
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater oxidation kinetics of nanoporous BiVO4 photoanodes functionalised with nickel/iron oxyhydroxide electrocatalysts†
Laia
Francàs‡§
 a,
Shababa
Selim‡
a,
Sacha
Corby
a,
Dongho
Lee
b,
Camilo A.
Mesa¶||
a,
Ernest
Pastor**
a,
Kyoung-Shin
Choi
b and
James R.
Durrant
*a
a,
Shababa
Selim‡
a,
Sacha
Corby
a,
Dongho
Lee
b,
Camilo A.
Mesa¶||
a,
Ernest
Pastor**
a,
Kyoung-Shin
Choi
b and
James R.
Durrant
*a
aDepartment of Chemistry, Centre for Processable Electronics, Imperial College London, White City Campus, London W12 0BZ, UK. E-mail: j.durrant@imperial.ac.uk
bDepartment of Chemistry, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA
First published on 15th April 2021
Abstract
In this work, spectroelectrochemical techniques are employed to analyse the catalytic water oxidation performance of a series of three nickel/iron oxyhydroxide electrocatalysts deposited on FTO and BiVO4, at neutral pH. Similar electrochemical water oxidation performance is observed for each of the FeOOH, Ni(Fe)OOH and FeOOHNiOOH electrocatalysts studied, which is found to result from a balance between degree of charge accumulation and rate of water oxidation. Once added onto BiVO4 photoanodes, a large enhancement in the water oxidation photoelectrochemical performance is observed in comparison to the un-modified BiVO4. To understand the origin of this enhancement, the films were evaluated through time-resolved optical spectroscopic techniques, allowing comparisons between electrochemical and photoelectrochemical water oxidation. For all three catalysts, fast hole transfer from BiVO4 to the catalyst is observed in the transient absorption data. Using operando photoinduced absorption measurements, we find that water oxidation is driven by oxidised states within the catalyst layer, following hole transfer from BiVO4. This charge transfer is correlated with a suppression of recombination losses which result in remarkably enhanced water oxidation performance relative to un-modified BiVO4. Moreover, despite similar electrocatalytic behaviour of all three electrocatalysts, we show that variations in water oxidation performance observed among the BiVO4/MOOH photoanodes stem from differences in photoelectrochemical and electrochemical charge accumulation in the catalyst layers. Under illumination, the amount of accumulated charge in the catalyst is driven by the injection of photogenerated holes from BiVO4, which is further affected by the recombination loss at the BiVO4/MOOH interface, and thus leads to deviations from their behaviour as standalone electrocatalysts.
Introduction
Artificial photosynthesis employing photoelectrochemical or photocatalytic systems is attracting increasing interest for the sustainable synthesis of molecular fuels and chemicals.1–4 Metal oxides are widely studied as light absorbing materials for such solar-to-fuel systems, particularly for the challenging water oxidation reaction.5,6 This interest is derived particularly from their stability under the highly oxidising conditions required to drive this reaction. However, the performance of such metal oxide-based photoelectrodes is generally limited by recombination losses; in particular, competing surface recombination losses (also known as back electron/hole recombination) due to the slow rate of water oxidation, can be very significant.7–12 One widely used approach to reduce such losses is the addition of water oxidation catalyst layers on the photoanode surface, which can lead to an enhancement in performance.12–16 However, the selection of suitable catalysts is challenging as the requirements for efficient photoanode performance is likely to be different from the requirements for efficient, dark electrocatalysis. This is because, unlike electrocatalytic systems, driven by a voltage source, photoanodes utilise photogenerated holes, which are subject to electron–hole recombination losses at the photoanode/electrocatalyst/electrolyte interfaces, substantially affecting water oxidation currents.11,13,17–19 Thus, even for a well-understood water oxidation catalyst used as an electrocatalyst, understanding the catalyst–photoanode interaction is critical to enhance photocurrent yields in metal oxide/electrocatalyst photoanodes.Several earth abundant and inexpensive electrocatalysts have garnered extensive attention for water oxidation, such as cobalt based CoPi,20 and Co–Fe Prussian Blue.14 Transition metal oxyhydroxides (referred to herein as MOOH) like NiOOH and FeOOH have also been coupled with BiVO4 and some of these combinations have resulted in the construction of state-of-the-art BiVO4 photoanodes,13,15,21 exhibiting remarkable photocurrent densities of 5.87 mA cm−2 at 1.23 VRHE.15 However, while the presence of MOOH considerably enhances the water oxidation performance of BiVO4, most notably for nanoporous electrodes, the origin of this enhancement has not been clearly elucidated to date. Crucially, it has also been observed that the performance enhancement of the BiVO4/MOOH system using either FeOOH or NiOOH for water oxidation does not correlate with the performance of FeOOH or NiOOH, respectively, when used as electrocatalysts on FTO.13 This discrepancy complicates the optimisation of photoelectrochemical devices and highlights the need to improve our understanding of photoanode/electrocatalyst interface.13
In this study, transient optical and spectroelectrochemical techniques are utilised to: (1) compare performances of FeOOH, NiOOH, FeOOHNiOOH in terms of when they are used as an electrocatalyst on a conducting substrate in neutral electrolyte; and (2) investigate how their performance changes when they are placed on BiVO4 nanoporous photoanodes and need to utilise photogenerated holes for water oxidation under the same electrolyte conditions. We employ a population model to study the kinetics and the charge accumulation of our systems as it offers a direct pathway to compare electrocatalysts and photoelectrodes, and highlights the role of charge accumulation in driving reaction kinetics.22,23 This model has previously been employed to study water oxidation,24–28 proton reduction,29 and organic oxidations30 across electrocatalysts and photoelectrodes. The results obtained from this study provide insight into the role of MOOH on BiVO4 in enhancing water oxidation photocurrent and in the performance differences of the three MOOH catalysts on a conducting substrate and on a BiVO4 photoelectrode.
Results and discussion
Nanoporous BiVO4 photoanodes used in this study were prepared using previously reported methods with one modification (see ESI† for details),13 which was decreasing the film thickness (from ∼750 to ∼450–500 nm). This was necessary to increase the transparency of the BiVO4 photoanodes, which is critical for the spectroscopic methods used in this study. As a result, the photocurrent obtained in this study with thinner BiVO4 photoanodes is lower than that previously reported.13 However, this decrease did not affect our comparison of the interactions of BiVO4 with various MOOH electrocatalysts.The FeOOH, NiOOH, FeOOHNiOOH electrocatalysts were synthesised as previously reported by anodic deposition on FTO to examine electrocatalytic function27 and on BiVO4 to examine photoelectrocatalytic function.13 The trend in the photoelectrochemical enhancements between the three electrocatalysts upon deposition on nanoporous BiVO4 has previously been reported.13 The thickness of the thinnest possible NiOOH layer that could be reproducibly deposited on FTO with a uniform coverage was ∼80 nm. Therefore, the same thickness was used to prepare FeOOH on FTO. The thickness of FeOOHNiOOH, which was prepared by sequential deposition of FeOOH and NiOOH layers, was not ∼160 nm, but ∼120 nm.27 This is because the underlying FeOOH is not dense but porous, allowing for the facile infiltration of NiOOH into the FeOOH layer and resulting in an atomic-level mixing of FeOOH and NiOOH.27 The thickness of the catalyst layer on BiVO4 was <5 nm for FeOOH or NiOOH and <10 nm for FeOOHNiOOH. These thicknesses were optimised to maximise photocurrent generation of BiVO4 prepared as thin photoelectrodes suitable for transient absorption spectroscopy studies. (The use of a catalyst layer that is thicker than the optimal thickness on a photoelectrode results in an increase in electron–hole recombination and is not favourable for photocurrent generation.13) We note that it would have been more ideal if the catalyst layers of the same thickness could be used on FTO and BiVO4. However, the thickness of the catalysts on FTO cannot be further reduced, and using thick catalyst layers on BiVO4 for the sake of using the same catalyst thickness makes it difficult to study optimal BiVO4–catalyst interactions. Thus, we chose to use catalysts with an optimised thickness on BiVO4 to obtain a more meaningful and useful understanding of the system. We confirmed using electrochemical impedance spectroscopy that the catalysts on FTO are still thin enough not to crease a serious resistivity issue (see ESI† for details). We note that NiOOH in this study was deposited, as previously, in an electrolyte that was not rigorously free of Fe impurities, and the NiOOH contained a trace amount of Fe (∼1 at%).27 Thus, hereafter, NiOOH will be denoted as Ni(Fe)OOH.
All investigations presented herein were performed at pH 7 as the current work aims to better understand the high performance of BiVO4/MOOH photoanodes reported previously at this pH.13 While MOOH electrocatalysts are typically used in basic conditions, MOOH-coupled photoanodes that are not stable in strongly basic conditions, such as BiVO4, are often used in (near) neutral conditions to optimise system stability.
At pH 7, the three MOOH electrocatalysts studied herein exhibit similar electrochemical water oxidation performances, as can be seen in the steady-state current voltage (J–V) plots shown in Fig. 1a. The similar overall performance at pH 7 differs from that observed under alkaline media (pH 13), where the catalytic onset potential for FeOOH is anodically shifted by 70 mV compared with Ni(Fe)OOH and FeOOHNiOOH.27 All the presented studies for NiOOH and FeOOHNiOOH and their photoanode counterparts were obtained after the in situ regeneration of NiOOH from Ni(OH)2, using an activation procedure detailed previously,27 as Ni(OH)2 forms over time when the electrodes are not in use (see ESI† for details). This activation process is referred to herein as the (0/+) oxidation process.
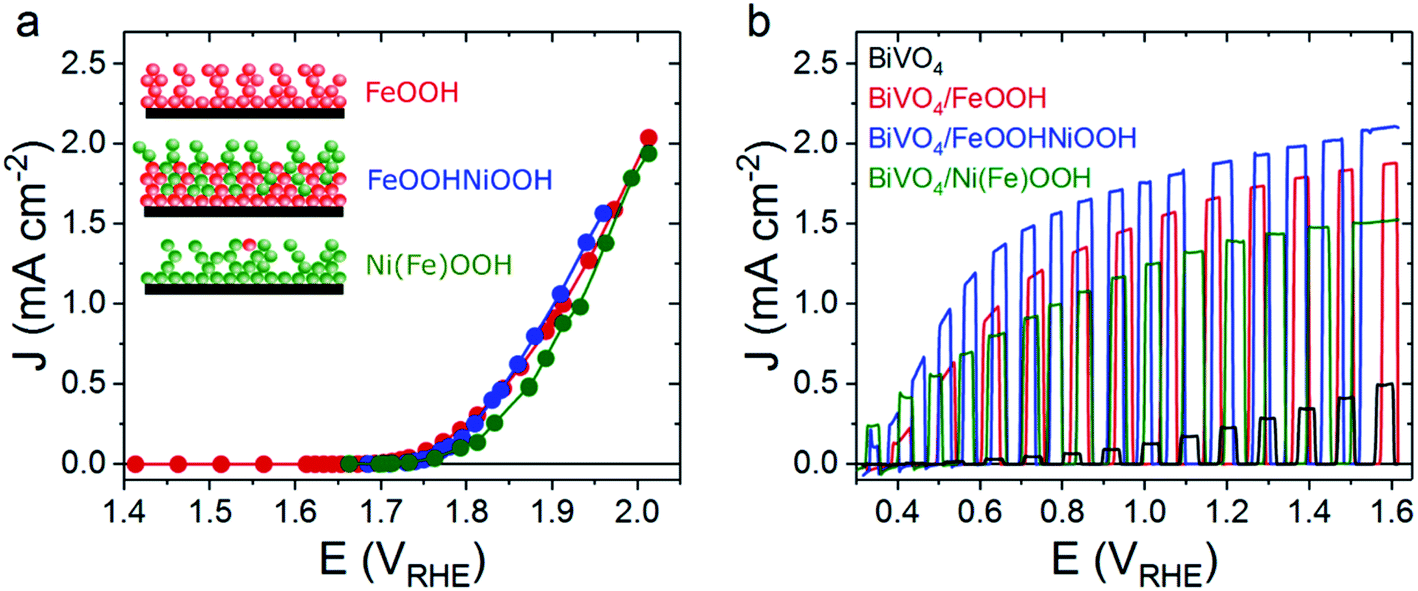 | ||
| Fig. 1 Electrochemical and photoelectrochemical performance of FeOOH, FeOOHNiOOH and Ni(Fe)OOH as electrocatalyst and when functionalised on BiVO4 photoanodes at pH 7. (a) Steady state J–V curves for FeOOH (red), FeOOHNiOOH (blue) and Ni(Fe)OOH (green); these were recorded by measuring the steady state current at different applied potential under atmospheric conditions. Inset: schematic representation of the different electrocatalyst samples studied in this work. Red represents a Fe-based group, and green represents a Ni-based group. (b) Linear Sweep Voltammetry (LSV) under chopped light (1 sun equivalent, 365 nm LED, back irradiation) for BiVO4/FeOOH (red), BiVO4/FeOOHNiOOH (blue), BiVO4/Ni(Fe)OOH (green) and un-modified BiVO4 (black). All measurements were conducted in 0.1 M phosphate buffer. The J–V curves were recorded using a 10 mV s−1 scan rate. | ||
When these catalysts are deposited on top of BiVO4 photoanodes remarkable enhancements in the photoelectrochemical water oxidation performance are observed in comparison to the unmodified nanoporous BiVO4 photoanode (Fig. 1b). To understand the origin of this performance enhancement upon MOOH addition, transient optical and spectroelectrochemical techniques were employed to compare the water oxidation and recombination kinetics of the Ni/Fe oxyhydroxide water oxidation electrocatalysts when used either as electrocatalyst or when deposited on BiVO4 photoanodes.
Firstly, the optical and kinetic characteristics of the electrocatalysts when deposited on FTO were analysed. The optical changes during the sample activation (0/+) oxidation process to convert Ni(OH)2 to NiOOH for both Ni(Fe)OOH and FeOOHNiOOH electrocatalyst samples are shown in Fig. S2.†27,31–33 It is observed that Ni(Fe)OOH accumulated more (0/+) compared with FeOOHNiOOH. Since Ni(Fe)OOH and FeOOHNiOOH both contain similar amounts of NiOOH,27 this result suggests that as the Fe content increases, the oxidation of Ni(OH)2 to NiOOH becomes more difficult. This is consistent with previous literature where the shift in the oxidation of Ni(OH)2/NiOOH to more anodic potentials has been correlated with the increase in Fe content.32,34
Fig. 2a shows the optical response of the electrocatalysts. At applied potentials greater than the water oxidation catalytic onset, growth of new spectral features can be observed for each of the electrocatalysts, as shown in Fig. 2a (and Fig. S3† for spectra as a function of applied potential). These spectral features are assigned to the generation of oxidised states that accumulate under water oxidation catalysis, referred to as MOOH(++) herein. As can be observed in Fig. 2a (blue and red traces), the spectra of the FeOOH(++) and the FeOOHNiOOH(++) species are almost indistinguishable, with an absorption maximum between 400–425 nm, suggestive of the similar nature of these oxidised states. In contrast, the Ni(Fe)OOH(++) species shown in Fig. 2a (green trace) exhibit a peak centred at ∼650 nm, indicative of a different nature of the oxidised state that forms in Ni(Fe)OOH. A similar trend for the respective spectral features of the MOOH(+) and MOOH(++) species of these three electrocatalysts, is also present at pH 13,27 suggesting that the catalytically active species observed under neutral and alkaline conditions are similar in nature. It is worth noting that the Ni(Fe)OOH samples were found to be relatively unstable when potentials greater than 2 VRHE were applied, leading to a loss in the optical signal and performance. This effect can be associated with the gradual dissolution of NiOOH at pH 7 under anodic bias required for water oxidation (see ESI for more details, Fig. S5 and S6†).35
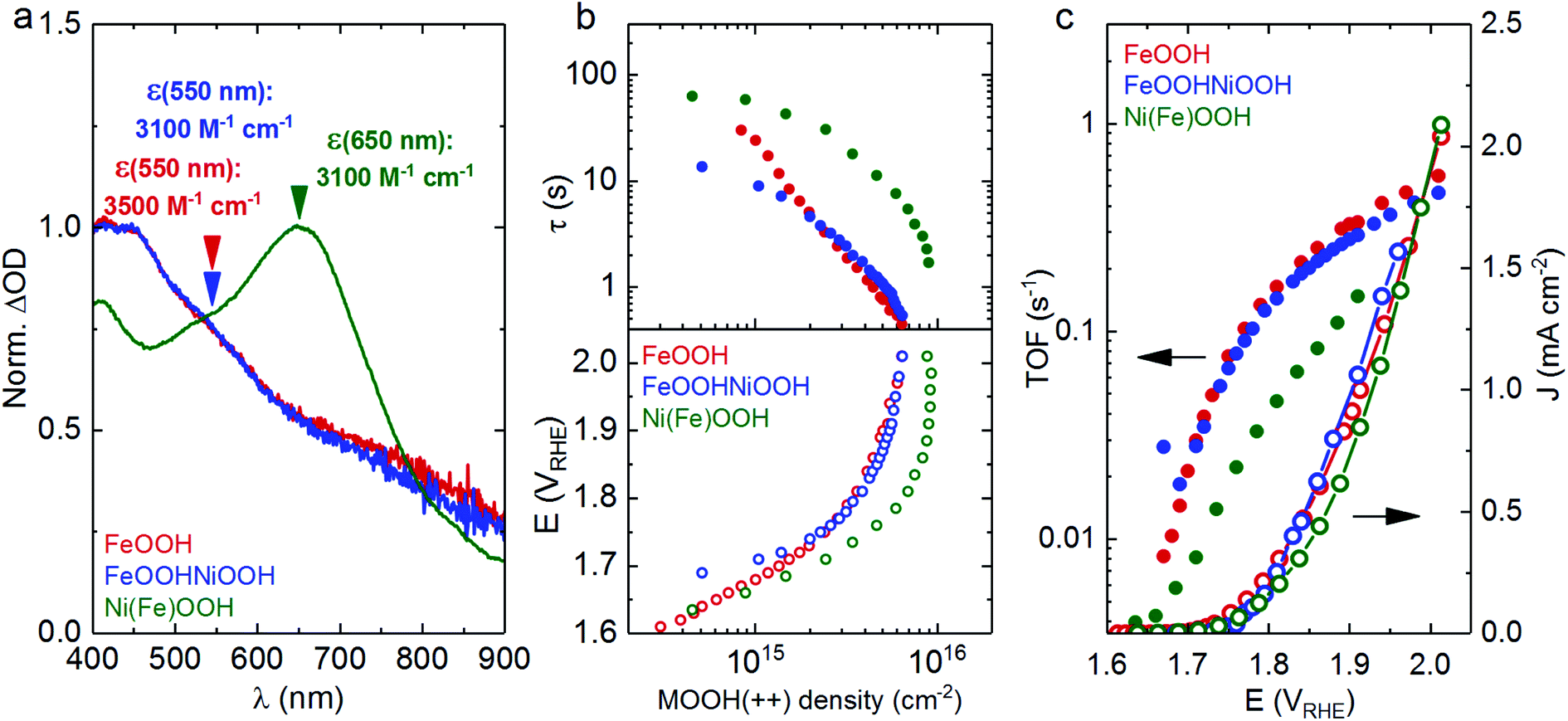 | ||
| Fig. 2 Kinetics of water oxidation under electrochemical conditions by the FeOOH, FeOOHNiOOH and Ni(Fe)OOH electrocatalysts. (a) Normalised ΔOD spectra of the MOOH(++) species and their corresponding extinction coefficient for FeOOH (red), FeOOHNiOOH (blue) and Ni(Fe)OOH (green). The spectra are obtained by subtracting the spectra after activation from the spectrum at 1.81 VRHE. (b) Water oxidation reaction time constants (τ, solid circles, top panel) and the applied potential (bottom panel) as a function of the accumulated MOOH(++) density. (c) The TOF for O2 evolution (solid circles, left axis) against the applied potential, plotted together with the current density (open circles, right axis). All measurements were taken in 0.1 M phosphate buffer (pH 7). Same colours used in (a), (b) and (c). | ||
To allow quantitative comparison of the oxidation of these electrocatalysts and the associated water oxidation kinetics, the extinction coefficients (ε) of the MOOH(++) oxidised species for each of the catalysts shown in Fig. 2a were estimated using step-potential spectroelectrochemistry (SP-SEC), (see Section D in ESI† for description of the method and additional data). Extinction coefficient (ε) values of ∼3500 and ∼3100 M−1 cm−1 were obtained at wavelengths of 550 nm for FeOOH and FeOOHNiOOH, and a value of ∼3100 M−1 cm−1 was obtained at 650 nm for Ni(Fe)OOH. Using the estimated values of ε for each of the samples, the optical signal (ΔOD) shown in Fig. S3† can be correlated with the density of accumulated MOOH(++) species as a function of applied potential (Fig. S4†). When combined with current densities, these data yield kinetic information on the water oxidation process, allowing the estimation of reaction time constant (τ) or turn over frequency (TOF) for O2 evolution, as a function of MOOH(++) species. These analyses have been described in Section G in the ESI† and have been reported previously.27 The water oxidation kinetic data are plotted as a function of MOOH(++) accumulated species (Fig. 2b) and also as a function of applied potential (Fig. 2c).
As can be observed in Fig. 2b (empty circles, bottom panel), the density of the MOOH(++) accumulated species increases with the applied potential for each of the electrocatalysts, resulting in an acceleration of the water oxidation τ by two orders of magnitude (solid circles, top panel). For a given potential, it is also observed that Ni(Fe)OOH accumulates more MOOH(++) species than FeOOH and FeOOHNiOOH. Despite the difference in thickness between the FeOOH and FeOOHNiOOH electrodes, both electrocatalyst electrodes display negligible differences in charge accumulation densities during catalysis. Whilst it is apparent that both FeOOH and FeOOHNiOOH exhibit similar τ, Ni(Fe)OOH(++) states oxidise water circa an order of magnitude more slowly, when compared at equivalent densities of MOOH(++) species. The slower water oxidation kinetics of Ni(Fe)OOH(++) is also reflected in the plot of TOF (s−1) vs. applied potential (Fig. 2c) which shows that at a given potential, FeOOH(++) and FeOOHNiOOH(++) both oxidise water at a similar rate, whereas the TOF (s−1) for Ni(Fe)OOH(++) is approximately halved. Overall, while Ni(Fe)OOH(++) is able to accumulate a greater density of MOOH(++) species than FeOOH and FeOOHNiOOH, this gain is offset by slower water oxidation kinetics. As a result, the electrocatalytic performance for water oxidation is relatively invariant for all three electrocatalyst as shown in Fig. 1a and 2c. These trends are markedly different to those observed at pH 13 where Ni(Fe)OOH shows the fastest water oxidation kinetics amongst the three and, in comparison to FeOOHNiOOH, FeOOH starts accumulating charge at more anodic potentials, consistent with a later catalytic onset.27 However the origin of these differences with pH is beyond the scope of this study.
Next we look at the interaction between these electrocatalyst and BiVO4 photoanodes. The poor performance of the un-modified nanoporous BiVO4 has previously been assigned to holes accumulating at the BiVO4 surface undergoing extensive losses due to water oxidation kinetics on BiVO4 being slower than surface recombination.13 However it was not clear if the performance enhancement observed after addition of MOOH results primarily from faster water oxidation kinetics or slower surface recombination kinetics.
The enhancement observed in Fig. 1b is consistent with that reported previously for these BiVO4/MOOH combinations13,15 and is much greater than that typically found when electrocatalysts (e.g. CoPi) are deposited on dense planar BiVO4 electrodes;11,36 such comparisons between CoPi and MOOH electrocatalysts are discussed later. Interestingly, while the three MOOH electrocatalysts show comparable performances, the BiVO4/MOOH photoelectrodes exhibit differences in their water oxidation photocurrents (Fig. 1b). In order to unravel the origin of the differences in performance enhancement upon the addition of MOOH electrocatalysts to BiVO4, two different time-resolved optical and photoelectrochemical techniques are employed: transient absorption spectroscopy (TAS) employing short laser pulse excitation, and spectroelectrochemical photo-induced absorption spectroscopy (PIAS), employing quasi-steady state LED excitation.
To investigate if charge transfer occurs following bandgap excitation of BiVO4, TAS was employed under an applied potential of 1.4 VRHE. Fig. 3a and b presents the TA spectra for BiVO4 and BiVO4/Ni(Fe)OOH and a comparison of the normalised charge carrier kinetics between un-modified BiVO4 and the BiVO4/Ni(Fe)OOH photoanodes. Equivalent TA spectra for the other BiVO4/MOOH configurations are included in the ESI (Fig. S8†). The data for BiVO4 are similar to that reported previously, and are associated with holes accumulated at the surface.37 The optical signal observed at shorter wavelengths (∼<500 nm) across all BiVO4 and BiVO4/MOOH photoanodes is dominated by a short-lived transient bleach and is indicative of the trapping of conduction band electrons into oxygen vacancy states as reported previously (we note that the magnitude of this bleaching varies between samples, mostly likely resulting from differences in oxygen vacancy density).38 The recovery of the bleach in the optical signal on the 1 ms timescale is concomitant with the electron extraction from the photoanodes, after which only the longer lived optical signal for BiVO4 holes or MOOH(++) species can be observed.38
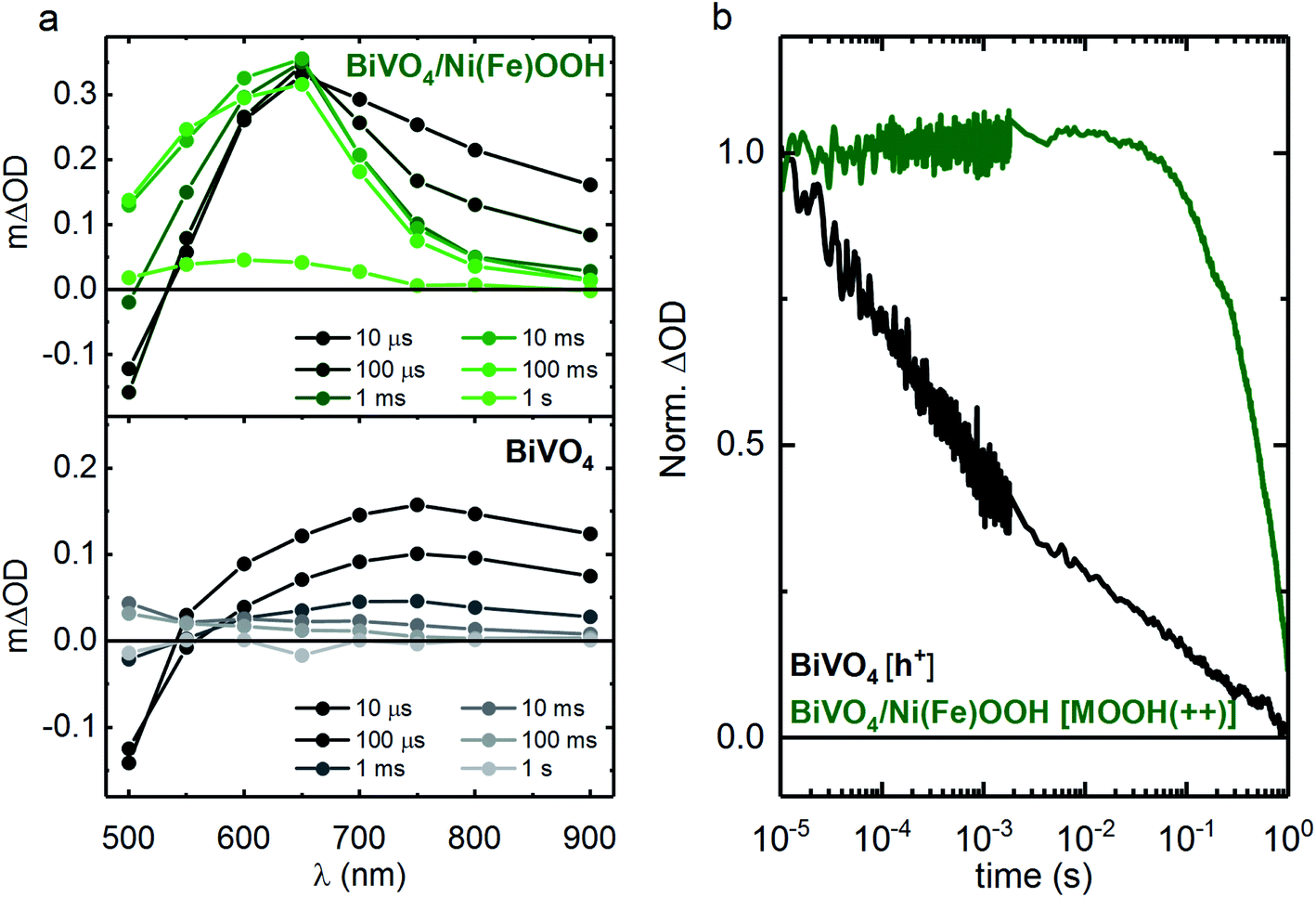 | ||
| Fig. 3 Transient absorption spectra and kinetics of BiVO4 and BiVO4/Ni(Fe)OOH photoanodes. (a) The transient absorption spectra showing the spectral signature of Ni(Fe)OOH(++) species in BiVO4/Ni(Fe)OOH (top panel) and BiVO4 (bottom panel). (b) Normalised transient absorption kinetics of BiVO4 (black, 650 nm, h+ in BiVO4) and BiVO4/Ni(Fe)OOH (green, 650 nm, Ni(Fe)OOH(++)). All data were obtained under an applied potential of 1.4 VRHE, using 355 nm excitation (300 μJ cm−2), in 0.1 M phosphate buffer. | ||
In the case of BiVO4/Ni(Fe)OOH (Fig. 3a, top panel), a peak at 650 nm can be observed from the 10 μs TA spectra onwards, which is reminiscent of the Ni(Fe)OOH(++) species observed under electrocatalytic conditions shown in Fig. 2a. This similarity in the spectral features at 650 nm suggest that both electrical and photo-induced oxidation (via surface holes from BiVO4) generate similar Ni(Fe)OOH(++) species. The observation of this feature in the spectrum obtained at 10 μs is indicative of a significant fraction of fast hole transfer from BiVO4 to the MOOH catalyst, which occurs within the time resolution of the measurements (<10 μs). The spectral shape in Fig. 3a (top panel) is clearly distinct from un-modified BiVO4 (Fig. 3a, bottom panel) at all timescales, indicating that holes primarily reside in the Ni(Fe)OOH catalyst layer over the timescales studied herein. Comparison of the kinetic traces presented in Fig. 3b shows that Ni(Fe)OOH(++) species have a much longer lifetime than the holes in BiVO4. Following hole transfer from BiVO4 to the Ni(Fe)OOH layer, the accumulated Ni(Fe)OOH(++) species are spatially separated from photogenerated electrons in BiVO4 and therefore, surface recombination in BiVO4/Ni(Fe)OOH is reduced in comparison to un-modified BiVO4. Analogous conclusions can be drawn for the cases of BiVO4/FeOOH and BiVO4/FeOOHNiOOH, where the spectral profiles of the MOOH(++) species can be observed in the TA spectra (millisecond onwards), although charge transfer is less clear than the case of BiVO4/Ni(Fe)OOH due to the similarity in the spectral signatures of holes in BiVO4 and MOOH(++) species in FeOOH and FeOOHNiOOH within the probed region. In addition, the optical signature of MOOH(++) species for FeOOH and FeOOHNiOOH at early timescales (μs to ms) also overlaps with the transient bleach observed due to electron trapping in BiVO4, further obscuring the detection of charge transfer at early timescales for these two cases. Nonetheless, all three catalyst-functionalised samples exhibit much longer lived signals than BiVO4 alone (as seen in the TA spectra, Fig. 3 and S8 in the ESI†), indicative of catalyst deposition resulting in a substantial extension of the lifetime of photogenerated charges, attributed to slower recombination kinetics.
At early timescales (between 10 μs and 1 ms), an absorption tail at longer wavelengths (∼700–900 nm) is present in all samples (Fig. 3a and S8†). As this spectral signal is also present in the un-modified BiVO4 photoanode, it may be indicative of photogenerated charges within BiVO4. In the case of the catalyst modified samples (BiVO4/MOOH), this tail absorption decays by ∼1 ms without any significant concomitant change in the signal observed for the MOOH(++) species, most clearly visible for the case of BiVO4/Ni(Fe)OOH (Fig. 3a). Since the extinction coefficients of the MOOH(++) species are ∼7–8 times larger than the hole ε of BiVO4 (420 M−1 cm−1),36 any further hole transfer from BiVO4 to the catalyst layer within 10 μs and 1 ms would be expected to give rise to a much larger signal observed for the MOOH(++) species. Since no increase in signal is observed, this may be indicative of recombination of photogenerated species in BiVO4 that have not transferred to the catalyst layer by ∼10 μs.
It is intriguing that the FeOOHNiOOH functionalised BiVO4 photoanode performs better than the FeOOH counterpart (Fig. 1b), despite exhibiting almost invariant kinetics for water oxidation under electrocatalytic conditions (Fig. 2b). From the TA spectra (Fig. S8†), it appears that the signal amplitude for BiVO4/FeOOHNiOOH is slightly larger than it is for BiVO4/FeOOH. Taking the respective extinction coefficients of the MOOH(++) species into consideration, this would indicate that a greater amount of holes successfully transfer from BiVO4 to FeOOHNiOOH in comparison to FeOOH, thus suggestive of FeOOHNiOOH being able to accumulate a greater density of MOOH(++) in comparison to FeOOH alone, when deposited on BiVO4. On the other hand, the lower performance of BiVO4/Ni(Fe)OOH can be explained by the slower kinetics of the Ni(Fe)OOH(++) species (as shown in Fig. 2b) for the same accumulated charges. For electrochemical water oxidation, this is compensated by the accumulation of a greater density of MOOH(++) species. However, under illumination when BiVO4/Ni(Fe)OOH is used, the maximum number of charges accumulated on the Ni(Fe)OOH layer will be determined primarily by the number of photogenerated holes that are transferred to the catalyst layer. As observed in the TA spectra at early timescales, the amount of MOOH(++) accumulated on the MOOH layer is within the same order of magnitude between the three catalysts. Since water oxidation is slower on Ni(Fe)OOH in comparison to the other two catalysts for a given MOOH(++) density, there is increased scope for competing recombination processes at the BiVO4/Ni(Fe)OOH interface, as reported previously.18
To examine the photoelectrochemical processes under steady-state conditions, spectroelectrochemical photoinduced absorption spectroscopy (PIAS) was employed. Unfortunately, the instability of BiVO4/Ni(Fe)OOH samples (Fig. S10†) under oxidative conditions over prolonged periods of time required for PIAS measurements precludes them from these analyses. The normalised PIAS spectra of BiVO4, BiVO4/FeOOH and BiVO4/FeOOHNiOOH under an applied potential of 1.4 VRHE and 1 sun illumination are shown in Fig. 4, along with the normalised spectra of the respective MOOH(++) species obtained under electrochemical conditions. The resemblance of the spectral shapes indicate that similar species accumulate under the two catalytic conditions, which, in conjunction with the TAS data discussed above, further confirms that photoexcitation of BiVO4 leads to the oxidation of the catalyst, also under PEC operation. Similar to the TA spectra, the difference in signal between PIA and SEC at shorter wavelengths (<550 nm) is most likely due to electron trapping within BiVO4.38
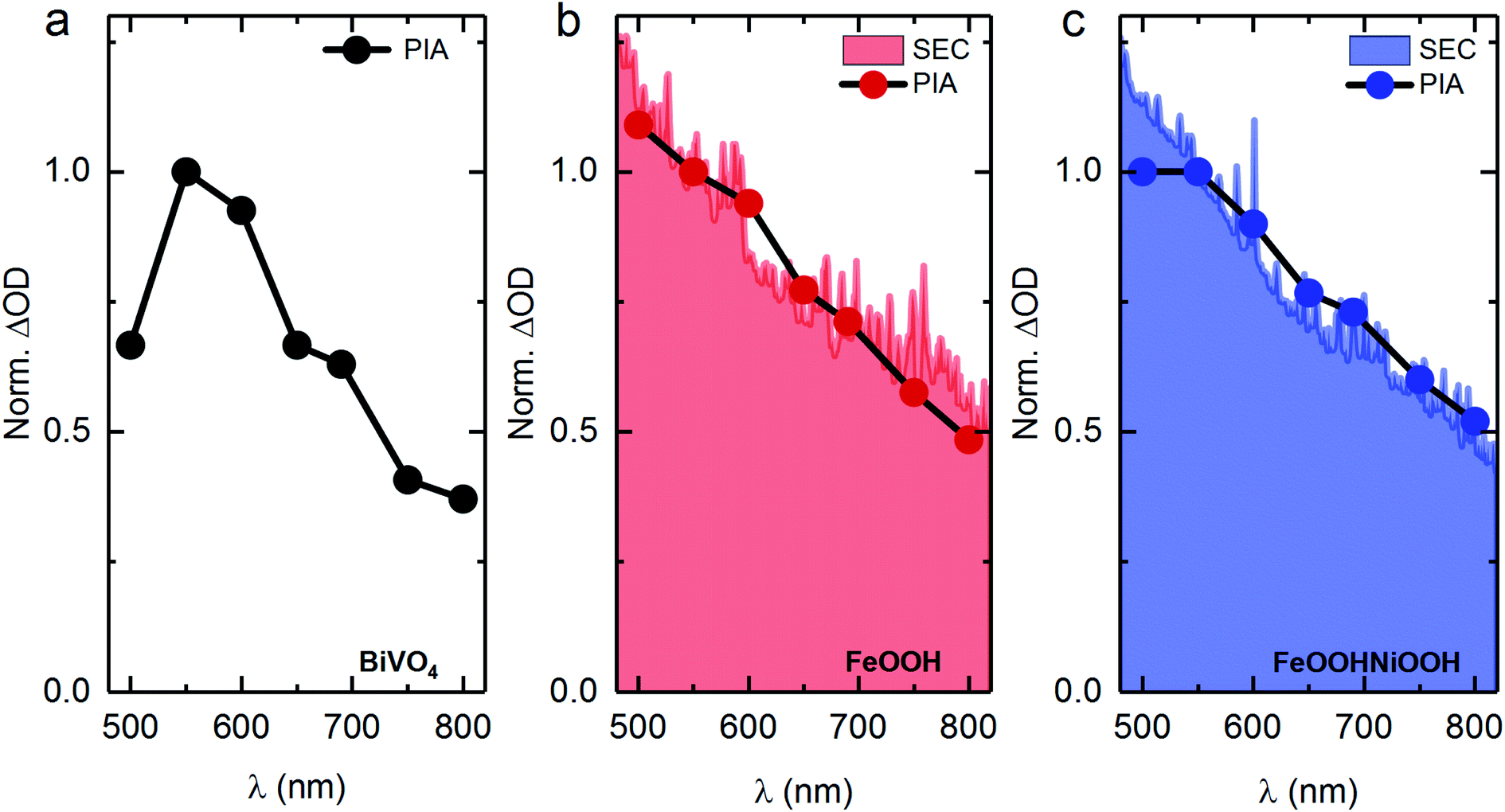 | ||
| Fig. 4 Comparison of normalised spectra of MOOH(++) species under electrochemical (EC) and photoelectrochemical (PEC) conditions. (a) The PIA spectrum of BiVO4. (b) The SEC spectrum of FeOOH electrocatalyst (red, shaded) and the PIA spectrum of BiVO4/FeOOH (solid red circle, black line). (c) The SEC spectrum of FeOOHNiOOH electrocatalyst (blue, shaded) and the PIA spectrum of BiVO4/FeOOHNiOOH (solid blue circle, black line). All SEC spectra under electrochemical conditions were obtained under an applied potential of 2 VRHE, and all PIA spectra were obtained under 1 sun equivalent illumination (365 nm LED) under 1.4 VRHE. All spectra were normalised at 550 nm. All measurements were taken in 0.1 M phosphate buffer (pH 7). | ||
The optical signals obtained through PIAS and SP-SEC for the photoanodes and electrocatalyst electrodes, respectively, under catalytic conditions, were converted to the amount of oxidising equivalents for water oxidation, i.e. holes in BiVO4 and MOOH(++) species in the case of the catalysts. By relating the optical signal to the concomitant current transient signals, kinetic information associated with water oxidation reaction can be analysed using rate law plots according to the following equation:
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J = logkWO + βlog(MOOH(++)) J = logkWO + βlog(MOOH(++)) | (1) |
Firstly, from Fig. 5a, we can observe that photogenerated holes in BiVO4 follow a reaction order (β) of 3 for water oxidation, consistent with previous reports,25,26 whereas both FeOOH and FeOOHNiOOH operate with a β value of ∼2 under both photoelectrochemical and electrochemical conditions at pH 7. A reaction order of ∼2 for these electrocatalysts differs from that observed under alkaline conditions (β ∼ 4 at pH 13),27 and is indicative of differences in reaction mechanisms in neutral and alkaline media. Surface protonation as well as the reactivity and stability of reaction intermediates can result in different reaction mechanisms being observed at neutral and alkaline pH.41 The effect of pH on reaction order has also been observed for TiO2.42 More importantly, at pH 7, the different reaction orders between BiVO4 and BiVO4/MOOH photoanodes indicates that water oxidation proceeds via different active species and consequently through different mechanisms. Whilst in un-modified BiVO4 photoanodes water oxidation is driven directly by photogenerated BiVO4 holes, in the case of BiVO4/MOOH(++) photoanodes, the oxidised MOOH(++) species drive water oxidation in the catalyst layer, following hole transfer from BiVO4. Furthermore, it is apparent that similar orders of magnitude of MOOH(++) species are observed optically under both electrochemical (EC) and photoelectrochemical (PEC) conditions, consistent with the MOOH(++) species driving water oxidation in both systems, although differences in film morphology between the EC and PEC samples prevent quantitative comparison. A comparison of the reaction time constants for PEC water oxidation, shown in Fig. 5b for BiVO4 and BiVO4/MOOH photoelectrodes, indicate that no significant enhancement in water oxidation timescale is gained upon electrocatalyst deposition.
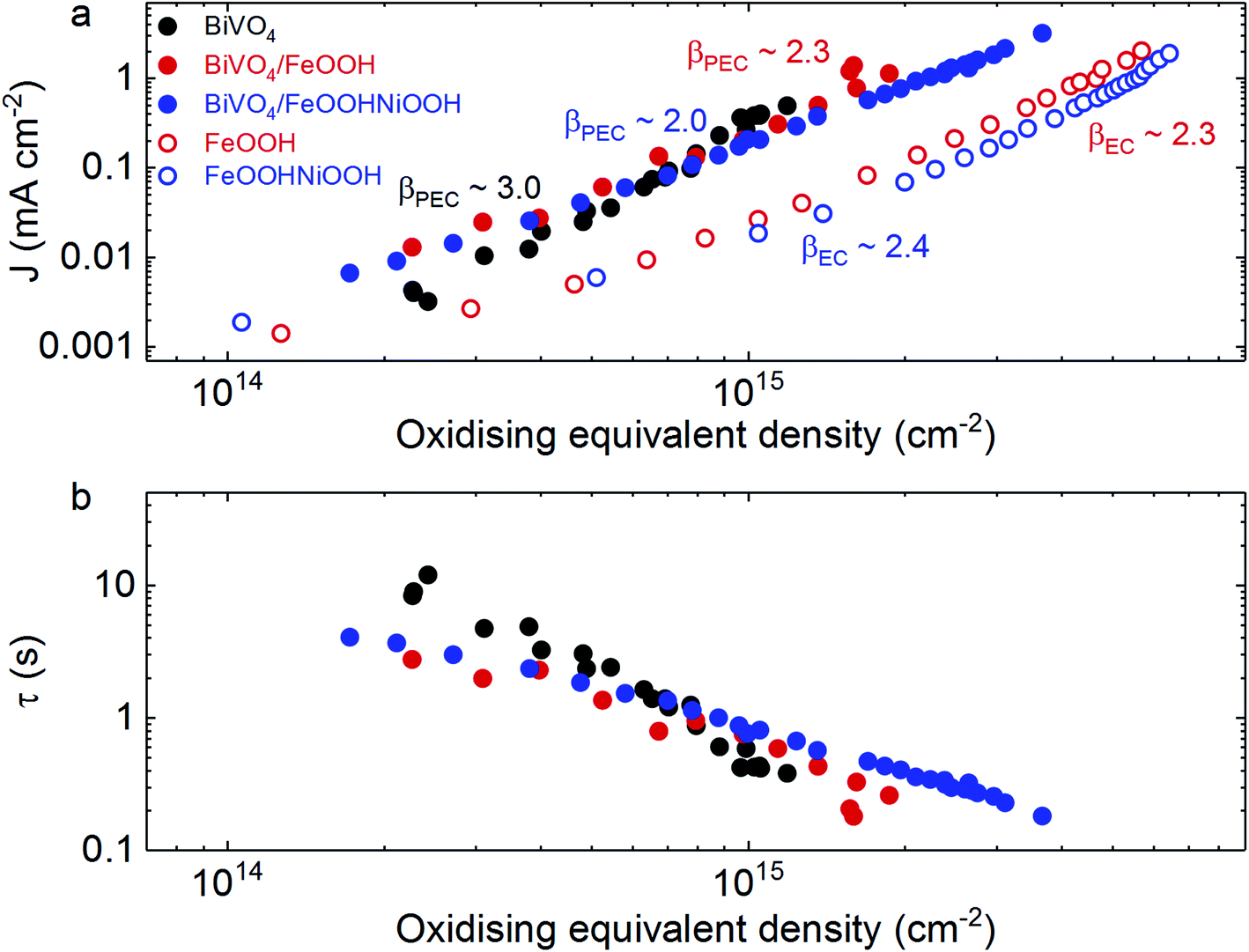 | ||
| Fig. 5 Comparison of kinetics under electrochemical and photoelectrochemical conditions. (a) The density of oxidising equivalents (h+ for BiVO4, or MOOH(++) species for catalysts) under catalytic conditions plotted against the (photo)current obtained. The reaction orders (β) for the respective electrochemical and photoelectrochemical water oxidation reactions are also shown (determined from the slope). (b) The density of oxidising equivalents plotted against the water reaction time constants (τ) under photoelectrochemical conditions for BiVO4 and BiVO4/MOOH photoanodes. Same colours used in (a) and (b): BiVO4 (black solid circles); BiVO4/FeOOH (red solid circles); BiVO4/FeOOHNiOOH (blue solid circles); FeOOH (red empty circles); FeOOHNiOOH (blue empty circles). All measurements were taken in 0.1 M phosphate buffer. | ||
It is also striking that catalysts often employed for water oxidation, where water oxidation is observed to take place via oxidised states in the catalyst (i.e. Co–Fe based Prussian Blue), display time constants on the order of seconds.43 Even the popular Ni(Fe)OOH catalysts investigated under optimal alkaline conditions27 operate with time constants that are comparable to water oxidation by metal oxides such as BiVO4, and slower than that observed for WO3.25,44 We note however that these electrocatalysts operate at much lower overpotentials for matched charge densities. Therefore, it appears that the significant enhancement in photoelectrochemical performance of BiVO4 photoanodes decorated with these electrocatalysts does not originate from faster kinetics of water oxidation relative to BiVO4. Rather, the results obtained in this study suggest that the role of the catalyst layer is to decrease the kinetics of surface recombination and effectively decrease the recombination losses via fast hole transfer from BiVO4 to the catalyst layer. This spatially separates charges, and retards surface recombination losses that compete with water oxidation.
The same observations were found for Co–Fe Prussian Blue, where fast hole transfer to the catalyst layer followed by fast water oxidation kinetics led to enhanced performance.43 This is however, markedly different to CoPi, which exhibits water oxidation time constants (τ) that are two to three orders of magnitude slower than what is observed for BiVO4 alone, where the enhancement in performance has been attributed to increased band-bending within the BiVO4 that drives charge separation.17,36 It is observed that the amount of charge accumulated in CoPi under PEC conditions (when deposited on BiVO4) is significantly lower (by two orders of magnitude) than that observed in the electrocatalyst under electrochemical water oxidation conditions at matched current densities.36 Therefore, it is concluded that the amount of charge that accumulates in CoPi from BiVO4 is insufficient to drive water oxidation. This conclusion is also supported by intensity-modulated photocurrent spectroscopy (IMPS) studies which indicate that CoPi mainly serves to passivate surface defects, with no significant enhancement of water oxidation kinetics when deposited on BiVO4 or α-Fe2O3.9,11 However, as conflicting studies based on dual working electrode measurements that assign water oxidation to the CoPi catalyst overlayers also have been reported,45 this makes the role of CoPi as a co-catalyst overlayer on semiconductor photoelectrodes difficult to determine. We note that factors such as the porosity of the semiconductor and the ion permeability of catalysts can also affect the nature of the interface between the semiconductor/catalyst.46 For example, the ability of some catalysts to form Schottky junctions has been proposed to increase the efficiency of a semiconductor/catalyst system.46 The nature of the interface playing a critical role is further demonstrated by IrOx electrocatalysts, where despite IrOx being an excellent water oxidation electrocatalyst, it has been reported as an ineffective co-catalyst when deposited on BiVO4.47
For the case of the MOOH electrocatalysts studied herein, although a slight discrepancy in the amount of MOOH(++) species is also observed between the electrocatalysts and the BiVO4/MOOH photoelectrodes, this difference (within an order of magnitude) is less dramatic than the case of CoPi,36 and is more similar to the case of RuOx catalysts operating as electrocatalysts and co-catalysts on photoelectrochemical systems.29 The charge transfer observed from BiVO4 to the catalyst layer to generate MOOH(++) species is seen both in the TA spectra and the photo-induced absorption spectra under steady-state conditions. Additionally, as the reaction order for water oxidation for BiVO4/MOOH photoanodes also differs from that of BiVO4 photoanodes, it strongly indicates that water oxidation in the catalyst-decorated photoanodes studied herein is driven via MOOH(++) species accumulated in the catalyst layer (as illustrated in Scheme 1).
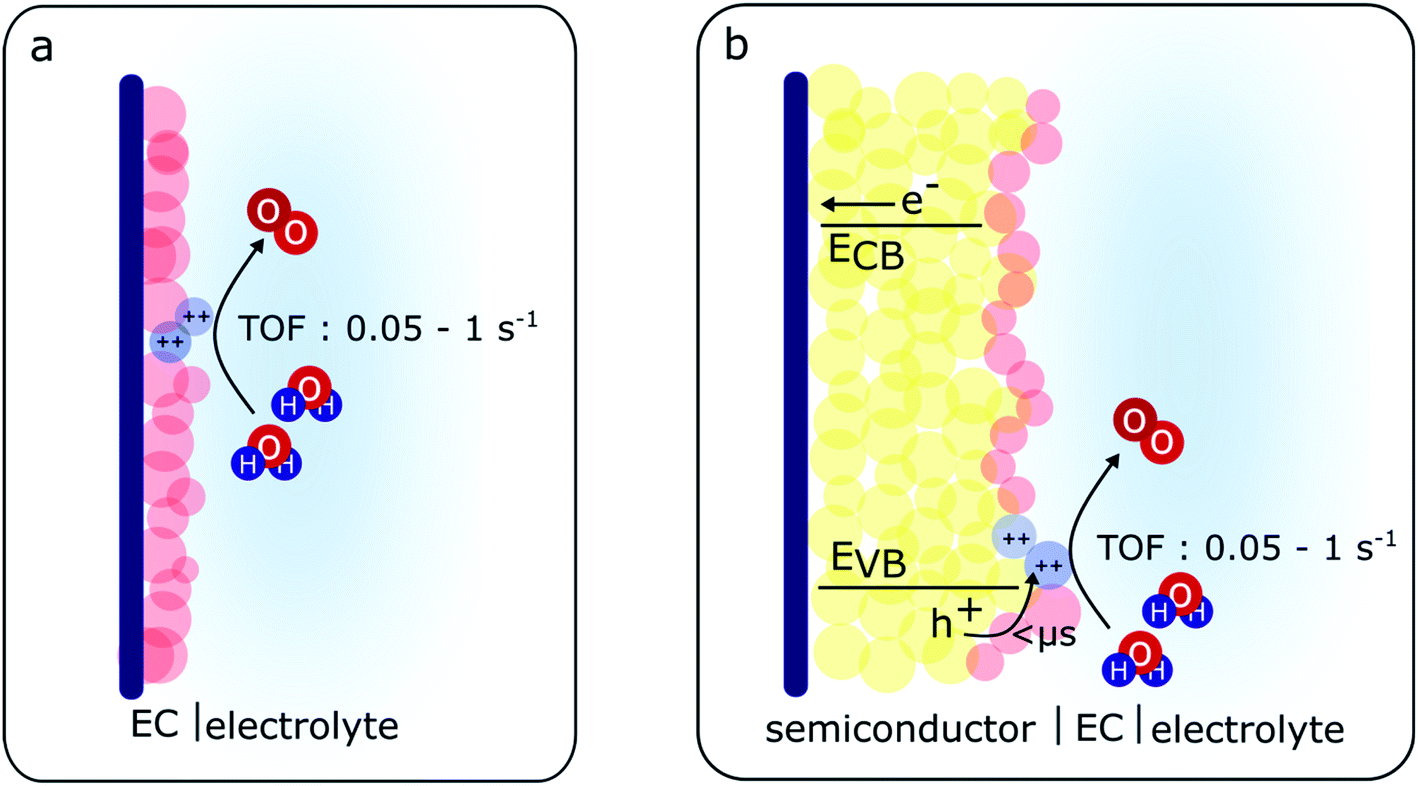 | ||
| Scheme 1 Schematic illustration of electrochemical and photoelectrochemical water oxidation in MOOH electrodes and BiVO4/MOOH photoelectrodes. (a) The electrochemically generated MOOH(++) species (blue spheres) in the activated electrocatalyst accumulate and drive water oxidation. (b) The photogenerated holes in the BiVO4 transfer to the activated catalyst, generating MOOH(++) species (blue spheres) which then drive water oxidation. The electrons in the conduction band are collected at the back contact. | ||
Conclusions
In this work, the electrochemical and photoelectrochemical performance of FeOOH, FeOOHNiOOH, and Ni(Fe)OOH catalysts are studied at pH 7 as standalone electrocatalysts and when added on the surface of BiVO4 photoanodes. Our results demonstrate that electrocatalytic performance results from both the intrinsic reaction kinetics and the potential dependence of charge accumulation, where the latter consideration is not important under photoelectrochemical conditions. We show that while FeOOH and FeOOHNiOOH exhibit faster kinetics for water oxidation than Ni(Fe)OOH, this nickel-based catalyst is able to accumulate a larger density of oxidative MOOH(++). As a result, the three catalyst show a similar electrocatalytic water oxidation performances. This trade-off between accumulation and reaction kinetics hinders the selection of electrocatalysts to make junctions with photoanodes, through standard J–V characterisations.Once deposited onto the surface of nanoporous BiVO4 photoanodes, substantial improvements in the photoelectrochemical performance are observed with respect to un-modified BiVO4, attributed to fast charge transfer (<10 μs) from BiVO4 to the catalyst layer, and the resultant suppressed recombination losses. However, rather surprisingly, no significant enhancement in the water oxidation time constant is observed for the BiVO4/MOOH photoanodes in comparison to BiVO4. Moreover, differences between the catalysts emerge in the BiVO4/MOOH photoanodes, stemming from the charge accumulation within the catalyst layer being governed by the transfer of photogenerated holes from BiVO4. This significantly impedes the ability of Ni(Fe)OOH to accumulate a greater amount of Ni(Fe)OOH(++) needed to offset its slower water oxidation kinetics, which leads to a lower performance of BiVO4/Ni(Fe)OOH photoanodes in comparison to the other two configurations. On the other hand, when NiOOH is added to FeOOH as a dual-layer catalyst on BiVO4, it helps accumulate more MOOH(++) species within the catalyst layer than FeOOH alone, resulting in a greater enhancement in the photocurrent generated by BiVO4/FeOOHNiOOH photoanodes. Overall, this study highlights the two different factors that govern the performance BiVO4/MOOH photoanodes and need to be considered when selecting electrocatalysts for applications in photoelectrochemical systems: (1) the number of charges that can be accumulated within the catalyst layer; and (2) how fast these charges can oxidise water to compete with recombination processes.
Author contributions
J. D. and K.-S. C. contributed to the conceptualisation. L. F., S. S., S. C. and D. L. contributed to the investigation and formal analysis. C. A. M. and E. P. contributed to the development of methodology and analysis.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
J. R. D. and E. P. acknowledges financial support from the European Research Council (project Intersolar 291482). This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement 732840-A-LEAF. C. A. M. thanks COLCIENCIAS (now Ministry of Science, Technology and Innovation, call 568) for funding, L. F. thanks the EU for a Marie Curie fellowship (658270), S. S. thanks the EPRSC for a DTP scholarship and S. C. thanks Imperial College London for a Schrödinger Scholarship. K.-S. C. acknowledges financial support by the National Science Foundation under CHE-1764399.References
- J. Barber and P. D. Tran, From Natural to Artificial Photosynthesis, J. R. Soc., Interface, 2013, 10(81), 20120984, DOI:10.1098/rsif.2012.0984.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Artificial Photosynthesis: From Molecular Catalysts for Light-Driven Water Splitting to Photoelectrochemical Cells, Photochem. Photobiol., 2011, 87(5), 946–964, DOI:10.1111/j.1751-1097.2011.00966.x.
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci., 2006, 103(43), 15729–15735, DOI:10.1073/pnas.0603395103.
- K. Sivula and R. van de Krol, Semiconducting Materials for Photoelectrochemical Energy Conversion, Nat. Rev. Mater., 2016, 1(2), 15010, DOI:10.1038/natrevmats.2015.10.
- Y. Yang, S. Niu, D. Han, T. Liu, G. Wang and Y. Li, Progress in Developing Metal Oxide Nanomaterials for Photoelectrochemical Water Splitting, Adv. Energy Mater., 2017, 7(19), 1–26, DOI:10.1002/aenm.201700555.
- M. Xiao, B. Luo, Z. Wang, S. Wang and L. Wang, Recent Advances of Metal-Oxide Photoanodes: Engineering of Charge Separation and Transportation toward Efficient Solar Water Splitting, Sol. RRL, 2020, 1900509, DOI:10.1002/solr.201900509.
- A. J. Cowan, J. Tang, W. Leng, J. R. Durrant and D. R. Klug, Water Splitting by Nanocrystalline TiO2 in a Complete Photoelectrochemical Cell Exhibits Efficiencies Limited by Charge Recombination, J. Phys. Chem. C, 2010, 114(9), 4208–4214, DOI:10.1021/jp909993w.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, Back Electron–Hole Recombination in Hematite Photoanodes for Water Splitting, J. Am. Chem. Soc., 2014, 136(6), 2564–2574, DOI:10.1021/ja412058x.
- C. Y. Cummings, F. Marken, L. M. Peter, A. A. Tahir and K. G. U. Wijayantha, Kinetics and Mechanism of Light-Driven Oxygen Evolution at Thin Film α-Fe2O3 Electrodes, Chem. Commun., 2012, 48(14), 2027–2029, 10.1039/C2CC16382A.
- L. M. Peter, K. G. U. Wijayantha and A. A. Tahir, Kinetics of Light-Driven Oxygen Evolution at α-Fe2O3 Electrodes, Faraday Discuss., 2012, 155, 309–322, 10.1039/C1FD00079A.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Photocurrent of BiVO4 Is Limited by Surface Recombination, Not Surface Catalysis, Chem. Sci., 2017, 8(5), 3712–3719, 10.1039/C7SC00363C.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Photoelectrochemical and Impedance Spectroscopic Investigation of Water Oxidation with “Co-Pi”-Coated Hematite Electrodes, J. Am. Chem. Soc., 2012, 134(40), 16693–16700, DOI:10.1021/ja306427f.
- T. W. Kim and K.-S. Choi, Nanoporous BiVO4 Photoanodes with Dual-Layer Oxygen Evolution Catalysts for Solar Water Splitting, Science, 2014, 343(6174), 990–994, DOI:10.1126/science.1246913.
- F. S. Hegner, I. Herraiz-Cardona, D. Cardenas-Morcoso, N. López, J. R. Galán-Mascarós and S. Gimenez, Cobalt Hexacyanoferrate on BiVO4 Photoanodes for Robust Water Splitting, ACS Appl. Mater. Interfaces, 2017, 9(43), 37671–37681, DOI:10.1021/acsami.7b09449.
- S. Wang, P. Chen, Y. Bai, J.-H. Yun, G. Liu and L. Wang, New BiVO4 Dual Photoanodes with Enriched Oxygen Vacancies for Efficient Solar-Driven Water Splitting, Adv. Mater., 2018, 30(20), 1800486, DOI:10.1002/adma.201800486.
- D. Wang, R. Li, J. Zhu, J. Shi, J. Han, X. Zong and C. Li, Photocatalytic Water Oxidation on BiVO4 with the Electrocatalyst as an Oxidation Cocatalyst: Essential Relations between Electrocatalyst and Photocatalyst, J. Phys. Chem. C, 2012, 116(8), 5082–5089, DOI:10.1021/jp210584b.
- Y. Ma, F. Le Formal, A. Kafizas, S. R. Pendlebury and J. R. Durrant, Efficient Suppression of Back Electron/Hole Recombination in Cobalt Phosphate Surface-Modified Undoped Bismuth Vanadate Photoanodes, J. Mater. Chem. A, 2015, 3(41), 20649–20657, 10.1039/C5TA05826K.
- H. Hajibabaei, A. R. Schon and T. W. Hamann, Interface Control of Photoelectrochemical Water Oxidation Performance with Ni1–xFexOy Modified Hematite Photoanodes, Chem. Mater., 2017, 29(16), 6674–6683, DOI:10.1021/acs.chemmater.7b01149.
- D. K. Zhong, S. Choi and D. R. Gamelin, Near-Complete Suppression of Surface Recombination in Solar Photoelectrolysis by “Co-Pi” Catalyst-Modified W:BiVO4, J. Am. Chem. Soc., 2011, 133(45), 18370–18377, DOI:10.1021/ja207348x.
- M. W. Kanan and D. G. Nocera, In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+, Science, 2008, 321(5892), 1072–1075, DOI:10.1126/science.1162018.
- D. K. Lee, D. Lee, M. A. Lumley and K. S. Choi, Progress on Ternary Oxide-Based Photoanodes for Use in Photoelectrochemical Cells for Solar Water Splitting, Chem. Soc. Rev., 2019, 48(7), 2126–2157, 10.1039/C8CS00761F.
- S. W. Boettcher and Y. Surendranath, Heterogeneous Electrocatalysis Goes Chemical, Nat. Catal., 2021, 4, 4–5, DOI:10.1038/s41929-020-00570-1.
- H. Gerischer, The Impact of Semiconductors on the Concepts of Electrochemistry, Electrochim. Acta, 1990, 35(11–12), 1677–1699, DOI:10.1016/0013-4686(90)87067-C.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, Rate Law Analysis of Water Oxidation on a Hematite Surface, J. Am. Chem. Soc., 2015, 137(20), 6629–6637, DOI:10.1021/jacs.5b02576.
- C. A. Mesa, L. Francàs, K. R. Yang, P. Garrido-Barros, E. Pastor, Y. Ma, A. Kafizas, T. E. Rosser, M. T. Mayer, E. Reisner, M. Grätzel, V. S. Batista and J. R. Durrant, Multihole Water Oxidation Catalysis on Haematite Photoanodes Revealed by Operando Spectroelectrochemistry and DFT, Nat. Chem., 2020, 12(1), 82–89, DOI:10.1038/s41557-019-0347-1.
- Y. Ma, C. A. Mesa, E. Pastor, A. Kafizas, L. Francàs, F. Le Formal, S. R. Pendlebury and J. R. Durrant, Rate Law Analysis of Water Oxidation and Hole Scavenging on a BiVO 4 Photoanode, ACS Energy Lett., 2016, 1(3), 618–623, DOI:10.1021/acsenergylett.6b00263.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Spectroelectrochemical Study of Water Oxidation on Nickel and Iron Oxyhydroxide Electrocatalysts, Nat. Commun., 2019, 10(1), 5208, DOI:10.1038/s41467-019-13061-0.
- J. Li, W. Wan, C. A. Triana, H. Chen, Y. Zhao, C. K. Mavrokefalos and G. R. Patzke, Reaction Kinetics and Interplay of Two Different Surface States on Hematite Photoanodes for Water Oxidation, Nat. Commun., 2021, 12(1), 1–9, DOI:10.1038/s41467-020-20510-8.
- E. Pastor, F. Le Formal, M. T. Mayer, S. D. Tilley, L. Francàs, C. A. Mesa, M. Grätzel and J. R. Durrant, Spectroelectrochemical Analysis of the Mechanism of (Photo)Electrochemical Hydrogen Evolution at a Catalytic Interface, Nat. Commun., 2017, 8, 14280, DOI:10.1038/ncomms14280.
- C. A. Mesa, A. Kafizas, L. Francàs, S. R. Pendlebury, E. Pastor, Y. Ma, F. Le Formal, M. T. Mayer, M. Grätzel and J. R. Durrant, Kinetics of Photoelectrochemical Oxidation of Methanol on Hematite Photoanodes, J. Am. Chem. Soc., 2017, 139(33), 11537–11543, DOI:10.1021/jacs.7b05184.
- M. W. Louie and A. T. Bell, An Investigation of Thin-Film Ni-Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2013, 135(33), 12329–12337, DOI:10.1021/ja405351s.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, Tracking Catalyst Redox States and Reaction Dynamics in Ni-Fe Oxyhydroxide Oxygen Evolution Reaction Electrocatalysts: The Role of Catalyst Support and Electrolyte PH, J. Am. Chem. Soc., 2017, 139(5), 2070–2082, DOI:10.1021/jacs.6b12250.
- Z. K. Goldsmith, A. K. Harshan, J. B. Gerken, M. Vörös, G. Galli, S. S. Stahl and S. Hammes-Schiffer, Characterization of NiFe Oxyhydroxide Electrocatalysts by Integrated Electronic Structure Calculations and Spectroelectrochemistry, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(12), 3050–3055, DOI:10.1073/pnas.1702081114.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation, J. Am. Chem. Soc., 2014, 136(18), 6744–6753, DOI:10.1021/ja502379c.
- M. Görlin, M. Gliech, J. F. De Araújo, S. Dresp, A. Bergmann and P. Strasser, Dynamical Changes of a Ni-Fe Oxide Water Splitting Catalyst Investigated at Different PH, Catal. Today, 2016, 262, 65–73, DOI:10.1016/j.cattod.2015.10.018.
- Y. Ma, A. Kafizas, S. R. Pendlebury, F. Le Formal and J. R. Durrant, Photoinduced Absorption Spectroscopy of CoPi on BiVO4: The Function of CoPi during Water Oxidation, Adv. Funct. Mater., 2016, 26(27), 4951–4960, DOI:10.1002/adfm.201600711.
- Y. Ma, S. R. Pendlebury, A. Reynal, F. Le Formal and J. R. Durrant, Dynamics of Photogenerated Holes in Undoped BiVO4 Photoanodes for Solar Water Oxidation, Chem. Sci., 2014, 5(8), 2964–2973, 10.1039/C4SC00469H.
- S. Selim, E. Pastor, M. García-Tecedor, M. R. Morris, L. Francàs, M. Sachs, B. Moss, S. Corby, C. A. Mesa, S. Gimenez, A. Kafizas, A. A. Bakulin and J. R. Durrant, Impact of Oxygen Vacancy Occupancy on Charge Carrier Dynamics in BiVO4 Photoanodes, J. Am. Chem. Soc., 2019, 141(47), 18791–18798, DOI:10.1021/jacs.9b09056.
- C. A. Mesa, L. Steier, B. Moss, L. Francàs, J. E. Thorne, M. Grätzel and J. R. Durrant, Impact of the Synthesis Route on the Water Oxidation Kinetics of Hematite Photoanodes, J. Phys. Chem. Lett., 2020, 11(17), 7285–7290, DOI:10.1021/acs.jpclett.0c02004.
- C. A. Mesa, R. R. Rao, L. Francàs, S. Corby and J. R. Durrant, Reply to: Questioning the Rate Law in the Analysis of Water Oxidation Catalysis on Haematite Photoanodes, Nat. Chem., 2020, 12(12), 1099–1101, DOI:10.1038/s41557-020-00570-5.
- T. Takashima, K. Hashimoto and R. Nakamura, Mechanisms of PH-Dependent Activity for Water Oxidation to Molecular Oxygen by MnO2 Electrocatalysts, J. Am. Chem. Soc., 2012, 134(3), 1519–1527, DOI:10.1021/ja206511w.
- A. Kafizas, Y. Ma, E. Pastor, S. R. Pendlebury, C. Mesa, L. Francàs, F. Le Formal, N. Noor, M. Ling, C. Sotelo-Vazquez, C. J. Carmalt, I. P. Parkin and J. R. Durrant, Water Oxidation Kinetics of Accumulated Holes on the Surface of a TiO2 Photoanode: A Rate Law Analysis, ACS Catal., 2017, 7(7), 4896–4903, DOI:10.1021/acscatal.7b01150.
- B. Moss, F. S. Hegner, S. Corby, S. Selim, L. Francàs, N. López, S. Giménez, J.-R. Galán-Mascarós and J. R. Durrant, Unraveling Charge Transfer in CoFe Prussian Blue Modified BiVO4 Photoanodes, ACS Energy Lett., 2018, 337–342, DOI:10.1021/acsenergylett.8b02225.
- S. Corby, L. Francàs, S. Selim, M. Sachs, C. Blackman, A. Kafizas and J. R. Durrant, Water Oxidation and Electron Extraction Kinetics in Nanostructured Tungsten Trioxide Photoanodes, J. Am. Chem. Soc., 2018, 140(47), 16168–16177, DOI:10.1021/jacs.8b08852.
- F. A. L. Laskowski, M. R. Nellist, J. Qiu and S. W. Boettcher, Metal Oxide/(Oxy)Hydroxide Overlayers as Hole Collectors and Oxygen-Evolution Catalysts on Water-Splitting Photoanodes, J. Am. Chem. Soc., 2019, 141(4), 1394–1405, DOI:10.1021/jacs.8b09449.
- T. J. Mills, F. Lin and S. W. Boettcher, Theory and Simulations of Electrocatalyst-Coated Semiconductor Electrodes for Solar Water Splitting, Phys. Rev. Lett., 2014, 112(14), 1–5, DOI:10.1103/PhysRevLett.112.148304.
- H. Ye, H. S. Park and A. J. Bard, Screening of Electrocatalysts for Photoelectrochemical Water Oxidation on W-Doped BiVO4 Photocatalysts by Scanning Electrochemical Microscopy, J. Phys. Chem. C, 2011, 115(25), 12464–12470, DOI:10.1021/jp200852c.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06429g |
| ‡ Authors contributed equally. |
| § Current address: Departament de Química, Universitat Autònoma de Barcelona, Barcelona, Spain. |
| ¶ Current address: Institute of Advanced Materials (INAM), Universitat Jaume I, Castelló, Spain. |
| || Current address: Departamento de Ingeniería Electrónica, Universidad Central, Bogotá, Colombia. |
| ** Current address: ICFO-Institut de Ciències Fotòniques, The Barcelona Institute of Science and Technology, Castelldefels, 08860, Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2021 |