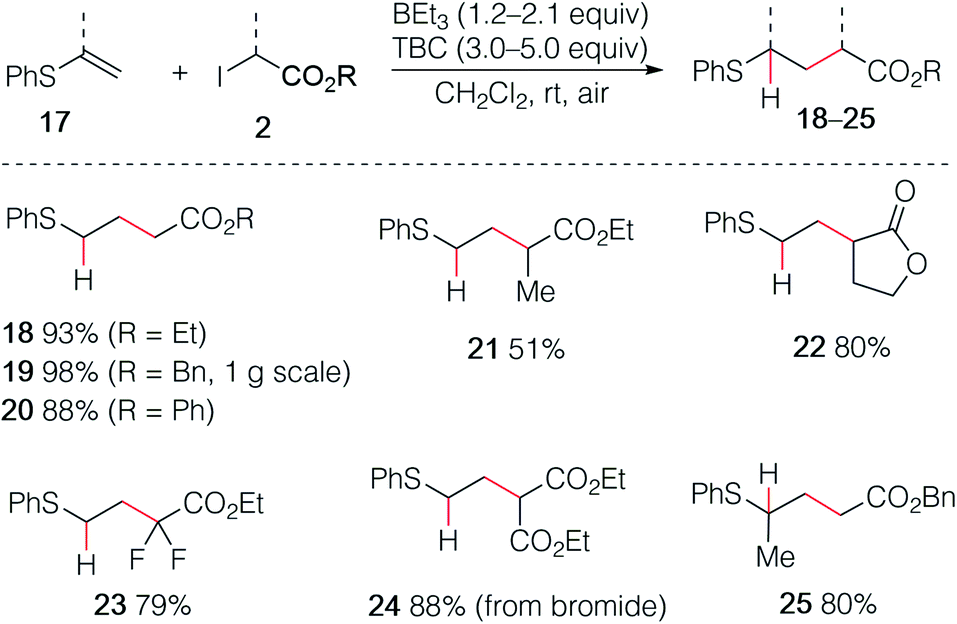
The reaction was then extended with success to alkenyl sulfides 17, affording the corresponding sulfides 18–25 in good to excellent yields (Scheme 4). The sulfide 19 was easily prepared by using this procedure on gram scale. Interestingly, the diethyl malonate derivative 24 was prepared in high yield using the corresponding bromomalonate radical precursor.
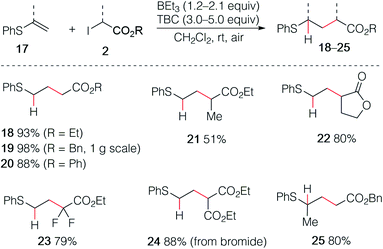 |
| | Scheme 4 Hydroalkylation of alkenyl sulfides. | |
Hydroalkylation of enol ethers
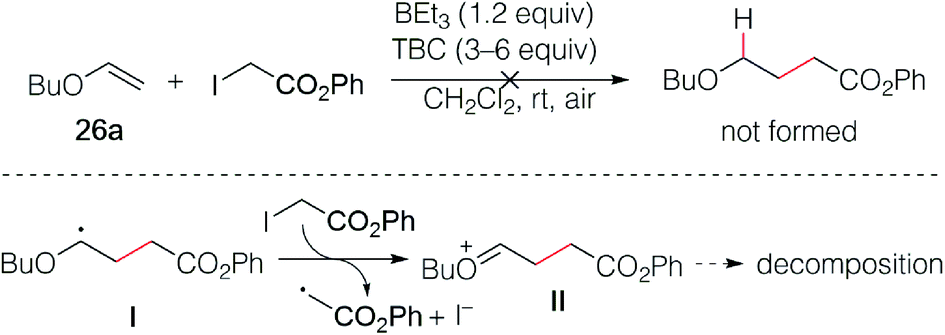
The reaction of butyl vinyl ether (26a) and phenyl iodoacetate was then attempted but led to decomposition products (Scheme 5). This was attributed to a fast electron transfer between radical adduct I and the starting iodoester 2c, leading to the formation of the oxonium ion II that decomposes presumably by oligomerization processes involving the starting vinyl ether. Similar reactions have been reported by Giese in his seminal work.13
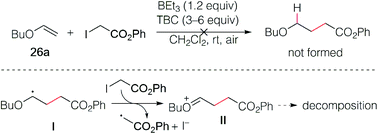 |
| | Scheme 5 Reaction of phenyl iodoacetate with butyl vinyl ether. | |
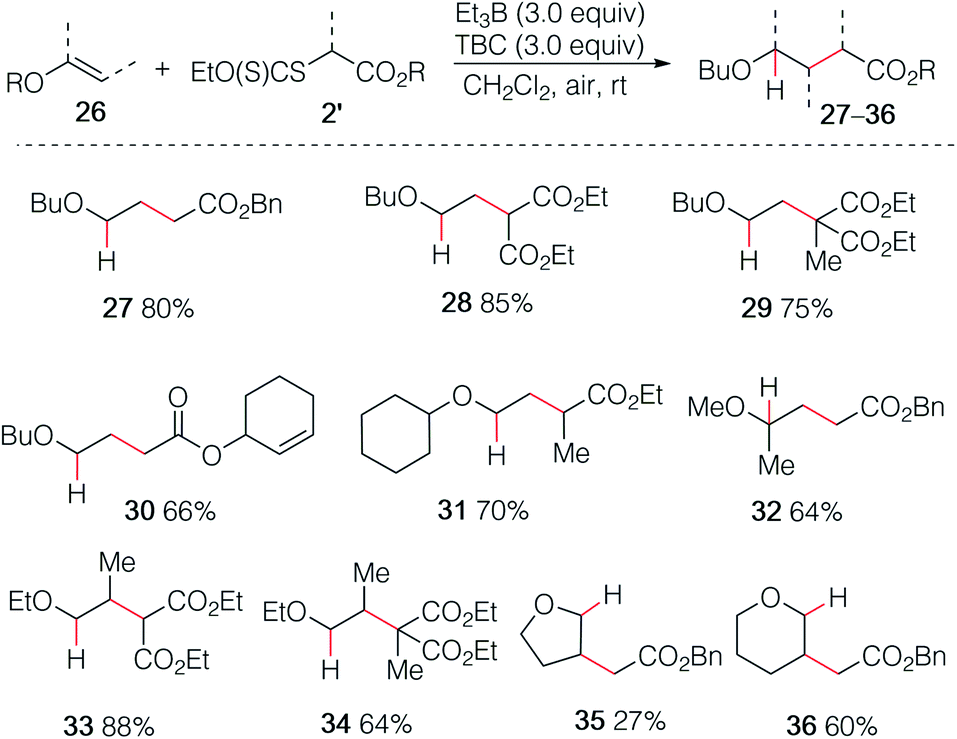
By employing xanthate radical precursors 2′ that are less prone to single electron transfer reduction than iodides,18,28–30 the hydroalkylation of enol ethers 26 could be successfully performed (Scheme 6). For instance, reaction of vinyl ethers with various xanthates afforded the hydroalkylated products 27–31 in 70–85% yield. Noteworthy, the reaction between the unsaturated cyclohex-2-en-1-yl acetate xanthate and butyl vinyl ethers 26a led to product 30 resulting from intermolecular addition in 66% yield, while no cyclized product was detected.31 Similar result was obtained for 32 starting from 2-methoxypropene. Interestingly, non-terminal 1-ethoxypropene also reacted efficiently to deliver the corresponding adducts 33 and 34 in 88% and 64% yield, respectively. Cyclic enol ethers such as 2,3-dihydrofuran and 3,4-dihydro-2H-pyran did not react cleanly at room temperature and better results were obtained by running the reaction at 0 °C (35 27%, 36 60%).
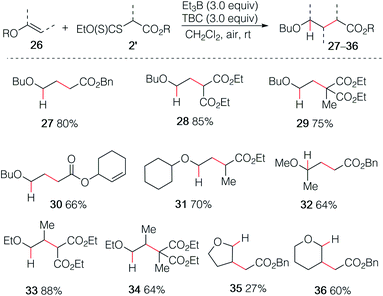 |
| | Scheme 6 Radical hydroalkylation of enol ethers with xanthates. | |
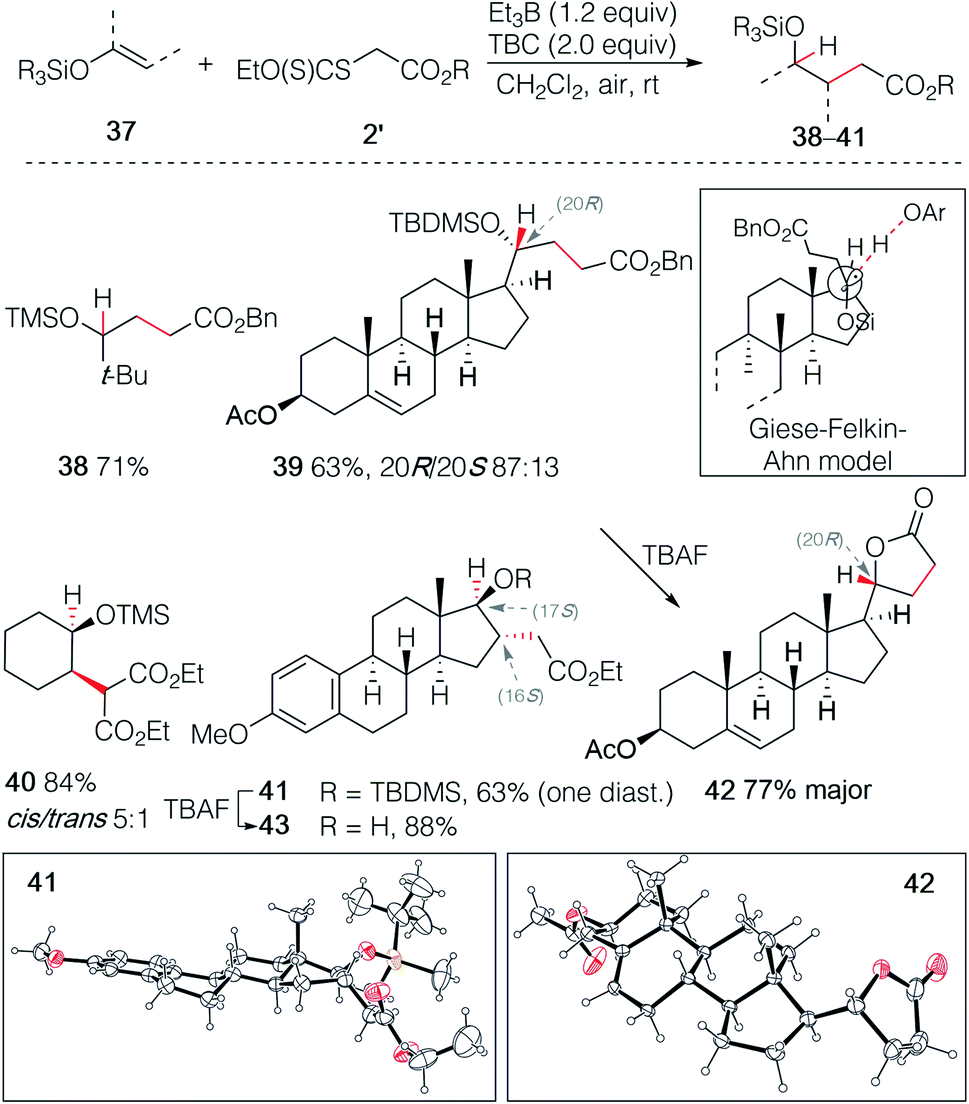
The hydroalkylation of terminal silyl enol ethers was examined next. Terminal silyl enol ethers derived from tert-butyl methyl ketone and pregnenolone acetate gave the desired γ-silyloxy esters 38 and 39 in 71% and 63% yield, respectively (Scheme 7). The silyl ether 39 was obtained with a good control of the stereochemistry (20R/20S 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13). The stereochemical outcome is rationalized by the Felkin–Ahn type model introduced by Giese for 1-alkoxysubstituted radicals.32 This example illustrates also the high regioselectivity of this hydroalkylation process. Indeed, the double bond in ring B of pregnenolone that can be hydroalkylated in 65% yield under similar reaction conditions26 remains untouched, demonstrating further the critical importance of polar effects in this reaction. Upon deprotection of the tert-butyldimethylsilyl (TBDMS) ether with TBAF, spontaneous lactonization affording 42 was observed. The major diastereomer of 42 was obtained in 77% yield and its (R) configuration at C(20) was confirmed by single crystal X-ray crystallography (Scheme 7).33–37 Similar results were obtained with the non-terminal silyl enol ethers derived from cyclohexanone and estrone methyl ether that gave the γ-silyloxy esters 40 and 41 in 84% (cis/trans mixture 5:1) and 59% (single diastereomer) yield, respectively. The relative configuration at C(16) and C(17) of 41 was established by single crystal X-ray crystallography (Scheme 7),33–37 indicating that both the stereochemical outcome of the radical addition and of the hydrogen atom transfer are controlled by the axial C(18)-methyl group. Deprotection of the silyl ether of 41 gave the stable trans-γ-hydroxy ester 43 and no lactonization could be achieved even under acidic treatment. Interestingly, the hydroalkylation of Me3SiO-cyclohexene reported by Baran and co-workers23,24 using an iron catalyzed process and by Shenvi using a dual manganese/nickel catalyzed process delivered adducts with the opposite regioselectivity.25
:13). The stereochemical outcome is rationalized by the Felkin–Ahn type model introduced by Giese for 1-alkoxysubstituted radicals.32 This example illustrates also the high regioselectivity of this hydroalkylation process. Indeed, the double bond in ring B of pregnenolone that can be hydroalkylated in 65% yield under similar reaction conditions26 remains untouched, demonstrating further the critical importance of polar effects in this reaction. Upon deprotection of the tert-butyldimethylsilyl (TBDMS) ether with TBAF, spontaneous lactonization affording 42 was observed. The major diastereomer of 42 was obtained in 77% yield and its (R) configuration at C(20) was confirmed by single crystal X-ray crystallography (Scheme 7).33–37 Similar results were obtained with the non-terminal silyl enol ethers derived from cyclohexanone and estrone methyl ether that gave the γ-silyloxy esters 40 and 41 in 84% (cis/trans mixture 5:1) and 59% (single diastereomer) yield, respectively. The relative configuration at C(16) and C(17) of 41 was established by single crystal X-ray crystallography (Scheme 7),33–37 indicating that both the stereochemical outcome of the radical addition and of the hydrogen atom transfer are controlled by the axial C(18)-methyl group. Deprotection of the silyl ether of 41 gave the stable trans-γ-hydroxy ester 43 and no lactonization could be achieved even under acidic treatment. Interestingly, the hydroalkylation of Me3SiO-cyclohexene reported by Baran and co-workers23,24 using an iron catalyzed process and by Shenvi using a dual manganese/nickel catalyzed process delivered adducts with the opposite regioselectivity.25
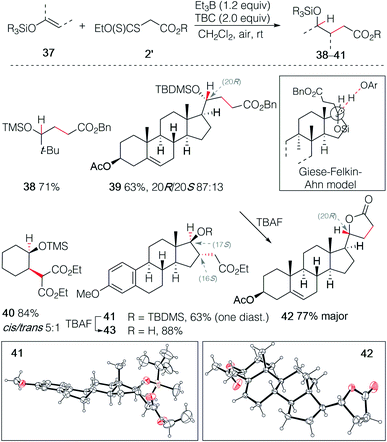 |
| | Scheme 7 Radical hydroalkylation of silyl enol ethers with xanthates. Single crystal X-ray structures of 42 and 43 (ellipsoids drawn at 50% probability). | |
Hydroalkylation of enamides and enecarbamates
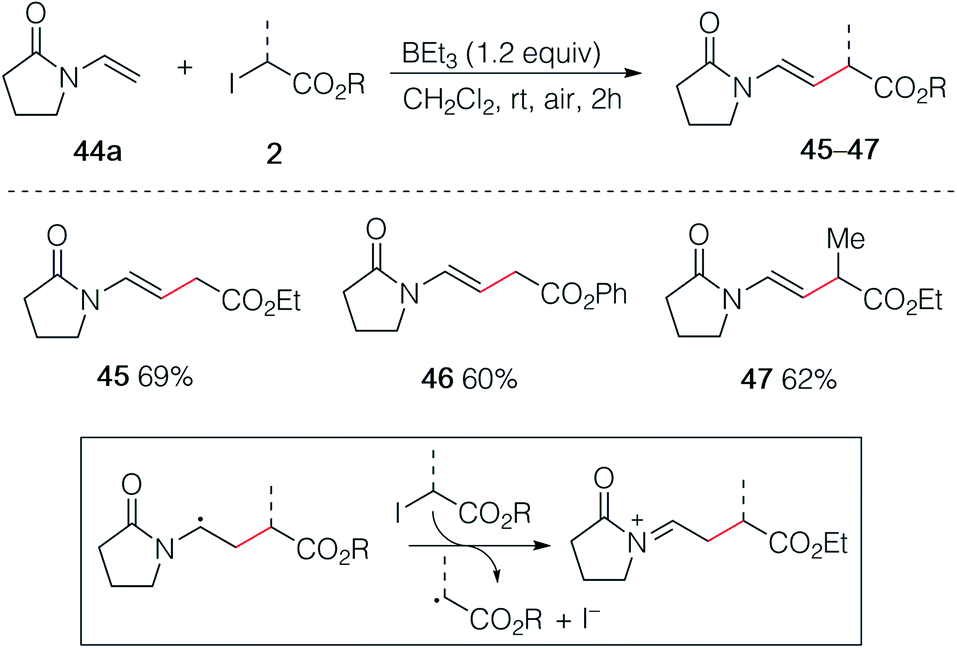
In an early attempt to run the hydroalkylation of 1-vinylpyrrolidin-2-one (44a) using ethyl iodoacetate, no trace of the hydroalkylated product was observed. Instead, the alkylated enamide 45 resulting from a non-reductive process was isolated. Rapid optimization of this process showed, as expected, that TBC was not necessary for this transformation and good yields of 45, 46 and 47 were obtained upon simple treatment of 44a with the correspond α-iodoesters in the presence of triethylborane which is presumably acting as a radical initiator in the presence of air and as a scavenger for HI generated during the process (Scheme 8). A similar non-reductive alkylation has already been reported by Friestad and Wu but required the use of a stoichiometric amount of tin hydride and a tertiary amine base.38 The reaction proceeds via formation of an acyliminium ion resulting most probably from a single electron transfer process between the radical adduct and the starting iodide 2 followed by elimination of a proton (Scheme 8, frame).39
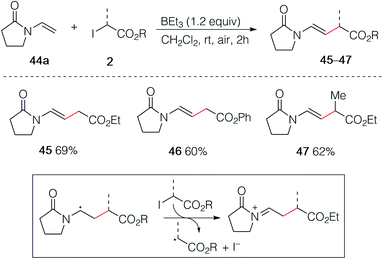 |
| | Scheme 8 Non-reductive alkylation of 1-vinylpyrrolidin-2-one 44a with iodoesters 2. | |
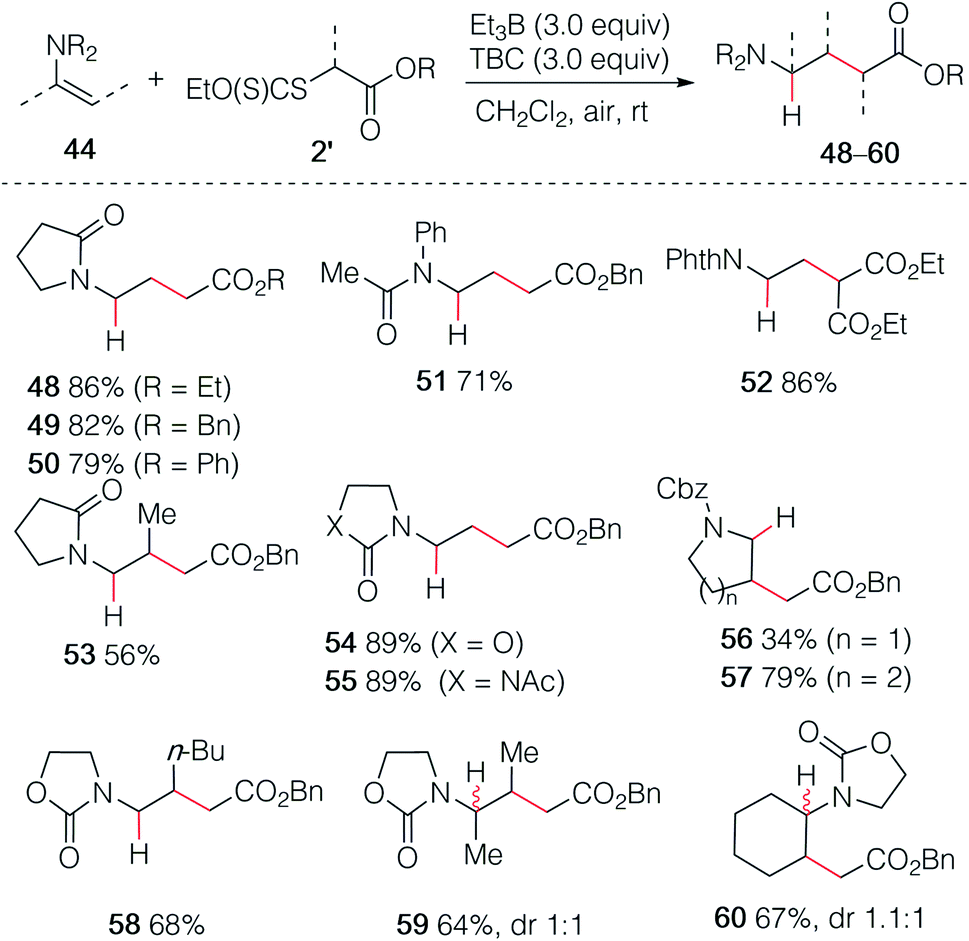
As already demonstrated for the enol ethers, the use of xanthate radical precursors 2′ allows to suppress the single electron transfer step and favor the hydroalkylation process.40–45 The hydroalkylation of enamides was examined first. Terminal enamides afforded the desired hydroalkylated products 48–52 in excellent yields (Scheme 9). Reaction involving a non-terminal enamide led to the hydroalkylated products 53 in satisfactory yields. Similar results were obtained with terminal (54 and 55) and non-terminal enecarbamates (56–60). These results diverges from the one obtained by Gillaizeau et al. who have obtained the product of non-reductive alkylation by performing the reaction between xanthates and enamides in the presence of dilauroyl peroxide acting as a radical initiator and oxidant,46 demonstrating the mildness and non-oxidative character of the triethylborane-involved initiation process.
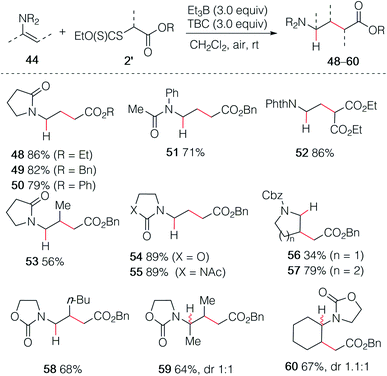 |
| | Scheme 9 Hydroalkylation of enamides and enecarbamates. | |
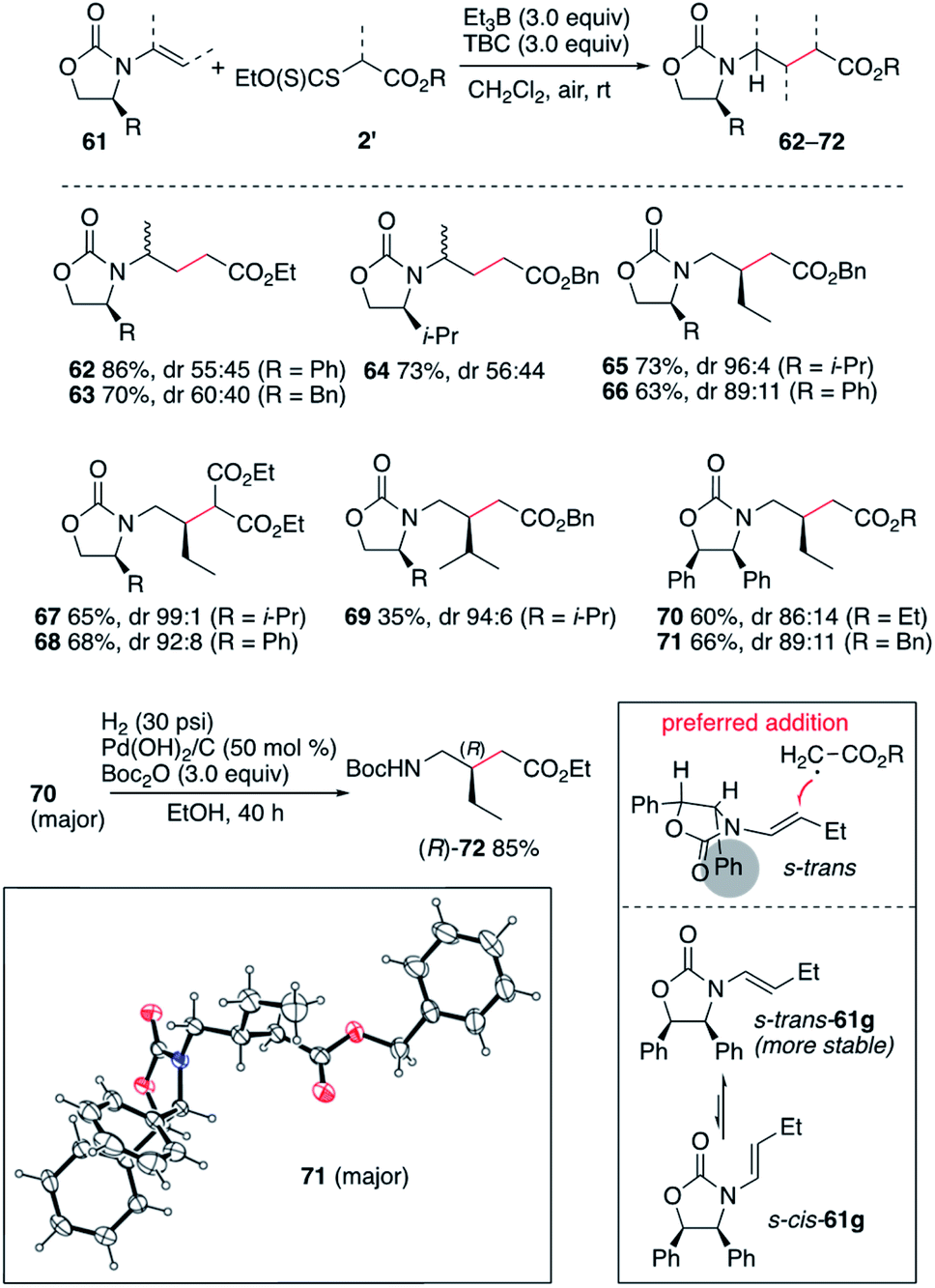
The efficient hydroalkylation of N-alkenyloxazolidinones reported in Scheme 9 offers the possibility of controlling the stereoselectivity of the process by using enecarbamates 61 derived from chiral oxazolidinones.47–51 Reactions involving the terminal 1-substituted N-prop-1-en-2-yloxazolidinones provided compounds 62–64 in high yield but poor stereocontrol. Fortunately, reactions involving the non-terminal enecarbamates proceeded with good to high diastereocontrol as illustrated by the formation of compounds 65–71. These results are in agreement with results obtained for the hydroamination of similar enecarbamates.48 The highest diastereoselectivity being observed for the 4-isopropyloxazolidin-2-ones leading to 65, 67, and 69 with dr ranging from 94:6 to 99:1. Reactions involving 4-phenyloxazoldin-2-one provided 66, 68, 70 and 71 with slightly lower diastereoselectivities varying from 86:14 to 92:8. The relative configuration of 71 was confirmed by single crystal X-ray crystallography of the major diastereomer (Scheme 10).33–37 The stereochemical outcome of the process can be rationalized by radical addition from the less hindered face (anti to the 4-phenyl substituent) of the enecarbamate lying in its most stable s-trans conformation as proposed by Studer and coworkers for the related hydroamination process.48,52,53 Interestingly, the diphenyloxazolidinone derivative 70 was easily converted to the corresponding enantiomerically pure protected γ-amino acid (R)-72 under mild hydrogenolysis conditions.54
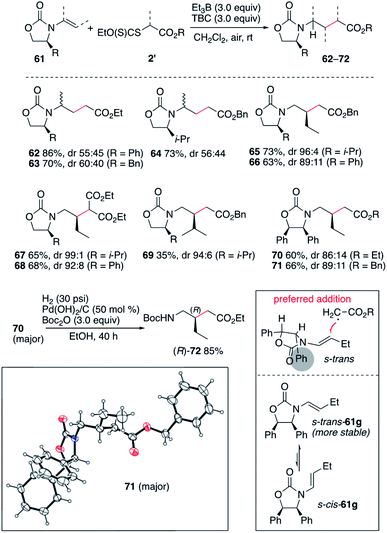 |
| | Scheme 10 Stereoselective radical hydroalkylation of chiral enecarbamates. Single crystal X-ray structure of 71 (ellipsoids drawn at 50% probability). | |
Conclusions
We have developed a general and operationally simple radical chain process for the hydroalkylation of electron-rich terminal and non-terminal alkenes with α-iodo- and α-xanthylesters. The reaction is initiated with triethylborane and air while the inexpensive and non-toxic TBC is used as a source of hydrogen atom. Highly diastereoselective hydroalkylation was also achieved by using chiral enecarbamates, providing access to chiral γ-amino acid derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Swiss National Science Foundation (Project 200020_172621) and the University of Bern are gratefully acknowledged for financial support. SRS was supported by the State Secretariat for Education and Innovation (SERI) via a Swiss Government Excellence Scholarships for Foreign Scholars and Artists. The X-ray crystal structure determination service unit of the Department of Chemistry and Biochemistry of the University of Bern (Prof. Piero Macchi, Dr Simon Grabowsky and Dr Michal Andrzejewski) is acknowledged for measuring, solving, refining and summarizing the structures of compounds 41, 42, and 71. The Synergy diffractometer was partially funded by the Swiss National Science Foundation within the R'Equip programme (project number 206021_177033).
Notes and references
-
A. Börner and R. Franke, Hydroformylation: fundamentals, processes, and applications in organic synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2016 Search PubMed.
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 7200–7204 CrossRef CAS.
-
B. Giese, Radicals in organic synthesis: formation of carbon-carbon bonds, Pergamon Press, Oxford, New York, 1st edn, 1986 Search PubMed.
- D. P. Curran, Synthesis, 1988, 1988, 417–439 CrossRef.
- D. P. Curran, Synthesis, 1988, 1988, 489–513 CrossRef.
-
P. Renaud, M. P. Sibi and Wiley InterScience (Online service), Radicals in organic synthesis, Wiley-VCH, Weinheim; New York, 2001 Search PubMed.
-
C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley, Chichester, 2012 Search PubMed.
- B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753–764 CrossRef.
- B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef.
- G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef CAS.
- T. Kawamoto and I. Ryu, Org. Biomol. Chem., 2014, 12, 9733–9742 RSC.
- J. Streuff and A. Gansäuer, Angew. Chem., Int. Ed., 2015, 54, 14232–14242 CrossRef CAS.
- B. Giese, H. Horler and M. Leising, Chem. Ber., 1986, 119, 444–452 CrossRef CAS.
- P. Renaud and S. Schubert, Angew. Chem., Int. Ed. Engl., 1990, 29, 433–434 CrossRef.
- S. Schubert, P. Renaud, P.-A. Carrupt and K. Schenk, Helv. Chim. Acta, 1993, 76, 2473–2489 CrossRef CAS.
- M. He, C. Qu, B. Ding, H. Chen, Y. Li, G. Qiu, X. Hu and X. Hong, Eur. J. Org. Chem., 2015, 2015, 3240–3250 CrossRef CAS.
- L. Anthore-Dalion, Q. Liu and S. Z. Zard, J. Am. Chem. Soc., 2016, 138, 8404–8407 CrossRef CAS.
- S. Z. Zard, Acc. Chem. Res., 2018, 51, 1722–1733 CrossRef CAS.
- H.-S. Dang, M. R. J. Elsegood, K.-M. Kim and B. P. Roberts, J. Chem. Soc., Perkin Trans. 1, 1999, 2061–2068 RSC.
- S. Sumino, A. Fusano and I. Ryu, Org. Lett., 2013, 15, 2826–2829 CrossRef CAS.
- M. Nakajima, Q. Lefebvre and M. Rueping, Chem. Commun., 2014, 50, 3619 RSC.
- E. Fava, M. Nakajima, M. B. Tabak and M. Rueping, Green Chem., 2016, 18, 4531–4535 RSC.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS.
- S. A. Green, T. R. Huffman, R. O. McCourt, V. van der Puyl and R. A. Shenvi, J. Am. Chem. Soc., 2019, 141, 7709–7714 CrossRef CAS.
- G. Povie, S. R. Suravarapu, M. P. Bircher, M. M. Mojzes, S. Rieder and P. Renaud, Sci. Adv., 2018, 4, eaat6031 CrossRef CAS.
- S. R. Suravarapu, B. Peter and P. Renaud, Sci. China: Chem., 2019, 62, 1504–1506 CrossRef CAS.
- S. Z. Zard, Angew. Chem., Int. Ed. Engl., 1997, 36, 672–685 CrossRef.
- B. Quiclet-Sire and S. Z. Zard, Pure Appl. Chem., 2010, 83, 519–551 Search PubMed.
- B. Quiclet-Sire and S. Z. Zard, Chem.–Eur. J., 2006, 12, 6002–6016 CrossRef CAS.
- F. Barbier, F. Pautrat, B. Quiclet-Sire and S. Z. Zard, Synlett, 2002, 2002, 811–813 CrossRef.
- W. Damm, J. Dickhaut, F. Wetterich and B. Giese, Tetrahedron Lett., 1993, 34, 431–434 CrossRef CAS.
- CCDC 2031380, 2031383, 2031378 contain the supplementary crystallographic data for compound 41, 42, and 71. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif.
-
CrysAlisPro (Version 1.171.40.37a), Oxford Diffraction Ltd, Yarnton, Oxfordshire, UK, 2018 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. K. Friestad and Y. Wu, Org. Lett., 2009, 11, 819–822 CrossRef CAS.
- T. Zhu, S. Xie, P. Rojsitthisak and J. Wu, Org. Biomol. Chem., 2020, 18, 1504–1521 RSC.
- F. Gagosz and S. Z. Zard, Org. Lett., 2003, 5, 2655–2657 CrossRef CAS.
- B. Quiclet-Sire and S. Z. Zard, Synlett, 2016, 27, 680–701 CrossRef CAS.
- S. Han and S. Z. Zard, Org. Lett., 2014, 16, 5386–5389 CrossRef CAS.
- B. Quiclet-Sire, G. Revol and S. Z. Zard, Tetrahedron, 2010, 66, 6656–6666 CrossRef CAS.
- B. Quiclet-Sire, G. Revol and S. Z. Zard, Org. Lett., 2009, 11, 3554–3557 CrossRef CAS.
- V. Liautard, F. Robert and Y. Landais, Org. Lett., 2011, 13, 2658–2661 CrossRef CAS.
- S. Bertho, I. Dondasse, P. Retailleau, C. Nicolas and I. Gillaizeau, New J. Chem., 2020, 44, 7129–7141 RSC.
- G. K. Friestad, Eur. J. Org. Chem., 2005, 2005, 3157–3172 CrossRef.
- J. Guin, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 779–782 CrossRef CAS.
- A. K. Mourad and C. Czekelius, Top. Heterocycl. Chem., 2020, 1–44 CAS.
- P. Renaud and S. Schubert, Synlett, 1990, 1990, 624–626 CrossRef.
- T. Courant, G. Dagousset and G. Masson, Synthesis, 2015, 47, 1799–1856 CrossRef CAS.
- O. Tamura, M. Hashimoto, Y. Kobayashi, T. Katoh, K. Nakatani, M. Kamada, I. Hayakawa, T. Akiba and S. Terashima, Tetrahedron Lett., 1992, 33, 3487–3490 CrossRef CAS.
- Z. Song, T. Lu, R. P. Hsung, Z. F. Al-Rashid, C. Ko and Y. Tang, Angew. Chem., Int. Ed., 2007, 46, 4069–4072 CrossRef CAS.
- C. Palomo, J. M. Aizpurua, M. Legido, A. Mielgo and R. Galarza, Chem.–Eur. J., 1997, 3, 1432–1441 CrossRef CAS.
Footnotes |
| † Dedicated with admiration and friendship to Professor Ilhyong Ryu for his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and full characterization of all new compounds including copies of 1H- and 13C-NMR spectra. CCDC 2031378, 2031380 and 2031383. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06341j |
| § Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*