
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free C(sp3)–H functionalization of sulfonamides via strain-release rearrangement†
Jiefeng
Hu
 *a,
Xianyu
Yang
a,
Shasha
Shi
a,
Bo
Cheng
a,
Xiaoling
Luo
*c,
Yu
Lan
*d and
Teck-Peng
Loh
*ab
*a,
Xianyu
Yang
a,
Shasha
Shi
a,
Bo
Cheng
a,
Xiaoling
Luo
*c,
Yu
Lan
*d and
Teck-Peng
Loh
*ab
aInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, Nanjing 211816, China. E-mail: iasjfhu@njtech.edu.cn
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
cChongqing Key Laboratory of Inorganic Functional Materials, College of Chemistry, Chongqing Normal University, Chongqing 401331, China
dCollege of Chemistry, Institute of Green Catalysis, Zhengzhou University, Zhengzhou, Henan 450001, China
First published on 22nd January 2021
Abstract
With the increasing awareness of sustainable chemistry principles, the development of an efficient and mild strategy for C(sp3)–H bond activation of nitrogen-containing compounds without the utilization of any oxidant and metal is still highly desired and challenging. Herein, we present a metal-free reaction system that enables C–H bond functionalization of aliphatic sulfonamides using DABCO as a promoter under mild conditions, affording a series of α,β-unsaturated imines in good yields with high selectivities. This protocol tolerates a broad range of functionalities and can serve as a powerful synthetic tool for the late-stage modification of complex compounds. More importantly, control experiments and detailed DFT calculations suggest that this process involves [2 + 2] cyclization/ring-cleavage reorganization, which opens up a new platform for the establishment of other related reorganization reactions.
Introduction
Nitrogen-containing compounds are present widely in a broad range of natural, pharmaceutical and synthetic compounds. In the past ten years, considerable effort has been directed toward the exploration of their organic synthesis methodologies. Among the various methods reported, direct activation of unreactive C–H bonds in amine derivatives for coupling reactions is one of the most effective and important strategies. In this context, early research has focused primarily on α-C–H bond functionalization of amines using transition metal catalysts with stoichiometric amounts of oxidants.1 In recent years, activation of more remote C–H bonds in compounds containing nitrogen has attracted great attention and interest. For example, γ-C–H activation of amines was achieved by Yu's group using a palladium complex as the catalyst in combination with a directing group.2 On the other hand, δ-C–H bond functionalization in sulfonamides has been accomplished through 1,5-hydrogen atom transfer of amidyl radicals in the presence of a copper catalyst.3 Given the medicinal value of β-substituted amine derivatives,4 some methods for β-C–H functionalization have been developed based on an oxidative cross-coupling pathway (Scheme 1A).5,6 These reactions often require transition metals (Ru, Ir, Pd and Fe),7–10 oxidizing reagents11 and high temperature.12 In contrast, transition metal-free protocols are very rare in this field. In 2014, Seidel et al. reported a redox-neutral α,β-difunctionalization cascade reaction of cyclic amines under microwave irradiation at 200 °C.13 In 2018, Chang et al. presented a B(C6F5)3-catalyzed β-C–H silylation of N-aryl piperidines with hydrosilanes at 120 °C.14 Notably, Seidel et al. used organolithium reagents to achieve multi-C–H functionalization of alicyclic amines. And a key intermediate, the in situ-generated endocyclic 1-azaallyl anion, was proposed in this report.15 Despite these advances in cyclic amines, the development of a mild and metal-free approach for the functionalization of C–H bonds in aliphatic amides is extremely challenging.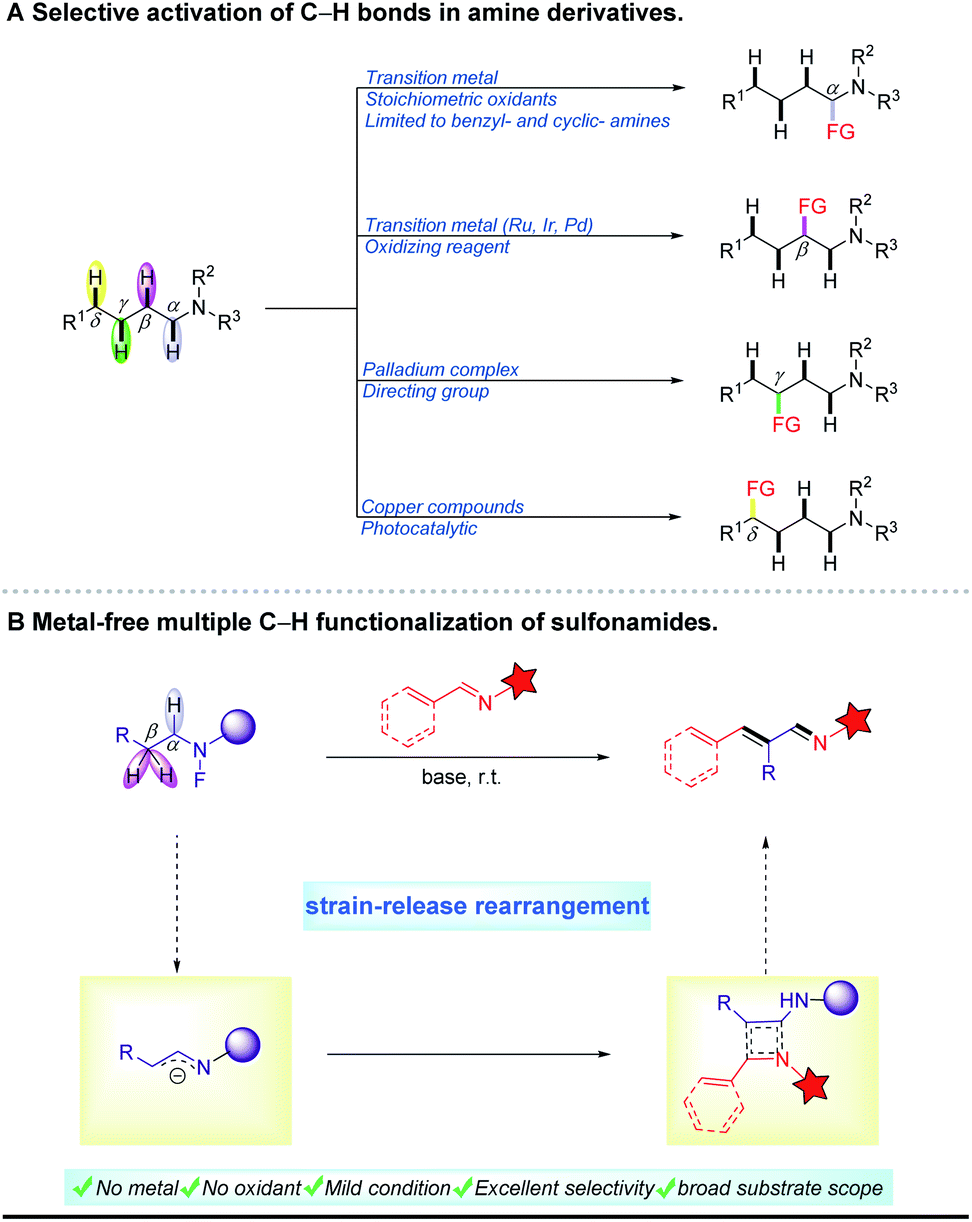 | ||
| Scheme 1 Development of a novel process. FG, functional group. | ||
Imines are ubiquitous and valuable intermediates in the synthesis of nitrogen-containing compounds, pharmaceuticals and natural products.16–19 Among many different imines, α,β-unsaturated imines are a unique class of difunctionalized compounds and play key roles in the synthesis of heterocycles and pharmaceuticals.20 Traditionally, these compounds are synthesized through aldol-type condensation of aldehydes, followed by dehydrative condensation with amines.21 Unfortunately, these condensation reactions usually suffer from harsh acidic conditions and high temperatures, and require dehydrating apparatus. On the other hand, some aldehydes, as reaction substrates, are not easily available and unstable, especially due to aerial oxidation. In addition, the conjugated imines obtained through this strategy are very difficult to purify because they are frequently mixed with other isomers. Thus, the widespread application of α,β-unsaturated imines is extremely hindered. Notably, in recent years significant progress has been made in the selective oxidation of amines for the synthesis of imines based on biocatalysts,22–25 complex metal catalysts26–32 or oxidizing reagents33,34. However, their substrates are limited to benzylamines and cyclic amines.35 Actually, the oxidation of amides is more difficult than that of amines because their nitrogen atoms have much lower electron density.36 Moreover, it is very important37 and challenging to directly synthesize unsaturated imines through dehydrogenation of amine derivatives. Hence, we report an efficient metal-free system for the synthesis of α,β-unsaturated imines with high geometrical selectivities through multiple C–H bond functionalization of N-fluoro sulfonamides under mild conditions (Scheme 1B). It is noteworthy that this pathway proceeded via a four-membered ring reorganization process.
Results and discussion
Initial studies involved the evaluation of the selective cross-coupling reaction of the N-fluorotosylamide derivative 1a with N-tosylbenzaldimine 1b (Table 1). The reaction was found to be feasible in the presence of K2CO3 (2.0 equiv.) in DMF at room temperature, providing the N-((E)-2-((E)-benzylidene)butylidene)-4-methylbenzenesulfonamide (1c) in an isolated yield of 29% (entry 1). Other inorganic bases, such as K3PO4 and CsOAc, also showed low yields (entries 2 and 3). Strong bases (KOH) inhibited the progress of the reaction (entry 4). To our delight, organic bases have high reactivity for this cross-coupling reaction (entries 5–7). When 1.0 equiv. DABCO was added as a base in this system, the yield of 1c was dramatically increased to 81% and no other isomers were detected. It is important to note that the high yield of imine products could still be maintained even when the DABCO loading was decreased to 30 mol% (entry 8). As expected, removal of the base resulted in no reaction (entry 9). This result illustrates that DABCO is an essential driving force for this transformation. It is noteworthy that when 4 Å molecular sieves were removed, the product was obtained in a slightly lower yield of 72% (entry 10). Additionally, reactions carried out in other polar aprotic solvents, such as MeCN, THF and DCE, furnished the product in lower yields (entries 11–13). Finally, no products were observed using toluene as the solvent (entry 14).|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), 1b (0.26 mmol), 1.0 equiv. base and 150 mg 4 Å molecular sieves in solvent (1.0 mL), 24 h, at room temperature, under Ar. b Isolated yield. c Using 30 mol% DABCO. d After removing molecular sieves. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, DABCO = 1,4-diazabicyclo[2.2.2]octane, DMF = N,N-dimethylformamide, Ts = 4-toluenesulfonyl. | |||
| 1 | K2CO3 | DMF | 29 |
| 2 | CsOAc | DMF | 26 |
| 3 | K3PO4 | DMF | 31 |
| 4 | KOH | DMF | Trace |
| 5 | Et3N | DMF | 65 |
| 6 | DBU | DMF | 53 |
| 7 | DABCO | DMF | 81 |
| 8c | DABCO | DMF | 80 |
| 9 | — | DMF | N |
| 10d | DABCO | DMF | 72 |
| 11 | DABCO | MeCN | 58 |
| 12 | DABCO | THF | 39 |
| 13 | DABCO | DCE | 38 |
| 14 | DABCO | Toluene | N |
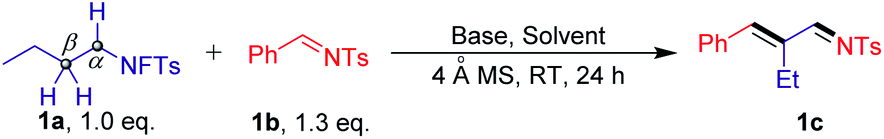
With this exciting initial result in hand, we proceeded to investigate the scope of the reactions using various N-fluorotosylamides, prepared from the corresponding alkyl amines (Scheme 2). As shown, N-fluorotosylamide substrates with different carbon chains were efficiently transformed into the corresponding α,β-unsaturated imines in moderate to excellent yields (2c–7c). These results revealed that the products were obtained in lower yields with sterically bulky substrates. The products with substituted alkyl ethers, phenolic ethers and silyl ethers were formed in good yields (8c–12c). It should be noted that the reaction was not affected by halo substituents such as F, CF3, Cl and Br on the substrates (13c–19c), highlighting its potential in combination with the subsequent cross-coupling transformations. Under our conditions, ester-containing substrate 20c also worked well. In particular, the substrates 21c–23c, which contained an alkenyl motif, were also found to be compatible with the reaction conditions. Excitingly, naphthalene (24c) and benzofuran (25c) were also shown to exhibit high levels of reactivities. To showcase the utility of the transformation for diversifying natural product amines, we prepared a diverse collection of N-fluorotosylamide derivatives and subjected them to the optimized reaction conditions. For example, our strategy has been applied in the modification of N-fluorotosylamide 26a with two cis-alkene groups from linolenyl alcohol, generating product 26c in 67% yield. The steroid derivative 27a was also readily transformed into the α,β-unsaturated imine 27c in 62% yield. Moreover, the N-fluorotosylamide 28a, which was synthesized from stigmasterol derivatives by a series of synthetic methods, could also be converted into the desired product 28c in 63% yield.
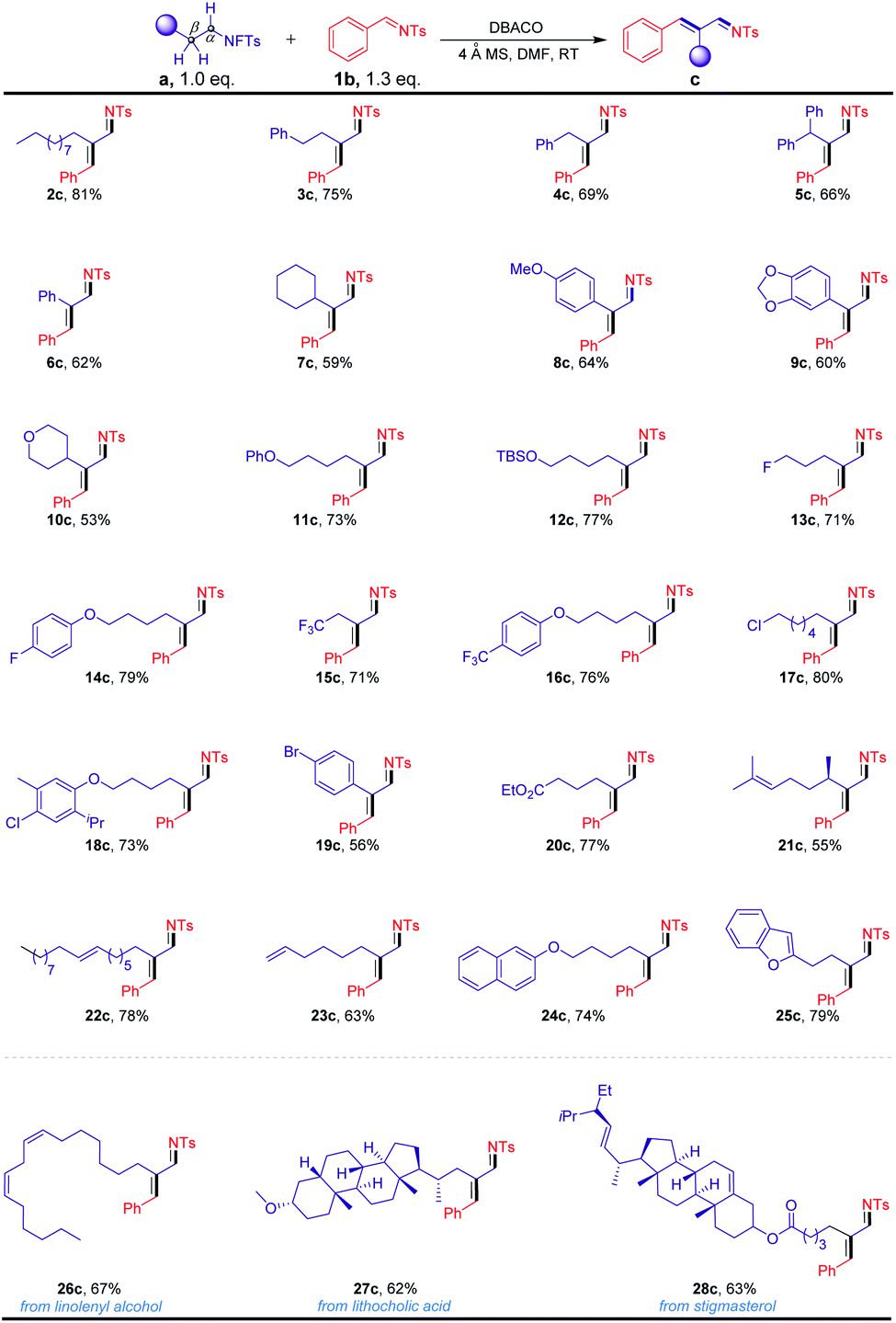 | ||
| Scheme 2 Substrate scope of alkylamine derivatives. Reaction conditions: a mixture of N-fluorotosylamides a (0.20 mmol), N-tosylbenzaldimine 1b (0.30 mmol), 150 mg 4 Å molecular sieves and DABCO (0.20 mmol) in DMF (1 mL) was stirred for 24 h at room temperature under Ar, isolated yield after chromatography. TBS = tert-butyldimethylsilyl. | ||
The scope of this transformation was further evaluated using N-fluorotosylamide (1a) in conjunction with a more diverse pool of arylimines (b) as shown in Scheme 3. A wide range of substituted imines that possess electron-neutral, electron-donating, and electron-withdrawing substituents were readily tolerated (29c–51c). Among them, the substrate bearing a CN group afforded the desired product in 67% yield (42c). Furthermore, the presence of oxygen-, sulfur- and nitrogen-containing heterocycles (44c–47c) did not interfere with the reactions. Notably, the structure of one of the products obtained, 46c, was characterized by X-ray crystallographic analysis. We were also pleased to observe that this method can be applied to some more challenging substrates. For instance, a series of π-extended systems could also participate in the transformation to afford the conjugated imines 48c–51c in moderate yields.
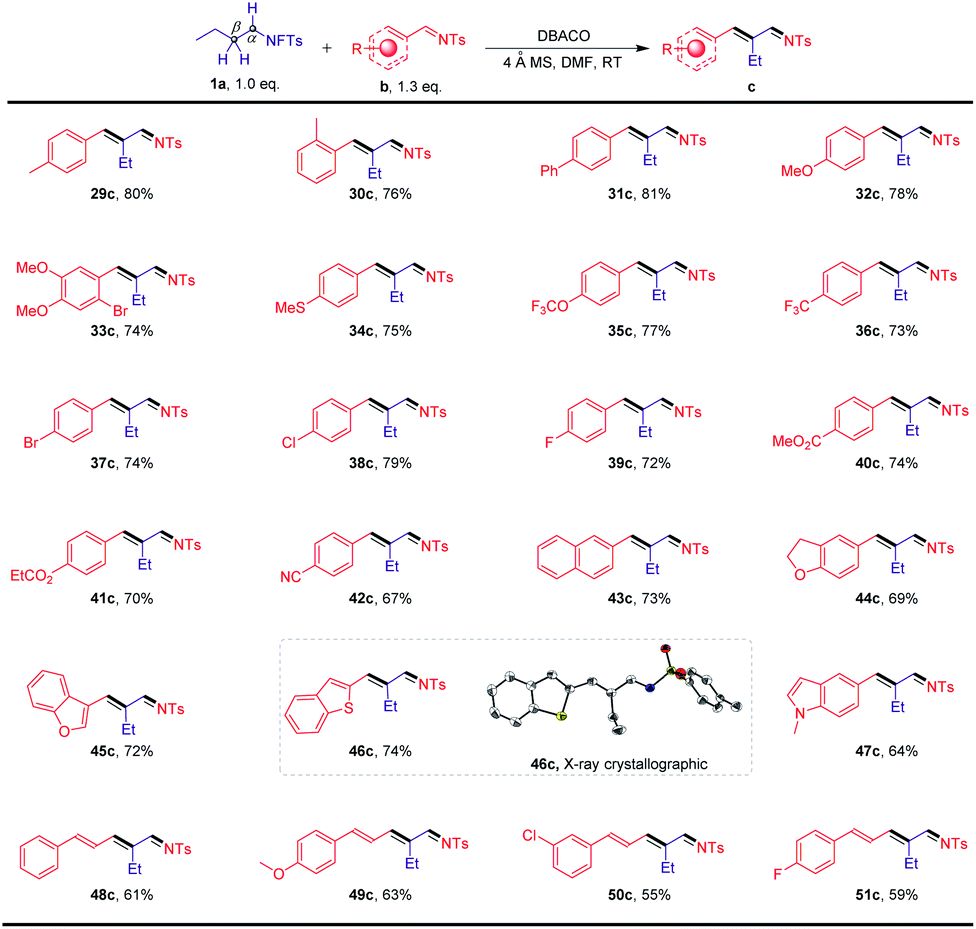 | ||
| Scheme 3 Substrate scope of arylimines. Reaction conditions: a mixture of N-fluorotosylamide 1a (0.20 mmol), N-tosylbenzaldimines b (0.26 mmol), 150 mg 4 Å molecular sieves and DABCO (0.20 mmol) in DMF (1 mL) was stirred for 24 h at room temperature under Ar, isolated yield after chromatography. | ||
We next investigated the potential of α,β-unsaturated imines in the subsequent synthetic transformations. For example, allylamine compounds can be formed in the presence of DIBAL-H at −78 °C. And we also explored the metal-mediated allylation of 1c in THF, obtaining the corresponding product with a yield of 80%. Under the catalysis of FeCl3, the formed imines can undergo a cyclization reaction to provide indenamine derivatives. Interestingly, the synthetic utility of the formed unsaturated imine was further demonstrated through Prins cyclization reactions (Scheme 4).
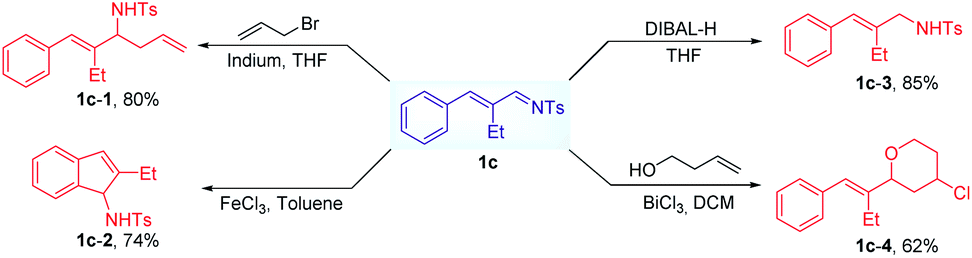 | ||
| Scheme 4 Utility of the formed α,β-unsaturated imines. | ||
Several experiments were conducted to provide insight into the potential mechanism of this cross-coupling reaction. (1) When using TEMPO and BHT as free radical inhibitors, this reaction was not completely suppressed. These results imply that the free radical pathway was probably not involved (Scheme 5A). (2) When the reaction was carried out in the absence of 1b under the standard reaction conditions, 52c was formed through the self-coupling of 1a, and no imine 1d was detected (Scheme 5B). Next, the progress of the experiment was monitored by GC-MS; we detected the formation of the corresponding imine 1e with 29a as the substrate (Scheme 5C). Furthermore, control experiment using N-tosylimine 1d instead of the N-fluoro sulfonamides also resulted in the formation of the desired product with Et3N·HF as a promoter (Schemes 5D and E). On the other hand, when using N-chloro sulfonamide 1f instead of the N-fluoro sulfonamides, no desired product was obtained. These observations illustrated that the imine may be a key intermediate and the fluorine atom was a necessary driving force for this C–H functionalization (Scheme 5F). (3) To rule out the possibility of the involvement of the classical aldol condensation reaction, we carried out cross-over experiments by addition of an external aldehyde. The corresponding imine with the aldehyde fragment was not detected (Schemes 5G and H). (4) To gain deeper insights into the mechanism, 30a–35a with different substituents were treated with 1b under our conditions, and the same product 1c was obtained in good yields (Scheme 5I). More excitingly, using 25b as the substrate instead of 1b, 54c was formed in 71% yield in this system (Scheme 5J). These results suggest that the sulfonamide substituent of the product (c) should be derived from the arylimine substrate (b). And a possible rearrangement process is proposed as shown in Scheme 5K.
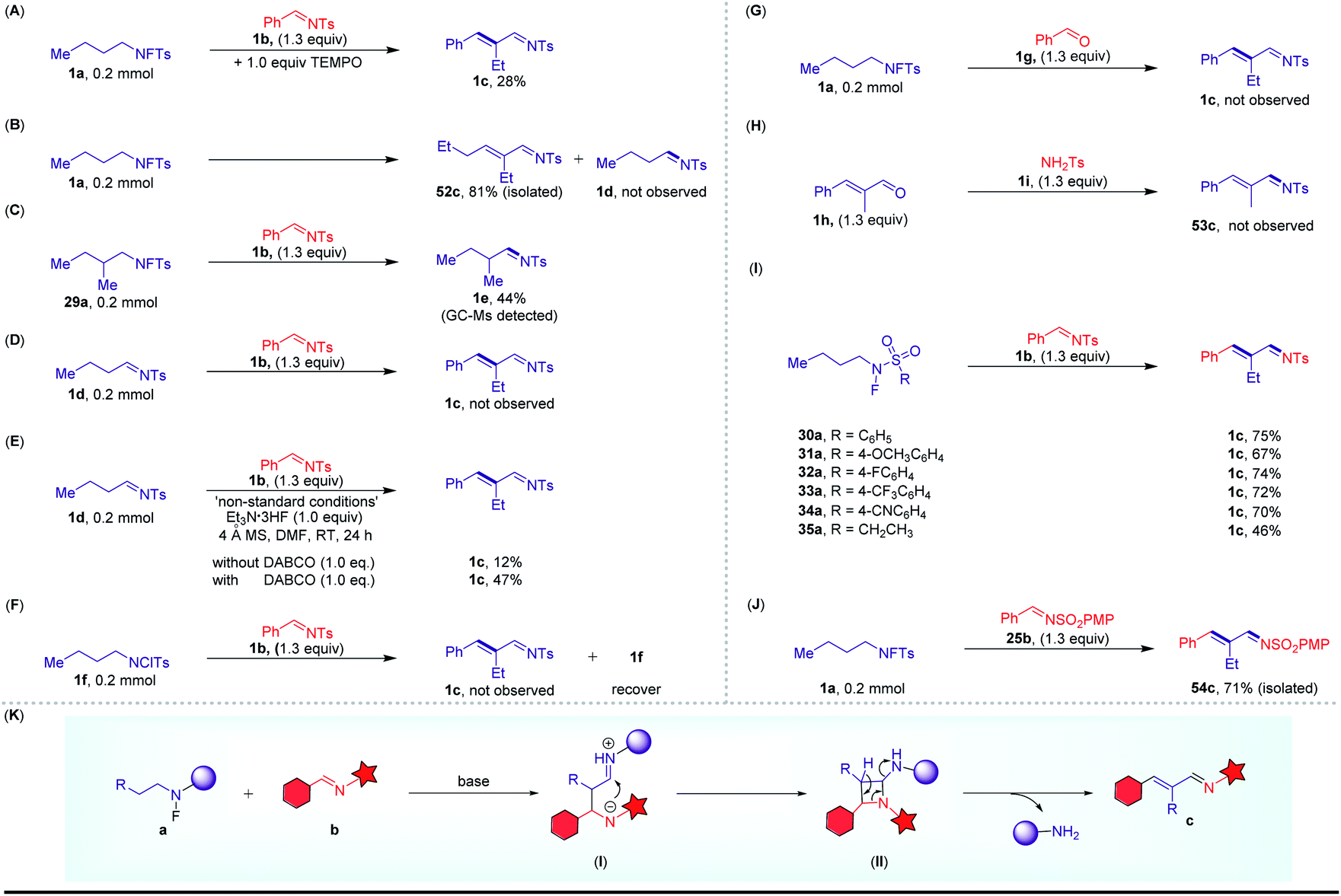 | ||
| Scheme 5 Mechanistic studies of the metal-free C–H functionalization strategy. (A–J) control experiments; (K) a possible reaction pathway. | ||
To further reveal the details of this transformation, we performed a detailed DFT investigation. The calculated free energy profile of the favored path is shown in Scheme 6 (the detailed information on the other paths is given in the ESI†). The reaction starts with a base-assisted HF elimination from N-fluorotosylamide 1avia transition state 55-ts with an energy barrier of 22.9 kcal mol−1, while (E)-imine 1d is generated irreversibly with an exergonic 69.2 kcal mol−1. Then an imine/enamine tautomerization38 takes place via transition state 56-ts with an energy barrier of 18.1 kcal mol−1 in the presence of DABCO to afford a hydrogen bond complex 57. Enamine 57 serves as a nucleophile, which can react with arylimine 1b through an intermolecular nucleophilic attack39via transition state 58-ts to form a new C–C covalent bond. Interestingly, the nucleophile 57 is activated by the formation of a hydrogen bond with DABCO as well as the electrophilic partner imine 1b is activated by another hydrogen bond formation with HF. Concomitantly, the generated zwitterionic intermediate 59 is stabilized by two hydrogen bonds with DABCO and HF, respectively. The overall activation free energy for this step is 26.5 kcal mol−1, which would be considered as the rate limiting step in the whole pathway. When zwitterionic intermediate 59 is formed, an intramolecular nucleophilic addition can occur via transition state 60-ts with an energy barrier of 13.4 kcal mol−1 to achieve the stepwise [2 + 2] cycloaddition. After the release of DABCO and HF, an azetidine intermediate 62 is generated reversibly. Intermediate 62 also exhibits amine characteristics, and therefore, a Brønsted acid assisted deamination can occur via transition state 64-ts with an energy barrier of 20.4 kcal mol−1 in the presence of HF. The generated iminium intermediate 65 can be further deprotonated by DABCO via transition state 66-ts to form the corresponding enamine intermediate 67. The ring strain of that intermediate results in a 4π-electrocyclic ring-opening process via transition state 68-ts to afford product 1c. To summarise, the ring-closing/ring-opening rearrangement of N-fluorotosylamide with aldimines is constituted by base-assisted HF elimination to an imine, base-assisted imine/enamine tautomerization, step-wise [2 + 2] cyclization to generate azetidine, Brønsted acid prompted deamination of the aminal moiety, and deprotonated ring-opening.
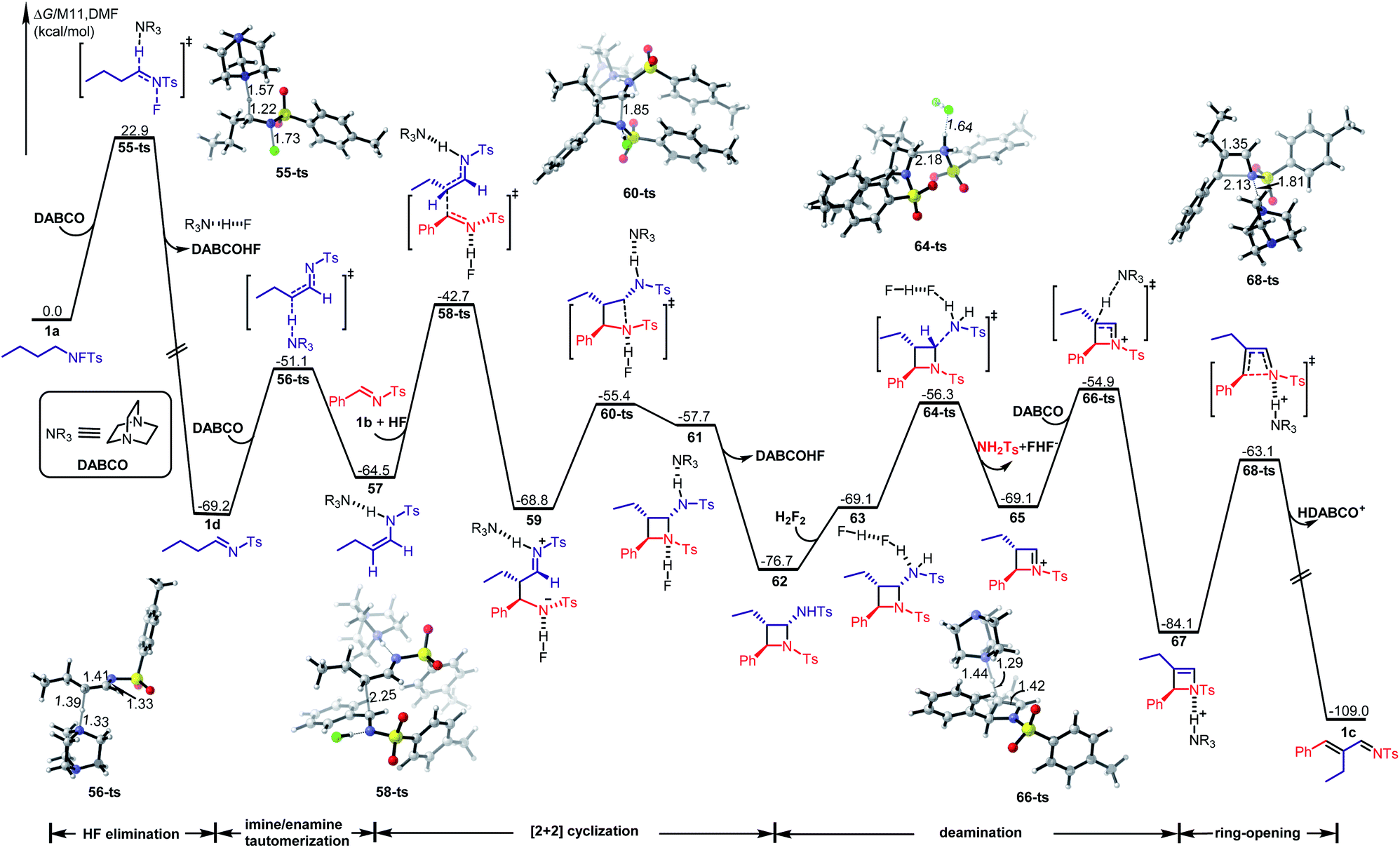 | ||
| Scheme 6 DFT calculations for the reaction of 1a with 1b at the M11/6-311+G(d,p)/SMD//B3LYP-D3/6-31+G(d)/SMD level of theory. The energies are in kcal mol−1 and represent the relative free energies in DMF. The bond distances are in angstroms. | ||
Conclusions
In summary, we have developed a robust system that is capable of the cross-coupling of N-fluoro sulfonamides and arylimines without the need to use metals and oxidants, giving a series of α,β-unsaturated imines. This transformation showed exceptional functional group tolerance, high efficiency, and excellent chemoselectivity. More importantly, control experiments and DFT calculations indicate that the process involved ring-closing/ring-opening reorganization. We envision that this methodology will not only open up new avenues for synthesizing complex bioactive molecules but also inspire the discovery of more novel rearrangement systems.Author contributions
J. H. conceived the project and directed the research. Y. L. and T.-P. L. supervised the mechanistic study. J. H. and T.-P. L. wrote the paper. S. S., X. Y. and B. C. performed the experiments. X. L. performed the DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21901114), the Natural Science Foundation of Jiangsu Province (BK20180685), the Natural Science Foundation of Jiangsu Provincial Department of Education (18KJB150017), and the Start-up Grant from Nanjing Tech University (38274017102 and 39837101). The SICAM Fellowship from the Jiangsu National Synergistic Innovation Center for Advanced Materials is also acknowledged.Notes and references
- (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (b) D. Seidel, Acc. Chem. Res., 2015, 48, 317–328 CrossRef CAS; (c) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS.
- (a) H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef CAS; (b) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef; (c) Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS; (d) Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS.
- (a) Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288–13292 CrossRef CAS; (b) Z. Liu, H. Xiao, B. Zhang, H. Shen, L. Zhu and C. Li, Angew. Chem., Int. Ed., 2019, 58, 2510–2513 CrossRef CAS; (c) A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405–18410 CrossRef CAS; (d) D. Bafaluy, H. Munoz-Molina, I. Funes-Ardoiz, S. Herold, A. J. D. Aguirre, H. Zhang, F. Maseras, T. R. Belderrain, P. J. Perez and K. Muniz, Angew. Chem., Int. Ed., 2019, 58, 8912–8916 CrossRef CAS; (e) Y. Qin, Y. Han, Y. Tang, J. Wei and M. Yang, Chem. Sci., 2020, 11, 1276–1282 RSC.
- (a) L. I. Willems and A. P. IJzerman, Med. Res. Rev., 2010, 30, 778–817 CrossRef CAS; (b) L. Jia, R. D. Simpson, J. Yuan, Z. Xu, W. Zhao, S. Cacatian, C. M. Tice, J. Guo, A. Ishchenko, S. B. Singh, Z. Wu, B. M. McKeever, Y. Bukhtiyarov, J. A. Johnson, C. P. Doe, R. K. Harrison, G. M. McGeehan, L. W. Dillard, J. J. Baldwin and D. A. Claremon, ACS Med. Chem. Lett., 2011, 2, 747–751 CrossRef CAS.
- (a) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS; (b) S. Ye, W. Yang, T. Coon, D. Fanning, T. Neubert, D. Stamos and J.-Q. Yu, Chem.–Eur. J., 2016, 22, 4748–4752 CrossRef CAS.
- (a) B. F. V. Steijvoort, N. Kaval, A. A. Kulago and B. U. W. Maes, ACS Catal., 2016, 6, 4486–4490 CrossRef; (b) M. Maetani, J. Zoller, B. Melillo, O. Verho, N. Kato, J. Pu, E. Comer and S. L. Schreiber, J. Am. Chem. Soc., 2017, 139, 11300–11306 CrossRef CAS; (c) D. Antermite, D. P. Affron and J. A. Bull, Org. Lett., 2018, 20, 3948–3952 CrossRef CAS.
- (a) B. Sundararaju, Z. Tang, M. Achard, G. V. M. Sharma, L. Toupet and C. Bruneau, Adv. Synth. Catal., 2010, 352, 3141–3146 CrossRef CAS; (b) B. Sundararaju, M. Achard, G. V. M. Sharma and C. Bruneau, J. Am. Chem. Soc., 2011, 133, 10340–10343 CrossRef CAS; (c) G.-Q. Xu, J.-T. Xu, Z.-T. Feng, H. Liang, Z.-Y. Wang, Y. Qin and P.-F. Xu, Angew. Chem., Int. Ed., 2018, 130, 5204–5208 CrossRef.
- (a) B. Su, T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 18032–18038 CrossRef CAS; (b) K. Yuan, F. Jiang, Z. Sahli, M. Achard, T. Roisnel and C. Bruneau, Angew. Chem., Int. Ed., 2012, 124, 9006–9010 CrossRef.
- (a) A. Millet, P. Larini, E. Clot and O. Baudoin, Chem. Sci., 2013, 4, 2241–2247 RSC; (b) D. P. Affron, O. A. Davis and J. A. Bull, Org. Lett., 2014, 16, 4956–4959 CrossRef CAS.
- N. Takasu, K. Oisaki and M. Kanai, Org. Lett., 2013, 15, 1918–1921 CrossRef CAS.
- (a) X.-Z. Shu, X.-F. Xia, Y.-F. Yang, K.-G. Ji, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2009, 74, 7464–7469 CrossRef CAS; (b) R. J. Griffiths, W. C. Kong, S. A. Richards, G. A. Burley, M. C. Willis and E. P. A. Talbot, Chem. Sci., 2018, 9, 2295–2300 RSC; (c) Q. Gao, M. Wu, K. Zhang, N. Yang, M. Liu, J. Li, L. Fang, S. Bai and Y. Xu, Org. Lett., 2020, 22, 5645–5649 CrossRef CAS.
- L. Ma, A. Paul, M. Breugst and D. Seidela, Chem.–Eur. J., 2016, 22, 18179–18189 CrossRef CAS.
- W. Chen, Y. Kang, R. G. Wilde and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5179–5182 CAS.
- J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209–13213 CrossRef CAS.
- W. Chen, A. Paul, K. A. Abboud and D. Seidel, Nat. Chem., 2020, 12, 545–550 CrossRef CAS.
- (a) A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS; (b) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS.
- (a) R. M. Borzilleri, S. M. Weinreb and M. Parvez, J. Am. Chem. Soc., 1995, 117, 10905–10913 CrossRef CAS; (b) J. Jin and S. M. Weinreb, J. Am. Chem. Soc., 1997, 119, 5773–5784 CrossRef CAS; (c) O. Garcia-Mancheno, R. Gomez-Arrayas and J. C. Carretero, J. Am. Chem. Soc., 2004, 126, 456–457 CrossRef.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS.
- B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2017, 117, 8326–8419 CrossRef CAS.
- (a) X. Yu, K. Chen, Q. Wang, S. Guo, S. Zha and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5222–5226 CrossRef CAS; (b) I. Chogii, P. Das, J. S. Fell, K. A. Scott, M. N. Crawford, K. N. Houk and J. T. Njardarson, J. Am. Chem. Soc., 2017, 139, 13141–13146 CrossRef CAS; (c) C. L. Hugelshofer, V. Palani and R. Sarpong, J. Am. Chem. Soc., 2019, 141, 8431–8435 CrossRef CAS; (d) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 36, 354–401 RSC.
- (a) G. K. S. Prakash, M. Mandal and G. A. Olah, Org. Lett., 2001, 3, 2847–2850 CrossRef CAS; (b) K. Y. Lee, C. G. Lee and J. N. Kim, Tetrahedron Lett., 2003, 44, 1231–1234 CrossRef CAS; (c) J. H. Babler, M. C. Atwood, J. E. Freaney and A. R. Viszlay, Tetrahedron Lett., 2007, 48, 7665–7667 CrossRef CAS.
- M. Largeron and M. B. Fleury, Science, 2013, 339, 43–44 CrossRef.
- V. Köhler, K. R. Bailey, A. Znabet, J. Raftery, M. Hel-liwell and N. J. Turner, Angew. Chem., Int. Ed., 2010, 122, 2228–2230 CrossRef.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS.
- M. R. Chapman, S. C. Cosgrove, N. J. Turner, N. Kapur and A. J. Blacker, Angew. Chem., Int. Ed., 2018, 57, 10535–10539 CrossRef CAS.
- H. Yuan, W.-J. Yoo, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2012, 134, 13970–13973 CrossRef CAS.
- T. Sonobe, K. Oisaki and M. Kanai, Chem. Sci., 2012, 3, 3249–3255 RSC.
- M. Largeron and M. B. Fleury, Angew. Chem., Int. Ed., 2012, 51, 5409–5412 CrossRef CAS.
- H. A. Ho, K. Manna and A. D. Sadow, Angew. Chem., Int. Ed., 2012, 51, 8607–8610 CrossRef CAS.
- M. L. Deb, S. S. Dey, I. Bento, M. T. Barros and C. D. Maycock, Angew. Chem., Int. Ed., 2013, 125, 9973–9977 CrossRef.
- N. Li, X. Lang, W. Ma, H. Ji, C. Chen and J. Zhao, Chem. Commun., 2013, 49, 5034–5036 RSC.
- X. Cui, W. Li, K. Junge, Z. Fei, M. Beller and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 7501–7507 CrossRef CAS.
- A. E. Wendlandt and S. S. Stahl, Org. Lett., 2012, 14, 2850–2853 CrossRef CAS.
- M. Rueping, C. Vila, A. Szadkowska, R. M. Koenigs and J. Fronert, ACS Catal., 2012, 2, 2810–2815 CrossRef CAS.
- (a) X. J. Lang, H. W. Ji, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS; (b) X. J. Lang, W. H. Ma, Y. B. Zhao, C. C. Chen, H. W. Ji and J. C. Zhao, Chem.–Eur. J., 2012, 18, 2624–2631 CrossRef CAS; (c) S. Naya, K. Kimura and H. Tada, ACS Catal., 2012, 3, 10–13 CrossRef.
- K. Moriyama, M. Kuramochi, K. Fujii, T. Morita and H. Togo, Angew. Chem., Int. Ed., 2016, 55, 14546–14551 CrossRef CAS.
- (a) S. Mandal, S. Mahato and C. K. Jana, Org. Lett., 2015, 17, 3762–3765 CrossRef CAS; (b) Y. Huang, H.-L. Fang, Y.-X. Huang, J. Sun and C.-G. Yan, J. Org. Chem., 2019, 84, 12437–12451 CrossRef CAS.
- X. Jie, Y. Shang, Z.-N. Chen, X. Zhang, W. Zhuang and W. Sun, Nat. Commun., 2018, 9, 5002–5011 CrossRef.
- X. Bian, D. Zhang, Z. Huang and Y. Qin, Synlett, 2006, 20, 3419–3422 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2025337. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06603f |
| This journal is © The Royal Society of Chemistry 2021 |